

Abstract
Biotin-thiamine responsive basal ganglia disease (BTRBGD) is an autosomal recessive neurometabolic disorder with poor genotype-phenotype correlation, caused by mutations in the SLC19A3 gene on chromosome 2q36.6. The disease is characterized by three stages: stage 1 is a sub-acute encephalopathy often triggered by febrile illness; stage 2 is an acute encephalopathy with seizures, loss of motor function, developmental regression, dystonia, external ophthalmoplegia, dysphagia, and dysarthria; stage 3 is represented by chronic or slowly progressive encephalopathy. Clinical and biochemical findings, as well as the magnetic resonance imaging (MRI) pattern, resemble those of Leigh's syndrome, so that BTRBGD can be misdiagnosed as a mitochondrial encephalopathy.Here we report the clinical and radiological phenotypes of two siblings diagnosed with BTRBGD in which a novel SLC19A3 mutation (NM_025243.3: c.548C > T; p.Ala183Val) was found by whole exome sequencing (WES) of the family members.
Keywords: biotin-thiamine responsive basal ganglia disease, biotin, thiamine, encephalopathy, Leigh's syndrome
Introduction
Biotin-thiamine responsive basal ganglia disease or biotin-thiamine responsive encephalopathy type 2 (BTRBGD, MIM607483) is a rare autosomal recessive neurometabolic and degenerative disorder of the brain caused by mutations in the SLC19A3 gene. 1 It is characterized by the variable combination of severe encephalopathy, seizures, external ophthalmoplegia, dysphagia, and sometimes coma and death. 2 Early molecular diagnosis and treatment with a lifelong supplementation of high doses of biotin and thiamine influences disease outcome and life expectancy of these patients.
The disease was first described in 10 patients of Arab ancestry by Ozand et al and to date it has been diagnosed in 95 patients (50 females and 45 males). 3 It is a panethnic disease, affecting patients with prevailing Saudi origin, with onset in childhood. 4 The natural history of this illness is characterized by different stages. The first is represented by an initial subacute encephalopathy, often triggered by a febrile illness, characterized by vomiting and confusion. In the later stages of acute encephalopathy seizures, loss of motor function (quadriparesis or quadriplegia 5 ), regression of developmental milestones, dystonia, external ophthalmoplegia, and dysphagia can be observed. In a proportion of cases it can evolve in a chronic (or slowly progressive) encephalopathy, characterized by akinetic (mute) state, loss of speech, and eventually coma and death. 6
BTRBGD is caused by mutations in the SLC19A3 gene on chromosome 2q36.6, encoding human thiamine (vitamin B1) transporter 2 (hTHTR2). Phenotypic heterogeneity has been described, and clinical presentation may include lactic acidosis with encephalopathy, infantile epileptic spasms or recurrent coma and encephalopathy triggered by illness or trauma. 7 Moreover, genotype–phenotype correlation is poor. 8 Pathogenetic SLC19A3 mutations include (in decreasing order of frequency): c.1264G > A, c.68G > T, c.74dupT, c.980–38dupA, c.980–14A > G. In the brain, SLC19A3 is localized in the basement membrane and is expressed by the perivascular pericytes of the cerebral vessels and the choroid plexus where it transports thiamine across the blood–brain barrier. 4 SLC19A3 mutations are associated with low thiamine levels in cerebrospinal fluid (CSF) but not in blood, suggesting impaired transport to central nervous system (CNS). 1 Exclusively in the brain, the two thiamine transporters ( SLC19A3 and SLC19A2 ) are polarized (the first at the basement membrane and the latter on the luminal side) so that both transporters are required to get thiamine across the blood brain barrier. 9 It may explain why individuals harboring SLC19A3 gene mutations develop neurological symptoms and pathology, without having a systemic thiamine deficiency. 10 Furthermore, the effect of thiamine and thiamine diphosphate on a poorly-myelinated developing brain is very different form the clinical picture of thiamine deficiency in the mature brain. Glutamate excitotoxicity has been proposed as a major cause of thiamine deficiency-related neurodegeneration but selective neuronal vulnerability may occur in different stages of the brain maturation. 11
Brain imaging in the acute phase shows severe swelling and diffuse T2-hyperintensity of the basal ganglia, thalamus, cortex, cerebral, and cerebellar white matter, with multiple areas of restricted diffusion. 12 13 The postacute phase is characterized by partial resolution of the white matter swelling with rarefaction and/or cystic degeneration of the subcortical white matter, basal nuclei, and thalami. The intermediate phase instead consists of a variable degree of atrophy, involving both deep gray and white matter, including the corpus callosum; it can be associated with extensive thinning of the cerebral cortex and in some cases, presence of cystic-like structures in cerebral white matter, thalami, basal ganglia, and cerebellar white matter. 4 The end stage is characterized by severe global atrophy. 14
Laboratory investigations are usually normal, except for lactic acid serum level that may be high during acute phase. 4 Moreover, elevated lactate levels in affected white and gray matter were found by magnetic resonance spectroscopy. 4 The magnetic resonance imaging (MRI) pattern and clinical and biochemical findings resemble those of Leigh's syndrome, so that BTRBGD can be misdiagnosed as a mitochondrial encephalopathy. 15
Here we report the clinical and radiological phenotype of two siblings diagnosed with BTRBGD in which a novel SLC19A3 mutation (NM_025243.3: c.548C > T; p.Ala183Val) was found by whole exome sequencing (WES).
