

Abstract
The kallikrein/kinin system, an intravascular biochemical pathway that includes several proteins involved in the contact activation system of coagulation, renin-angiotensin activation, and inflammation, may or may not play a role in venous thromboembolism (VTE) occurrence. Within a large prospective population-based study in the United States, we conducted a nested case-cohort study to test the hypothesis that higher plasma levels of high molecular weight kininogen (HK) or prekallikrein are associated with greater VTE incidence. We related baseline ELISA measures of HK and prekallikrein in 1993–95 to incidence VTE of the lower extremity (n=612) through 2015 (mean follow-up = 18 years). We found no evidence that plasma HK or prekallikrein was associated positively with incident VTE. HK, in fact, was associated inversely and significantly with VTE in most proportional hazards regression models. For example, the hazard ratio of VTE per standard deviation higher HK concentration was 0.88 (95% confidence interval = 0.81, 0.97), after adjustment for several VTE risk factors. Our findings suggest that plasma levels of these factors do not determine the risk of VTE in the general population.
Keywords: high molecular weight kininogen, prekallikrein, prospective study, pulmonary embolus, venous thrombosis
Introduction
Aberrant hemostasis pathways play key roles in the etiology of deep vein thrombosis (DVT) and pulmonary embolism (PE) (or, together, venous thromboembolism--VTE). Genomic studies in the general population consistently show that common and low-frequency variants in hemostasis pathway genes are associated with VTE risk.1 Likewise, epidemiological studies have demonstrated that higher plasma levels of coagulation markers, such as factors VIII and XI, von Willebrand factor, and D-dimer, and shorter activated partial thromboplastin time (aPTT), are associated with increased incidence of VTE.2,3
The kallikrein/kinin system, an intravascular biochemical pathway that includes several proteins involved in the contact activation system of coagulation, renin-angiotensin activation, and inflammation,4 also might play a role in VTE occurrence. High molecular weight kininogen (HK), produced by the gene KNG1, serves as a cofactor of factor XIIa in the activation of prekallikrein to kallikrein, an activation that is also stimulated by prolylcarboxypeptidase. Kallikrein cleaves HK to liberate bradykinin, which binds to receptors that modulate vascular permeability and vasodilation. In the contact activation system, HK helps accelerate factor XIIa formation, which activates factor XI in the coagulation cascade. Kallikrein also helps activate plasminogen to plasmin in the fibrinolytic system.
Although the contact system is essential for in vitro surface-initiated coagulation, as exemplified by the aPTT, the degree to which the contact system has a role in initiating physiologic in vivo coagulation or in the etiology of VTE is unclear. Yet, growing experimental evidence suggests that the contact system may make a contribution to thrombosis.5 Knockout mice deficient in factor XII, prekallikrein, or HK are protected against artificially induced thrombosis.6–8 Agents inhibiting the contact pathway also can prevent thrombosis in animals in both venous and arterial vascular beds.9
Genomic studies have consistently linked a functional KNG1 missense variant (rs710446) to VTE occurrence1 and to the aPTT.10 This suggests that the kallikrein/kinin system may be involved in VTE through contact activation or the kallikrein/kinin system’s other effects. Although higher plasma levels of prekallikrein have been associated with greater arterial cardiovascular disease in a few studies,11,12 to our knowledge, only one retrospective study has examined the association of plasma HK and prekallikrein with VTE. It found that mean HK was higher in VTE patients than in controls, whereas mean prekallikrein was not different.13 However, the VTE patients studied were taking vitamin K antagonists, which might have influenced results.
To more fully test the hypothesis that higher plasma levels of HK or prekallikrein are associated with greater VTE incidence in the general population, we conducted a nested case-cohort study within the prospective Atherosclerosis Risk in Communities (ARIC) cohort.
Methods
Study design and sample
We reported the overall ARIC study design, methods, and VTE incidence rates in detail elsewhere.14,15 In brief, 15,792 men and women aged 45 to 64 years enrolled in the ARIC study in 1987–1989 (visit 1). ARIC performed subsequent examinations in 1990–92 (visit 2), 1993–95 (visit 3), 1996–98, 2011–13, 2016–2017, as well as annual or semi-annual telephone contact. At each visit, risk factors were measured, and plasma was obtained and stored at −80 degrees C. The institutional review committees at each study center approved the methods, and ARIC staff obtained informed participant consent.
For this report of the association of HK and prekallikrein with VTE, we employed a nested case-cohort study within the ARIC cohort.16 This is a prospective study design in which the baseline biomarker concentrations are measured and compared between all cohort members who develop VTE and a subcohort selected randomly from the entire cohort. ARIC visit 3 (1993–95) served as the baseline, because citrated plasma from the previous two ARIC visits was in short supply. Following the case-cohort method, we included the incident VTE cases that occurred in the full ARIC cohort between the 1993–95 visit and 2015, along with a subcohort selected as a simple random sample from the full ARIC cohort examined at visit 3. We then measured plasma HK and prekallikrein in stored visit 3 samples from the cases and the subcohort sample.
Figure 1 summarizes our case-cohort study design and exclusions. Of the 12,887 ARIC cohort participants examined at visit 3, 12,825 had citrate plasma samples remaining for assay in 2018. Among these, we selected for assay all participants who had developed VTE in ARIC and 4,195 for the random sample subcohort. The laboratory pulled the visit 3 citrate samples and performed the assays, but 32 participants had insufficient plasma to assay either HK or prekallikrein. From those with results, we excluded 148 whose first VTE occurred before visit 3 (because of our interest in incident VTE after visit 3), 13 who were of race groups other than black or white, 443 who had a history of cancer prior to visit 3, 43 who were taking anticoagulants at visit 3, and 110 missing information on visit 3 covariates used in modeling. This left for analysis 612 incident VTE cases and a subcohort sample of 3,530.
Figure 1.
