

Abstract
Ataxia with oculomotor apraxia type 4 (AOA4) is a rare autosomal recessive, PNKP -related disorder delineated in 2015 in Portugal. We diagnosed AOA4 by next generation sequencing (NGS) followed by Sanger's sequencing in three boys from two unrelated Belarusian families. In both families, one of the heterozygous PNKP mutations was c.1123G>T, common in Portuguese patients; biallelic mutations, c.1270_1283dup14 and c.1029+2T>C, respectively, were novel. These are the first reported AOA4 Slavic cases and the first with a “Portuguese” PNKP mutation outside Portugal. Distinction in two brothers was microcephaly but their disease was not severe in contrast to PNKP -related “microcephaly, seizures, and developmental delay” and reported cases with features of both phenotypes.
Keywords: ataxia with oculomotor apraxia type 4, microcephaly, mutation frequency
Introduction
The PNKP gene (MIM*605610, locus 19q13.33) encodes polynucleotide kinase 3′-phosphatase, a protein involved in DNA damage repair, and it is associated with two autosomal recessive disorders: ataxia with oculomotor apraxia type 4 (AOA4; MIM#616267) recognized in Portuguese families 1 and “microcephaly, seizures, and developmental delay” (MCSZ; MIM#613402) delineated in families of various ethnicity. 2 Though different, the diseases have crosspoints, and mixed phenotypes have been reported. 3 4 PNKP -related cases with predominant congenital or adult-onset axonal polyneuropathy and later-appearing mild AOA4 features adjoin AOA4. 5 6 Some PNKP mutations are shared by different phenotypes. The PNKP missense mutation c.1123G>T (p.Gly375Trp) was common in a Portuguese AOA4 group 1 but not found elsewhere. We diagnosed AOA4 in two Belarusian families with compound heterozygosity for c.1123G>T and novel mutations. These cases and our data on frequency of the detected mutations in a population-matched control are presented.
Families and Methods
The two families are of Belarusian ethnicity, nonconsanguineous, unrelated, and come from different Belarus regions.
DNA testing in patients 1 and 2–1 was performed by targeted next generation sequencing (NGS) panel Illumina TruSight One (clinical exome sequencing, CES) performed on an Illumina NextSeq 500 instrument in 2×151 bp paired-end mode in the Laboratory of Molecular Pathology (Genomed Ltd., Moscow). The bioinformatics pipeline of NGS data analysis was described previously. 7 Further filtering was performed by functional consequences, population frequencies, and clinical relevance. All variants were named according to the NM_007254.3( PNKP _v001) reference transcript variant. NGS findings of definite/probable diagnostic value were verified by Sanger's sequencing in the patients and their parents.
For the population study, peripheral blood samples were collected from 116 unrelated healthy individuals of Belarusian origin. The population frequency of detected PNKP mutations was studied in the control sample by PCR-RFLF for c.1123G>T (by restriction with Bss T1I endonuclease) and c.1029+2T>C (by restriction with Hph I) and by AFLP for c.1270_1283dup14. Primer sequences are listed in Supplementary Table S1 (available in online version only).
Informed consent of the families for obtaining blood samples and for publication was obtained. The study was approved by the Ethics Committee of the Research Centre for Medical Genetics.
Results
Clinical Findings
The case of Patient 1, a 9-year-old boy, was partially reported (for pedigree see Fig. 1A ). 8 Early development was normal except for non-progressing dysarthria. He walked at 10 months, at 2 years moderate unsteadiness appeared and was stable up to 8 years, initial diagnosis was mild cerebral palsy. At 8 years, ataxia progressed and hyperkinesia became evident. Brain magnetic resonance imaging (MRI), which was previously normal, revealed cerebellar atrophy ( Fig. 1C ). Electroneuromyography (ENMG) revealed axonal polyneuropathy. Examination showed normal height and occipitofrontal circumference (OFC), vertical ophthalmoparesis, oculomotor apraxia (OA), peroneal paresis (inability to stand on heels), low arm and patellar reflexes, Achilles areflexia, normal sensation, unsteadiness in Romberg's position, slow unsupported gait with features of paretic and ataxic, moderate generalized dystonic hyperkinesia (particularly in hands), and dysarthria; there was impression of mild/moderate cognitive deficit. Additional information was received from parents 1.5 years later: neurological disorders progressed, the boy moved with a support indoors and in a wheel-chair outdoors. He had learning difficulties at school for children with special needs but managed reading, writing, and simple arithmetic. Psychological and psychiatric examination showed moderate total mental underdevelopment, particularly in verbal tests, and stiffness of thinking with no behavioral disturbances. AOA type 1 (MIM#20820, gene APTX ) characterized by average onset at 4 to 5 years, ataxia, dysarthria, OA, axonal polyneuropathy, cerebellar atrophy on MRI and some facultative signs (hyperkinesia, cognitive deficiency, hypoalbuminemia, and hypercholesterolemia) seemed most probable, and NGS was performed.