Patients and Methods
The two siblings (one male and one female) described in our paper were born from consanguineous Egyptian parents (first cousins). The family history was negative for neurological diseases. The siblings had a first-born healthy sister.
Patient 1
Patient 1 is the second-born daughter and was born by normal delivery after a full-term normal pregnancy. Apgar's score was 10/10 after 1 minute and early psychomotor development was normal. At 2 months of age, she began to refuse feeding and she experienced a first generalized tonic seizure. Electroencephalography (EEG) was therefore performed, showing low-amplitude activity with focal abnormalities in the centro-temporo-occipital regions. Cranial ultrasonography (USG) showed symmetric diffuse alteration of the periventricular parenchyma and basal ganglia. Brain MRI was characterized by bilateral symmetrical increased T2 and decreased T1 signal intensity at the basal ganglia, thalami, and brainstem ( Fig. 1 ), with restricted diffusion. The same abnormalities were evident in cerebral cortex and subcortical white matter (leukoencephalopathy with areas of cavitation) and cerebellar hemispheres. The imaging pattern was compatible with Leigh's syndrome. In the suspicion of mitochondrial encephalopathy, vitamin supplementation was started, including thiamine 2000 mg/day, riboflavin 300 mg/day, ubidecarenone 150 mg/day, and creatine 1,500 mg/day. Clinical conditions globally improved but epilepsy worsened until epileptic spasms appeared. Anticonvulsant therapy with vigabatrin 1,500 mg/day was therefore started at 4 months, with good clinical response.
Fig. 1.
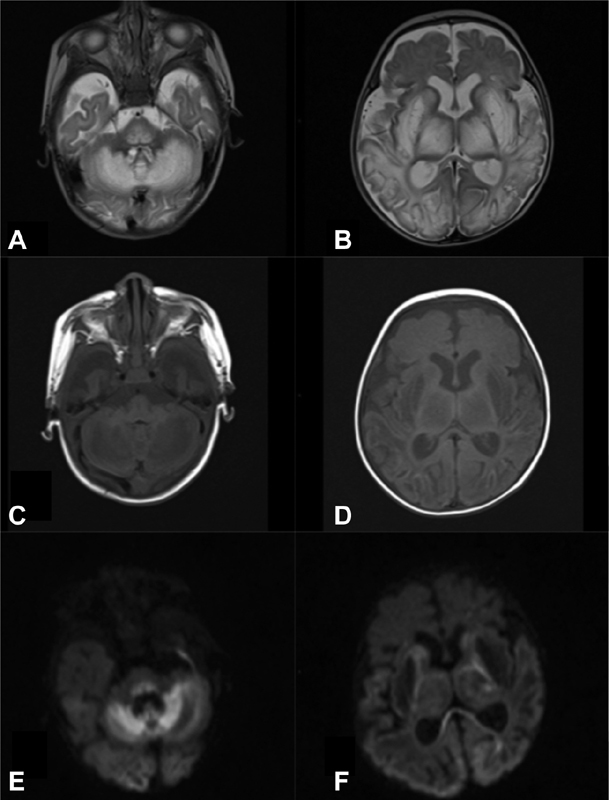
Brain MRI (patient 1, at the age of 2 months): Bilateral and symmetrical increased signal intensity in axial T2-weighted images ( A,B) with decreased signal intensity in T1-weighted images ( C, D ) in basal ganglia, thalamus, midbrain, dorsal pons, medulla oblongata, frontotemporal-parieto-occipital cortex, and subcortical white matter and cerebellum. Diffusion weighted imaging: b1000 images ( E, F ) showing extensive areas of restricted diffusion (hyperintensity) in cerebral white and gray matter structures (splenium of corpus callosum, cortex, cerebellum, dorsal pons, and midbrain). Notice the ventricular enlargement and corpus callosum hypoplasia. MRI, magnetic resonance imaging.
CSF analysis documented hyperglychorrachia and high levels of proteins and lactate (37.6 mg/dL–normal values (n.v.) 0–30 mg/dL). Also plasma lactate was slightly elevated (2.5 mmol/L–n.v. 0.5–1 mmol/L). Respiratory-chain enzymes activity on skin and muscle biopsy, urinary amino acids, and organic acids were normal.
At 4 months of age, she presented with generalized status epilepticus and spastic paraparesis. Gradually, she developed a tetraventricular hydrocephalus ( Fig. 2 ), and an external ventricular drainage was applied.
Fig. 2.
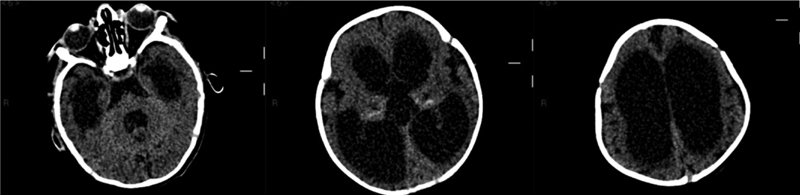
Brain CT (patient 1, at the age of 10 months): tetraventricular hydrocephalus. CT, computed tomography.
Abdominal imaging showed hepatomegaly and dolichocolon. An X-ray study of the skeleton showed thoracolumbar scoliosis. At the age of 4 years further disease progression was observed, as she was macrocephalic (head circumference 56 cm) with loss of fixation and eye movements pursuit, distal spasticity, and severe developmental delay with axial hypotonia (she was unable to sit at the age of 4 years; Fig. 3 ).