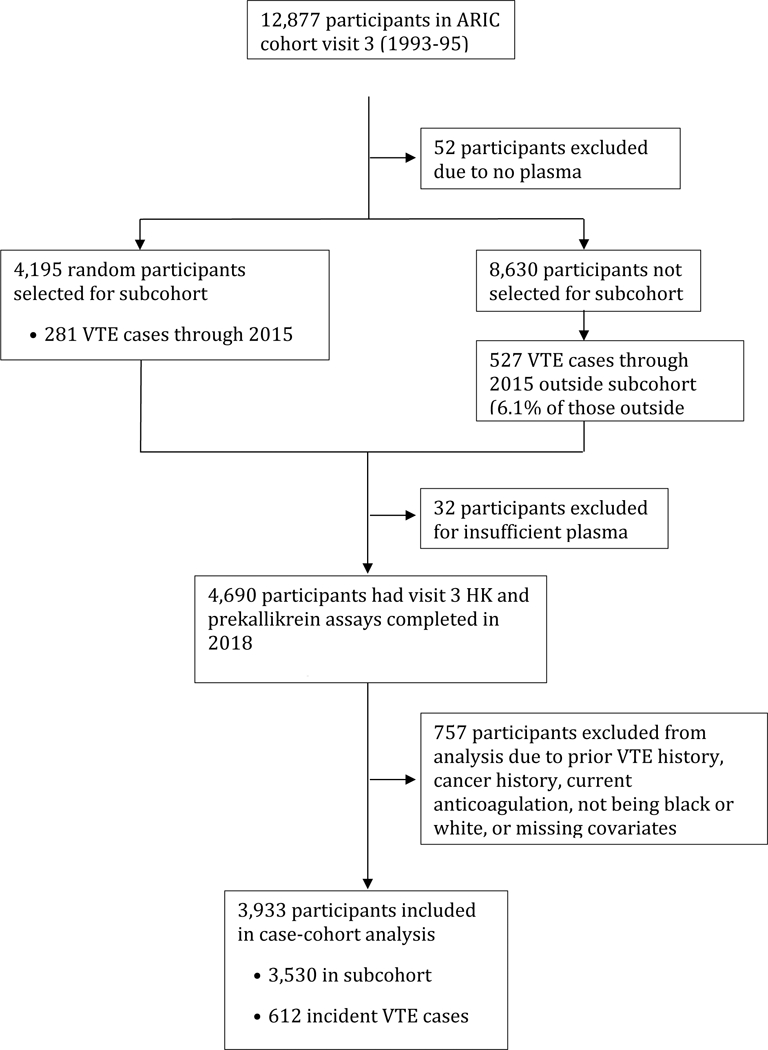
Nested case-cohort study design and exclusions, ARIC. Abbreviations: ARIC = Atherosclerosis Risk in Communities; HK = high molecular weight kininogen; VTE = venous thromboembolism.
VTE occurrence
ARIC staff contacted participants annually or semi-annually by telephone and asked about all hospitalizations in the previous year. Staff then retrieved hospital records for possible VTE events through 2015. To validate VTE events, two physicians reviewed the records using standardized criteria,15 requiring positive imaging tests for diagnosis of DVT and PE. The reviewers sub-classified VTEs as unprovoked (no obvious cause) or provoked (associated with cancer, major trauma, surgery, marked immobility). For this report, we restricted DVTs to those in the lower extremity or vena cava, because upper extremity DVTs were relatively few and almost always the result of venous catheters.
Measurement of hemostatic factors at ARIC visit 3
The Laboratory for Clinical Biochemistry Research at the University of Vermont measured several hemostatic factors. In 2014, the laboratory thawed a citrated plasma sample from 1993–95 twice to measure factor XI and D-dimer, which proved to be VTE risk factors.3,17 The assay for factor XI was a sandwich ELISA using affinity-purified polyclonal antibodies from Affinity Biologicals (Ancaster, Ontario, CAN), with a coefficient of variation for control samples of 9.6%. D-dimer was measured with an immuno-turbidimetric assay (Liatest D-DI; Diagnostica Stago, Parsippany, NJ), with an analytical coefficient of variation of 4–16%. Blind analysis of 73 pairs of ARIC samples split at the time of blood draw and stored until 2014 yielded an intra-class reliability coefficients of 0.81 for factor XI and 0.92 for D-dimer.
In 2018, the laboratory thawed the samples again for measurement of HK (third thaw) and prekallikrein (fourth thaw). Pilot studies demonstrated that prekallikrein was stable across multiple thaws (mean value for one to four thaws in four samples: 18.8, 20.6, 18.4, and 20.1 μg/mL). In contrast, HK declined 16% across four repeated thaws (mean value for one to four thaws in six samples: 29.4, 26.2, 23.5, and 24.8 μg/mL), but we proceeded with analysis on the assumption that the thaw effect would be uniform across samples.
The laboratory used a single reagent lot for prekallikrein and HK measurements in this report. The assay for prekallikrein was an ELISA using monoclonal antibodies from LifeSpan Biosciences (Seattle, WA) with an average coefficient of variation for control samples of 8.9%. The measurement of HK used the Simple Step ELISA® from Abcam (Eugene, OR), which required a high dilution (1:40,000) and produced an average CV for HK of 16.9%. The Abcam kit suggests a “normal range” for HK based on 10 individuals of 42.8–69.5 μg/mL. This is lower than an average normal value based on 10 subjects of 80 μg/mL, reported by Schmaier et al.18
As a further check on repeatability, ARIC staff split a number of EDTA specimens at the time of blood draw, processed the split samples separately, and froze them. When the laboratory analyzed 93 paired, similarly thawed samples randomly and blindly in the batches for this study, the reliability (intraclass correlation) coefficients were r=0.56 for HK and r=0.49 for prekallikrein. The Pearson correlation for prekallikrein reliability was r=0.60.
