

Abstract
Ion channels play fundamental roles in both excitable and non-excitable tissues and therefore constitute attractive drug targets for a myriad of neurological, cardiovascular and metabolic diseases as well as for cancer and immunomodulation. However, achieving selectivity for specific ion channel subtypes with small molecule drugs has been challenging and there currently is a growing trend to target ion channels with biologics. One approach is to improve the pharmacokinetics of existing or novel venom derived peptides. In parallel, after initial studies with polyclonal antibodies demonstrated the technical feasibility of inhibiting channel function with antibodies, multiple preclinical programs are now using the full spectrum of available technologies to generate conventional monoclonal and engineered antibodies or nanobodies against extracellular loops of ion channels. After a summary of the current state of ion channel drug discovery, this review discusses recent developments using the purinergic receptor channel P2X7, the voltage-gated potassium channel KV1.3 and the voltage-gated sodium channel Nav1.7 as examples of targeting ion channels with biologics.
Introduction
Ion channels are pore-forming transmembrane proteins that allow the regulated flow of cations or anions across membranes. The IUPHAR (International Union of Basic and Clinical Pharmacology) Guide to Pharmacology1 currently lists 145 genes for voltage-gated-like ion channels2, 82 genes for ligand-gated ion channels3, and 52 genes for “so-called” other channels4 like aquaporins, connexins or store-operated channels in humans. However, since many ion channels form homo- or heteromers of two, three, four, or five subunits - which additionally may interact with auxiliary proteins - the total number of possible ion channels that can be assembled from these genes to serve very specific physiological functions is much larger. Ion channels currently constitute important drug targets for the treatment of type-2 diabetes, hypertension, epilepsy, cardiac arrhythmia, and anxiety, and many of the classical drugs on the WHO’s list of essential medicines, like nifedipine (Cavl.x inhibitor), amiodarone (mixed KV channel inhibitor), phenytoin (Nav inhibitor), or diazepam (GABAA activator) are ion channel modulators. A recent comprehensive analysis of molecular targets5 estimated that 18% of small-molecule drugs exert their therapeutic effects through ion channels (see Supplementary Table1 for a complete list of clinically used drugs targeting human ion channels). It is noteworthy that these clinically successful examples are all small molecule drugs that were developed long before their molecular targets were identified, solely by exploiting ex vivo/in vivo approaches. Following the initial registrations of these “classics” between the early 50s and early 80s the number of ion channel targeting drugs increased quickly, arguably often because of “me too” drug development around the original chemical structure and “incremental” property adjustments (most clearly illustrated by the dihydropyridine anti-hypertensives, the sulfonylurea type-2 antidiabetics, and the benzodiazepine anxiolytics shown in Supplementary Table 1).
Based on electrophysiological experiments, and especially with the introduction of patch-clamp technology6, the mode-of-actions of the most important ion channel targeting drug classes (Nav inhibitors, KATP inhibitors etc.) were quite well established around 19907. At the end of the cloning era in the late 90s, the majority of ion channels were identified revealing a complex picture with respect to subtypes, stoichiometry and pharmacology8. The exceptions were voltage-dependent H+ channels (Hv), Ca2+- and volume regulated Cl- channels (CACL, VRAC), the stretch- and voltage activated Piezo channel, and the Ca2+ release activated Ca2+-channel (Orai), which were only cloned a decade later. It specifically became apparent that even closely related subtypes like members of the KV1 family can have very different physiological functions, and thus can provide highly variable therapeutic and adverse effect profiles. This realization initiated an “explosion” in the number of ion channel drug discovery programs9, 10, 11,12, 13, dedicated towards development of subtype-selective small molecule drugs, tailormade for improving the therapeutic index (i.e.: Cav2.2/3 inhibitors for neurological indications like stroke or pain without effects on Cav1 and thus without cardiovascular side effects; GABAA α2/α3 selective activators for anxiety without effect on α1 containing channels and therefore without sedative properties). Hand-in-hand with this progress, cloning, molecular, and cell culturing techniques improved significantly, high-throughput automated ion channel assays were developed, and all large pharmaceutical companies upgraded (often to several millions chemical entities) and refined their chemical libraries14, 15 In the most streamlined versions a total screening campaign, including reporting on one or more subtypes, could be completed in just 2–3 months.
This tremendous and long-lasting effort across essentially the entire pharmaceutical industry should have been a leap into the future with respect to improved ion channel medicines, but very few registrations have resulted from these efforts. For example, out of the Kv channel modulator programs we reviewed in 200910 only one compound, the Kv7 activator retigabine (known as ezogabine in the USA) for treatment of pharmacotherapy-resistant partial epilepsies made it to market in 2011 and then was withdrawn in 2017 by GlaxoSmithKline because of skin discoloration and suspected eye toxicity related to slow accumulation of a colored metabolite. It is especially sobering that even recent registrations are either based on long-known compounds (e.g. the Kv channel blocker 4-aminopyridine for multiple sclerosis16, 2010, the GABAA activating allopregnanolone registered in 2018 for postpartum depression) or were discovered in low-throughput phenotypic screening by combining organ preparations with animal model work (the If (HCN) inhibitor ivabradine17, 2015). The only notable exception we could find is the CFTR potentiator ivacaftor (VX-770), which originated out of a high-throughput membrane potential assay screen and which increases the open probability of wild-type and mutant CFTR18. For a list of the ion channel targeting drugs approved in the last 10 years, see Table 1.
Table 1:
Ion channel targeting drugs approved in the last 10 years.
| Compound | Modality | Indication | Route of administration |
Target/Mechanism | Approval/ Withdrawal |
Company |
|---|---|---|---|---|---|---|
| Brexanolone | Neurosteroid | Post partum depression | PO | GABAA δ preferring PAM | 2018 | Sage Therapeutics |
| Clevidipine | Small molecule | Acute hypertension | IV | Cav1.x inhibitor | 2008 | The Medicines Company |
| Lacosamide | Small molecule | Epilepsy | PO | Nav inhibitor | 2008 | UCB |
| Amifampridine | Small molecule | Lambert-Eaton myasthenic syndrome | PO | Kv inhibitor | 2009 | BioMarin Pharmaceutical |
| Dronedarone | Small molecule | Arrhythmia | PO | Multi K channel inhibitor | 2009 | Sanofi-Aventis (Sanofi) |
| Eslicarbazepine | Small molecule | Epilepsy | PO | Nav inhibitor | 2009 | Bial |
| Fampridine | Small molecule | Multiple sclerosis | PO | Kv inhibitor | 2010 | Acorda Therapeutics |
| Retigabine | Small molecule | Epilepsy | PO | Kv7.2-Kv7.5 activator | 2011/2017 | GSK |
| Clobazam | Small molecule | Anxiety, epilepsy | PO | GABAA activator | 2011 | Maestretti Research Laboratories (Sanofi) |
| Clobazam | Small molecule | Anxiety, epilepsy | PO | GABAA activator | 2011 | Maestretti Research Laboratories (Sanofi) |
| Perampanel | Small molecule | Epilepsy | PO | GluA1–4 inhibitor | 2012 | Eisai |
| Ivacaftor | Small molecule | Cystic fibrosis | PO | CFTR potentiator | 2012 | Vertex; Cystic Fibrosis Foundation |
| Ivabradine | Small molecule | Heart failure | PO | If (HCN) inhibitor | 2005 EMA 2015 FDA | Servier Amgen |
| Lumacaftor/ ivacaftor | Small molecule | Cystic fibrosis | PO | CFTR chaperone plus potentiator | 2015 | Vertex |
Inspecting the reported clinical pipelines from major pharmaceuticals and biotech companies does not change the picture significantly (Table 2): One topical polyclonal antibody, three peptides and 33 small molecule clinical programs are currently reported; most are in phase I, a handful are in phase I/II, and only two, SAGE-217, a synthetic GABAa activating neurosteroid for major depression, and Gefapixant, a P2X3 inhibitor for chronic cough, are in phase III. One compound, mirogabalin, an improved gabapentenoid targeting Cav channels, is under registration in Japan for peripheral neuropathic pain. To underline the case further, two of the phase II examples are “repurposed” compounds (senicapoc, a Kca3.1 inhibitor, for hereditary xerocytosis and Alzheimer’s disease and gaboxadol, a δ-subunit preferring GABAa agonist for Angelman’s/Fragile-X syndrome), and XEN496 is retigabine now redeveloped for the orphan indication Kv7.2-mediated epileptic encephalopathy. A trend is that many of these development programs originate from small/intermediate pharmaceuticals and biotech companies, which may indicate a decline in the popularity of ion channels with the major pharmaceutical companies.
Table 2:
Ion channel targeting drug development candidates currently in clinical trials.
| compound | company | indication | Target or mechanism |
clinical phase |
| Granexin gel (αCT1) | FirstString Research | Diabetic foot ulcer, cutaneous radiation injury | Cx43 c-terminal mimetic peptide | III |
| Dalazatide (ShK- 186) | Kv1.3 Therapeutics | Psoriasis, IBM | Kv1.3 inhibitor | Ib/IIa |
| Tozuleristide (BLZ-100) |
Blaze Bioscience | Imaging of glioma (‘tumour paint’) | Metalloprotease inhibitor, possible chloride channel inhibitor | I/II |
| SOR-C13 | Soricimed Biopharma | Cancer | TRPV6 inhibitor | I |
| Antibodies | ||||
| BILO10t | Biosceptre | Cancer | Non-functional P2X7 | I/II |
| Small molecules and neurosteroids | ||||
| Mirogabalin | Daiichi Sankyo | Peripheral neuropathic pain | Cav inhibitor (α2δ subunit | Registration phase |
| XEN007 | Xenon | Hemiplegic migraine | Cav2.1 inhibitor | I |
| Tetrodotoxin (Halneuron) |
Wex Pharmaceuticals | Cancer and chemotherapy induced pain | Nav inhibitor | III |
| Vixotrigine (BIIB074) |
Biogen | Painful lumbosacral radiculopathy, trigeminal neuralgia | Nav1.7 inhibitor | II |
| BIIB095 | Biogen | Pain | Nav1.7 inhibitor | I |
| CC-8464 | Chromocell, Astellas Pharma | Neuropathic pain | Nav1.7 inhibitor | I |
| DSP-2230 | Sumitomi Dainippon Pharma | Neuropathic pain | Nav1.7, Nav1.8 inhibitor | I |
| DSP-3905 | Sumitomi Dainippon Pharma | Neuropathic pain | Nav1.7 inhibitor | I |
| RG-6029 (GDC- 0310) | Roche | Pain | Nav1.7 inhibitor | I |
| Xen901 | Xenon | Epilepsy | Nav1.6 inhibitor | I |
| Gefapixant | Merck, Roche | Chronic cough | P2X3 inhibitor | III |
| BAY-1902607 a | Bayer | Persistent chronic cough | P2X3 inhibitor | II |
| BAY-1817080 a | Bayer | Endometriosis, persistent chronic cough | P2X3 inhibitor | I |
| P2X4 inhibitor a | Bayer | Endometriosis | P2X4 inhibitor | I |
| JNJ-55308942 | Johnsson & Johnsson | Neuroinflammation, anhedonia |
P2X7 inhibitor | I |
| Intravenous glibenclamide (BIIB093) |
Biogen | Stroke | SUR1-TRPM4 inhibitor | III |
| Basmisanil (RG- 1662) | Roche | Cognitive impairment associated with schizophrenia | GABAA α5-subunit-targeting NAM | II |
| Sage-217 b | Sage Therapeutics | Major depression | GABAA δ-subunit-preferring PAM | II |
| Gaboxadol (OV- 101) | Ovid Therapeutics | Angelman syndrome, Fragile X syndrome | GABAA δ-subunit-preferring agonist | I/II (repurposed) |
| RG-7816 | Roche | Autism spectrum disorder | GABAA α5-subunit- targeting NAM | I |
| Evt201 | Evotec | Insomnia | GABAA PAM | II |
| BIIB104 | Biogen | Cognitive impairment associated with schizophrenia | AMPA activator | IIa |
| TAK-653 | Takeda | Depression | AMPA activator | II |
| PF-04958242 | Pfizer | Schizophrenia | AMPA activator | II |
| BAY-2253651 a | Bayer | Obstructive sleep apnoea | K2p3.1 inhibitor | II |
| ASP-0819 | Astellas Pharma | Fibromyalgia | KCa3.1 activator | II |
| Senicapoc | SpringWorks Therapeutics and University of California, Davis | Hereditary xerocytosis, Alzheimer’s disease | KCa3.1 inhibitor | II (repurposed) |
| AP-30663 | Acesion Pharma | Atrial fibrillation | KCa2 inhibitor | I/II |
| CAD-1883 | Cadent Therapeutics | Ataxia or essential tremors | KCa2 activator | I/II |
| XEN496 (retigabine) |
Xenon | KCNQ2 epileptic encephalopathy | Kv7 activator | II |
| GSK-2798745 | GlaxoSmithKline | Heart failure | TRPV4 inhibitor | II |
| SB705498 | GlaxoSmithKline | Rhinitis/Pain/Cough | TRPV1 inhibitor | II |
| Galicaftor (ABBV-2222) |
Abbvie | Cystic fibrosis | CFTR | II |
| GLPG-1837 | Galapagos, Abbvie | Cystic fibrosis | CFTR potentiator | II |
| ABBV-2451 | Abbvie | Cystic fibrosis | CFTR | I |
| ABBV-2737 | Abbvie | Cystic fibrosis | CFTR | I |
| ABBV-3067 | Abbvie | Cystic fibrosis | CFTR | I |
| Sage-324 a,b | Sage Therapeutics | Essential tremor Parkinson’s disease | GABAA PAM | I |
| Sage-718 a,b | Sage Therapeutics | NMDA hypofunction | NMDA activator | I |
| TrpC4/5 inhibitor |
Hydra/Boehringer Ingelheim |
Anxiety disorder, depression | TRPC4, TRPC5 inhibitor | I |
| Xen1101 | Xenon | Epilepsy | Kv7 activator | I |
| HBI-3000 | HUYA Bioscience International | Atrial fibrillation | Multi channel inhibitor | I |
| DWJ-208 | Daewoong Pharmaceutical |
Neuropathic and cancer pain | NaV1.7 | I |
Cx43, connexin 43; IBM, inclusion body myositis; NAM, negative allosteric modulator; PAM, positive allosteric modulator.