Fig. 1.
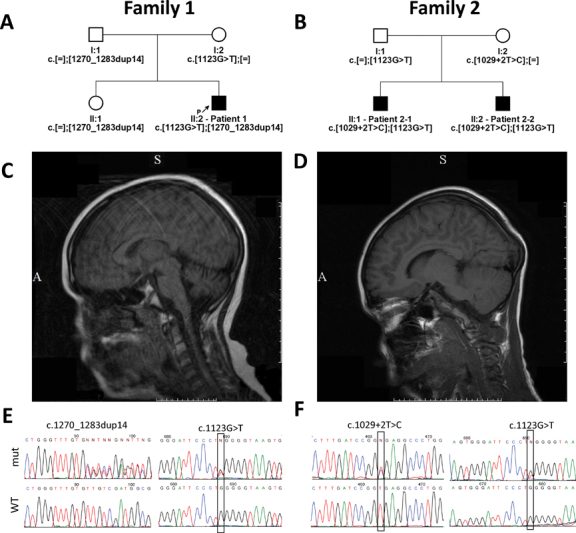
Pedigrees of families 1 and 2 are shown in ( A ) and ( B ), respectively. T1-weighted FLAIR mode sagittal slice images of brain MRI of patients 1 and 2–1 are shown in ( C ) and ( D ), respectively. Sequenograms of the identified mutations in patients 1 and 2–1 are shown in ( E ) and ( F ), respectively. FLAIR, fluid attenuated inversion recovery; MRI, magnetic resonance imaging.
Family 2 had two affected children, 2–1 and 2–2 (for pedigree see Fig. 1B ). We examined them twice: at 5 (2–1) and 3 (2–2) years, and at 8 (2–1) and 6 (2–2) years; additional information from parents was received 1 year later. Though patient 2–1 had pre-/perinatal complications, the boys developed similarly and achieved early milestones timely but ataxia was evident from the age of independent walking. At 5 to 6 years of age, moderate hyperkinesia appeared. Other signs were microcephaly, speech delay, and dysarthria. Examination revealed normal height, microcephaly (on second visit OFC 48 cm, under 3rd percentile, < − 2 standard deviation (SD) in both), OA, retained muscle power, reflexes, and sensation, moderate ataxia (unsteadiness in Romberg's position, intention tremor in limbs, unsupported ataxic gait), dysarthria, and moderate dystonic hyperkinesia. In addition, since 5.5 years, patient 2–1 had attacks of severe headache, abdominal pain, and retching, sometimes with a short phase of tonic tension, blurred (not lost) conscience, and incoherent stereotypical murmuring. They occurred after falling asleep and could be suppressed by instant analgesics injection. Over 2.5 years, there were only five attacks. Regular electroencephalography (EEG) registered no epileptic activity, epilepsy was rejected and complicated migraine was diagnosed. On recent information, with schooling onset at 8.5 years, the attacks provoked by fatigue or emotional stress occurred monthly, twice accompanied by urination, and once by defecation. Though there was no loss of conscience or epileptic EEG activity, epilepsy was suggested again. Over 4 months on levetiracetam the boy is free of attacks, yet their epileptic nature remains questionable.
Since 8.5 years (patient 2–1) and 7 years (patient 2–2) the boys began studying at school with sparing regimen. Results of neuropsychological examination performed at that time were similar. Both boys contacted adequately. Insufficiency of neurodynamic activities (split attention and failing concentration) was detected. Signs in emotional sphere were infantilism and emotional lability. Impairment of visuoconstructive and graphic praxis was detected by tests for recoding and head tests and simultaneous agnosia was a sign of fragmentary perception. There were difficulties in performing tests related to logical and grammatical constructions comprehension. Detected disturbances are of functional nature due to deprivation of mediobasal temporal regions, temporo-parieto-occipital (TPO) zone and limbic subcortex. Mental development is within normal limits, yet educational capacities need medical, pedagogical, and psychological corrections.