Measurement of other VTE risk factors
Except where noted, our analysis included VTE risk factors documented previously in ARIC and measured at ARIC visit 3. Participants reported race, smoking status, use of hormone replacement therapy (HRT), use of blood pressure medication, and history of cancer. ARIC staff measured sitting blood pressure thrice after a 5-minute rest via a random-zero sphygmomanometer, and we used the mean of the last two measures. Staff measured weight and height. We defined diabetes as a fasting blood glucose of 126 mg/dl or higher, non-fasting blood glucose of 200 mg/dl or higher, a self-reported physician diagnosis of diabetes, or use of antidiabetic medication in the past 2 weeks. ARIC isolated genomic DNA and measured five key variants important for VTE: F5 Leiden rs6025, F2 rs1799963, ABO rs8176719 (O vs. non-O groups), FGG rs2066865, and F11 rs2036914, and a weighted genetic risk score was created, as previously reported.19 ARIC also assessed the KNG1 missense variant (rs710446), previously associated in genomic consortia with VTE.1
At visit 1, ARIC had measured aPTT, factor VIII, von Willebrand factor, and protein C as previously described.20,21 At visit 2, ARIC estimated glomerular filtration rate (eGFR) from creatinine using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) algorithm.22 These were unavailable at ARIC visit 3.
Statistical methods
We used SAS (Statistical Analysis System, SAS Institute, Cary, NC) for analysis and accounted for the case-cohort design. We evaluated potential confounding variables by examining means or frequencies of VTE risk factors across quartiles of HK and prekallikrein in the subcohort random sample. We performed proportional hazards regressions to estimate the hazard ratio (HR) of VTE in relation to HK or prekallikrein (quartiles or continuous), employing Barlow’s method for variance estimation for the case-cohort design,23 which takes into account some VTE cases being in the subcohort and some not. Follow-up for VTE began at ARIC visit 3 and went until the date of first VTE occurrence, loss to follow-up, death, or else December 31, 2015. Model 1 adjusted for age, race, and sex. Model 2, our main model, adjusted for age, race, HRT-sex (women-current user, women-nonuser, women-unknown status, men), weight (continuous), height (continuous), diabetes (yes/no), smoking status (current, former, never), systolic BP (continuous), BP medications (yes no), and visit 2 eGFR (continuous). In supplementary analyses, Model 3 added visit 3 hemostatic factors (factor XI and D-dimer) and a weighted risk score comprising key genetic variants (F5, F2, ABO, F11, FGG), and Model 4 added ARIC visit 1 hemostatic factors (fVIII, VWF, aPTT, protein C). We also generated restricted cubic splines with knots at the 5th, 27.5th, 50th, 72.5th and 95th percentiles to explore the shape of the association of HK and prekallikrein with VTE risk after adjustment for age, race, and sex.
Results
Correlates of HK and prekallikrein
In the subcohort sample of ARIC participants in 1993–95 (visit 3), the mean value for plasma HK was 23.6 μg/mL (SD, 6.4) and for prekallikrein was 15.1 μg/mL (SD, 9.1). The Spearman correlation between the two was r = 0.30.
As shown in Table 1, higher HK was associated most strikingly in the subcohort sample with the following variables: female sex; greater prevalences of diabetes, HRT use, never smoking, and hypertensive medication use; and higher levels of Factor XI (with the Spearman correlation between HK and Factor XI being r = 0.34). Higher plasma prekallikrein demonstrated many of these associations with potential confounding variables (Table 2), but was also more common in whites than blacks. The Spearman correlation between prekallikrein and Factor XI was r = 0.32. In addition, both higher HK and prekallikrein in 1993–95 were associated with higher levels of Factor VIII and shorter aPTT at ARIC visit 1 in 1987–89.
Table 1.
Baseline venous thromboembolism risk factor levels (mean or %) by quartiles of plasma high molecular weight kininogen (HK), ARIC subcohort random sample, 1993−95
| Quartiles |
||||
|---|---|---|---|---|
| Q1 | Q2 | Q3 | Q4 | |
| n | 871 | 870 | 887 | 902 |
| HK range, μg/mL | 2.08−19.11 | 19.12−22.66 | 22.67−26.76 | 26.77−58.57 |
| Age, years | 60 | 60 | 60 | 59 |
| Sex, % | ||||
| Male | 54 | 47 | 44 | 34 |
| Female | 46 | 53 | 56 | 66 |
| Race, % | ||||
| White | 76 | 78 | 78 | 77 |
| African American | 24 | 22 | 22 | 23 |
| Height, cm | 170 | 169 | 168 | 167 |
| Weight, kg | 81 | 81 | 80 | 81 |
| Diabetes, % | 12 | 12 | 17 | 23 |
| Hormone replacement therapy, % (females only) | 14 | 17 | 18 | 21 |
| Smoking status, % | ||||
| Current | 20 | 17 | 19 | 16 |
| Former | 40 | 43 | 41 | 40 |
| Never | 40 | 40 | 40 | 44 |
| Systolic blood pressure, mmHg | 124 | 124 | 125 | 123 |
| Antihypertensive medication, % | 35 | 33 | 37 | 43 |
| Estimated glomerular filtration rate, mL/min/1.73 m2 2 | 97 | 96 | 97 | 97 |
| Factor XI, % | 102 | 111 | 117 | 125 |
| D-Dimer, μg/mL1 | 0.49 | 0.43 | 0.46 | 0.43 |
| Activated partial thromboplastin time, seconds1,3 | 29.6 | 29.2 | 28.9 | 28.5 |
| Factor VIII, %1,3 | 125 | 128 | 128 | 134 |
| Protein C, μg/mL1,3 | 3.0 | 3.1 | 3.2 | 3.3 |
| Von Willebrand factor, %1,3 | 114 | 113 | 114 | 117 |
| Weighted genetic risk score (F5, F2, ABO, F11, FGG variants)1 | 1.38 | 1.44 | 1.44 | 1.52 |
Sample size modestly smaller due to additional missing data.
From ARIC visit 2, 1990−92.
From ARIC visit 1, 1987−89.
Abbreviations: ARIC = Atherosclerosis Risk in Communities; HK = high molecular weight kininogen
Table 2.
