

Abstract
This is the first reported case of prosaposin ( PSAP ) mutation from India manifesting as an acute neuronal Gaucher disease-like condition. A 2-month-old male baby presented with encephalopathy, resistant tonic–clonic seizures, moderate hepatosplenomegaly, hypotonia, and cherry red spot in the retinae. The child had anemia, thrombocytopenia, elevated chitotriosidase, and normal activity of acid sphingomyelinase and low normal activity of β-glucosidase 1 (β-glucocerebrosidase 1, GBA). The child succumbed in the fourth month of life due to persistent respiratory distress and refractory seizures. The clinical phenotype, cherry red spots, elevated chitotriosidase, and lysosomal assays led to the suspicion of Gaucher disease. Exome sequencing revealed a homozygous stop codon mutation in the PSAP gene (c.G1228T, p.Glu410ter). Prenatal diagnosis in the next pregnancy revealed a carrier fetus, who was unaffected postnatally. The diagnosis of specific activator deficiency such as saposin C and saposin D deficiency (in the current study) should be considered and tested for when Gaucher disease is suspected in an infant with partially deficient or near normal GBA activity.
Keywords: prosaposin, specific activator deficiency, neuronopathic Gaucher disease
Introduction
The accumulation of glucocerebroside in the monocyte-macrophage system due to a deficiency of either β-glucosidase 1 (β-glucocerebrosidase 1, GBA) or saposin C results in classical Gaucher disease or a Gaucher-like clinical manifestation. While deficiency of β-glucosidase 1 is common, deficiency of saposin C is very rare. Glucocerebroside is catabolized to ceramide in the presence of β-glucosidase 1. In the absence of β-glucosidase activity, glucocerebroside accumulates in the reticuloendothelial cells to form Gaucher cells. These Gaucher cells are predominant in liver, spleen, bone marrow, and rarely in the lung, thus contributing to symptoms that can include hepatosplenomegaly, pancytopenia, bone complications, interstitial lung disease, and pulmonary hypertension. Gaucher disease manifests in non-neuronopathic (type 1) or neuronopathic (type 2 and type 3) forms. Hepatosplenomegaly is the hallmark of type 1 Gaucher. Type 2 is the infantile form of Gaucher disease with severe neurological symptoms with fatal outcome. Type 3 is a chronic neurological phenotype much less prevalent than type 1. In addition to the phenotypic presentation, biochemical and molecular evaluation, diffusion-weighted magnetic resonance imaging (MRI) of liver/spleen, brain, vertebra, and bone marrow 1 2 3 4 can be used as supporting evidence in the diagnosis of both the non-neuronopathic and neuronopathic types of Gaucher disease. Proton MR spectroscopy assists in the diagnosis of neuronopathic Gaucher disease based on choline/creatine ratio and the presence of a lipid peak. 5
Prosaposin (PSAP) is a precursor of saposin A, saposin B, saposin C, and saposin D, which are activators of sphingolipid degrading enzymes. PSAP is localized on chromosome 10q21 and spans 17 kb of genomic DNA with 15 exons and 14 introns. 6 Each saposin comprises ∼80 amino acid residues with six cysteine residues conferring heat stability and tertiary structure stabilization with three disulfide bridges. A total of 22 mutations comprising of missense, splicing, small deletions, and gross deletions have been reported in the PSAP gene in the Human Gene Mutation Database. To date, only seven patients with saposin C deficiency are reported, of whom five have type 3, and two have type 1 Gaucher disease. The saposin C deficiency was observed for the first time in a Caucasian female with severe accumulation of glucosylceramide in the spleen who was heterozygous for PSAP c.1154 G > T (p.Cys385Phe) and was suspected to have another mutation in the other PSAP allele. 7 A male child succumbed from variant Gaucher disease was found to be heterozygous for PSAP c .1144 T > G (p.Cys382Gly) inherited from his father and a presumed mutation inherited from his mother who had half of the normal amount of mRNA for PSAP in her cultured skin fibroblasts. 8 Further analysis revealed the maternal mutation to be PSAP c.1288 C > T (p.Gln340ter) mutation in the saposin D domain. 9 Other reported mutations include a homozygous c.803delG mutation in a patient who had histiocytic infiltration in many visceral organs and active demyelination in the brain. 10 Two siblings with compound heterozygous mutations in PSAP (c.1A > T/c.1046T > C p.Met1Leu/p.Leu349Pro) manifested non-neuronopathic Gaucher disease in adulthood. 11 Another individual with compound heterozygous mutations in PSAP (c.1A > G/c.943T > A p.Met1Val/p.Cys315Ser) had features of type 3 Gaucher disease, while an individual with a homozygous PSAP c.1024_1044 del TTTGACAAAATGTGCTCGAAG (p.Phe342_Lys348del) manifested as type 1 Gaucher disease. 12 A patient heterozygous for PSAP c.1133C > G (p.Pro378Arg) and deletion of exon 2 to 7 presented with hepatosplenomegaly, thrombocytopenia, and anemia. 13 Herein, we present the first reported case from India of an acute neuronopathic Gaucher-like disorder caused by homozygosity for a novel mutation in the PSAP gene.