Repeated ENMG and brain MRI in both boys were normal. MRI picture of patient 2–1 is shown in Fig. 1D . Tests for several neurometabolic disorders were negative. NGS was recommended with no concrete preliminary diagnosis.
Molecular Findings
NGS in patient 1 revealed two heterozygous PNKP mutations: c.1123G>T (p.Gly375Trp) in exon 16, common in Portuguese patients, and a novel small deletion c.1270_1283dup14 in exon 14 leading to frame shift, p.Ala429Gln fs *43 ( Fig. 1E ). According to Bras et al, 1 c.1123G>T is located in a potential ATP nucleotide-binding domain within the kinase region of PNKP . The latter variant is predicted to disrupt the second half of the kinase domain of PNKP protein and is thus regarded as probably pathogenic. Neither mutation was registered in the Genome Aggregation (gnomAD) database. Sanger's sequencing in the family detected biallelic mutations in the patient, heterozygous c.1123G>T in the mother, and heterozygous c.1270_1283dup14 in the father.
NGS in patient 2–1 also detected two heterozygous PNKP mutations: c.1123G>T and an intronic mutation, c.1029+2T>C (rs199919568), affecting an invariant donor splicing site dinucleotide of intron 11 ( Fig. 1F ), which was not previously described in association with human diseases and thus considered as probably pathogenic. Disrupting the donor splicing site should lead to entire exon 11 skipping as predicted by Human Splicing Finder v. 3.1 ( http://www.umd.be/HSF3/ ) and MutationTaster, which would result in in-frame deletion p.(Phe313_Pro343del) affecting the phosphatase domain of PNKP protein. The latter variant is present in the total gnomAD database with an allele frequency of 0.001067, but has never been found in the homozygous state. Sanger's sequencing in the family revealed biallelic mutations in patient 2–1, an identical PNKP genotype in patient 2–2, and heterozygosity in the parents: paternal c.1123G>T, maternal c.1029+2T>C. Thus, AOA4 was diagnosed in both families.
Population screening for the three detected mutations was performed: c.1123G>T and c.1270_1283dup14 were not detected in 232 population-matched control chromosomes tested, while c.1029+2T>C was found in one case.
Discussion
Families 1 and 2 are the first reported AOA4 Slavic cases. Features of the disease are summarized in Table 1 . Patient 1 presents typical AOA4 and vertical ophthalmoparesis is his only neurodegenerative sign, unreported in AOA4 previously. Family 2 has some distinctions from AOA4, namely microcephaly in patients 2–1 and 2–2 and probable nonsevere epilepsy in patient 2–1. Both features are signs of MCSZ but, in contrast to the disease in the brothers, MCSZ is characterized by progressing microcephaly, severe development delay, and infantile-onset refractory epilepsy, but no ataxia or OA. 2 9 10 In few reported cases of mixed MCSZ-AOA4 phenotypes, MCSZ features were predominated. 3 4 Though AOA4 with microcephaly and/or epilepsy was not reported previously, we consider the phenotype in the brothers as AOA4 variant. Absence of cerebellar atrophy on MRI and of polyneuropathy may be other distinctions of AOA4 in family 2, or these signs may appear later, like polyneuropathy in Swedish and Norwegian patients, 11 12 like cerebellar signs in Latin American cases with predominant polyneuropathy, 5 6 or like cerebellar atrophy in patient 1. In spite of some differences, the disease in the brothers is similar, but intrafamilial AOA4 variability may be more pronounced as in the Arab family. 13
Table 1. AOA4 features in families 1 and 2 and in other reported cases.