Baseline venous thromboembolism risk factor levels (mean or %) by quartiles of plasma prekallikrein, ARIC subcohort random sample, 1993–95
| Quartiles |
||||
|---|---|---|---|---|
| Q1 | Q2 | Q3 | Q4 | |
| n | 873 | 898 | 872 | 887 |
| Prekallikrein range, μg/mL | 2.08−10.59 | 10.60−12.80 | 12.81−15.70 | 15.71−95.79 |
| Age, years | 60 | 60 | 60 | 59 |
| Sex, % | ||||
| Male | 63 | 56 | 37 | 24 |
| Female | 37 | 44 | 63 | 76 |
| Race, % | ||||
| White | 67 | 78 | 83 | 80 |
| African American | 33 | 22 | 17 | 20 |
| Height, cm | 171 | 170 | 167 | 165 |
| Weight, kg | 81 | 82 | 80 | 79 |
| Diabetes, % | 13 | 16 | 17 | 18 |
| Hormone replacement therapy, % (females only) | 8 | 10 | 18 | 35 |
| Smoking status, % | ||||
| Current | 24 | 19 | 16 | 12 |
| Former | 43 | 42 | 39 | 40 |
| Never | 33 | 38 | 46 | 48 |
| Systolic blood pressure, mmHg | 125 | 123 | 124 | 125 |
| Antihypertensive medication, % | 37 | 34 | 37 | 41 |
| Estimated glomerular filtration rate, mL/min/1.73 m2 2 | 98 | 96 | 97 | 97 |
| Factor XI, % | 103 | 110 | 118 | 124 |
| D-Dimer, μg/mL1 | 0.49 | 0.42 | 0.37 | 0.54 |
| Activated partial thromboplastin time, seconds1,3 | 29.9 | 29.2 | 28.9 | 28.1 |
| Factor VIII, %1,3 | 129 | 126 | 129 | 132 |
| Protein C, μg/mL1,3 | 3.0 | 3.1 | 3.2 | 3.3 |
| Von Willebrand factor, %1,3 | 119 | 113 | 113 | 113 |
| Weighted genetic risk score (F5, F2, ABO, F11, FGG variants)1 | 1.43 | 1.42 | 1.46 | 1.48 |
Sample size modestly smaller due to additional missing data.
From ARIC visit 2, 1990−92.
From ARIC visit 1, 1987−89.
Abbreviations: ARIC = Atherosclerosis Risk in Communities
Greater numbers of risk alleles for KNG1 rs710446 were associated, as expected, with higher plasma concentrations of HK and prekallikrein in the subcohort sample. Participants with 0, 1, or 2 risk alleles had mean HK concentrations of 22.3, 23.9, and 25.1 μg/mL and mean prekallikrein of 14.1, 15.1, and 16.6 μg/mL, respectively.
Associations of HK and prekallikrein with incident VTE
Over a mean of 18 years of follow-up (maximum 23 years), 612 participants developed VTE, for a crude incidence rate of 3 per 1000 person-years. Weighted for the case-cohort sampling, we found no evidence that plasma HK or prekallikrein was associated positively with incident VTE (Figure 2, Table 3). HK, in fact, was associated inversely and significantly with VTE in most models (Figure 2, Table 3). These conclusions were unchanged by restricting follow-up for VTE to a maximum of 10 years after HK and prekallikrein measurement: the Model 2 hazard ratio for VTE per standard deviation increment of HK being 0.88 (95% CI 0.81–0.97) for full follow-up (Table 3) versus 0.82 (0.69–0.97) for the first 10 years of follow-up (data not shown). Tests of multiplicative interactions of HK or prekallikrein with sex, HRT, factor XI, and D-dimer in relation to VTE were non-significant (p.0.05, data not shown).
Figure 2.
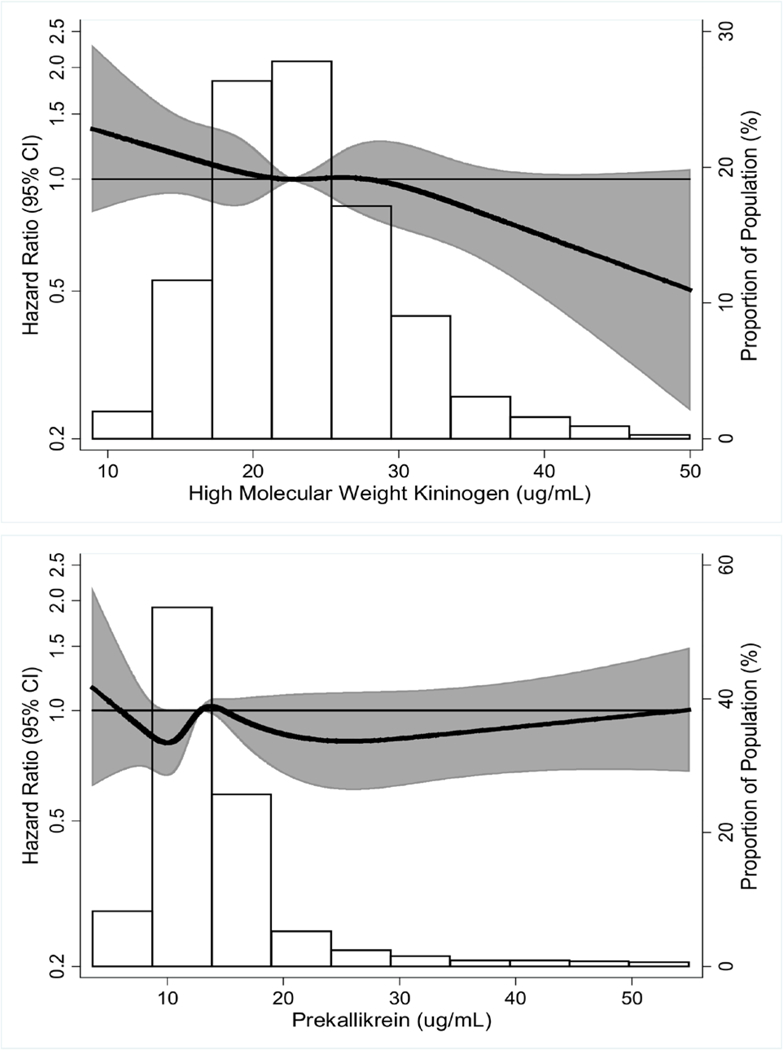
Hazard ratio of venous thromboembolism in relation to plasma high molecular weight kininogen (top) and prekallikrein (bottom).