Fig. 3.
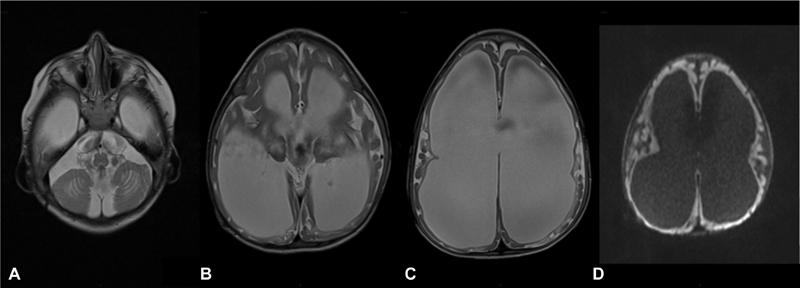
Brain MRI (patient 1, at the age of 4 years): Axial T2-weighted images ( A–C ): Progressive worsening of tetra ventricular hydrocephalus and advanced atrophy of the cerebral white matter, basal nuclei, thalami and brainstem. Axial DWI b1000 images ( D ) showing persistent diffusion restriction of the cortex. DWI, diffusion weighted imaging; MRI, magnetic resonance imaging.
At this time, the parents referred themselves to another Centre for a second opinion, due to a temporary worsening of epilepsy occurring after a viral illness. During this hospitalization, vitamin supplementation was stopped because a non-mitochondrial encephalopathy was suspected. The girl died 4 months later.
Patient 2
Patient 2 is the third-born son, born full-term after a normal pregnancy. Apgar's score was 6/10 after 1 minute and hypertonia with hyperreflexia were present after birth. At 1 month of age, he was brought to our attention because of vomiting after each feeding. Hypertrophic pyloric stenosis was excluded. During observation, he presented clonic seizures of the four limbs, associated with episodes of apnea and desaturation, needing ICU (intensive care unit) admission for stabilization. Severe psychomotor delay and episodes of generalized hypotonia, dysphagia and inefficient sucking reflex were present. In addition frequent generalized tonic-clonic seizures arose. EEG recorded generalized epileptic activity ( Fig. 4A and B ).
Fig. 4.
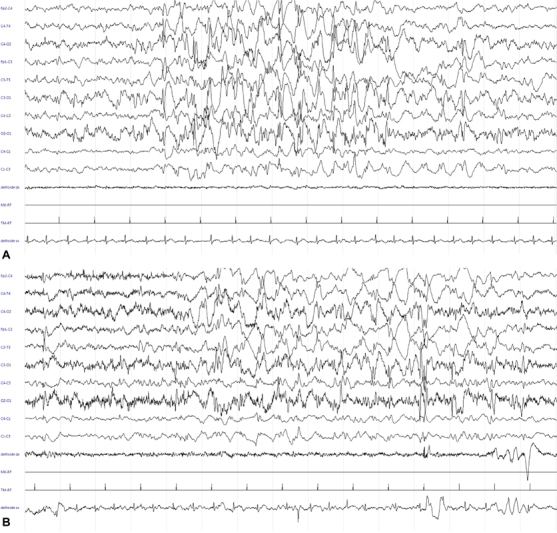
( A ) EEG: 30 mm/sec, 14 µV/mm. Burst of generalized polispikes-wave with maximal amplitude over the right central regions, alternating with period of poor low-voltage activity (burst-suppression pattern). ( B ) EEG: 30 mm/sec, 20 µV/mm. Subcontinuouspolispike activity with inter-burst periods of less than 10 seconds, characterized by asynchronous, low-voltage slow activity and multifocal spikes
As for his older sister, CSF analysis evidenced hyperglychorrachia (84 mg/dL–n.v. 40–80 mg/dL), high protein levels (96 mg/dL–n.v. 15–60 mg/dL), and high lactate levels (29.1 mg/dL). Similarly, skin and muscle biopsy for respiratory chain enzymes activity, urinary amino acids, and organic acids were not diagnostic; however, muscle biopsy revealed myopathy with hypotrophy and fiber-disproportion. Brain MRI showed several areas of bilateral symmetric abnormal signal intensity, especially in the basal ganglia, internal capsule, thalami, and brainstem with increased T2 and decreased T1 signal intensity ( Fig. 5 ). The clinical and radiologic pattern could be referred to Leigh's syndrome or to other types of mitochondrial encephalopathy. Vitamin treatment was therefore started, with thiamine 900 mg/day, riboflavin 100 mg/day, ubidecarenone 200 mg/day, creatine 1,000 mg/day; vigabatrin therapy (500 mg/day) was effective against seizures.
Fig. 5.