Clinical Phenotype
A 2-month-old male baby, the first child to first cousin parents, presented with encephalopathy. The child was delivered by cesarian section and birth weight was 2.9 kg. At the age of 1 month, the baby presented with bronchopneumonia and respiratory distress. By the second month, tonic–clonic seizures, poor feeding, abdominal distension, moderate hepatosplenomegaly, hypotonia of all limbs, and sluggish deep tendon reflexes were evident. The response to antiepileptic medication was poor and he continued to be encephalopathic. Due to recurrent respiratory distress, the child was put on ventilator support. Brain MRI was normal; however, fundus evaluation revealed bilateral cherry red spots. An electroencephalogram was abnormal with generalized epileptiform discharges. Biochemically, the child had anemia, thrombocytopenia, and abnormal liver transaminases. Plasma chitotriosidase was elevated and lysosomal sphingomyelinase and β-glucosidase 1 activities were normal. Due to persistent respiratory distress and refractory seizures, the child succumbed by the fourth month of life.
Methods
Plasma chitotriosidase activity was determined using artificial fluorogenic substrate 4-methylumbelliferyl β-D-N, N′, N′′-triacetylchitotrioside, 14 while β-glucosidase and sphingomyelinase activities were determined in leukocyte extract by fluorimetry using 4-methylumbelliferyl-β-d-glucopyranoside 15 and 6-hexadecanoyiamino-4-methylumbelliferyl-phosphoryl-choline 16 as substrates, respectively.
Genomic DNA was extracted using standard methods (QIAcube Qiagen, Valencia, California, United States) and quantified by nanodrop and Qubit. The Agilent Sure Select Human All Exon V2 50Mb Illumina Paired-End Sequencing Library Prep protocol was used for the library preparation and exome capture. A50-bp paired-end run sequencing was performed using Illumina Nextseq500 which resulted in 60 million independent paired reads. The Genome Reference Consortium Human Build 37 human reference genome was used for sequence alignment. INDEL realignment and base quality recalibration were performed using the Genome Analysis Toolkit. Variant annotation was performed using Variant Annotator X. The depth of coverage was calculated using the Genome Analysis Toolkit. The My Structured Query Language 5.2 database was used for filtering annotated variants.
Results
Plasma chitotriosidase was elevated to 944 nmol/h/mL (Ref range: 8–87 nmol/h/mL). Acid sphingomyelinase was normal (15.1 nmol/17 h/mg, Ref range: 5.8–55 nmol/17h/mg), and β-glucosidase 1 was low normal (5.2 nmol/h/mg, Ref range: 4–24 nmol/h/mg). Whole exome sequencing revealed a novel homozygous stop gain mutation in PSAP c.G1228T (p.Glu410ter) in exon 11 ( Fig. 1 ). This variant is not reported in 1000 genome and exome aggregation consortium databases and is predicted to be pathogenic by both Scoring Intolerant from Tolerant (SIFT) and Mutation Taster. Both the parents were heterozygous for this variant ( Fig. 1 ). Prenatal diagnosis in the subsequent pregnancy revealed a carrier fetus ( Fig. 1 ) .
Fig. 1.