Footnote: Hazard ratios (dark line) and 95 percent confidence interval (shaded area) were derived from restricted cubic spline models with knots at the 5th, 27.5th, 50th, 72.5th, and 95th percentiles of the distributions (histograms). For ease of presentation, we omitted depiction of the curve at extreme values: HK <5 μg/mL (n=2) or >50 μg/mL (n=12) and prekallikrein <3 μg/mL (n=3) or >55 μg/mL (n=45).
Table 3.
Hazard ratios (95% confidence intervals) of venous thromboembolism (VTE) by quartiles or SD increment of high molecular weight kininogen and prekallikrein, ARIC, 1993–95 through 2015
| Quartiles |
|||||
|---|---|---|---|---|---|
| Q1 | Q2 | Q3 | Q4 | Per SD increment2 |
|
| High molecular weight kininogen, μg/mL | 2.08−19.11 | 19.12−22.66 | 22.67−26.76 | 26.77−58.57 | |
| n at risk | 983 | 983 | 984 | 983 | 3,933 |
| Incident VTE, n | 172 | 161 | 141 | 138 | 612 |
| Model 1 HR (95% CI) | 1 (Reference) |
0.91 (0.72−1.15) | 0.85 (0.67−1.08) | 0.79 (0.62−1.01) | 0.89 (0.82−0.97) |
| Model 2 HR (95% CI) | 1 (Reference) |
0.90 (0.71−1.14) | 0.84 (0.66−1.07) | 0.77 (0.60−0.99) | 0.88 (0.81−0.97) |
| Model 3 HR (95% CI)1 | 1 (Reference) |
0.93 (0.72−1.21) | 0.79 (0.60−1.04) | 0.68 (0.51−0.90) | 0.83 (0.75−0.92) |
| Model 4 HR (95% CI)1 | 1 (Reference) |
0.93 (0.71−1.21) | 0.80 (0.61−1.06) | 0.67 (0.50−0.90) | 0.83 (0.75−0.92) |
| Prekallikrein, μg/mL | 2.08−10.59 | 10.60−12.80 | 12.81−15.70 | 15.71−95.79 | |
| n at risk+ | 983 | 983 | 985 | 982 | 3,933 |
| Incident VTE, n | 156 | 147 | 163 | 146 | 612 |
| Model 1 HR (95% CI) | 1 (Reference) |
0.96 (0.75−1.22) | 1.17 (0.92−1.49) | 0.99 (0.77−1.27) | 1.00 (0.91−1.09) |
| Model 2 HR (95% CI) | 1 (Reference) |
0.97 (0.76−1.24) | 1.14 (0.89−1.46) | 0.95 (0.73−1.23) | 0.99 (0.91−1.09) |
| Model 3 HR (95% CI)1 | 1 (Reference) |
1.03 (0.78−1.36) | 1.20 (0.91−1.59) | 1.05 (0.79−1.41) | 0.95 (0.86−1.06) |
| Model 4 HR (95% CI)1 | 1 (Reference) |
1.03 (0.78−1.36) | 1.18 (0.89−1.57) | 1.04 (0.78−1.40) | 0.95 (0.85−1.05) |
Model 1: Adjusted for age, race, and sex
Model 2: Adjusted for age, race, sex-hormone replacement therapy, weight, height, diabetes, smoking status, systolic blood pressure, antihypertensive medications, and estimated glomerular filtration rate
Model 3: Model 2 + adjustment for factor XI, log(D-dimer), and weighted genetic risk score (F5, F2, ABO, F11, FGG variants).
Model 4: Model 3 + adjustment for factor VIII, activated partial thromboplastin time, protein C, and Von Willebrand factor.
Models 3 and 4 have a reduced sample size of 3,352 at risk and 514 VTE events due to missing values.
Standard deviation increments were 6.4 μg/mL for HK and 9.1 μg/mL for prekallikrein.
Abbreviations: ARIC = Atherosclerosis Risk in Communities; CI = confidence interval; HK = high molecular weight kininogen; HR = hazard ratio; SD= standard deviation; VTE = venous thromboembolism
Examining the outcomes of DVT or PE separately (Table 4) or provoked versus unprovoked VTE (Table 5), there still was no positive association with either plasma marker. Rather, HK was inversely associated with all VTE outcomes—statistically significantly so for DVT alone and provoked VTE.
Table 4.