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor. Cav, voltage-gated calcium channel. CFTR, cystic fibrosis transmembrane conductance regulator. GABA, γ-aminobutyric acid. GluA, ionotropic glutamate receptor. HCN, hyperpolarization-activated cyclic nucleotide-gated channel. K2P, two-pore domain potassium channel. Kv, voltage-gated potassium channel. KCa, calcium-activated potassium channel. Nav, voltage-gated sodium channel. NMDA, N-methyl-D-aspartate receptor. P2X, purinoreceptor. SUR, sulfonylurea receptor. TRPC, transient receptor potential canonical channel. TRPM, transient receptor potential melastatin channel. TRPV, transient receptor potential vanilloid channel.
Presumed to be small molecules.
Neurosteroids.
Even though small molecules will continue to be important for ion channel drug development in years to come, we are convinced that the role of ion channel targeting biologics will increase and help “revitalize” ion channels as targets for drug development. Following on the heels of the G-protein coupled receptor field19, where several dozen antibody programs have advanced into clinical trials and CCR4 and CGRP targeting antibodies are on the market, the ion channel field is presently particularly excited about antibody approaches. This article will discuss the current state of research into antibodies and peptides that target this complex and diverse group of molecules.
Challenges in targeting ion channels
Ion channels have long been regarded as difficult drug targets and the reasons that have traditionally been given for this with respect to small molecule drug discovery10 were 1) the technical difficulties in ion channel high-throughput screening; 2) the lack of crystal structures enabling “true” structure based drug design; 3) the challenges of achieving subtype selectivity, also taking into account stoichiometry of heteromeric channels and possible interactions with auxiliary subunits (a point often ignored in drug discovery), and ideally state/conformation selectivity. All these issues are also applicable to ion channel targeted biologics. The first issue has been somewhat addressed with the development of ultra-high-throughput membrane potential or flux assay systems and automated electrophysiology platforms that are currently capable of running high quality giga seal electrophysiological recordings in 48, 96 and 384 wells. However, we would like to posit that the specialized biophysical expertise that is necessary to expertly execute and analyze ion channel screens constitutes a real or sometimes perceived barrier to committing to an ion channel drug discovery program, which together with the fact that overall more resources in industry and academia have been dedicated to GPCRs than to ion channels, is probably responsible for the overall slower progress of the ion channel than the GPCR field. The 2nd point, the lack of structures, is a deficiency that is currently being addressed by advances in structural biology (see Box 1), but again somewhat more slowly than in the GPCR field. The 3rd point, obtaining relevant subtype and state/confirmation selectivity, remains a challenge for both the ion channel and the GPCR field (although voltage-clamp electrophysiology with its high temporary resolution and voltage control combined with fast application should give ions channels an advantage), and requires very detailed understanding of the exact role and context of the target protein in the chosen disease indication.
Text Box 1.
Recent progress and challenges for ion channel structure-based drug design
In the last 5 years an increasing number of ion channel structures has become available through advances in cryoelectron microscopy (cryoEM) such as improvements in microscope design and imaging hardware, and enhanced image processing which allow the reconstruction of 3D structures from a large number of single particle 2D projection images even even in the presence of structural and conformational heterogeneity183. While many cryo-EM structures are not high enough resolution for “true” structure- based drug design because they do not allow the possible location of hydrogen bonds or salt bridges to be seen, Rosetta computational structure refinement can be used to improve atomic details of cryoEM structures with 3–5 Å resolution184. For example, in the Nav channel field both traditional x-ray and cryoEM structures now provide highresolution structural templates for structure-based design of novel small molecules, peptides, and antibodies. However, while many of the structures contain fully resolved extracellar loops, a remaining challenge that needs to be further explored is how to stabilize the extracellular loop regions in native conformations for immunization without having to produce large quantities of full-length protein.
An x-ray structure of a human Nav1.7– bacterial NavAb chimera31 with a picomolar affinity drug bound to the domain IV (DIV) voltage sensor constitutes a template for targeting the voltage sensor, which plays a key role in stabilizing Nav channels in an inactivated state, with small molecules or peptides.
All human Nav channels have unique sequences in the extracellular loop regions within the voltage-sensing and pore-forming domains. CryoEM structures of the human Nav1.441 and American cockroach NavPaS185with a small molecule and or a peptide toxin bound have all extracellular loops resolved in the pore and voltage sensor domains.
The CryoEM structure of the electric eel Nav1.440 has all extracellular loops in the pore domain resolved.
Target specificity
Since nearly all efforts within ion channel drug discovery/development previously focused on small molecules, it is worth considering the one key challenge for the field in addition to the general issues all small molecule medicinal chemistry programs face, namely optimizing pharmacokinetic properties20, avoiding toxicity due to toxic metabolites21, and finding chemical matter that ideally allows one to obtain “strong” composition of matter patents. Obtaining selectivity within ion channel families can be difficult to achieve, since subtypes are often highly homologous. This is especially challenging for small molecule drugs due to their small sizes and therefore limited number of interaction points with the target. The field of Nav inhibitors is particularly instructive: Antiepileptics, class I antiarrhythmics, and local anesthetics target voltage-gated Nav channels at a highly conserved site within the pore lumen formed by transmembrane segments S6 in the third and fourth domains22. Thus, despite many years of drug-development by many companies, all classical Nav drugs are essentially unselective and their therapeutic value as systemically administered drugs relies solely on their strong state- or use-dependency, which favors binding to channels in pathologically depolarized or excessively firing cells.
State-dependence in this context means that a drug preferentially binds to one of the conformational states (closed, open, inactivated) that an ion channel undergoes during its gating cycle (Fig. 1A). For voltage-gated channels like NaV, Cav and KV channels the occupancy of these states strongly depends on the membrane potential which means that drug development programs focused on targeting neuronal ion channels must decide which state of the channel it would be most desirable to target. For example, for NaV channels that is generally assumed to be the open or inactivated state, while it might be better to target the closed state when developing a KV channel opener. When neuronal ion channels are expressed in HEK293 or CHO cells, their voltage-sensor domains (VSD) are exposed to more depolarized resting membrane potentials (around −40 mV) instead of the more hyperpolarized resting membrane potential (around −70 mV) they would be experiencing in a neuron. This means that in drug screening assays such as flux or binding assays, that do not allow to control the membrane potential, the VSDs will tend to be in the thermodynamically favored “up” state that is present in open or inactivated states of the channel, which makes it difficult to capture the “down” state of the VSD that is present in the closed state (Fig. 1A). One of the major advantages of using manual or HTS patch-clamp electrophysiology for ion channel drug screens is that the membrane potential can be precisely controlled to capture the desired state of the channel.
Figure 1.
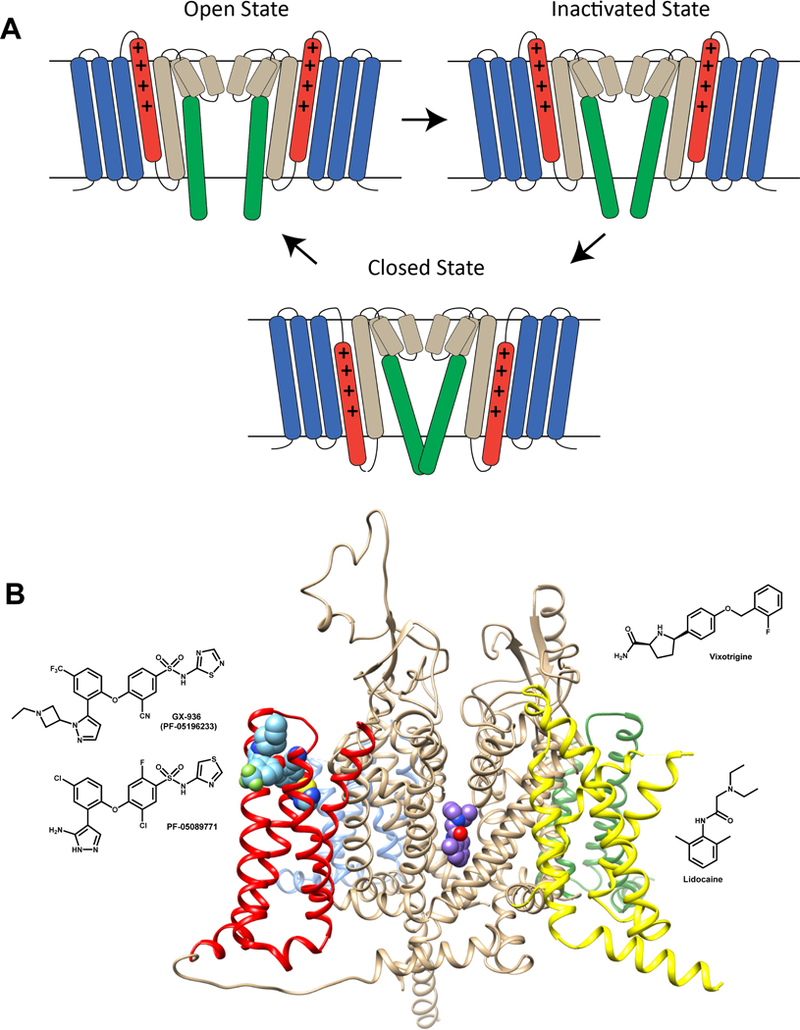
A) Cartoon representation of the Nav channel gating cycle in closed, open and inactivated states. The voltage sensing domain (VSD) segments are colored blue (S1-S3) and red (S4). The pore-forming domain segments are colored sand (S5, P1-helix, P2-helix, and N-terminal part of S6) and green (C-terminal part of S6). The gating charge carrying arginines in the S4 segment are represented by “+” signs. Key conformational changes in the Nav channel during gating between closed, open, and inactivated states are highlighted by transmembrane movement of the S4 segment (colored in red) and lateral movement of the C-terminal part of S6 segment (colored in green). Inactivation in Nav channels can be either fast and involve one of the cytoplasmic inter-repeat loops “plugging” the inner vestibule8 or slow and involve a rearrangement of the selectivity filter8. B) Small molecule receptor sites within Nav channels. Transmembrane view of Nav channel interaction with pore bound (lidocaine) and voltage-sensor bound (GX-936) small molecule drugs. The Nav channel structure is based on the electric eel Nav1.4 channel structure40 (pdb: 5XSV) and is shown in ribbon representation. The pore domain is shown in beige and the voltage-sensors are shown in blue (domain I), green (domain II), yellow (domain III), and red (domain IV). Lidocaine is shown in space-filling representation and colored purple. GX-936 is shown in space-filling representation and colored light blue. The chemical structures of vixotrigine, lidocaine, GX-936, and PF-05089771 are shown in 2D representation next to the channel.