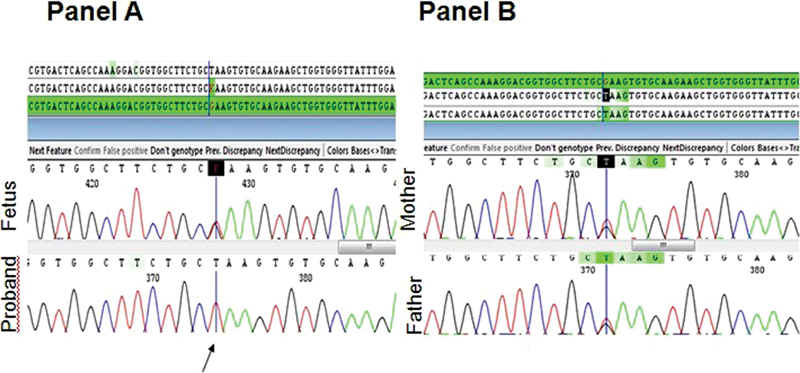
Sanger sequencing of the proband, sibling, and parents for PSAP c.1228 G > T. ( A ) Upper sequence depicts a heterozygous fetus, while the lower sequence depicts the affected proband. ( B ) Upper sequence (maternal) and the lower sequence (paternal) indicate carrier status of parents for PSAP c.1228 G > T.
Discussion
The finding presented in this article is the first reported case from India of an acute neuronal Gaucher like phenotype with the presence of cherry red spots in the retina due to saposin C deficiency. Sphingolipid activator proteins are needed for the degradation of glycosphingolipids. The PSAP post-translationally cleaves into four proteins—saposin A, B, C, and D. Saposin C is required for glucosylceramide degradation and its deficiency results in Gaucher disease. A systematic approach is essential to diagnose specific activator protein deficiency involving ophthalmological, biochemical, and molecular tools. The clinical and biochemical phenotype of our patient posed a challenge for diagnosis. The presence of “cherry red spots” is often a sign of infantile neurodegenerative disorder, such as a gangliosidosis or Niemann–Pick disease (rather than Gaucher disease). Elevation of chitotriosidase is particularly suggestive of either Gaucher disease or Niemann–Pick disease; however, the plasma level of chitotriosidase is elevated in many other lysosomal storage disorders. The degree of elevation is higher in Gaucher (40–326-fold) and Niemann–Pick A/B (7–22-fold) diseases. 17 In our patient, the chitotriosidase elevation was ∼11-fold compared with normal. The enzyme activities for these two disorders were normal, although the β-glucosidase 1 activity was at the lower limit of the normal. The nondeficient β-glucosidase 1 activity and remarkably elevated chitotriosidase level prompted us to conduct PSAP gene testing. A stop-gain mutation c.G1228T (p.Glu410ter) in exon 11 of the PSAP gene in the child confirmed the diagnosis of saposin C deficiency. This mutation is novel and pathogenic as it is not reported in 1000 genome and exome aggregation consortium databases and segregates in the family. The prediction of deleterious effect by SIFT and Mutation Taster and the patient's phenotype is highly specific for Gaucher disease and the variant is classified as pathogenic following the American College of Medical Genetics and Genomics/Association of Molecular Pathology sequence variant pathogenicity classification guidelines. The premature termination of the protein as a result of this mutation not only leads to saposin C deficiency but also to saposin D deficiency.
The importance of saposin D in the lysosomal degradation of ceramide was evident as its addition to the cultured fibroblasts of patients with deficiency of all saposins (PSAP deficiency) stimulated lysosomal degradation of ceramide in vivo. 18 Saposin D deficiency is likely to contribute toward ceramidosis. 19 Hence, a combined deficiency of saposin C and D together may favor the appearance of cherry red spots that was observed in our patient. White blood cell β-glucosidase 1 activity was very close to the lower limit of normal. It is known that in the absence of saposins, this enzyme has reduced activity, 20 particularly in combined saposin C and D deficiency. 21 Such combined deficiencies of saposin C and D were shown to contribute to a neuronopathic phenotype in mice with ataxia, kyphotic posturing, and hind limb paralysis; and cause loss of Purkinje cells. 21 The importance of exon 11 of PSAP was shown by a knock-in point mutation (cysteine-to-proline) in this exon in a mouse model, which resulted in weakness of the hind limbs, progressive ataxia, decreased motor activity, impaired hippocampal long-term potentiation, progressive loss of cerebral Purkinje cells, atrophy of cerebellar granule cells, and impaired axonal integrity due to inclusion bodies. 22 The proband in the current study had hypotonia of all limbs and recurrent seizures. The earlier reported case with combined deficiency of saposin C and D was a 15-year-old male who died with evidence of glucosylceramide storage in the spleen with normal GBA activity. 8 9
Figure 2 illustrates the mutation spectrum of saposin C-deficient cases reported to date with four homozygous mutants and three compound heterozygous mutants. Biochemical and functional characterization of five mutations, p.Cys315Ser, p.342_348FDKMCSKdel, p.Leu349Pro, p.Cys382Gly, and p.Cys382Phe, revealed that diverse aberrant disulfide bridge rearrangements in these mutants result in diminished half-life of saposin C and rapid degradation by autophagy. 23
Fig. 2.