Hazard ratios (95% confidence intervals) of deep vein thrombosis (DVT) alone and pulmonary embolism (PE, with or without DVT) by quartiles or SD increment of high molecular weight kininogen and prekallikrein, ARIC, 1993–95 through 2015
| Quartiles |
|||||
|---|---|---|---|---|---|
| Q1 | Q2 | Q3 | Q4 | Per SD increment2 |
|
| High molecular weight kininogen, μg/mL | 2.08–19.11 | 19.12–22.66 | 22.67–26.76 | 26.77–58.57 | |
| n at risk1 | 983 | 983 | 984 | 983 | 3,933 |
| Incident DVT, n | 89 | 80 | 56 | 60 | 285 |
| Model 2 HR (95% CI) | 1 (Reference) |
0.87 (0.63–1.20) | 0.66 (0.46–0.93) | 0.67 (0.47–0.94) | 0.81 (0.71–0.93) |
| Model 3 HR (95% CI) | 1 (Reference) |
0.89 (0.63–1.27) | 0.61 (0.41–0.92) | 0.58 (0.39–0.87) | 0.76 (0.65–0.89) |
| Incident PE, n | 83 | 81 | 85 | 78 | 327 |
| Model 2 HR (95% CI) | 1 (Reference) |
0.92 (0.67–1.27) | 1.03 (0.75–1.41) | 0.88 (0.63–1.22) | 0.95 (0.85–1.06) |
| Model 3 HR (95% CI) | 1 (Reference) |
0.96 (0.68–1.37) | 0.97 (0.68–1.39) | 0.78 (0.53–1.14) | 0.89 (0.79–1.01) |
| Prekallikrein, μg/mL | 2.08–10.59 | 10.60–12.80 | 12.81–15.70 | 15.71–95.79 | |
| n at risk1 | 983 | 983 | 985 | 982 | 3,933 |
| Incident DVT, n | 84 | 67 | 76 | 58 | 285 |
| Model 2 HR (95% CI) | 1 (Reference) |
0.86 (0.62–1.20) | 1.10 (0.78–1.54) | 0.81 (0.56–1.17) | 0.92 (0.78–1.08) |
| Model 3 HR (95% CI) | 1 (Reference) |
0.97 (0.66–1.40) | 1.24 (0.84–1.81) | 0.89 (0.59–1.33) | 0.92 (0.78–1.09) |
| Incident PE, n | 72 | 80 | 87 | 88 | 327 |
| Model 2 HR (95% CI) | 1 (Reference) |
1.10 (0.78–1.54) | 1.19 (0.86–1.66) | 1.10 (0.78–1.54) | 1.04 (0.93–1.16) |
| Model 3 HR (95% CI) | 1 (Reference) |
1.11 (0.76–1.62) | 1.18 (0.80–1.73) | 1.19 (0.81–1.76) | 0.97 (0.86–1.09) |
Model 2: Adjusted for age, race, sex-hormone replacement therapy, weight, height, diabetes, smoking status, systolic blood pressure, antihypertensive medications, and estimated glomerular filtration rate
Model 3: Model 2 + adjustment for factor XI, log(D-dimer), and weighted genetic risk score (F5, F2, ABO, F11, FGG variants).
Model 3 has a reduced sample size due to missing values (see Table 3). Model 1 findings were similar to Model 2 and Model 4 findings were similar to Model 3 and thus not shown.
Standard deviation increments were 6.4 μg/mL for HK and 9.1 μg/mL for prekallikrein.
Abbreviations: ARIC = Atherosclerosis Risk in Communities; CI = confidence interval; DVT = deep vein thrombosis; HK = high molecular weight kininogen; HR = hazard ratio; PE = pulmonary embolism; SD = standard deviation.
Table 5.
Hazard ratios (95% confidence intervals) of provoked and unprovoked venous thromboembolism (VTE) by quartiles or SD increments of high molecular weight kininogen and prekallikrein, ARIC, 1993–95 through 2015
| Quartiles |
|||||
|---|---|---|---|---|---|
| Q1 | Q2 | Q3 | Q4 | Per SD increment2 |
|
| High molecular weight kininogen, μg/mL | 2.08–19.11 | 19.12–22.66 | 22.67–26.76 | 26.77–58.57 | |
| n at risk1 | 983 | 983 | 984 | 983 | 3,933 |
| Incident Provoked VTE, n | 102 | 104 | 84 | 77 | 367 |
| Model 2 HR (95% CI) | 1 (Reference) |
0.97 (0.73–1.30) | 0.84 (0.62–1.14) | 0.71 (0.52–0.98) | 0.86 (0.77–0.96) |
| Model 3 HR (95% CI) | 1 (Reference) |
1.00 (0.73–1.37) | 0.78 (0.55–1.11) | 0.64 (0.44–0.93) | 0.82 (0.73–0.94) |
| Incident Unprovoked VTE, n | 70 | 57 | 57 | 61 | 245 |
| Model 2 HR (95% CI) | 1 (Reference) |
0.79 (0.54–1.13) | 0.85 (0.59–1.23) | 0.86 (0.60–1.24) | 0.93 (0.81–1.06) |
| Model 3 HR (95% CI) | 1 (Reference) |
0.82 (0.54–1.23) | 0.80 (0.52–1.21) | 0.74 (0.49–1.12) | 0.85 (0.73–0.99) |
| Prekallikrein, μg/mL | 2.08–10.59 | 10.60–12.80 | 12.81–15.70 | 15.71–95.79 | |
| n at risk1 | 983 | 983 | 985 | 982 | 3,933 |
| Incident Provoked VTE, n | 103 | 87 | 96 | 81 | 367 |
| Model 2 HR (95% CI) | 1 (Reference) |
0.87 (0.65–1.18) | 1.03 (0.76–1.41) | 0.80 (0.58–1.11) | 0.96 (0.84–1.09) |
| Model 3 HR (95% CI) | 1 (Reference) |
0.90 (0.64–1.25) | 1.11 (0.79–1.57) | 0.86 (0.59–1.25) | 0.92 (0.80–1.05) |
| Incident Unprovoked VTE, n | 53 | 60 | 67 | 65 | 245 |
| Model 2 HR (95% CI) | 1 (Reference) |
1.16 (0.79–1.70) | 1.34 (0.92–1.95) | 1.24 (0.84–1.82) | 1.04 (0.91–1.18) |
| Model 3 HR (95% CI) | 1 (Reference) |
1.34 (0.86–2.11) | 1.40 (0.90–2.18) | 1.47 (0.96–2.26) | 1.00 (0.86–1.15) |
Model 2: Adjusted for age, race, sex-hormone replacement therapy, weight, height, diabetes, smoking status, systolic blood pressure, antihypertensive medications, and estimated glomerular filtration rate
Model 3: Model 2 + adjustment for factor XI, log(D-dimer), and weighted genetic risk score (F5, F2, ABO, F11, FGG variants).
Model 3 has a reduced sample size due to missing values (see Table 3). Model 1 findings were similar to Model 2 and Model 4 findings were similar to Model 3 and thus not shown.
Standard deviation increments were 6.4 μg/mL for HK and 9.1 μg/mL for prekallikrein.