The NaV channel family contains 9 functional subtypes23, NaV1.1 to NaV1.9. NaV1.1 is expressed in fast spiking interneurons of the brain, where loss-of-function causes Dravet syndrome23, and has recently been shown to regulate the mechanical excitability of visceral nerves in the gut24, 25 Nav1.2 is the predominant brain isoform and, together with Nav1 .6, is a major target for anti-epileptics. NaV1.5 is the main cardiac channel and the target for class I antiarrhythmics, whereas the analgesic effects of local anesthetics are mediated by several channels, including NaV1.7, NaV1.8, and possibly NaV1.9, which are preferentially expressed in sensory nerves26. NaV1.7 and NaV1.8 have clear pain phenotypes in humans as loss-of-function and gain-of-function mutations in NaV1.7 result in congenital insensitivity to pain27 and primary erythromelalgia28, respectively, whereas gain-of-function mutations of Nav1.8 may cause painful peripheral neuropathy29. Despite convincing preclinical effects of unselective NaV blockers in animal pain models and the effective use of the antiepileptic carbamazepine in trigeminal neuralgia pain30 existing NaV channel inhibiting drugs are not optimal for general treatment of severe pain disorders due to their unselective mode-of-action. Several companies (Genentech, Biogen, Amgen, Pfizer, Bayer) are or have been developing highly subtype selective and potent small molecule inhibitors of NaV1.7 by targeting binding to a site in the domain IV voltage sensor responsible for channel inactivation rather than the classical local anesthetic site in the pore lumen31, 32, 33 Based on the currently published structures, this is achievable with a series of close analogues all with a conserved aryl-sulfonamide core structure, often dramatically called the “warhead”. For example, Genentech has published an x-ray structure31 of the arylsulfonamide GX-936 bound to a receptor site within the NaV1.7 fourth voltage-sensing domain (Fig. 1B). Unfortunately, one of these small molecules, the clinical phase II compound PF-05089771 (Fig. 1B), recently failed in patients suffering from painful diabetic neuropathy34, which led Pfizer to stop further development activities. Bankar et al. suggested this failure possibly reflected insufficient NaV1.7 targeting in the clinical study and recommend the evaluation of acyl-sulfonamides with better physiochemical and pharmacological properties35, including a longer residence time on the target36. Since PF-05089771 is intentionally peripherally restricted, compounds with improved blood brain barrier (BBB) penetrability in humans, might better engage NaV1.7 channels expressed close to or on the bouton of dorsal root ganglion (DRG) neuron dorsal horn synapse. Another reason for the apparent failure of some NaV1.7 blockers could be interaction with β-subunits for the native channel in DRG neurons, which was not recaptured in heterologous screening systems using the “naked” α-subunit alone. Sokolov et al. recently demonstrated that co-expression of NaV1.7 with the glycosylated form of the sodium channel β3, an auxiliary subunit that is upregulated in injured human sensory neurons37 and in DRGs in rat pain models38, makes NaV1.7 less sensitive to several state-dependent NaV blockers39. Whether this would also be the case for the aryl-sulfonamides was not investigated in the study but it is interesting to speculate based on the recently solved structures of the human and electric eel NaV1.4-β1 complexes40, 41, that β-subunits could possibly prevent the binding of small molecules or antibodies to NaV1.7. Finally, it is possible that a slightly broader selectivity comprising also the other DRG channels, NaV1.8 and Nav1.9, may be needed. The Biogen compound, vixotrigine (a.k.a Raxatrigine), which is currently still active in Phase II, despite a recent failure in painful lumbosacral radiculopathy, has a different structure (Fig. 1B), passes the BBB, and is reportedly less subtype selective than the aryl-sulfonamides42.
An interesting and highly challenging “spin-out” of the selectivity issues with small molecules is that considerable domain homology also exists between even remotely related ion channel families, which can lead to the apparently paradoxical finding that highly subtype-selective compounds may still have off-target effects on other ion channels like the cardiac delayed rectifier Kv11.1 (hERG), inhibition of which may cause ventricular fibrillation and sudden death43, 44 Unfortunately, hERG is remarkably promiscuous with respect to binding of many different small molecule chemotypes43, 45 and thousands of otherwise useful drug candidates have been “filtered out” due to hERG activity43, 45
Another aspect that is worth mentioning, when discussing the problem of selectivity, is species specificity. While it has been incredibly challenging to achieve subtype selectivity between closely related human channels like within the NaV or CaV family or for certain GABAa or nACh receptor subtypes, species selective small molecules seem to be more easily attainable as demonstrated by the extremely low acute mammalian toxicity of neuroactive insecticides like the Nav channel blocking pyrethroids, the nicotinic receptor channel activating neonicotinoids and the GABAa channel blocking polychlorocyclohexanes and fiproles46. Examples of species specific ion channel modulators used in human medicine are the antivirals amantadine and rimantadine, which target the M2 proton channel, a member of the so-called viroporin ion channel family specific to viruses, with very little homology to prokaryotic or eukaryotic ion channels47. However, species differences in sensitivity to drugs among mammals can also be very problematic for translational drug discovery, as exemplified by the insensitivity of the rat isoform of NaV1.7 towards inhibition by the aryl-sulfonamide inhibitor, AMG-837933.
Biologics versus small molecules
Even though biologics have their own well-known challenges, such as poor membrane permeability and the risk of triggering adverse immune reactions, they constitute an attractive alternative to small molecules, first-of-all because of their much higher binding selectivity (approaching true specificity), both with respect to subtypes and off-targets, than is generally possible to achieve with small molecules. Additionally, antibodies and peptides are metabolized as part of the body’s normal protein dynamics and thus do not show the drug-drug interactions, and metabolism-mediated toxicity that are always a risk with small molecules. Taken together, these two advantages are often considered to be responsible for the overall higher success rate of new molecular entity biologics (13.2%) versus small molecules (7.6%) for progression from Phase-1 to gaining FDA approval48.
The feasibility of targeting ion channels with biologics has been amply demonstrated by Nature. Venomous animals have developed a myriad of highly potent and selective peptides that can both inhibit and activate ion channels and that have been incredibly useful in probing ion channel structure-function relationships. Similarly, autoantibodies developed by a patient’s own immune system can inhibit ion channel function and acutely transfer disease to experimental animals. For example, most cases of myasthenia gravis, a disease leading to skeletal muscle weakness, are caused by pathogenic antibodies that bind to nicotinic acetylcholine (nACh) receptors at the neuromuscular junction49, while a related condition, Lambert-Eaton myasthenic syndrome is triggered by antibodies against P/Q-type voltage-gated Ca2+ channels on presynaptic nerve terminals50. In both cases, some autoantibody clones have been shown to directly block the respective channels in electrophysiological experiments, while other antibodies can activate complement or induce channel internalization49, 50 Another instance of a neurological disease characterized by the presence of ion channel specific autoantibodies is neuromyelitis optica, where most patients test positive for aquaporin-4 antibodies51.
Ion channel-targeted peptides as therapeutics
Since the development of exenatide52, a 39-amino acid glucagon-like peptide-1 agonist isolated from the venom of a lizard, and its approval in 2005 as an injectable treatment for type-2 diabetes, peptides are increasingly being considered as viable therapeutics. The FDA has approved over 60 peptide drugs (predominantly targeting GPCRs for the treatment of metabolic disease and for oncology53), around 140 peptides are in clinical trials, and another 500 are in pre-clinical development 53 The global peptide therapeutic market is estimated to be close to US$25 billion in 201854.
Venoms are a rich source of bioactive peptides with therapeutic potential 55, 56, 57, 58 Over eons, more than 100,000 venomous creatures – arthropods (scorpions, spiders, bees, centipedes, wasps), cnidarians (sea anemones, jellyfish), mollusks (cone snails), annelids (fire worms, parasitic worms) and vertebrates (snakes, lizards, frogs, mammals) – have used their toxic cocktails to engage important biological targets. These cocktails are deployed as weapons to immobilize or kill prey, or as defense to deter predators or microbial invaders. Over 10 million bioactive peptides and proteins are estimated to be present in animal venoms, and their immense chemical diversity is unrivalled by synthetic libraries. The ability of these animals to kill with tiny amounts of powerful venom has inspired both fascination and fear in humans, and snakes and scorpions were deified in the ancient world59. Traditional medicines in China, India, Greece and the Middle East have for centuries used this vast bioactive resource for medicines. In modern times, there has been an increasing interest in exploiting this extensive and relatively untapped pharmacopeia using proteomic and genomic approaches55, 58 As of today, six venom derived peptides, including the CaV2.2 blocking Ziconotide, have been approved by the FDA, about a dozen are in clinical trials, and several more are in pre-clinical development55, 58 Below we will discuss the ion channel targeting peptides in more detail by focusing on peptides that have entered clinical trials or started preclinical development in the last 10 years.
Ion channels are frequently targeted by peptide toxins. Starting in the 1960s, animal toxins were used as molecular tools to investigate ion channels60, 61,62 Many of these toxins have disulfide-constrained architectures that enhance their stability and protease resistance and permit tremendous variations in primary sequence without perturbation of the three-dimensional fold (Supplementary Figure 1). These characteristics make peptides with disulfide-rich scaffolds attractive as therapeutics. Figure 2A highlights the channel-modulating peptides that are discussed below. Many peptides are pore blockers56, 57, 58 that bind at the extracellular entrance to the channel’s pore (external vestibule) and occlude the ion conduction pathway (Fig. 2B). Others bind to the voltage-sensor of the channel and impact channel-gating (Fig. 2B).
Figure 2.
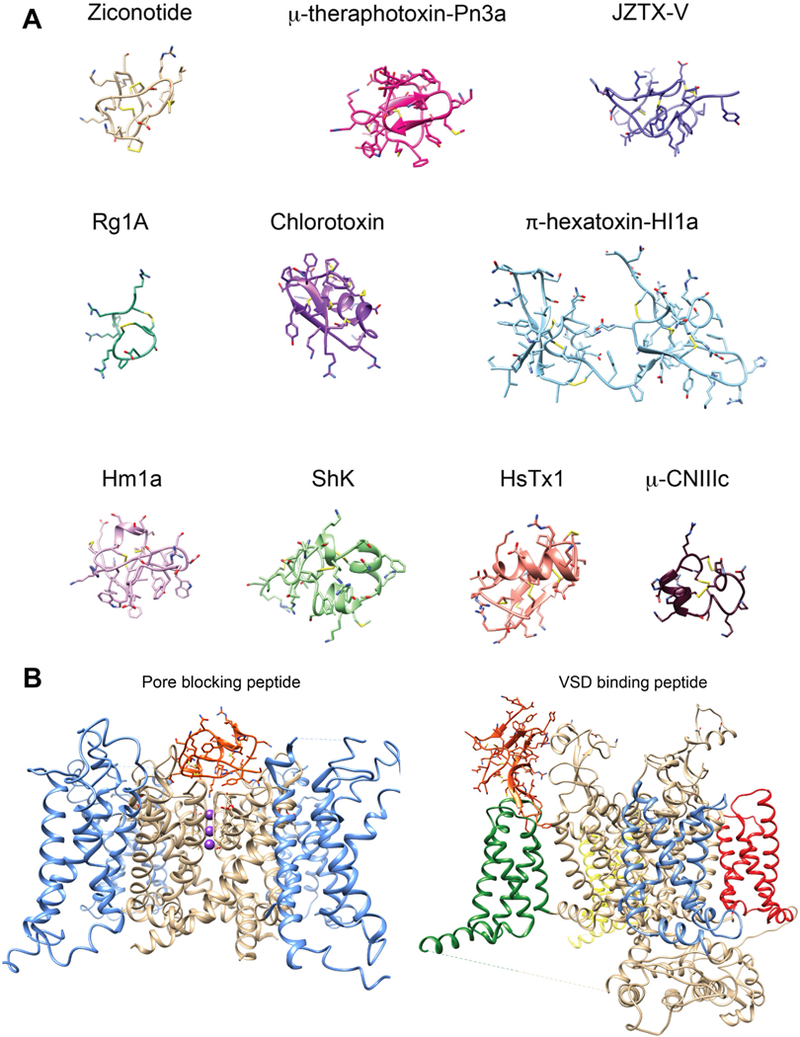
A) Structures of peptide toxins targeting ion channels. Ribbon representation of peptide toxin structures colored individually and labeled. Disulfide bonds are colored in yellow. Ziconotide (pdb: 1TTK), μ-theraphotoxin-Pn3a (pdb: 5T4R_A), JZTX-v (pdb: 6CGW), Rg1A (pdb: 2JUT), chlorotoxin (pdb: 5L1C), π-hexatoxin-HI1a (pdb: 2N8F), Hm1a (pdb: 2N6O), ShK (pdb:2K9E_A), HsTx1 (pdb: 1QUZ_A) μ-CNIIIc (pdb: 2YEN). B) Peptide toxin receptor sites on ion channels. Left, Pore blocking peptide receptor site in a Kv channel illustrated by the transmembrane view of charybdotoxin in the pore of the Kv1.2-Kv2.1 chimera structure196 (pdb: 2R9R) and shown in ribbon representation. The pore is shown in beige and the voltage-sensors are shown in blue. Charybdotoxin is shown in orange. Potassium ions within the selectivity filter region are shown in sphere representation and colored purple. Right, voltage sensor binding peptide receptor site in a Nav channel illustrated by the transmembrane view of the voltagesensor bound peptide toxin Dc1a. The Nav structure is the American cockroach NavPaS structure197 (pdb: 6A90) and shown in ribbon representation. The pore is shown in beige and voltage-sensors are shown in blue (domain I), green (domain II), yellow (domain III), and red (domain Iv). The Dc1a toxin is shown in orange.