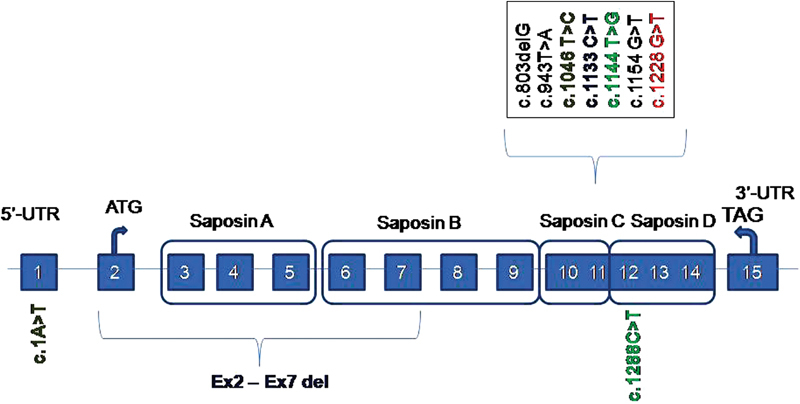
The spectrum of mutations reported in saposin C deficiency. Reported cases of saposin C deficiency homozygous for c.803delG, c.943T > A, c.1154G > T, c.1228 G > T (current) mutations and compound heterozygous cases with the following mutations: c.1A > T/c.1046 T > C; c.1133C > T/ex2-ex7 del; c.1144T > G/c.1288C > T.
Functional studies on two truncating PSAP mutations, that is, c.895G > T (p.Glu299*) and c.834_835delGA (p.Glu278Aspfs*27), revealed that these variants lead to complete lack of processed transcript. 14 As a result, globotriaosylsphingosine and glucosylsphingosine are elevated and autophagosomes accumulate due to a decreased autophagic flux. 24
At present, no specific treatment is available for saposin C deficiency. The expression and distribution of cathepsins were reported to be significantly altered in neuronopathic forms of Gaucher disease and other sphingolipidoses. 25 Ceramide is an activator of cathepsin D, leading to its proteolytic activation in the lysosomes and thus contributing to proapoptotic functions. 26 Saposin C-deficient fibroblasts were shown to exhibit delayed degradation of autolysosomes due to reduced levels and activities of cathepsins B and D, while glucosylceramide-deficient fibroblasts have normal level and activity of cathepsin D; and double the activity of cathepsin B. 27 Hence, restoration of cathepsin B and D was shown to facilitate complete recovery of autophagic flux in saposin C-deficient fibroblasts and maintain lysosome homeostasis 28 . In vitro studies showed that synthetic saposin C increased β-glucosidase activity. 29 Further studies are required to justify its utility as an enzyme replacement therapy in Gaucher disease. Saposin C serves as an activator, stabilizer, and protector of glucocerebrosidase activity. 29
In conclusion, we report a case of specific activator deficiency with a biallelic PSAP mutation from India adding to the very limited number of individuals reported with this disorder. The cherry red spots observed in our patient are often a sign of an infantile neurodegenerative disorder, such as a gangliosidosis or Niemann–Pick disease (rather than Gaucher disease). The observed elevation of chitotriosidase was suggestive of Gaucher disease and Niemann–Pick disease. Specific lysosomal enzyme assays were normal for Niemann–Pick disease type A and low normal for Gaucher disease. The diagnosis of PSAP -related disease was made using whole exome analysis. Future studies are warranted to investigate whether this mutation induces disulfide bridge rearrangement in saposins and thus influences the half-life of saposin C and saposin D. The diagnosis of specific activator deficiency should be considered and tested for when Gaucher disease is suspected in an infant with elevated chitotriosidase and partially deficient GBA activity.
Footnotes
Conflict of Interest None declared.