| Family 1 | Family 2 |
Reported cases
a
(17 patients/13 families) |
||
|---|---|---|---|---|
| Patient 1 | Patient 2–1 | Patient 2–2 | ||
| Gender | Male | Male | Male | 6 males, 11 females |
| Family ethnicity | Belarusian | Belarusian | Portuguese (8), Swedish, Norwegian, German, Brazilian, Arabic (one each) | |
| Age at diagnosis (y) | 9 | 8 | 6 | Variable, up to 50 |
| Age at onset (y) | 2 | 1 (since age of walking) | Average, 4–5 (range, 1–14) | |
| First sign | Ataxia | Ataxia | Variable: hyperkinesia, ataxia, OA, polyneuropathy | |
| Ataxia | + b | + c | + | |
| Dysarthria | + | + | + | |
| Oculomotor apraxia | + | + | ± (absent in 3 patients) | |
| Axonal polyneuropathy | + | − | ++ | |
| Hyperkinesia | + | + | + | |
| Independent walking | + | + | Loss in 2nd–3rd decades | |
| Cognitive impairment | + (mild) | + (mild) | ± | |
| Microcephaly | − | + | − | |
| Other signs | Vertical ophthalmoparesis | Epilepsy ( ?,e ) | − | Edema (1); pilocytic astrocytoma (1) |
| MRI: cerebellar atrophy | + d | − | + | |
| MRI: brainstem atrophy | − | − | ± | |
| Obesity | − | − | ± | |
| Hypercholesterolemia | + (6.6 mg/dL; n < 5.2) | − | ± | |
| Hypoalbuminemia | – | Not tested | ± | |
| Serum α -fetoprotein | ↑(17.7 ME/mL; n < 7.3) | Normal | Normal/raised (rarely) | |
| PNKP mutations | c.[1123G>T];[1270_1283dup14] | c.[1123G>T];[1029+2T>C] | Ten reported mutations; c.1123G>T common in Portugal | |
Abbreviations: AOA4, Ataxia with oculomotor apraxia type 4; MCSZ, microcephaly, seizures, and developmental delay; MRI, magnetic resonance imaging.
MCSZ cases with some AOA4 signs (Poulton et al 3 , Taniguchi-Ikeda et al 4 ) and Costa Rican cases regarded as hereditary neuropathy (Leal et al 6 ) are not included.
Deterioration at 8 years.
No deterioration.
Appeared at 8 years.
To our knowledge, families 1 and 2 are the first reported cases with PNKP mutation c.1123G>T outside Portugal, where it was found homozygous in four and heterozygous in two of eight AOA4 families. 1 Belarusian origin of both families is of interest (the more as families from Belarus are infrequent among our patients) but may be coincidental.
Another mutation in the Portuguese AOA4 group, с.1253_1269dup17 (p.Thr424fs), was reported also in MCSZ, 2 in mixed MCSZ-AOA4 phenotype, 3 and in a German patient with AOA4 and pilocytic astrocytoma. 14 The mutation together with mutation с.1270_1283dup14 (p.Ala429fs), identified also in Family 1, was detected by CES in our third PNKP- related case. This 1-year-old Russian boy had typical MCSZ: congenital microcephaly (OFC; 29 cm at birth, 39 cm in 1 year), profound mental and motor delay, severe epilepsy since 4 months, and no malformations on brain computed tomography. As the parents' DNA was unavailable for genotype verification, the family was not reported yet. Thus, both mutations can be found in AOA4 as well as in MCSZ. Another example of identical PNKP mutation in differing phenotypes is c.1221_1223delCAC (p.Thr408del) found in typical AOA4 1 and in cases with predominant polyneuropathy. 5 6
The phenotype of the brothers in family 2 is in line with the opinion that MCSZ and AOA4 present a clinical continuum rather than independent phenotypes. 4 PNKP mutations shared by both disorders support this opinion. Genetic and/or epigenetic modifiers are most likely reasons for inter- and intrafamilial variability of PNKP -related disorders. 15
Conclusion
The two reported Belarusian families have several points of interest: they are the first Slavic AOA4 cases; the heterozygous PNKP mutation c.1123G>T, found in both families, proves that the mutation is not only “Portuguese”; two novel PNKP mutations were detected; microcephaly in affected brothers and probable nonsevere epilepsy in one of them are novel AOA4 features expanding its phenotype and adding to the opinion of absence of strict borders between PNKP -related disorders.
Acknowledgments
We thank the families for their cooperation. We thank Dr. Richard H. Lozier, Prof. Lyubov A. Troitskaya, and Dr. Inna V. Sharkova for interest in our work and useful assistance. Population screening was performed with support of the Russian Science Foundation grant no. 17–15–01051.
Conflict of Interest None declared.