Channel-modulating peptides for the management of pain
The opioid crisis in the United States of America has spurred efforts to develop non-opioid drugs for chronic pain, but many of these efforts have failed. A notable exception is Ziconotide (Prialt®), a peptide derived from the venom of fish-hunting cone snails63, which was FDA-approved in 2004. In the 1980s, ω-conotoxin GVIA from Conus geographus and ω-conotoxin MVIIA from Conus magus (Fig. 2A) were shown to block voltage-gated calcium channels in the nervous system but not muscle64, 65, 66 The neuronal calcium channel was later defined as the N-type CaV2.2 channel. These peptides decrease neurotransmitter release from nociceptive afferents that terminate in the dorsal horn of the spinal cord. The peptide that advanced to the clinic was native MVIIA (Ziconotide)63. It is administered intrathecally through an implanted pump because it does not cross the BBB. Ziconotide is efficacious when administered as a single therapeutic, but is increasingly combined with an intrathecal opioid for the management of refractory chronic and cancer pain. Recent guidelines also recommend Prialt® as a first-line agent for neuropathic and nociceptive pain. Prialt®’s wider clinical use is impeded by its relatively high cost, narrow therapeutic window, and by the requirement for an intrathecal pump63, 67
Another ion channel that has been suggested as a pain target based on genetic studies is the sodium channel NaV1.727, 28 μ-theraphotoxin-Pn3a, a peptide from the South American tarantula Pamphobeteus nigricolor (Fig. 2A) blocks Nav1.7 with picomolar affinity and selectivity over other NaV channels68. The peptide is analgesic in rodent pain models when co-administered with sub-therapeutic doses of opioids, but displays no analgesic activity when administered on its own68. A broad lack of analgesic activity was also found for the selective NaV1.7 inhibitors PF-04856264 and phlotoxinI68. Amgen recently engineered AM-8145 and AM-0422, two peptides based on JzTx-V toxin that selectively inhibit NaV1.769. As mentioned above, in a recent human trial the peripherally restricted, NaV1.7-selective small molecule PF-05089771 did not significantly reduce pain scores compared to placebo34. Taken together, these results might suggest that selective NaV1.7 inhibitors may need to be administered with a sub-therapeutic dose of an opioid to achieve adequate analgesia70, 71, 72
The α9α10 nictonic acetylcholine (nACh) receptor is also considered a therapeutic target for pain73. The α-conotoxins Vc1.1 and RgIA from the cone snails Conus victoriae and Conus regius respectively (Fig. 2A), antagonize α9α10 nACh receptors74, 75 Agonism of GABAB receptors and resulting inhibition of CaV2.2 channels via G-protein βγ interaction is a second mechanism of action that has been suggested for these peptides76. Vc1.1 was effective in rodent models of pain77, and advanced to human phase-2 trials where it failed, possibly because it was less potent on human than rat α9α10 nACh receptors58, 63, 78, another example of the translational challenges of mammalian species differences. RgIA-4, an analogue of RgIA with high affinity for both rodent and human α9α10 nACh receptors and no activity on GABAB receptors, suppresses rodent cancer chemotherapy-induced neuropathic pain58, 63, 79 and is currently in pre-clinical development.
P2X3 receptors are being targeted with small molecules for endometriosis-associated pain (Table 2). These receptors are also inhibited by purotoxin-1 (PT1), a 35-amino acid residue peptide from the venom of the wolf spider Geolycosa sp., that slows recovery from desensitization following channel activation with ATP and reduces hyperalgesia in rat models of inflammatory pain80. Analogues of PT1 or related peptides, if optimized for specificity, potency and stability, could be advanced for pain indications.
Channel-modulating peptides for cancer therapy
Chlorotoxin (Fig. 2A), a peptide from the death stalker scorpion Leiurus quinquestriatus hebraeus, was initially reported to inhibit small-conductance chloride currents in epithelial cells, astrocytomas and gliomas81, 82, 83, 84 but later found to also bind to matrix metalloproteinase-2 (MMP2) on glioma cells85 and not block volume-, cATP- or Ca2+- activated chloride channels86. The molecular identity of the putative chloride channel blocked by chlorotoxin is currently undetermined. Human phase-1 trials a decade ago showed that a 131I-radiolabeled analog was safe following intravenous administration for high-grade glioma, but the compound was not advanced further (NCT00733798). However, there are currently efforts underway to use chlorotoxin-based fluorescent dye conjugates as so-called “tumor paint” to help visualize gliomas87, and one analog, BLZ-100 88, was tested in a Phase-1 study (NCT02234297) in 2016, with a new Phase-2/3 study about to start (NCT03579602) in pediatric patients with CNS tumors where fluorescent-labeled chlorotoxin will be assessed with an imaging system.
TRPV6 channels are over-expressed in ovarian, breast, prostate, colon and thyroid cancers, and have been implicated in tumor progression89. SOR-C13, a C-terminal truncation of a longer, 54-residue paralytic peptide called Soricidin (accession number POC2P6) from the short-tailed shrew (Blarina brevicauda), blocks TRPV6 at low nanomolar concentrations and suppresses tumors in xenograft models of ovarian and breast cancer58, 90 Fluorescent-labeled and super-paramagnetic iron oxide-conjugated analogues are able to visualize ovarian tumors in vivo in mouse models90. In a phase-1 trial in 23 patients with cancers of epithelial origin, SOR-C13 stabilized disease suggesting antitumor activity91 but caused grade 2–3 dose-related hypocalcemia and atrial fibrillation in a quarter of patients91. Soricimed, the company developing SOR-C13, currently seems to be focusing on 2nd generation peptide drug conjugates.
Channel-modulating peptides for neurological diseases
The acid-sensing ion channel 1a (ASIC1a), a key mediator of acidosis-mediated neuronal damage in cerebral ischemia, is widely regarded as a potential therapeutic target for the treatment of ischemic stroke92. π-hexatoxin-Hi1a, a peptide from the Australian funnel-web spider Hadronyche infensa (Fig. 2A), delays ASIC1a channel-activation by binding to an acidic pocket critical for proton gating of the channel93. In rodent stroke models, Hi1a attenuates brain damage and improves behavioral outcomes even when administered intracerebroventricularly 8 hours after stroke onset93. Intravenous and intranasal routes are currently being trialed in mice.
Nav1.1 is localized in fast-spiking inhibitory neurons in the brain, and epilepsy in Dravet’s syndrome, a loss of function mutation of Nav1.1, is thought to be due to reduced inhibitory neurotransmission. The peptide Hm1a from the spider Heteroscodra maculate (Fig. 2A), activates and slows inactivation of Nav1.1 at nanomolar concentrations24. In mice carrying the human R1407X nonsense mutation, Hm1a rescued the collapse of action potentials in inhibitory interneurons without affecting excitatory neurons94. Intracerebroventricular delivery of Hm1a reduced seizures and post-ictal mortality in Dravet syndrome mice94. Further development of Hm1a would require microinfusion pumps like those used for Prialt® delivery or microfluidic ion pumps that electrophoretically pump ions across an ion exchange membrane and thereby deliver the peptide “dry” without fluid95.
Channel-modulating peptides for treatment of autoimmune diseases
Potassium channels were discovered in T lymphocytes in 198496, 97 Two potassium channels, the voltage-gated KV1.3 and the calcium-activated KCa3.1 channel, promote calcium Ca2+ entry into lymphocytes through store-operated CRAC (Orai/Stim) by providing a counterbalancing cation efflux98, 99 Blockade of these channels therefore suppresses T lymphocyte activation. Differential expression of these channels allows preferential suppression of terminally-differentiated effector memory T cells (TEM), which contribute to the pathogenesis of many different autoimmune diseases, with specific KV1.3 inhibitors100. The ShK peptide from the sea anemone Stichodactyla helianthus (Fig. 2A) blocks KV1.3 with picomolar affinity but also displays high affinity for neuronal potassium channels101,102 An extensive structure-activity-relationship program led to the development of ShK-186 (Dalazatide), an analogue with picomolar affinity for KV1.3 and 100–1000-fold selectivity over related channels103, 104. Sustained high picomolar levels of ShK-186 are achieved in plasma following subcutaneous injection. Due to its long circulating-half-life, ShK-186 is effective in rodent models of multiple sclerosis, rheumatoid arthritis and atopic dermatitis when administered once every 2–3 days104,105. In phase-1 trials in healthy human volunteers, no ECG changes were observed, and no severe or life threatening adverse effects were noted. In a Phase-1b trial in patients with plaque psoriasis, Dalazatide administered twice weekly by subcutaneous injection significantly reduced the psoriasis area and severity index in nine of ten patients106. It caused temporary mild grade 1 hypoesthesia and paresthesia involving the hands, feet, or perioral area in the majority of patients106, possibly because Dalazatide is cleaved into a product with decreased KV1.3-specificity, 103,104.Plans for a phase-2 trial in the orphan disease inclusion body myositis107 are on hold due to financial constraints.
Other efforts have focused on improving in vivo pharmacokinetic properties of Kv1.3-blocking peptides. PEGylation of the scorpion peptide HsTx1[R14A] prolonged plasma circulating half-life in rodents and resulted in sustained efficacy in rodent models of multiple sclerosis and rheumatoid arthritis108. By screening a combinatorial ShK peptide library, novel analogues were identified that, when fused to the C-termini of IgG1-Fc, retained picomolar potency, effectively suppressed in vivo delayed-type hypersensitivity and exhibited a prolonged circulating half-life109. Other approaches included the engineering of a scorpion toxin into a humanized antibody to achieve picomolar affinity for Kv1.3 and long plasma half-life110, and the development of novel formulations that achieved satisfactory blood levels of peptide inhibitors following buccal or pulmonary delivery111,112. Scientists at Amgen generated the derivative ShK[Q16K] by “brute-force” structure-activity analoging and then demonstrated sustained inhibition of plasma cytokine levels in primates with weekly administration of a PEG-conjugated version as well as efficacy in a rat model of multiple sclerosis113, while a group at Janssen created half-life-extending Fc or albumin fusion proteins114 with the α-KTx3 scorpion toxin OsK1 (α-KTx3.7) and tested them in minipigs. However, both companies subsequently seem to have dropped KV1.3 as a target based on the previously made observation200, that the efficacy of KV1.3 blockers depends on the strength of T-cell stimulation and that KV1.3 inhibition is therefore immunomodulatory rather than immunosuppresssive115,116
Other channel-modulating peptides
The μ-conotoxin CncIIIC from the cone snail Conus consors (Fig. 2A), a blocker of Nav1.4 sodium channels with myorelaxant and analgesic properties117, is marketed by Activen as XEP-018, a topical cosmetic cream to reduce periocular wrinkles58, 118 SYN-AkeTM, an analogue of the peptide Waglerin-1 from the Southeast Asian Temple viper Tropidolaemus wagleri, blocks muscle nicotinic acetylcholine receptors and modulates GABAA receptors119, and is being developed as an alternative dermaceutical to BoTox™ for wrinkles58.
Lastly, αCTI (ACT1), a peptide based on the last nine amino acids of the C-terminus of connexin 43, a gap junction protein often found at the edge of wounds and associated with poor wound healing102, has been shown in multiple Phase-2 trials to reduce mean ulcer area for chronic neuropathic foot121 and venous leg ulcers122. Granexin® gel is currently being tested in a Phase-3 trial (NCT02667327) for diabetic foot ulcers, which affect roughly 15% of the diabetic population globally.