References
- 1.Razek A AKA, Abdalla A, Barakat T, El-Taher H, Ali K. Assessment of the liver and spleen in children with Gaucher disease type I with diffusion-weighted MR imaging. Blood Cells Mol Dis. 2018;68:139–142. doi: 10.1016/j.bcmd.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Razek A A, Abdalla A, Gaber N A et al. Proton MR Spectroscopy of the brain in children with neuronopathic Gaucher's disease. Eur Radiol. 2013;23(11):3005–3011. doi: 10.1007/s00330-013-2924-9. [DOI] [PubMed] [Google Scholar]
- 3.Razek A A, Abdalla A, Fathy A, Megahed A. Apparent diffusion coefficient of the vertebral bone marrow in children with Gaucher's disease type I and III. Skeletal Radiol. 2013;42(02):283–287. doi: 10.1007/s00256-012-1464-8. [DOI] [PubMed] [Google Scholar]
- 4.Razek A AKA, Abdalla A, Barakat T, El-Taher H, Ali K. Multi-parametric MR imaging using apparent diffusion coefficient and fat fraction in quantification of bone marrow in pediatrics with Gaucher disease. Clin Imaging. 2018;51:318–322. doi: 10.1016/j.clinimag.2018.06.011. [DOI] [PubMed] [Google Scholar]
- 5.Abdel Razek A A, Abd El-Gaber N, Abdalla A, Fathy A, Azab A, Rahman A A. Apparent diffusion coefficient vale of the brain in patients with Gaucher's disease type II and type III. Neuroradiology. 2009;51(11):773–779. doi: 10.1007/s00234-009-0548-1. [DOI] [PubMed] [Google Scholar]
- 6.Cormand B, Montfort M, Chabás A, Vilageliu L, Grinberg D. Genetic fine localization of the beta-glucocerebrosidase (GBA) and prosaposin (PSAP) genes: implications for Gaucher disease. Hum Genet. 1997;100(01):75–79. doi: 10.1007/s004390050468. [DOI] [PubMed] [Google Scholar]
- 7.Schnabel D, Schröder M, Sandhoff K. Mutation in the sphingolipid activator protein 2 in a patient with a variant of Gaucher disease. FEBS Lett. 1991;284(01):57–59. doi: 10.1016/0014-5793(91)80760-z. [DOI] [PubMed] [Google Scholar]
- 8.Rafi M A, de Gala G, Zhang X L, Wenger D A. Mutational analysis in a patient with a variant form of Gaucher disease caused by SAP-2 deficiency. Somat Cell Mol Genet. 1993;19(01):1–7. doi: 10.1007/BF01233949. [DOI] [PubMed] [Google Scholar]
- 9.Diaz-Font A, Cormand B, Santamaria R, Vilageliu L, Grinberg D, Chabás A.A mutation within the saposin D domain in a Gaucher disease patient with normal glucocerebrosidase activity Hum Genet 2005117(2–3):275–277. [DOI] [PubMed] [Google Scholar]
- 10.Hulková H, Cervenková M, Ledvinová J et al. A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum Mol Genet. 2001;10(09):927–940. doi: 10.1093/hmg/10.9.927. [DOI] [PubMed] [Google Scholar]
- 11.Tylki-Szymańska A, Czartoryska B, Vanier M T et al. Non-neuronopathic Gaucher disease due to saposin C deficiency. Clin Genet. 2007;72(06):538–542. doi: 10.1111/j.1399-0004.2007.00899.x. [DOI] [PubMed] [Google Scholar]
- 12.Vaccaro A M, Motta M, Tatti M et al. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum Mol Genet. 2010;19(15):2987–2997. doi: 10.1093/hmg/ddq204. [DOI] [PubMed] [Google Scholar]
- 13.Kang L, Zhan X, Ye J et al. A rare form of Gaucher disease resulting from saposin C deficiency. Blood Cells Mol Dis. 2018;68:60–65. doi: 10.1016/j.bcmd.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Guo Y, He W, Boer A M et al. Elevated plasma chitotriosidase activity in various lysosomal storage disorders. J Inherit Metab Dis. 1995;18(06):717–722. doi: 10.1007/BF02436762. [DOI] [PubMed] [Google Scholar]
- 15.Daniels L B, Glew R H, Diven W F, Lee R E, Radin N S. An improved fluorometric leukocyte β-glucosidase assay for Gaucher's disease. Clin Chim Acta. 1981;115(03):369–375. doi: 10.1016/0009-8981(81)90251-5. [DOI] [PubMed] [Google Scholar]