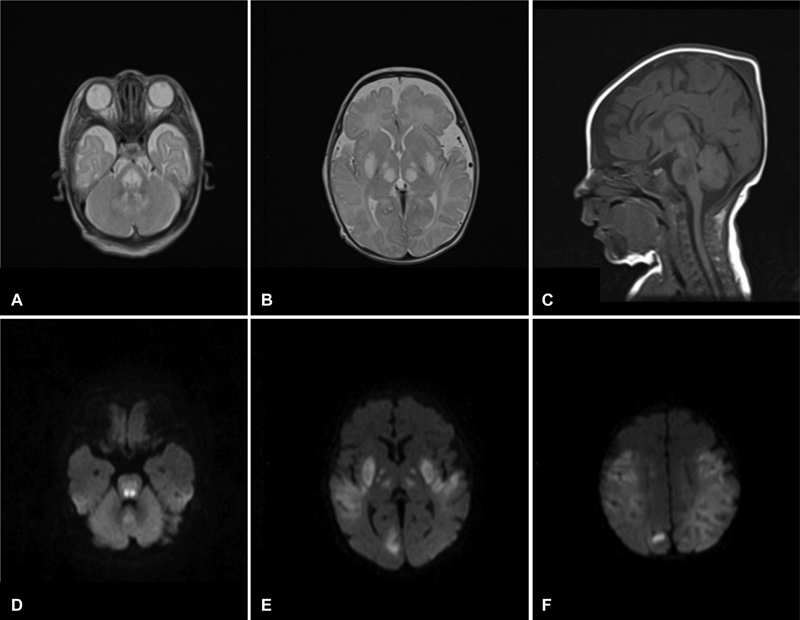
Brain MRI (patient 2, at the age of 1 month): Axial T2-weighted images ( A,B ) showing several areas of bilateral symmetric abnormal signal intensity, with diffusion restriction ( D–F ) in the lentiform nuclei, internal capsule, thalami, brainstem, dentate nuclei, and cortical and subcortical regions. Sagittal T1-w images (C) show hypointensity in the affected areas and a thin corpus callosum.
At 3 months of age, gastroesophageal reflux was diagnosed and because of a lack of growth, a percutaneous endoscopic gastrostomy feeding tube was placed. Neuroradiological features slowly progressed, along with the clinical regression, as shown in Fig. 6 . At 2 years, he showed severe psychomotor delay, microcephalia, generalized hypertonia and dystonia with axial hypotonia. X-ray showed dorsal scoliosis.
Fig. 6.
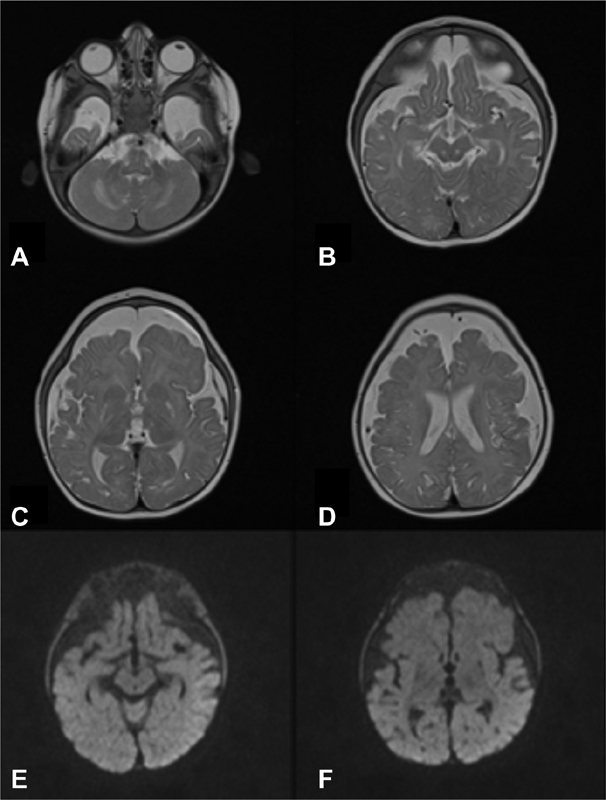
Brain MRI (patient 2, at the age of 6 months): Axial T2-weighted images ( A–D ): atrophy of the brainstem, thalami, basal ganglia, and cerebral white matter (especially in the frontotemporal regions), corpus callosum hypoplasia, and extensive thinning of the cerebral cortex. No diffusion restriction can be noted in DWI ( E,F ). DWI, diffusion weighted imaging; MRI, magnetic resonance imaging.
At the age of 2 years, together with his sister, he was referred to a different Centre where thiamin supplementation was stopped. He died 24 hours apart from his sister.
Genetic Results
Array-comparative genomic hybridization (CGH) was performed in both siblings, leading to normal results. Next Generation Sequencing (NGS) analysis was therefore performed, upon acquisition of written informed consent of the parents. DNA was extracted from peripheral blood and WES was performed. Based on the family pedigree, we assumed that the causative mutation was recessively inherited. No novel/rare (minor allele frequency < 1%) plausible segregating compound heterozygous variants were identified in the family WES data. Homozygosity mapping was performed, identifying two regions of homozygosity shared between the affected individuals, including chromosome 2 (226516429–236579317) and chromosome 3 (101575882–122345870). Variants within these regions were filtered for rare mutations that were described as deleterious in silico protein prediction programmes and were present as homozygous in the Probands and heterozygous in unaffected parents. We identified an homozygous mutation (NM_025243.3: c.548C > T; p.Ala183Val, confirmed by Sanger sequencing; Fig. 7 ) in SLC19A3 (MIM 60152), a gene associated to autosomal recessive biotin- or thiamine-responsive encephalopathy type 2 (MIM 607483). This mutation was predicted as deleterious by Sorting Intolerant From Tolerant (SIFT) and damaging by polyphen-2. The missense mutation affects a conserved residue (genomic evolutionary rate profiling (GERP) + + 5.77) of the transmembrane helical domain of the protein encoded by SLC19A3 . Within the homozygous block on chromosome 3 (101575882–122345870) WES identified a homozygous missense mutation (NM_175056.1: c.823C > G; p.Pro275Ala) in ZPLD1 (MIM 615915), a gene encoding a Zonapellucida-like domain, mostly expressed in placenta, and a mutation (NM_016298.3: c.863C > T; p.Ala288Val) in FBXO40 (MIM 609107), a gene encoding a F-box only protein of unknown function. The homozygous mutation in SLC19A3 emerged as the most fitting with the clinical phenotype of our Probands. Sanger's sequencing segregation analysis confirmed segregation and identified the variant as heterozygous in the unaffected sister.