Extending the plasma half-life of peptides
One weakness of peptide-based drugs is their short in vivo half-life due to their instability and their rapid renal elimination. The first drawback can be overcome by increasing the effective molecular mass of the peptide via conjugation to create so called “biobetters”123. As discussed for several examples above, ion channel-targeting peptides have been conjugated to polyethylene glycol (PEG), to large proteins (e.g. human serum albumin), or protein domains (e.g. antibody Fc domain) or engineered into the complementary-determining region of humanized antibodies to generate fusion proteins129. Strategies to enhance peptide stability include peptide backbone cyclization, disulfide-bridge modification, residue substitutions, peptide stapling125, and computational design of hyper-stable constrained peptide scaffolds126. Backbone cyclization of the α-conotoxin RgIA enhanced its stability in human serum without perturbing the structure or function of the peptide127,128,129,130. However, cyclization of APETx2 decreased its activity on the ASIC3 channel while enhancing stability131. Modification of disulfide bridges is another approach to enhance stability. In the α-conotoxin Vc1.1, disulfide bridges have been replaced by non-reducible dicarba linkages, or have been eliminated and the core of the peptide stabilized by residue-substitutions near the removed bridges132,133. Both strategies improved stability and oral bioavailability of Vc1.1, but reduced activity. Stapling technologies have also been successful for stabilizing peptides that target N-methyl-D-aspartic acid receptors134. Thus, several post-translational methods can be used to augment stability, improve circulating half-life, and even achieve oral bioavailability. The need for half-life extension, however, will depend on a number of factors, including the route of administration, nature of the disease, and whether the drug is intended for acute or chronic use. Some venom peptides are exceedingly stable in plasma, while others depot after cutaneous injection (e.g. Dalazatide104). For chronic diseases such as diabetes and persistent pain, daily injectables are likely to be tolerated by patients and therefore half-life may not be as critical. Exenatide with an elimination half-life of 140 minutes requires twice daily injection in diabetic patients and has peak sales approaching $1 billion/year.
Using peptide-channel interactions to guide the design of peptidomimetics or small molecule inhibitors
Many groups have exploited the understanding of peptide-channel interactions to design channel-modulating peptidomimetics. The earliest attempt to design a peptidomimetic of the sea anemone ShK peptide resulted in a million-fold loss in potency against KV1.3135. More recently developed peptidomimetic 4-arm ethylene glycol-conjugated star polymers based on scorpion toxins have achieved nanomolar potency against KV1.3136. Peptidomimetics have also been designed to target connexins, neuronal CaV2.2 channels137,138 and the Cavβ2 subunit of cardiac L-type (CaV1.2) calcium channels139.
In the 1990s, high-throughput screens using [125I]-charybdotoxin competitive binding assays led to the discovery of several small molecule inhibitors (WIN-17317–3, CP-339,818) with nanomolar potency against the KV1.3 channel140,141,142. However, while charybdotoxin bound in the outer vestibule of the channel, these small molecules likely bound in the inner chamber below the selectivity filter. Displacement of the radiolabeled peptide by these small molecules was due to a transpore effect. Similar transpore inhibition of [125I]-charybdotoxin competitive binding to the large-conductance KCa1.1 channel was seen with indole diterpenes143. Radiolabeled-peptide toxin binding assays have subsequently fallen out of favor for screening and more recent high throughput screens have used thallium flux or membrane potential- or calcium-sensing dyes.
Ion channel targeted antibodies and nanobodies
There are currently more than 60 approved therapeutic antibodies on the market, predominantly in the areas of oncology, autoimmunity and inflammatory disease, and approximately 550 antibodies are estimated to be in various stages of clinical development144. However, ion channel antibody discovery and development has lagged far behind with only one antibody, a sheep polyclonal (BILO10t) targeting a non-functional form of P2X7 from Biosceptre formulated as a topical ointment for the treatment of basal cell carcinoma having reached human clinical trials thus far145.
While venom peptides have long been recognized to be superior to small molecules in terms of their potency and selectivity for ion channels, targeting channels with equally selective antibodies has more recently generated intense interest, mostly driven by the highly desirable pharmacokinetic characteristics of immunoglobulins compared to peptides resulting in a much lower dosing frequency. However, antibodies also have unique properties in terms of their mode of action. Small molecules and peptides usually act by directly blocking ion flux through the open channel or by interfering with the gating processes, which can be both positive (activators) or negative (inhibitors), and therefore affect channel function on the micro- to millisecond time scale (e.g. use-dependent inhibition of NaV channels by classical small molecule anti-epileptics). Antibodies can also work by occlusion of the ion channel pore or induce allosteric-induced gating effects, but in addition they can be engineered to either lack or possess enhanced Fc-mediated functions, such as antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and FcRn-mediated transcytosis. Antibodies can further carry toxic or radioactive payloads to target disease cells144,146 Thus, antibodies can potentially exert their modulating effect through one, or a combination of mechanistic actions, including, Fc-mediated depletion, internalization of ion channel-antibody complexes147, and/or modulation of ion channel behavior with a conjugated peptide or small molecule toxin - features that may also be enhanced by bivalent avidity effects.
The above mentioned role of anti-ion channel autoantibodies in autoimmune diseases148 and success in generating functional polyclonal antibodies against multiple ion channel targets149 provided strong evidence for the feasibility of generating functional monoclonal antibodies (mAbs). However, overall progress in ion channel mAb discovery has been slow due to significant technical challenges that appear to be somewhat target specific. Indeed, ion channels and other multi-pass membrane proteins have recently been ranked among the “high-hanging fruit” in terms of antibody drug discovery144. For example, while many ion channels share common structural motifs based around transmembrane and pore-forming domains, the surface topography of ion channels where antibodies would be predicted to bind and exert their effect (the extracellular epitope target area) can be quite different amongst ion channel families. For example, members of the acid sensing ion channel (ASIC) and the purinoceptor P2X family contain extracellular amino acids that constitute approximately 68% or 48%, respectively, of the total protein3. Conversely, voltage-gated ion channels2 such as Nav family members have far fewer amino acids exposed to the extracellular space (approximately 15%). Therefore, it is not surprising that reports of successful monoclonal antibody programs have tended to describe the discoveries of antibodies targeting the former and not the latter. Further complicating antibody discovery against these targets is the relative lack of immunogenicity of small surface loops, conservation amongst orthologs leading to tolerance in host immune animals and a relative lack of robust sources of recombinant ion channel protein to enable large scale antibody discovery programs.
Nevertheless, several strategies have been successful in generating monoclonal antibodies that inhibit ion channel function with the simplest approaches using antigenic peptides derived from ion channel extracellular (ECL) domains. A particularly popular tactic has been targeting the pore-forming E3 loop of 6-transmembrane domain KV, NaV, or TRP channels using peptide antigens with the rationale that antibody binding to this region will inhibit ion flow. This approach has been mostly successful in generating polyclonal antibodies that block channel function149 although it has also been used to generate functionally blocking mAbs targeting KV10.1150 or the T cell calcium influx channel Orai1. Using a peptide from the second extracellular loop of Orai1 a group at Amgen generated while a group at Novo Nordisk produced mAbs that, while a group at Novo Nordisk produced mAbs that 151inhibit calcium influx in T cells, suppress T cell proliferation and cytokine production, and showed efficacy in a graft-versus-host disease mouse model induced by human T cell transfer presumably by inducing Orai1 internalization125. Channel internalization also is the mechanism of action of a mAb generated against purified peptide antigens from the first ECL domain of hK2P9.1 (KCNK9) that inhibits tumor growth and metastasis in mouse cancer xenograft models153. However, there appear to be clear limitations for isolating mAbs using peptide-derived strategies presumably due to the fact, that peptide antigens, while abundant and inexpensive, typically are not representative of native structures. This challenge can be mitigated by employing immunization strategies that use ion channel antigens that contain a native fold. For example, mAbs with blocking properties have been generated by immunizing host animals with DNA (ASIC1154, KV1.3155), cells expressing target ion channels (P2X7156, Orai1151), ion channel containing virus like particles (P2X3157), and recombinant purified ion channels (P2X3147, KV1.3158). In an alternative approach, Quiang et al. generated a mAb that blocks ASIC1a currents with an IC50 of 85 nM and reduces infarction in a rodent stroke model by panning a human scFv combinatorial antibody phage library with nanodiscs containing reconstituted truncated ASIC1a protein and then converting into a full-length IgG1 form159.
The difficulty in discovering clinically relevant conventional ion channel mAbs has led academic and industry investigators to explore alternative modalities. For example, the variable domain of heavy chain only camelid antibodies, or nanobodies160, are small, 12–15 kDa, modular immunoglobulins that can offer distinct advantages over conventional mAbs such as access to epitopes that otherwise would be difficult to reach with conventional immunoglobulins161,162 Additionally, the modular nature of their small singular domains allows for relatively straightforward engineering161 of homo- and heteromeric molecules that can increase their potency, avidity, bispecificity and lead to half-life extension (HLE). Several examples of ion channel modulating nanobodies, including bispecific ones, have now been described including those targeting P2X7 and Kv1.3 (see below for further details).
Other modalities that are garnering considerable interest include KnotBodies, which incorporate cystine-knot proteins like venom toxins110 into the complementary-determining region (CDR) of the variable domain of an antibody light chain and antibodies complexed with warheads or small molecule moieties. While these fusion molecules differ from traditional antibodies where target binding is solely CDR driven, they nevertheless, represent a step-forward in the development of potential ion channel therapeutics by combining the potency of toxins with the therapeutic advantages afforded by mAbs. Another, largely unexplored group of biologics in terms of targeting and modulating ion channel function are various non-immunoglobulin protein-binding scaffolds characterized by their relatively small mass (~6–20 kDa). These include DARPins (designed ankyrin repeat proteins), affilins (ubiquitin), anticalins (lipocalin), atrimers (C-type lectin), monobodies (fibronectin type 111), Kunitz domains (serine protease inhibitor) and affibodies (staphylococcal protein A domain Z)163.As of 2015 more than 20 scaffold-derived candidates were in preclinical or clinical development with one product derived from a Kunitz domain, the kallikrein inhibitor Ecallantide (Kalbitor®), on the market for the treatment of hereditary angioedema163. While none of the identified clinical candidates target ion channels, there is evidence suggesting that these large and diverse scaffold libraries could provide fertile ground for identifying novel ion channel binders. For example, monobodies selected from combinatorial libraries were shown to bind and block two bacterial Fluc-type fluoride channels164, while high-affinity DARPins have been co-crystallized in complex with AcrB, the inner membrane pump of the E. coli multi-drug resistance tripartite complex AcrAB-TolC165.
Case study: P2X7 as a target for cancer therapy and inflammation
The lone ion channel targeting immunoglobulin formulation that is currently in clinical development is BIL010t145 from Biosceptre. BIL010t is a topical therapy for basal cell carcinoma (BCC) that contains sheep polyclonal antibodies directed against a non-functional form of the purinergic receptor channel P2X7. This form, called nfP2X7, represents a distinct conformation with a non-functional pore that is upregulated in response to high-ATP concentrations in tumor micro-environments and is required for tumor cell survival166. Moreover, the P2X7 E200 peptide sequence (G200HNYTTRNILPGLNITC216) whose conformation is distinct and exposed in nfP2X7 but not in WT P2X7 has enabled the generation of polyclonal and monoclonal antibodies that selectively bind nfP2X7 on the surface of tumor cells166. A PEG-based topical ointment containing polyclonal antibodies purified from sheep repeatedly immunized with the E200 peptide conjugated to keyhole limpet hemocyanin caused significant reduction in B16F10 tumor growth in an orthotopic mouse model of melanoma145. In a Phase-I clinical trial (NCT02587819) an ointment containing 10% BIL010t was applied to primary BCC lesions twice daily for 28 days. The treatment was well-tolerated, did not result in systemic penetration of sheep polyclonal antibodies and resulted in a decrease in lesion size in 65% of the patients with 20% showing no change and 15% showing an increase in size145. In addition to BIL010t, Biosceptre is developing BIL03s, an anti-nfP2X7 human monoclonal antibody for treatment of solid and hematological tumors.
A monoclonal antibody targeting human P2X7 was generated following immunization of mice with a mouse myeloma cell line, XS63, expressing P2X7 and screening hybridoma cell line supernatants by flow cytometry using transfected and non-transfected XS63 cells156.. One mAb that specifically reacted with HEK cells expressing P2X7 but not cells expressing P2X1 or P2X4 also recognized native P2X7 in human monocytes that had been differentiated into macrophages with LPS or γ-IFN. The anti-P2X7 mAb was shown to block BzATP-induced inward currents in HEK cells transfected with human P2X7 (IC50~5 nM), but not mouse or rat orthologues156.