Abbreviations: ARIC = Atherosclerosis Risk in Communities; CI = confidence interval; HK = high molecular weight kininogen; HR = hazard ratio; SD = standard deviation; VTE = venous thromboembolism
Associations of KNG1 rs710446 variant with VTE, with and without adjustment for HK
Given no association of plasma prekallikrein with VTE, it could not mediate the association of KNG1 rs710446 with VTE.1 Similarly, given a possible inverse association of plasma HK with VTE, it seemed highly unlikely that HK could mediate the association of KNG1 rs710446 with VTE1 either, but we ran a “mediation” model to verify this. In a proportional hazards model, adjusted for age, race and sex but not HK, the HRs (95% CI) of VTE were 1 (reference), 1.12 (0.90, 1.39), and 1.24 (0.96, 1.61) for 0, 1, or 2 risk alleles for KNG1, and the HR was 1.11 (0.98, 1.27) per each KNG1 risk allele treated as an ordinal variable. When HK was added as a continuous variable to the model, these HRs of VTE changed little [1 (reference), 1.16 (0.93, 1.44), and 1.31 (1.00, 1.71) for 0, 1, or 2 KNG1 risk alleles; 1.15 (1.00, 1.31) per each risk allele]. Thus, plasma HK did not mediate the association of KNG1 rs710446 with incident VTE.
Discussion
This prospective study showed no significant positive association of plasma levels of HK or prekallikrein with incident VTE in the general population. If anything, HK was inversely associated with VTE. This finding was contrary to our hypothesis of a positive association derived from several lines of evidence: animal studies of contact system factors and thrombosis,5–9 genome-wide association studies linking a KNG1 missense variant (rs710446) with VTE,1 and a single previous study showing a positive association of HK with VTE.13 Although we measured just two factors in the kallikrein/kinin system, our findings do not support that higher levels of these two factors in the general population influence the occurrence of VTE. Our findings further suggest that plasma levels of HWMK or prekallikrein do not explain the documented association of KNG1 rs710446 with VTE.
We have no firm explanation for the graded inverse association between HK and VTE. There is very little prior literature on whether HK and prekallikrein are correlated with other VTE risk factors, which might confound associations with VTE. We, in fact, found HK and prekallikrein associated with several factors (Table 1), but adjustment for them did not greatly change the inverse association of HK and prekallikrein. Nevertheless, there still may have been unidentified confounding, or the inverse association may have been a chance finding, so it certainly warrants replication. In any case, we can speculate about some possible mechanisms. Since HK is a multifunctional protein,24 an inverse association with VTE could be mediated through pathways other than the contact coagulation system. We observed that D-dimer was lower with increasing HK, supporting the notion that higher HK would be inversely associated with VTE insofar as higher D-dimer is a VTE risk factor. An in vitro study reported HK can inhibit thrombin-induced platelet aggregation by impairing thrombin binding to platelets.25 Another in vitro study showed that cleaved HK (i.e., the kinin-free form) could interfere with the function of plasminogen activator inhibitor-1,26 an important regulator of the fibrinolytic system. Experimental rat models of spontaneous kininogen deficiency, due to a point mutation in kininogen, have reported kininogens exert antithrombotic effects in arterial thrombosis.27,28
In addition, kininogens are inhibitors of cysteine cathepsins, a group of cysteine proteases.24 Falanga et al reported that cancer cells expressed a cysteine protease that can directly activate factor X in the absence of factor VII.29 Cancer is a major contributor to provoked VTE. Other cysteine cathepsins are involved in a variety of physiological function and pathological conditions, including cancer, atherosclerosis and inflammatory diseases,30 which might contribute to VTE.
Although our study was large, population based, and prospective, it had some drawbacks. Firstly, samples were stored approximately 24 years at −80˚C and previously thawed. As described in Methods, laboratory variability for both HK and prekallikrein was high and pilot studies suggested that HK concentrations in citrate plasma fall somewhat with freeze-thaw cycles. The laboratory imprecision would tend to obscure associations with VTE, but drift from multiple thaws might be expected to have limited impact on associations, as all samples were handled similarly during freeze-thaw cycles. Secondly, the mean value of HK was much lower than a previously established mean value from healthy volunteers of 80 μg/mL.18 The Abcam assay we chose seems to run low (see Methods), but alternatively ARIC samples may have deteriorated while frozen for approximately 14 years. Assuming the degree of underestimation of concentrations would be comparable in those who did versus did not develop VTE, systematic underestimation of HK should have led to little bias in the hazard ratios.Nevertheless, our findings require replication.
Thirdly, we had only single measures of HK and prekallikrein. During several decades of follow-up, ARIC participants’ usual plasma levels of these biomarkers likely changed, but we have no follow-up measures to verify how much. Such within-person variability likely would be random with respect to VTE occurrence, and thus would tend to obscure any real association between the factors and VTE. Fourthly, we identified hospitalized VTE patients only, but pilot data suggest that the vast majority of first VTEs in ARIC were hospitalized.
In conclusion, plasma concentrations of HK and prekallikrein were not positively associated with incidence of VTE, but rather there was an inverse association for HK. Our study suggests that plasma levels of these factors do not determine the risk of VTE in the general population.
Summary Table.
What is known about this topic?
Whether the kallikrein/kinin and contact activation systems play a role in venous thromboembolism (VTE) is uncertain, although a KNG1 mutation is associated with VTE.
A few epidemiological studies have provided mixed evidence that higher plasma concentrations of high molecular weight kininogen (HK) or prekallikrein may increase VTE risk.
What does this paper add?
Our large prospective study found that higher concentrations of HK and prekallikrein were not associated with greater VTE risk.
In fact, HK was associated inversely with VTE.
Our findings suggest that plasma levels of these factors do not determine the risk of VTE in the general population.
Acknowledgements
We thank the ARIC participants and staff for their important contributions to ARIC research, and Elaine Cornell and Dr. Russell Tracy for assistance with laboratory measurements.
Funding
The National Heart, Lung, and Blood Institute provided support for venous thromboembolism identification via R01 HL059367 and for the Atherosclerosis Risk in Communities Study via contracts HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I, and HHSN268201700005I.
Footnotes
Conflicts of Interest
None.