Fig. 7.
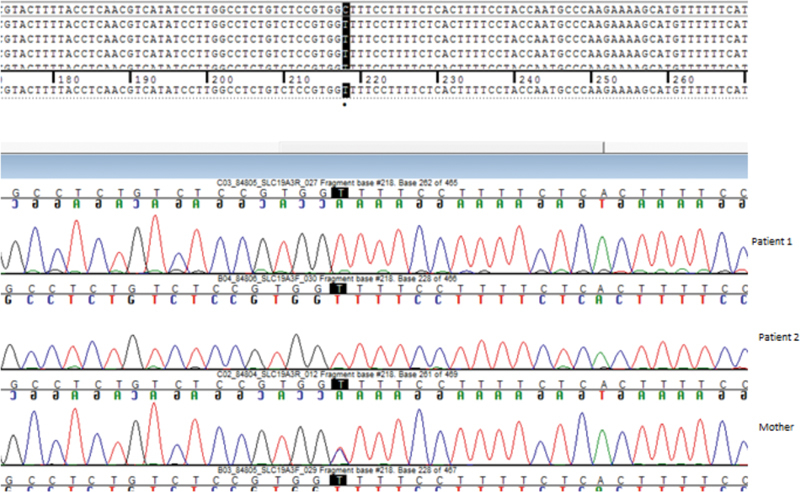
−c.548C > T; p.Ala183 Val in the gene SLC19A3 , Sanger's sequencing confirmation.
Discussion
Biotin-Thiamine-Responsive Basal Ganglia Disease is a rare panethnic encephalopathy, affecting patients with prevailing Saudi origin (57/95, 60%). Other cases have been reported in Japan (5), 14 16 India (4) 15 17 18 Germany (3), 7 19 Norway (3), 20 Portugal (2), 21 Spain (2), 22 and Sweden (2). 23 Our reported siblings had Egyptian origin and they are the first cases reported in Italy with an onset before 3 months. The first Italian report of BTRBGD was made in 2015 by Sechi et al. 24
More than 50% (54/95) of cases reported are represented by affected siblings. In many cases (11), BTRBGD has been diagnosed in patients with a positive family history for early, undefined encephalopathy leading to death. 13 25
BTRBGD has an autosomal recessive inheritance and it should be suspected in families with recurrent child death caused by undefined encephalopathy and consanguineous parents, particularly first or second relatives. Sixty-seven patients of 95 (70%), parents were consanguineous.
The average age at diagnosis is 8 years (range: 2–15 years) and the age at onset spaces between 6 days of life and 15 years. Debs et al 21 , 2010 reported two siblings with onset in adult age (33 and 29 years) and Sechi et al 24 reported the encephalopathy in a 21-year-old woman. BTRBGD usually starts as a subacute encephalopathy characterized by confusion, stupor and coma, loss of language, and acquired neurodevelopment stages, ataxia, vomiting, and seizures (usually generalized tonic–clonic [BinSaeedan and Dogar]). 26 In early onset forms (<12 months), symptoms include lethargy, hypotonia, dysphagia, psychomotor delay, dystonia, and seizures. We hereby described two patients with an early onset presentation: patient 1 presented at 2 months of age with decreased level of consciousness, dysphagia, and tonic seizures, while patient 2 presented at 1 month of age with clonic jerks, apneas, desaturations, and bradycardia. They are the first cases reported in Italy with an onset before 3 months. Severe psychomotor delay, axial hypotonia, dysphagia, inefficient sucking reflex, and dystonia were also present. Increased CSF levels of lactate induced suspicion of a metabolic disease.
After thiamine supplementation, significant improvement was noted, with response to tactile and pain stimuli, spontaneous limbs movements, and weak cry.
All the 12 patients reported to date with an early onset were characterized by hypotonia, psychomotor delay, and seizures. 14 17 20 23 27 28 Moreover, it has been analyzed that patients with early onset disease tend to have worse outcome and to develop tonic-clonic seizures with generalized burst-suppression EEG pattern and multifocal epileptiform discharges, 28 as in patient 2. Epilepsy may evolve to atypical infantile spasm, 14 or difficult-to-treat generalized epilepsy, dystonia, and myoclonus.
The MRI pattern was typical in the two siblings and it could lead to prompt consideration of a possible thiamine deficiency. 29 Alterations are usually absent in the globus pallidi: although rare, this involvement (like in patient 1) has been described in other patients. 28 Hydrocephalus had never been reported before, except for a ventricular dilatation in a 5-year-old girl reported by Ozand et al. 13 . In both patients, neuroradiological evolution was dramatically characterized by a diffuse cerebral and cortical atrophy with dilatation of the ventricular system, as already described in other patients. 4 12
WES analysis identified a homozygous mutation in both probands in SLC19A3 (MIM 60152), inherited by the two heterozygous unaffected parents (NM_025243.3: c.548C > T; p.Ala183Val). SLC19A3 is associated to autosomal recessive biotin- or thiamine-responsive encephalopathy type 2 (MIM 607483). This mutation had never been described in the literature.