Anti-mouse P2X7 nanobodies with antagonistic (13A7, IC50 = 12 nM) or potentiating activity (14D5, EC50 = 6 nM) were isolated from phage libraries derived from llamas immunized with either HEK cells stably expressing P2X7 or cDNA167. The potencies of the blocking and the enhancing nanobodies increased upon multimerization. A dimeric-HLE version of 13A7 that additionally contains the albumin-binding nanobody Alb8 to extend serum half-life was effective in ameliorating both allergic contact dermatitis and experimental glomerulonephritis in mice167. Additionally, another nanobody called Dano1, that specifically recognizes human P2X7, blocked ATP-induced Ca2+ influx and pore formation in P2X7-expressing HEK cells with dimerization leading to increased potency (IC50 = 0.2 nM). Dano1 also inhibited inflammasome assembly by LPS-primed human monocytes as well as the shedding of CD62L and the externalization of phosphatidylserine by T cells. Interestingly, Dano1 was significantly more potent (20–50 fold) than the previously described conventional anti-P2X7 mAb167.
Case study: Kv1.3 as a target for autoimmune disease
In addition to the validation accompanying KV1.3 as a therapeutic target for autoimmune disease100,106,168 its accessibility to antibodies in autoreactive effector memory T-cells makes it a particularly strong ion channel target for therapeutic intervention with immunoglobulins. This, may explain, in part, why KV1.3 has been a popular target for the discovery and development of immunoglobulin-based molecules amongst academic and biotech researchers with examples describing polyclonal and monoclonal (conventional and nanobody) antibodies as well as KnotBodies (Fig. 3), that inhibit ion channel function and alleviate T-cell mediated autoimmune disease in animal models. For example, a polyclonal antibody (E314) purified from the sera of rabbits immunized with a 14-amino acid E3 peptide antigen directed against the hKV1.3 S5-S6 loop specifically bound to HEK cells expressing KV1.3 but not cells expressing other KV family members169. The E314 antibody was also shown to inhibit KV1.3 currents by approximately 90% at a concentration of 300 nM in transfected HEK cells and Jurkat T cells but showed no significant effect on KV1.1, KV1.2, KV1.4, KV1.5 or KV11.1 (hERG)169. Interestingly, a KV1.3 vaccine was recently shown to induce high titers of anti-KV1.3 antibodies in mice and rats that lessened clinical symptoms and decreased pathological CNS damage in a model of experimental autoimmune encephalomyelitis-presumably through the action of polyclonal anti-KV1.3 antibodies170.
Figure 3.
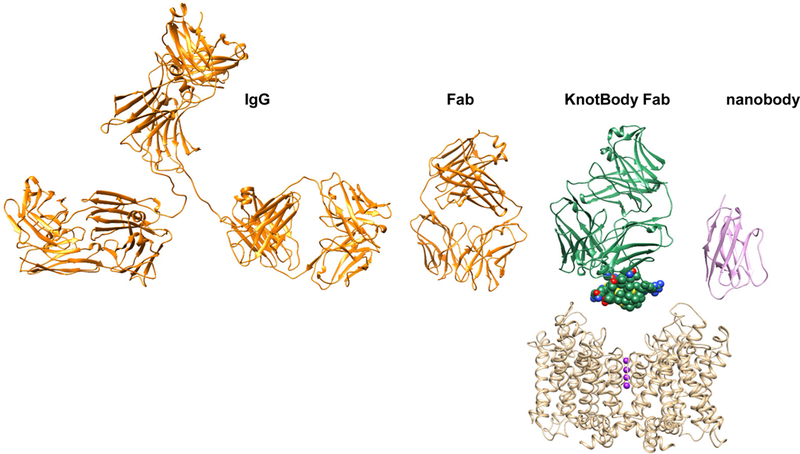
Figure illustrating the size relation of a full-length immunoglobulin G (IgG2a, pdb: 11GT, ribbon presentation) colored in orange, an antigen-binding fragment (Fab, pdb: 1K4C) colored in orange, a KnotBody Fab colored in green, and a nanobody (pdb: 6C5W) colored in pink. The Ecballium elaterium Trypsin Inhibitor (EETI-II) structure within the KnotBody is shown in space-filling representation. The Kv channel structure is based on the Kv1.2-Kv2.1 chimera196 (pdb: 2R9R) and shown in ribbon representation and colored in beige. Potassium ions within the selectivity filter region are shown in sphere representation and colored in purple.
Ablynx, now a Sanofi company, have described the generation of anti-KV1.3 nanobodies isolated from llamas using a genetic prime-boost immunization strategy. KV1.3 nanobodies were found to be selective (10,000-fold over Kv1 family members), state-dependent and demonstrated varying functional profiles. The fast onset of functional effects strongly indicated that they were due to channel inhibition and not to internalization mechanisms155. Construction of bivalent hetero- and homodimers resulted in molecules with mixed functional properties in the case of the former, namely increased potencies, target residence time, avidity effects and duration of blockade. Additionally, a trimeric nanobody displayed higher potency in T cell based assays. A nanobody comprising two identical anti-KV1.3 nanobody monomers fused to an anti-albumin moiety to increase half-life was effective in vivo in reducing ear thickness in a rat delayed hypersensitivity model155. More recently, conventional full-length anti-KV1.3 mAbs have been isolated from chickens and llamas using a similar prime-boost immunization and screening strategy158. Chickens were immunized and boosted with purified recombinant KV1.3 reconstituted into liposome formulations and anti-KV1.3 antibodies identified and cloned following B-cell screening using a gel encapsulated microenvironment (GEM) assay incorporating various KV1.3 containing formulations including liposomes and oriented channel adhered to magnetic beads158. In the case of llamas, initial immunizations were carried out with DNA and boosts with KV1.3 liposomes. Phage libraries derived from immunized llamas were screened using KV1.3 bound to magnetic beads. In all, 69 specific anti-KV1.3 scFv-Fc antibodies were isolated from both host-animal platforms with 10 antibodies (9 chicken and 1 llama) demonstrating functional block of KV1.3 current. Characterization of select scFv-Fc clones demonstrated that current block was time and concentration dependent with the most potent clone having an IC50 of 6 nM. Antibodies showed no activity against related family members (KV1.1, KV1.2, KV1.5), hERG or NaV1.5. Epitope binning analysis revealed that while most (7 of 10) functional antibodies segregated to one bin suggesting a dominant functional epitope, 3 antibodies segregated to different bins indicating that functional block could be achieved through distinct epitope binding events.
In an alternative approach IONTAS, a company developing a KnotBody platform based on incorporating peptide toxins with a conserved inhibitory cysteine knot (knottin) structural motif into peripheral antibody CDR loops (Fig. 3), have generated a ShK-KnotBody with a reported KV1.3 inhibitory IC50 of 8.6 nM171. Using a similar approach, a group at the Scripps Research Institute previously incorporated the KV1.3 inhibitory scorpion peptide toxins Moka1 and Vm24-toxin into the CDR3H domain of a humanized anti-lysozyme antibody with high structural similarity to a bovine antibody and into the CDr2h (Moka1) and CDR3L (Moka and Vm24) domains of the humanized RSV-neutralizing antibody Syn110. The syn-Vm24-CDR3L antibody fusion demonstrated subnanomolar potency (IC50 = 0.59 nM) in a K+ flux assay, inhibited activation of αCD3 stimulated TEM cells with an IC50 of 1 nM, displayed a long serum half-live of approximately 2 days, and showed dose-dependent inhibition of delayed type hypersensitivity reactions in vivo in rats.
Case study: NaV1.7 as a target for pain
A monoclonal antibody, SVmab1, with purported nanomolar potency (IC50 31 nM) was generated in mice using a peptide targeting the NaV1.7 S3-S4 extracellular loop region in the domain II (DII) voltage-sensor172. Using antibody purified from hybridoma cultures, Svmab1 exhibited state-dependent inhibition of NaV1.7, 400–1500-fold selectivity over most NaV1.X family members, efficacy in both inflammatory and neuropathic pain models in mice and reduced scratching in both acute and chronic itch models172. However, a subsequent study using a recombinant form of the antibody (rSVmab) generated from published light and heavy chain antibody sequences was unable to bind neither the S3-S4 antigen peptide, a soluble and purified DII voltage sensor protein nor the mammalian cell expressing NaV1.7. Neither was rSvmab able to specifically block Nav1.7 current in transfected HEK293 cells173. Interestingly, the discoverers of Svmab confirmed the apparent discrepancies between the functional hybridoma-derived and non-functional recombinant forms of the mAb and speculated that the difference may have resulted from incorrect disclosure of antibody sequences or possibly differences in post-translational modifications of antibodies produced in hybridomas compared to transfected mammalian cells174. It will be of considerable interest to learn if other researchers utilizing a similar DII voltage-sensor targeted strategy can identify mAbs with similar functional properties to Svmab.
In an approach distinct from the CDR-targeted KnotBody strategy110, a Nav1.7 toxin-antibody conjugate was recently described that maintained the selective potency of the toxin warhead while conferring significant half-life extension and biodistribution to nerve fibers175. In their study, Biswas et al. describe assembling a conjugate comprised of a nontargeting anti-2,4-dinitrophenol human IgG1 with a cysteine mutation at a surface residue (excluding CDR, effector binding domains, proline and glycine residues) attached to the tarantula venom GpTx-1 peptide toxin via a PEG11 linker175. In a combinatorial approach several factors including cysteine mutation sites of the carrier antibody, linker chain-length, conjugation chemistry and peptide loading were evaluated for their effect on Nav1.7 current inhibition. One antibody conjugated at substituted cysteine E384C exhibited a 30-fold loss in potency compared with naked peptide (IC50 250 nM vs 8.5 nM), but had a serum half-life of 80 h, about 130-fold longer than the naked peptide. Following intravenous administration, the GpTx-1 mAb conjugate biodistributed to mice dorsal root and sciatic nerve better than the parent mAb175.. Additionally, distribution of the conjugate to dorsal root and sciatic nerve was significantly more pronounced in WT mice compared to Nav1.7 knockout mice indicating that both the presence of Nav1.7 and the peptide toxin were required for distribution to peripheral nerve elements across the so-called blood nerve barrier (BNB). While the conjugate was not effective in vivo in a mouse histamine-induced pruritis model, most likely due to plasma concentrations that were not sufficiently greater than measured IC50s, the data demonstrate the effective combination of Nav inhibitory properties of a toxin with the desired pharmacokinetic characteristics of an antibody.
Ion channel antibodies going forward
In many respects the ion channel antibody field is playing “catch-up” with its GPCR cousin with the latter able to boast three marketed antibodies, the anti-CCR4 antibody mogamulizumab for the treatment of Adult and Peripheral T cell lymphoma and two anti- CGRP antibodies (erenumab and fremanezumab) for migraine; there are also at least fifteen other immunoglobulins in various phases of clinical development and more projects at the preclinical stage19. The challenges associated with developing GPCR antibodies have been described in detail elsewhere19, 176, 177; suffice it to say, that in cases where functional antibodies are desirable, the GPCR and the ion channel field face the same difficulties of developing antibodies that recognize specific conformational states of the target protein (e.g. the active versus the inactive state of a GPCR or the open versus the inactivated state of an ion channel, see Fig. 1A). However, attributing the discrepancy in the current development status of GPCR and ion channel antibodies to the comparative difficulty of targeting ion channels versus GPCRs would not sufficiently acknowledge the significant advancements made in the GPCR field. These include technologies like StaR® (Stabilized Receptor), where a small number of point mutations are introduced into a GPCR to improve its thermostability without disrupting its pharmacology 178, 179. Heptares Therapeutics is using this technology not only to generate protein for x-ray crystallography and small molecule screening, but also to enable purification of high-quality, functional and monodisperse protein that can be incorporated into effective antigen and screening formulations for both in vivo immunization and in vitro antibody discovery platforms. Instead, the proliferation of clinical antibody candidates during the last 10 years against a difficult class of membrane proteins like GPCRs should provide a level of optimism that similar advancements in ion channels are also within reach.