- 16.van Diggelen O P, Voznyi Y V, Keulemans J LM et al. A new fluorimetric enzyme assay for the diagnosis of Niemann-Pick A/B, with specificity of natural sphingomyelinase substrate. J Inherit Metab Dis. 2005;28(05):733–741. doi: 10.1007/s10545-005-0105-y. [DOI] [PubMed] [Google Scholar]
- 17.Kadali S, Kolusu A, Sunkara S, Gummadi M R, Undamatla J. Clinical evaluation of chitotriosidase enzyme activity in Gaucher and Niemann Pick A/B diseases: a retrospective study from India. Clin Chim Acta. 2016;457:8–11. doi: 10.1016/j.cca.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Klein A, Henseler M, Klein C, Suzuki K, Harzer K, Sandhoff K. Sphingolipid activator protein D (sap-D) stimulates the lysosomal degradation of ceramide in vivo. Biochem Biophys Res Commun. 1994;200(03):1440–1448. doi: 10.1006/bbrc.1994.1612. [DOI] [PubMed] [Google Scholar]
- 19.Matsuda J, Kido M, Tadano-Aritomi K et al. Mutation in saposin D domain of sphingolipid activator protein gene causes urinary system defects and cerebellar Purkinje cell degeneration with accumulation of hydroxy fatty acid-containing ceramide in mouse. Hum Mol Genet. 2004;13(21):2709–2723. doi: 10.1093/hmg/ddh281. [DOI] [PubMed] [Google Scholar]
- 20.Bradová V, Smíd F, Ulrich-Bott B, Roggendorf W, Paton B C, Harzer K. Prosaposin deficiency: further characterization of the sphingolipid activator protein-deficient sibs. Multiple glycolipid elevations (including lactosylceramidosis), partial enzyme deficiencies and ultrastructure of the skin in this generalized sphingolipid storage disease. Hum Genet. 1993;92(02):143–152. doi: 10.1007/BF00219682. [DOI] [PubMed] [Google Scholar]
- 21.Sun Y, Witte D P, Zamzow M et al. Combined saposin C and D deficiencies in mice lead to a neuronopathic phenotype, glucosylceramide and alpha-hydroxy ceramide accumulation, and altered prosaposin trafficking. Hum Mol Genet. 2007;16(08):957–971. doi: 10.1093/hmg/ddm040. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y, Ran H, Zamzow M et al. Specific saposin C deficiency: CNS impairment and acid beta-glucosidase effects in the mouse. Hum Mol Genet. 2010;19(04):634–647. doi: 10.1093/hmg/ddp531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motta M, Camerini S, Tatti M et al. Gaucher disease due to saposin C deficiency is an inherited lysosomal disease caused by rapidly degraded mutant proteins. Hum Mol Genet. 2014;23(21):5814–5826. doi: 10.1093/hmg/ddu299. [DOI] [PubMed] [Google Scholar]
- 24.Motta M, Tatti M, Furlan F et al. Clinical, biochemical and molecular characterization of prosaposin deficiency. Clin Genet. 2016;90(03):220–229. doi: 10.1111/cge.12753. [DOI] [PubMed] [Google Scholar]
- 25.Vitner E B, Dekel H, Zigdon H et al. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum Mol Genet. 2010;19(18):3583–3590. doi: 10.1093/hmg/ddq273. [DOI] [PubMed] [Google Scholar]
- 26.Samarani M, Loberto N, Soldà G et al. A lysosome-plasma membrane-sphingolipid axis linking lysosomal storage to cell growth arrest. FASEB J. 2018;32(10):5685–5702. doi: 10.1096/fj.201701512RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tatti M, Motta M, Di Bartolomeo S et al. Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts. Hum Mol Genet. 2012;21(23):5159–5173. doi: 10.1093/hmg/dds367. [DOI] [PubMed] [Google Scholar]
- 28.Tatti M, Motta M, Di Bartolomeo S, Cianfanelli V, Salvioli R. Cathepsin-mediated regulation of autophagy in saposin C deficiency. Autophagy. 2013;9(02):241–243. doi: 10.4161/auto.22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoneshige A, Muto M, Watanabe T, Hojo H, Matsuda J.The effects of chemically synthesized saposin C on glucosylceramide-β-glucosidase Clin Biochem 201548(16–17):1177–1180. [DOI] [PubMed] [Google Scholar]