Since its first description, BTRBGD has been characterized by a variable phenotypes. Differential diagnosis is difficult, especially regarding inborn errors of metabolism associated with basal ganglia degeneration (e.g., glutaricaciduria type I, metylmalonicacidaemia, 3-methyl-glutaconic aciduria). Neuroradiological alterations are highly suggestive of Leigh's syndrome: symmetric bilateral alterations in basal ganglia, thalami, and brainstem. 30 Interestingly, Leigh's syndrome was postulated to be a disorder of thiamine metabolism because of neuropathological similarities to Wernicke's encephalopathy. 31 32 33 Moreover, a novel Moroccan 34 founder SLC19A3 nonsense mutation has been described in a family with early-childhood fatal Leigh's syndrome 32 35 and recently, mutations in SLC19A3 have been proved to cause a “Leigh's-like” encephalopathy in Alaskan Husky dog. 36 37
It is important to stress that these patients might benefit from high doses of biotin and thiamine which would make this a partly treatable form of Leigh's syndrome. 38 In fact, in our patients the first hypothesis was Leigh's syndrome, furthermore supported by elevated lactate in CSF and serum and by neuroradiological findings. 39 Normal skin and muscle biopsy performed for respiratory chain enzymes activity, negative urinary amino acids, and organic acids made the diagnosis of mitochondrial disease less likely. However, the diagnosis of mitochondrial disease is challenging due to clinical, biochemical, and genetic heterogeneity. The only alteration was myopathy with hypotrophy and fiber-disproportion in patient 2, found in another reported case with a similar onset at 6 days of life. 27
Of the 95 cases described so far in the literature, we found that in 20% BTRBGD was misdiagnosed; it was diagnosed only with the help of WES. The majority of cases had an European origin, possibly due to the rarity of the disease in this area and the higher possibility to perform such expensive investigations, compared with lower-income regions. The most validated diagnosis was mitochondrial encephalopathy, particularly subacute necrotizing encephalomyelopathy (Leigh's syndrome). In one patient, 19 acute disseminated encephalomyelitis (ADEM) was suspected because of its insidious onset following a viral infection. Ygberg et al 23 reported typical signs of BTRBGD in a newborn who died at the age of 8 weeks and later proved this diagnosis by autoptic evidence. Finally, a late onset during adolescence and a possible association with ocular ptosis or ophtalmoplegia, 15 widens the differential diagnosis to Kearns–Sayre syndrome and Myasthenia gravis.
It is interesting to note that our cases showed other undescribedextraneurological clinical features: hepatomegaly and dolichocolon in patient 1, patent foramen ovale (PFO), and gastroesophageal reflux disease in patient 2, loss of visual acuity, and scoliosis in both patients.
Because of the severity of the disease and the dramatic response to therapy, early diagnosis has a crucial role in preventing death, brain damage, and neurological sequelae. Moreover, the survival rate is 85%, with a heterogeneous outcome even in the same family and with the same mutation, widening from complete remission to severe neurological sequelae. In particular, 62% (59/95) improved significantly with complete remission or minimal sequelae (mild dystonia and dysarthria not interfering with daily life); 11 of which were treated with biotin alone, while 48 patients with both biotin and thiamine. A randomized study by Tabarki et al, 40 compared combination of biotin plus thiamine versus thiamine alone and suggested that over 30 months of treatment, the combination is not superior to thiamine alone except for the duration of the acute crisis. In the last revision, 3 authors recommend the use of both vitamins (biotin 5 mg/kg/day and thiamine 10 to 40 mg/kg/day) to give the patients a benefit of doubt and to ensure possible better response. 41 Recently, Ortigoza-Escobar et al 42 find out that whole-blood thiamine quantification is a useful method for treatment monitoring. In the absence of a definite diagnosis, our patients were treated with thiamine alone as for mitochondrial disorders. This treatment was well tolerated and caused a significant stabilization of the clinical course in both of them. It is hard to speculate whether a combined therapy would have led to a better clinical response in our patients, namely, in terms of neurocognitive outcome and seizure control.
Conclusion
In conclusion, BTRBGD should be considered very early in the differential diagnosis of mitochondrial disease including Leigh's syndrome, since the clinical outcome is critically dependent on the time interval between the onset of symptoms and the start of the treatment. Atypical brain imaging findings (i.e., cortical signal alterations, cytotoxic edema) should alert clinicians to the differential diagnosis. Our patients' histories confirm the importance of starting vitamin supplementation early, without interruption of the treatment in each patient presenting neuroradiological typical features. Thiamine treatment can prevent the devastating progression of this condition and allow for appropriate genetic counseling.
Footnotes
Conflict of Interest None declared.