Overcoming the aforementioned challenges for advancing the ion channel antibody pipeline will undoubtedly involve integrating next generation platforms into the discovery process. For example, limitations of traditional approaches such as hybridoma screening, where non-efficient fusion events and subsequent loss of rare B cells can impact immune diversity180, can be mitigated by technologies that incorporate direct B-cell cloning allowing the identification of rare clones and the recovery of natively paired light and heavy chain genes119. Similarly, traditional display technologies such as phage and yeast-display are being further enhanced by next-generation sequencing that allows up to 10,000-fold more sequences than the Sanger method and is enabling deeper interrogation of library diversity and identification of rare clones181, 182. Parallel advances in recombinant ion channel production using alternative expression hosts (e.g. Tetrahymena thermophila), cell-free systems and non-detergent based purification methods and formulations leading to increases in the quality (native-fold) and quantity (>mg) of purified protein or following the development of protocols enabling the production of possibly even stable, state-specific channels will likely complement newer approaches to antibody discovery and therefore increase the chances of successfully recovering antibodies with desired properties. With nanobodies it might even be feasible to target intracellular ion channel domains (see Box 2).
Text Box 2.
Targeting ion channel cytoplasmic epitopes with nanobodies
Immunoglobulins are not able to passively access the cell cytoplasm. Therefore, ion channel antibody discovery programs, like those directed towards other cell surface molecules, are generally designed to identify antibodies that recognize extracellular epitopes to exert their functional effect, be it steric or allosteric block, or removal of a channel from the cell surface by internalization mechanisms. Consequently, the intracellular epitope space, which is considerably larger than that displayed on the surface for many voltage-gated like ion channels (e.g. ~70% of the sodium activated K+ channel KNa1.1), is left untargeted. In a recent review, Ingram et. al162 highlighted the attributes of nanobodies (Nbs) that may allow exploiting the cytosolic side of membrane proteins such as GPCRs or ion channels for basic research, immunodiagnostics and potentially therapeutic intervention.
Nbs are single domain antibodies comprised of the variable domain of VHH camelid IgGs. Nbs are small (~15kDa) and modular in nature, bind their targets with affinities similar to conventional antibodies186 and tend to recognize epitopes in clefts of protein surfaces or at protein-interaction interfaces187. Like conventional antibody light and heavy chain variable domains, Nb antigen binding is driven by three complementarity-determining regions (CDRs). However, unlike conventional antibodies that require disulfide-paired light and heavy chain variable domains to form a paratope188, this is achieved in Nbs in a single domain. Therefore, for Nbs where intrachain disulfide bonds are not necessary for maintaining target specificity this attribute imparts a significant tolerance, compared to conventional antibodies, to reducing environments like the cytoplasm where Nbs can be expressed in functional form, bind cytosolic epitopes and exert a functional effect.
Cytosolic expression of Nbs has been exploited in a number of phenotypic screens to identify functional hits and further explore cellular processes. In one example, a randomized VHH-CDR3 intracellular antibody library was introduced into highly metastatic human fibrosarcoma derived cells followed by iterative rounds of cell migration assays to identify clones associated with non-migrating cells189. Using this approach one nanobody clone was used to identify heterogenous nuclear ribonucleoprotein K (hnRNP-K) as having a role in metastasis189. In another instance VHH coding sequences derived from alpacas immunized with inactivated Influenza A virus (IAV) or vesicular stomatitis virus (VSV) were cloned into a lentiviral vector and used in a phenotypic screen to identify intracellularly expressed Nbs that protected human A549 cells from lethal infection with either IAV or VSV190. In addition to phenotypic screens, Nbs have been engineered to selectively degrade target molecules via ubiquitination by fusion to ubiquitin ligase F-box subunits191, 192 and have served as chaperones that bind to the intracellular surface of GPCRs to stabilize active conformations such as the p2-adrenergic and M2 muscarinic receptors that enabled structural determination by x-ray crystallography193,194
It remains to be determined whether targeting intracellular domains of ion channels with nanobodies will provide the same molecular insights into stabilized state- dependent structures as they have with GPCRs or whether therapeutic applications like gene therapy that result in the expression of anti-ion channel Nbs for functional disruption or degradation of targeted ion channel will move beyond speculation. Nevertheless, the propensity for Nbs to recognize epitopes in protein-protein interfaces raises the intriguing possibility of investigating the biological mechanisms of ion channel complexes. The alpha subunit comprising the ion channel pore typically represents only one of a number of regulatory and auxiliary proteins in a complex that influence channel function, trafficking, distribution and signaling. Indeed, several channelopathies are associated with mutations thast disrupt ion channel complex formation and cause cardiac arrhythmias195. Induced expression of Nbs raised against ion channel interaction domains may help de-convolute complex formation under varying physiological conditions, illuminate the precise temporal and spatial role of complex components and possibly identify novel targets for therapeutic intervention. New insight into the cytosolic face of ion channel biology may simply await the design of elegant phenotypic screens utilizing intracellular nanobody libraries as described above.
Overall perspective and concluding remarks
As seen from the foregoing paragraphs, ion channel drug discovery/development is approaching a cross road where future breakthroughs and new market introductions are likely to increasingly rely on biologic modalities comprising the plethora of technical opportunities based on peptides, antibodies and hybrid molecules. Given the diversity of solutions and the current speed of innovation across academia and the pharmaceutical industry, it is difficult to pinpoint exactly which therapeutic areas will gain the most and the fastest from this development, but a conservative view is that it will largely follow the accessibility of the cellular targets. Since injected peptides and antibodies usually have quite low “volumes of distribution”, meaning that they tend to stay in the blood stream rather than penetrating deeply into tissues and cells, it is likely that hematological diseases, certain cancer forms, and immune diseases are straightforward therapeutic areas, whereas CNS diseases are much more challenging due to the tight blood brain barrier. (This view is supported by a parallel development in the field of therapeutic antibodies targeting extracellular proteins, where peripheral anti-cytokine therapy is very established and successful for treatment of autoimmune diseases, whereas attempts to treat Alzheimer’s disease with antibodies against amyloid beta are lacking behind). That said, between these opposite poles there are many opportunities defined by no or less tight endothelial barriers towards other organs (e.g. bone marrow, liver), by accessibility via local administration (e.g. lungs, skin), by administration via specific routes (e.g. intrathecal) or even by uptake across disease damaged barriers, such as the blood brain barrier in stroke and the intestinal epithelium in inflammatory bowel diseases. Considering the ion channel targets themselves it is a common view that channels with prominent extracellular loops (like GABAA, nicotinic acetylcholine and P2X receptors or two pore domain K+ channels) are more tractable by antibodies than channels with restricted extracellular exposure, However, as discussed here it is indeed possible to generate function-blocking antibodies against ion channels with limited extracellular epitopes (e.g. Kv1.3 and Orai).
While we believe that the field of ion channel targeted biologics, which at the moment can only boast the intrathecally administered Cav2.2 blocking peptide Ziconotide in the clinic, and a polyclonal antibody and three peptides in clinical trials, is very likely to develop and expand in the next decade, it is currently hard to predict whether venom-derived peptides or antibodies will be more successful. Peptide design is currently benefitting tremendously from the advances in ion channel structural determination and molecular modeling which together are enabling structure-based or at least structure-assisted peptide design. However, “naked” peptides typically have short circulating half-lives and, depending on whether they are intended for acute or chronic use, might require some of the half-live extending modifications or formulation approaches described here to be considered suitable for clinical development. For traditional mAbs the main challenges in our opinion will be to select the most suitable ion channel targets and then tailor the antibody characteristics to the desired therapeutic effect (e.g. inhibition or activation of channel function, channel internalization, induction of target cell apoptosis) by engineering the antibody to fit the pathophysiology of the target disease. The above discussed BIL010t antibody against a non-functional form of P2X7 that is required for tumor cell survival166 is a perfect illustration of a very clever topical use of a polyclonal sheep antibody for basal cell carcinoma where the characteristic of the antibody seem to meet the medical need.
Supplementary Material
Acknowledgements
We thank Aneesh Karatt Vellatt at IONTAS for providing the pdb file of the x-ray coordinates of a KnotBody Fab for Figure 3.
This work was supported by the CounterACT Program, National Institutes of Health Office of the Director (NIH OD), and the National Institute of Neurological Disorders and Stroke (NINDS), Grant Number U54NS079202 and NS100294 (to H.W.); and NMRC-CS-IRG (NmRc/CIRG/1 427/2015), Singapore Ministry of Education Academic Research Fund Tier 1 (2015-T1–022-047) and Tier 2 (MOE2016-T2–2-032) (to K.G.C).
Footnotes
Competing interests statement
The authors declare competing financial interests: see web version for details.
Palle Christophersen is a full-time employee of Saniona A/S. Paul Colussi, is a full-time employee of TetraGenetics Inc.
References
- 1.Alexander SP et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Overview. Br. J. Pharmacol. 174 Suppl 1, S1–S16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander SP et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: voltage-gated ion channels. Br. J. Pharmacol. 174 Suppl 1, S160–S194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexander SP et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Ligand- gated ion channels. Br. J. Pharmacol. 174 Suppl 1, S130–S159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexander SP et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Other ion channels. Br. J. Pharmacol. 174 Suppl 1, S195–S207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santos R et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakmann B & Neher E Patch clamp techniques for studying ionic channels in excitable membranes. Ann. Rev. Physiol. 46, 455–472 (1984). [DOI] [PubMed] [Google Scholar]
- 7.Hille B Ionic channels of excitable membranes, 2nd edn Sinauer Associates: Sunderland, Mass, 1992. [Google Scholar]
- 8.Hille B Ion channels of excitable membranes. Sinauer Associates: Sunderland, MA, 2001. [Google Scholar]
- 9.Kaczorowski GJ, McManus OB, Priest BT & Garcia ML Ion channels as drug targets: the next GPCRs. J. Gen. Physiol. 131, 399–405 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wulff H, Castle NA & Pardo LA Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 8, 982–1001 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patapoutian A, Tate S & Woolf CJ Transient receptor potential channels: targeting pain at the source. Nat. Rev. Drug Discov. 8, 55–68 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verkman AS & Galietta LJ Chloride channels as drug targets. Nat. Rev. Drug Discov. 8, 153–171 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zamponi GW Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discov. 15, 19–34 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Bennett PB & Guthrie HR Trends in ion channel drug discovery: advances in screening technologies. Trends Biotechnol. 21, 563–569 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Dunlop J, Bowlby M, Peri R, Vasilyev D & Arias R High-throughput electrophysiology: an emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 7, 358–368 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Goodman AD et al. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet 373, 732–738 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Vilaine JP The discovery of the selective If current inhibitor ivabradine. A new therapeutic approach to ischemic heart disease. Pharmacol, Res, 53, 424–434 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Van Goor F et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 106, 18825–18830 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutchings CJ, Koglin M, Olson WC & Marshall FH Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat. Rev. Drug Discov. 16, 661 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Hann MM & Keseru GM Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov. 11, 355–365 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Stepan AF et al. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 24, 1345–1410 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Catterall WA Voltage-gated sodium channels at 60: structure, function and pathophysiology. J. Physiol. 590, 2577–2589 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catterall WA Forty Years of sodium channels: Structure, function, pharmacology, and epilepsy. Neurochem Res. 42, 2495–2504 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osteen JD et al. Selective spider toxins reveal a role for the Nay1.1 channel in mechanical pain. Nature 534, 494–499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salvatierra J et al. Nay1.1 inhibition can reduce visceral hypersensitivity. JCI Insight 3 (2018). Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Lera Ruiz M & Kraus RL Voltage-gated sodium channels: Structure, function, pharmacology, and clinical indications. J. Med. Chem. 58, 7093–7118 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Cox JJ et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cummins TR, Dib-Hajj SD & Waxman SG Electrophysiological properties of mutant Nay1.7 sodium channels in a painful inherited neuropathy. J. Neurosci. 24, 8232–8236 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faber CG et al. Gain-of-function Navy1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 109, 19444–19449 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feller L, Khammissa RAG, Fourie J, Bouckaert M & Lemmer J Postherpetic neuralgia and trigeminal neuralgia. Pain Res. Treat. 2017, 1681765 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahuja S et al. Structural basis of Nay1.7 inhibition by an isoform-selective small-molecule antagonist. Science 350, aac5464 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Alexandrou AJ et al. Subtype-selective small molecule inhibitors reveal a fundamental role for Nay1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PLoS ONE 11, e0152405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kornecook TJ et al. Pharmacologic characterization of AMG8379, a potent and selective small molecule sulfonamide antagonist of the voltage-gated sodium channel Nay1.7. J. Pharmacol. Exp. Ther. 362, 146–160 (2017). [DOI] [PubMed] [Google Scholar]