References
- 1.Morange PE, Suchon P, Trégouët DA. Genetics of venous thrombosis: update in 2015. Thromb Haemost 2015;114(5):910–919. [DOI] [PubMed] [Google Scholar]
- 2.Lijfering WM, Rosendaal FR, Cannnegieter SC. Risk factors for venous thrombosis – current understanding from an epidemiological point of view. Br J Haematol 2010;149(6):824–833. [DOI] [PubMed] [Google Scholar]
- 3.Folsom AR, Alonso A, George KM, Roetker NS, Tang W, Cushman M. Prospective study of plasma D-dimer and incident venous thromboembolism: The Atherosclerosis Risk in Communities (ARIC) Study. Thromb Res 2015;136(4):781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost 2016;14(1):28–39. [DOI] [PubMed] [Google Scholar]
- 5.Long AT, Kenne E, Jung R, Fuchs TA, Renné T. Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost 2016;14(3):427–437. [DOI] [PubMed] [Google Scholar]
- 6.Renné T, Pozgajová M, Grüner S, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med 2005;202(2):271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merkulov S, Zhang WM, Komar AA, et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood 2008;111(3):1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stavrou EX, Fang C, Merkulova A, et al. Reduced thrombosis in Klkb1−/− mice is mediated by increased Mas receptor, prostacyclin, Sirt1, and KLF4 and decreased tissue factor. Blood 2015;125(4):710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Y Contact pathway of coagulation and inflammation. Thromb J 2015;13:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Houlihan LM, Davies G, Tenesa A, et al. Common variants of large effect in F12, KNG1, and HRG are associated with activated partial thromboplastin time. Am J Hum Genet 2010;86(4):;626–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siegerink B, Govers-Riemslag JW, Rosendaal FR, Ten Cate H, Algra A. Intrinsic coagulation activation and the risk of arterial thrombosis in young women: results from the Risk of Arterial Thrombosis in relation to Oral contraceptives (RATIO) case-control study. Circulation 2010;122(18):1854–1861. [DOI] [PubMed] [Google Scholar]
- 12.Merlo C, Wuillemin WA, Redondo M, et al. Elevated levels of plasma prekallikrein, high molecular weight kininogen and factor XI in coronary heart disease. Atherosclerosis 2002;161(2):261–267. [DOI] [PubMed] [Google Scholar]
- 13.Gallimore MJ, Harris SL, Jones DW, Winter M. Plasma levels of factor XII, prekallikrein and high molecular weight kininogen in normal blood donors and patients having suffered venous thrombosis. Thromb Res 2004;114(2):91–96. [DOI] [PubMed] [Google Scholar]
- 14.The ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: Design and objectives. Am J Epidemiol 1989;129(4):687–702. [PubMed] [Google Scholar]
- 15.Cushman M, Tsai AW, Heckbert SR, White RH, Rosamond WD, Folsom AR. Deep vein thrombosis and pulmonary embolism in two cohorts: the Longitudinal Investigation of Thromboembolism Etiology (LITE). Am J Med 2004;117(1):19–25. [DOI] [PubMed] [Google Scholar]
- 16.Prentice R A case-cohort design for epidemiologic cohort studies and disease prevention trials. Biometrika 1986;73(1):1–11. [Google Scholar]
- 17.Folsom AR, Tang W, Roetker NS, Heckbert SR, Cushman M, Pankow JS. Prospective study of circulating factor XI and incident venous thromboembolism: the Longitudinal Investigation of Thromboembolism Etiology (LITE). Am J Hematol 2015;90(11):1047–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmaier AH, Zuckerberg A, Silverman C, Kuchibhotla J, Tuszynski GP, Colman RW. High-molecular weight kininogen. A secreted platelet protein. J Clin Invest 1983;71(5):1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Folsom AR, Tang W, Weng LC, et al. Replication of a genetic risk score for venous thromboembolism in whites but not in African Americans. J Thromb Haemost 2016;14(1):83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai AW, Cushman M, Rosamond WD, et al. Coagulation factors, inflammation markers, and venous thromboembolism: The Longitudinal Investigation of Thromboembolism Etiology (LITE). Am J Med 2002;113(8):636–642. [DOI] [PubMed] [Google Scholar]
- 21.Zakai NA, Ohira T, White R, Folsom AR, Cushman M. Activated partial thromboplastin time and risk of future venous thrombo embolism. Am J Med 2008;121(3):231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inker LA, Schmid CH, Tighiouart H, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med 2012;367(1):20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barlow WE. Robust variance estimation for the case-cohort design. Biometrics 1994;50(4):1064–1072. [PubMed] [Google Scholar]
- 24.Lalmanach G, Naudin C, Lecaille F, Fritz H. Kininogens: More than cysteine protease inhibitors and kinin precursors. Biochimie 2010;92(11):1568–1579. [DOI] [PubMed] [Google Scholar]
- 25.Puri RN, Zhou F, Hu CJ, Colman RF, Colman RW. High molecular weight kininogen inhibits thrombin-induced platelet aggregation and cleavage of aggreging by inhibiting binding of thrombin to platelet. Blood 1991;77(3):500–507. [PubMed] [Google Scholar]
- 26.Chavakis T, Pixley RA, Isordia-Salas I, Colman RW, Preissner KT. A novel antithrombotic role for high molecular weight kininogen as inhibitor of plasminogen activator-1 function. J Biol Chem 2002;277(36):32677–32682. [DOI] [PubMed] [Google Scholar]
- 27.Colman RW, White JV, Scovell S, Stadnicki A, Sartor RB. Kininogens are antithrombotic proteins in vivo. Arterioscler Thromb Vasc Biol 1999;19(9):2245–2250. [DOI] [PubMed] [Google Scholar]
- 28.Hassan S, Sainz IM, Khan MM, et al. Antithrombotic activity of kininogen is mediated by inhibitory effects of doman 3 during arterial injury in vivo. Am J Physiol Heart Circ Physiol 2007;292(6):H2959–H2965. [DOI] [PubMed] [Google Scholar]
- 29.Falanga A, Gordon SG. Isolation and characterization of cancer procoagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985;24(20):5558–5567. [DOI] [PubMed] [Google Scholar]
- 30.Patel S, Homaei A, El-Seedi HR, Akhtar N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed Pharmacother 2018;105:526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]