References
- 1.Zeng W Q, Al-Yamani E, Acierno J S, Jr. et al. Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. Am J Hum Genet. 2005;77(01):16–26. doi: 10.1086/431216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tabarki B, Alfadhel M, AlShahwan S, Hundallah K, AlShafi S, AlHashem A. Treatment of biotin-responsive basal ganglia disease: open comparative study between the combination of biotin plus thiamine versus thiamine alone. Eur J PaediatrNeurol. 2015;19(05):547–552. doi: 10.1016/j.ejpn.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Algahtani H, Ghamdi S, Shirah B, Alharbi B, Algahtani R, Bazaid A. Biotin-thiamine-responsive basal ganglia disease: catastrophic consequences of delay in diagnosis and treatment. Neurol Res. 2017;39(02):117–125. doi: 10.1080/01616412.2016.1263176. [DOI] [PubMed] [Google Scholar]
- 4.Tabariki B, Al-Hashem A, Alfadhel M . [Google Scholar]
- 5.Adhisivam B, Mahto D, Mahadevan S. Biotin responsive limb weakness. Indian Pediatr. 2007;44(03):228–230. [PubMed] [Google Scholar]
- 6.Aljabri M F, Kamal N M, Arif M, AlQaedi A M, Santali E YM. A case report of biotin-thiamine-responsive basal ganglia disease in a Saudi child: is extended genetic family study recommended? Medicine (Baltimore) 2016;95(40):e4819. doi: 10.1097/MD.0000000000004819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schänzer A, Döring B, Ondrouschek M et al. Stress-induced upregulation of SLC19A3 is impaired in biotin-thiamine-responsive basal ganglia disease. Brain Pathol. 2014;24(03):270–279. doi: 10.1111/bpa.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alfadhel M, Almuntashri M, Jadah R H et al. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J Rare Dis. 2013;8(83):83. doi: 10.1186/1750-1172-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subramanian V S, Marchant J S, Said H M. Biotin-responsive basal ganglia disease-linked mutations inhibit thiamine transport via hTHTR2: biotin is not a substrate for hTHTR2. Am J Physiol Cell Physiol. 2006;291(05):C851–C859. doi: 10.1152/ajpcell.00105.2006. [DOI] [PubMed] [Google Scholar]
- 10.Whitford W, Hawkins I, Glamuzina E et al. Compound heterozygous SLC19A3 mutations further refine the critical promoter region for biotin-thiamine-responsive basal ganglia disease . Cold Spring HarbMol Case Stud. 2017;3(06):a001909. doi: 10.1101/mcs.a001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hazell A S, Pannunzio P, Rama Rao K V, Pow D V, Rambaldi A. Thiamine deficiency results in downregulation of the GLAST glutamate transporter in cultured astrocytes. Glia. 2003;43(02):175–184. doi: 10.1002/glia.10241. [DOI] [PubMed] [Google Scholar]
- 12.Kevelam S H, Bugiani M, Salomons G Set al. Exome sequencing reveals mutated SLC19A3 in patients with an early-infantile, lethal encephalopathy Brain 2013136(Pt 5):1534–1543. [DOI] [PubMed] [Google Scholar]
- 13.Ozand P T, Gascon G G, Al Essa Met al. Biotin-responsive basal ganglia disease: a novel entity Brain 1998121(Pt 7):1267–1279. [DOI] [PubMed] [Google Scholar]
- 14.Yamada K, Miura K, Hara K et al. A wide spectrum of clinical and brain MRI findings in patients with SLC19A3 mutations. BMC Med Genet. 2010;11(01):171. doi: 10.1186/1471-2350-11-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fassone E, Wedatilake Y, DeVile C J, Chong W K, Carr L J, Rahman S. Treatable Leigh-like encephalopathy presenting in adolescence. BMJ Case Rep. 2013;2013:200838. doi: 10.1136/bcr-2013-200838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kohrogi K, Imagawa E, Muto Y et al. Biotin-responsive basal ganglia disease: a case diagnosed by whole exome sequencing. J Hum Genet. 2015;60(07):381–385. doi: 10.1038/jhg.2015.35. [DOI] [PubMed] [Google Scholar]
- 17.Bindu P S, Noone M L, Nalini A, Muthane U B, Kovoor J M. Biotin-responsive basal ganglia disease: a treatable and reversible neurological disorder of childhood. J Child Neurol. 2009;24(06):750–752. doi: 10.1177/0883073808329525. [DOI] [PubMed] [Google Scholar]
- 18.Schwarting J, Lakshmanan R, Davagnanam I. Teaching neuroimages: biotin-responsive basal ganglia disease. Neurology. 2016;86(17):e184–e185. doi: 10.1212/WNL.0000000000002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reihle C, von Kalle T, Severien Cet al. Biotin-responsive basal ganglia disease in a 10- year-old German girl without SLC19A3 gene mutationNeuropediatrics 2013;44. doi: 10.1055/s-0033-1337842
- 20.Flønes I, Sztromwasser P, Haugarvoll K et al. Novel SLC19A3 promoter deletion and allelic silencing in biotin-thiamine-responsive basal ganglia encephalopathy. PLoS One. 2016;11(02):e0149055. doi: 10.1371/journal.pone.0149055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Debs R, Depienne C, Rastetter A et al. Biotin-responsive basal ganglia disease in ethnic Europeans with novel SLC19A3 mutations. Arch Neurol. 2010;67(01):126–130. doi: 10.1001/archneurol.2009.293. [DOI] [PubMed] [Google Scholar]