Authors' Contribution
Authors G.E.R. and A.V.M. have designed the study; authors G.E.R., O.A.S., E.L.D., I.A.A., N.V.P. have collected data for the study; authors G.E.R., A.V.M., O.A.S., E.R.L., F.A.K. have done data analysis; and authors G.E.R., A.V.M. have prepared the manuscript.
Supplementary Material
References
- 1.Bras J, Alonso I, Barbot C et al. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet. 2015;96(03):474–479. doi: 10.1016/j.ajhg.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen J, Gilmore E C, Marshall C A et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet. 2010;42(03):245–249. doi: 10.1038/ng.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poulton C, Oegema R, Heijsman D et al. Progressive cerebellar atrophy and polyneuropathy: expanding the spectrum of PNKP mutations. Neurogenetics. 2013;14(01):43–51. doi: 10.1007/s10048-012-0351-8. [DOI] [PubMed] [Google Scholar]
- 4.Taniguchi-Ikeda M, Morisada N, Inagaki H et al. Two patients with PNKP mutations presenting with microcephaly, seizure, and oculomotor apraxia. Clin Genet. 2018;93(04):931–933. doi: 10.1111/cge.13106. [DOI] [PubMed] [Google Scholar]
- 5.Pedroso J L, Rocha C R, Macedo-Souza L I et al. Mutation in PNKP presenting initially as axonal Charcot-Marie-Tooth disease. Neurol Genet. 2015;1(04):e30. doi: 10.1212/NXG.0000000000000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leal A, Bogantes-Ledezma S, Ekici A B et al. The polynucleotide kinase 3′-phosphatase gene (PNKP) is involved in Charcot-Marie-Tooth disease (CMT2B2) previously related to MED25. Neurogenetics. 2018;19(04):215–225. doi: 10.1007/s10048-018-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lozier E R, Konovalov F A, Kanivets I V et al. De novo nonsense mutation in WHSC1 (NSD2) in patient with intellectual disability and dysmorphic features. J Hum Genet. 2018;63(08):919–922. doi: 10.1038/s10038-018-0464-5. [DOI] [PubMed] [Google Scholar]
- 8.Rudenskaya G E, Surkova E I, Konovalov F A. Ataxia with oculomotor apraxia type 4 detected by next-generation sequencing in Russian] Zh Nevrol Psikhiatr Im S S Korsakova. 2018;118(03):10–14. doi: 10.17116/jnevro20181183110-14. [DOI] [PubMed] [Google Scholar]
- 9.Nair P, Hamzeh A R, Mohamed M et al. Microcephalic primordial dwarfism in an Emirati patient with PNKP mutation. Am J Med Genet A. 2016;170(08):2127–2132. doi: 10.1002/ajmg.a.37766. [DOI] [PubMed] [Google Scholar]
- 10.Entezam M, Razipour M, Talebi S, Beiraghi Toosi M, Keramatipour M. Multi affected pedigree with congenital microcephaly: WES revealed PNKP gene mutation. Brain Dev. 2019;41(02):182–186. doi: 10.1016/j.braindev.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Paucar M, Malmgren H, Taylor M et al. Expanding the ataxia with oculomotor apraxia type 4 phenotype. Neurol Genet. 2016;2(01):e49. doi: 10.1212/NXG.0000000000000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tzoulis C, Sztromwasser P, Johansson S, Gjerde I O, Knappskog P, Bindoff L A. PNKP mutations identified by whole-exome sequencing in a Norwegian patient with sporadic ataxia and edema. Cerebellum. 2017;16(01):272–275. doi: 10.1007/s12311-016-0784-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schiess N, Zee D S, Siddiqui K A, Szolics M, El-Hattab A W.Novel PNKP mutation in siblings with ataxia-oculomotor apraxia type 4 J Neurogenet 201731(1–2):23–25. [DOI] [PubMed] [Google Scholar]
- 14.Scholz C, Golas M M, Weber R G et al. Rare compound heterozygous variants in PNKP identified by whole exome sequencing in a German patient with ataxia-oculomotor apraxia 4 and pilocytic astrocytoma. Clin Genet. 2018;94(01):185–186. doi: 10.1111/cge.13216. [DOI] [PubMed] [Google Scholar]
- 15.Dumitrache L C, McKinnon P J.Polynucleotide kinase-phosphatase (PNKP) mutations and neurologic disease Mech Ageing Dev 2017161(Pt. A):121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.