- 22.Serrano M, Rebollo M, Depienne C et al. Reversible generalized dystonia and encephalopathy from thiamine transporter 2 deficiency. Mov Disord. 2012;27(10):1295–1298. doi: 10.1002/mds.25008. [DOI] [PubMed] [Google Scholar]
- 23.Ygberg S, Naess K, Eriksson M et al. Biotin and thiamine responsive basal ganglia disease--a vital differential diagnosis in infants with severe encephalopathy. Eur J PaediatrNeurol. 2016;20(03):457–461. doi: 10.1016/j.ejpn.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 24.Sechi E, Addis A, Fadda G, Minafra L, Bravatà V, Sechi G. Teaching neuroimages: subacute encephalopathy in a young woman with THTR2 gene mutation. Neurology. 2015;85(14):e108–e109. doi: 10.1212/WNL.0000000000002002. [DOI] [PubMed] [Google Scholar]
- 25.Distelmaier F, Huppke P, Pieperhoff P et al. Biotin-responsive basal ganglia disease: a treatable differential diagnosis of leigh syndrome. JIMD Rep. 2014;13:53–57. doi: 10.1007/8904_2013_271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bin Saeedan M, Dogar M A. Teaching neuroimages: MRI findings of biotin-responsive basal ganglia disease before and after treatment. Neurology. 2016;86(07):e71–e72. doi: 10.1212/WNL.0000000000002372. [DOI] [PubMed] [Google Scholar]
- 27.El-Hajj T I, Karam P E, Mikati M A. Biotin-responsive basal ganglia disease: case report and review of the literature. Neuropediatrics. 2008;39(05):268–271. doi: 10.1055/s-0028-1128152. [DOI] [PubMed] [Google Scholar]
- 28.Sremba L J, Chang R C, Elbalalesy N M, Cambray-Forker E J, Abdenur J E. Whole exome sequencing reveals compound heterozygous mutations in SLC19A3 causing biotin-thiamine responsive basal ganglia disease . Mol Genet Metab Rep. 2014;1:368–372. doi: 10.1016/j.ymgmr.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sechi G, Sechi E, Fois C, Kumar N. Advances in clinical determinants and neurological manifestations of B vitamin deficiency in adults. Nutr Rev. 2016;74(05):281–300. doi: 10.1093/nutrit/nuv107. [DOI] [PubMed] [Google Scholar]
- 30.Savasta S, Comi G P, Perini M P et al. Leigh disease: clinical, neuroradiologic, and biochemical study of three new cases with cytochrome c oxidase deficiency. J Child Neurol. 2001;16(08):608–613. doi: 10.1177/088307380101600816. [DOI] [PubMed] [Google Scholar]
- 31.Cerase A, Rubenni E, Rufa A et al. CT and MRI of Wernicke's encephalopathy. Radiol Med (Torino) 2011;116(02):319–333. doi: 10.1007/s11547-011-0618-x. [DOI] [PubMed] [Google Scholar]
- 32.Gerards M, de Coo R, Smeets H.Reply: Infantile Leigh-like syndrome caused by SLC19A3 mutations is a treatable disease Brain 2014137(Pt 9):e296. [DOI] [PubMed] [Google Scholar]
- 33.Kono S, Miyajima H, Yoshida K, Togawa A, Shirakawa K, Suzuki H. Mutations in a thiamine-transporter gene and Wernicke's-like encephalopathy. N Engl J Med. 2009;360(17):1792–1794. doi: 10.1056/NEJMc0809100. [DOI] [PubMed] [Google Scholar]
- 34.Gerards M, Kamps R, van Oevelen Jet al. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome Brain 2013136(Pt 3):882–890. [DOI] [PubMed] [Google Scholar]
- 35.Haack T B, Klee D, Strom T Met al. Infantile Leigh-like syndrome caused by SLC19A3 mutations is a treatable disease Brain 2014137(Pt 9):e295. [DOI] [PubMed] [Google Scholar]
- 36.Vernau K M, Runstadler J A, Brown E A et al. Genome-wide association analysis identifies a mutation in the thiamine transporter 2 (SLC19A3) gene associated with Alaskan Husky encephalopathy. PLoS One. 2013;8(03):e57195. doi: 10.1371/journal.pone.0057195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vernau K, Napoli E, Wong S et al. Thiamine deficiency-mediated brain mitochondrial pathology in Alaskan Huskies with mutation in SLC19A3.1. Brain Pathol. 2015;25(04):441–453. doi: 10.1111/bpa.12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Knaap M S, Kevelam S H.Reply: infantile Leigh-like syndrome caused by SLC19A3 mutations is a treatable disease Brain 2014137(Pt 9):e297. [DOI] [PubMed] [Google Scholar]
- 39.Kassem H, Wafaie A, Alsuhibani S, Farid T. Biotin-responsive basal ganglia disease: neuroimaging features before and after treatment. Am J Neuroradiol. 2014;35(10):1990–1995. doi: 10.3174/ajnr.A3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabarki B, Al-Shafi S, Al-Shahwan S et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(03):261–267. doi: 10.1212/WNL.0b013e31827deb4c. [DOI] [PubMed] [Google Scholar]
- 41.Gerards M.Leigh syndrome: the genetic heterogeneity story continues Brain 2014137(Pt 11):2872–2873. [DOI] [PubMed] [Google Scholar]
- 42.Ortigoza-Escobar J D, Serrano M, Molero M et al. Thiamine transporter-2 deficiency: outcome and treatment monitoring. Orphanet J Rare Dis. 2014;9:92. doi: 10.1186/1750-1172-9-92. [DOI] [PMC free article] [PubMed] [Google Scholar]