- 34.McDonnell A et al. Efficacy of the Nay1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain 159, 1465–1476 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Focken T et al. Design of conformationally constrained acyl sulfonamide isosteres: Identification of N-([1,2,4]triazolo[4,3-a]pyridin-3-yl)methane-sulfonamides as potent and selective fiNav1.7 inhibitors for the treatment of Pain. J. Med. Chem. 61, 4810–4831 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Bankar G et al. Selective Nav1.7 antagonists with long residence time show improved efficacy against inflammatory and neuropathic pain. Cell Rep. 24, 3133–3145 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Casula MA et al. Expression of the sodium channel β3 subunit in injured human sensory neurons. Neuroreport 15, 1629–1632 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Berta T et al. Transcriptional and functional profiles of voltage-gated Na+ channels in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Mol. Cell. Neurosci. 37, 196–208 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Sokolov MV et al. Co-expression of β-subunits with the voltage-gated sodium channel Nav1.7: the importance of subunit association and phosphorylation and their effects on channel pharmacology and biophysics. J. Mol. Neurosci. (2018). Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 40.Yan Z et al. Structure of the Nav1.4-β1 complex from electric eel. Cell 170, 470–482 e411 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Pan X et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with β1. Science (2018). Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 42.Deuis JR et al. Analgesic effects of GpTx-1, PF-04856264 and CNV1014802 in a mouse model of Nav1.7-mediated pain. Toxins (Basel) 8, E78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanguinetti MC & Tristani-Firouzi M hERG potassium channels and cardiac arrhythmia. Nature 440, 463–469 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Wang W & MacKinnon R Cryo-EM Structure of the open human Ether-a-go-go-related K+ Channel hERG. Cell 169, 422–430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vandenberg JI, Perozo E & Allen TW Towards a structural view of drug binding to hERG K+ channels. Trends Pharmacol. Sci. 38, 899–907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casida JE & Durkin KA Neuroactive insecticides: targets, selectivity, resistance, and secondary effects. Annu. Rev. Entomol. 58, 99–117 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Nieva JL, Madan V & Carrasco L Viroporins: structure and biological functions. Nat. Rev. Microbiol. 10, 563–574 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hay M, Thomas DW, Craighead JL, Economides C & Rosenthal J Clinical development success rates for investigational drugs. Nat. Biotechnol. 32, 40–51 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Gilhus NE et al. Myasthenia gravis - autoantibody characteristics and their implications for therapy. Nat. Rev. Neurol. 12, 259–268 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Titulaer MJ, Lang B & Verschuuren JJ Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 10, 1098–1107 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Bradl M, Reindl M & Lassmann H Mechanisms for lesion localization in neuromyelitis optica spectrum disorders. Curr. Opin. Neurol. 31, 325–333 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holz GG & Chepurny OG Glucagon-like peptide-1 synthetic analogs: new therapeutic agents for use in the treatment of diabetes mellitus. Cur. Med. Chem. 10, 2471–2483 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fosgerau K & Hoffmann T Peptide therapeutics: current status and future directions. Drug Discov. Today 20, 122–128 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Al Musaimi O, Al Shaer D, de la Torre BG & Albericio F 2017 FDA peptide harvest. Pharmaceuticals (Basel) 11, E42 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.King GF Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin. Biol. Ther. 11, 1469–1484 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Robinson SD, Undheim EAB, Ueberheide B & King GF Venom peptides as therapeutics: advances, challenges and the future of venom-peptide discovery. Expert. Rev. Proteomics 14, 931–939 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Norton RS & Chandy KG Venom-derived peptide inhibitors of voltage-gated potassium channels. Neuropharmacology 127, 124–138 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Pennington MW, Czerwinski A & Norton RS Peptide therapeutics from venom: Current status and potential. Bioorg. Med. Chem. 26, 2738–2758 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Beeton C, Gutman GA & Chandy KG Targets and therapeutic properties of venom peptides In: Kastin AJ (ed). The Handbook of Biologically Active Peptides. Academic Press; 2006, pp 403–413. [Google Scholar]
- 60.Narahashi T, Anderson NC & Moore JW Tetrodotoxin does not block excitation from inside the nerve membrane Science 153, 765–767 (1966). [DOI] [PubMed] [Google Scholar]
- 61.McQuarrie C, Salvaterra PM, De Blas A, Routes J & Mahler HR Studies on nicotinic acetylcholine receptors in mammalian brain. Preliminary characterization of membrane-bound alpha-bungarotoxin receptors in rat cerebral cortex. J. Biol. Chem. 251, 6335–6339 (1976). [PubMed] [Google Scholar]
- 62.Ray R, Morrow CS & Catterall WA Binding of scorpion toxin to receptor sites associated with voltage-sensitive sodium channels in synaptic nerve ending particles. J. Biol. Chem. 253, 7307–7313 (1978). [PubMed] [Google Scholar]
- 63.Safavi-Hemami H, Brogan SE & Olivera BM Pain therapeutics from cone snail venoms: From Ziconotide to novel non-opioid pathways. J. Proteomics 190, 12–20. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kerr LM & Yoshikami D A venom peptide with a novel presynaptic blocking action. Nature 308, 282–284 (1984). [DOI] [PubMed] [Google Scholar]
- 65.Olivera BM et al. Peptide neurotoxins from fish-hunting cone snails. Science 230, 1338–1343 (1985). [DOI] [PubMed] [Google Scholar]
- 66.McCleskey EW et al. Omega-conotoxin: direct and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc. Natl. Acad. Sci. USA 84, 4327–4331 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidtko A, Lotsch J, Freynhagen R & Geisslinger G Ziconotide for treatment of severe chronic pain. Lancet 375, 1569–1577 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Deuis JR et al. Pharmacological characterisation of the highly Nav 1.7 selective spider venom peptide Pn3a. Sci. Rep. 7, 40883 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moyer BD et al. Pharmacological characterization of potent and selective Nav 1.7 inhibitors engineered from Chilobrachys jingzhao tarantula venom peptide JzTx-V. PLoS ONE 13, e0196791 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Minett MS et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav 1.7. Nat. Commun. 6, 8967 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Isensee J et al. Synergistic regulation of serotonin and opioid signaling contributes to pain insensitivity in Nav 1.7 knockout mice. Sci. Signal. 10, eaah4874 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pereira V et al. Analgesia linked to Nav 1.7 loss of function requires μ and δ-opioid receptors. Wellcome Open Res 3, 101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vincler M et al. Molecular mechanism for analgesia involving specific antagonism of a9a10 nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. USA 103, 17880–17884 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clark RJ, Fischer H, Nevin ST, Adams DJ & Craik DJ The synthesis, structural characterization, and receptor specificity of the α-conotoxin Vcl.1. J. Biol. Chem. 281, 23254–23263 (2006). [DOI] [PubMed] [Google Scholar]
- 75.Ellison M et al. α-RgIA: a novel conotoxin that specifically and potently blocks the α9α10 nAChR. Biochemistry 45, 1511–1517 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Callaghan B et al. Analgesic α-conotoxins Vcl.1 and RgIA inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J. Neurosci. 28, 10943–10951 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Castro J et al. α-Conotoxin Vcl.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABAB receptors. Gut 66, 1083–1094 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Azam L & McIntosh JM Molecular basis for the differential sensitivity of rat and human α9α10 nAChRs to α-conotoxin RgIA. J. Neurochem. 122, 1137–1144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Romero HK et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad Sci. USA 114, E1825–E1832 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grishin EV et al. Novel peptide from spider venom inhibits P2X3 receptors and inflammatory pain. Ann. Neurol. 67, 680–683 (2010). [DOI] [PubMed] [Google Scholar]
- 81.DeBin JA, Maggio JE & Strichartz GR Purification and characterization of chlorotoxin, a chloride channel ligand from the venom of the scorpion. Am. J. Physiol. 264, C361–369 (1993). [DOI] [PubMed] [Google Scholar]
- 82.Ullrich N, Gillespie GY & Sontheimer H Human astrocytoma cells express a unique chloride current. Neuroreport 7, 343–347 (1995). [PubMed] [Google Scholar]
- 83.Ullrich N & Sontheimer H Biophysical and pharmacological characterization of chloride currents in human astrocytoma cells. Am. J. Physiol. 270, C1511–1521 (1996). [DOI] [PubMed] [Google Scholar]
- 84.Soroceanu L, Manning TJ Jr. & Sontheimer H Modulation of glioma cell migration and invasion using Cl- and K+ ion channel blockers. J. Neurosci. 19, 5942–5954 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deshane J, Garner CC & Sontheimer H Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 278, 4135–4144 (2003). [DOI] [PubMed] [Google Scholar]
- 86.Maertens C, Wei L, Tytgat J, Droogmans G & Nilius B Chlorotoxin does not inhibit volume-regulated, calcium-activated and cyclic AMP-activated chloride channels. Br. J. Pharmacol. 129, 791–801 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stroud MR, Hansen SJ & Olson JM In vivo bio-imaging using chlorotoxin-based conjugates. Curr. Pharm. Des. 17, 4362–4371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parrish-Novak J et al. Nonclinical profile of BLZ-100, a tumor-targeting fluorescent imaging agent. Int. J. Toxicol. 36, 104–112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lehen’kyi V, Raphael M & Prevarskaya N The role of the TRPV6 channel in cancer. J. Physiol. 590, 1369–1376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bowen CV et al. In vivo detection of human TRPV6-rich tumors with anti-cancer peptides derived from soricidin. PLoS ONE 8, e58866 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fu S et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Invest. New Drugs 35, 324–333 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xiong ZG et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118, 687–698 (2004). [DOI] [PubMed] [Google Scholar]
- 93.Chassagnon IR et al. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 114, 3750–3755 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Richards KL et al. Selective Nav1.1 activation rescues Dravet syndrome mice from seizures and premature death. Proc. Natl. Acad. Sci. USA 115, E8077–E8085. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Proctor CM et al. Electrophoretic drug delivery for seizure control. Sci. Adv. 4, eaau1291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.DeCoursey TE, Chandy KG, Gupta S & Cahalan MD Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature 307, 465–468 (1984). [DOI] [PubMed] [Google Scholar]
- 97.Matteson DR & Deutsch C K channels in T lymphocytes: a patch clamp study using monoclonal antibody adhesion. Nature 307, 468–471 (1984). [DOI] [PubMed] [Google Scholar]
- 98.Cahalan MD & Chandy KG The functional network of ion channels in T lymphocytes. Immunol. Rev. 231, 59–87 (2009).. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Feske S, Wulff H & Skolnik EY Ion channels in innate and adaptive immunity. Ann. Rev. Immunol. 33, 291–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Beeton C et al. Kv1 .3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. USA 103, 17414–17419 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pennington M et al. Chemical synthesis and characterization of ShK toxin: a potent potassium channel inhibitor from a sea anemone. Int. J. Pept. Protein Res. 346, 354–358 (1995). [DOI] [PubMed] [Google Scholar]
- 102.Kalman K et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 273, 32697–32707 (1998). [DOI] [PubMed] [Google Scholar]
- 103.Beeton C et al. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol. Pharmacol. 67, 1369–1381 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tarcha EJ et al. Durable pharmacological responses from the peptide drug ShK-186, a specific Kv1.3 channel inhibitor that suppresses T cell mediators of autoimmune disease. J. Pharmacol. Exp. Ther. 342, 642–653. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chi V et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 59, 529–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tarcha EJ et al. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: A randomized phase 1b trial. PLoS ONE 12, e0180762 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mozaffar T, Wencel M, Goyal N, Philips C & Olsen C Kv1.3 expression on effector memory T cells in sporadic inclusion body myositis: potential for targeted immunotherapy with dalazatide. Neuromuscular Disorders 27, S158 (2017). [Google Scholar]
- 108.Tanner MR et al. Prolonged immunomodulation in inflammatory arthritis using the selective Kv1.3 channel blocker HsTX1[R14A] and its PEGylated analog. Clin. Immunol. 180, 45–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang HK et al. Autocrine-based selection of drugs that target ion channels from combinatorial venom peptide libraries. Angew. Chem. Int. Edit. 55, 9306–9310 (2016). [DOI] [PubMed] [Google Scholar]
- 110.Wang RSE et al. Rational design of a Kv1.3 channel-blocking antibody as a selective immunosuppressant. Proc. Natl. Acad. Sci. USA 113, 11501–11506 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jin L et al. Buccal mucosal delivery of a potent peptide leads to therapeutically-relevant plasma concentrations for the treatment of autoimmune diseases. J. Control. Release 199, 37–44 (2015). [DOI] [PubMed] [Google Scholar]
- 112.Jin L et al. Pulmonary delivery of the Kv1.3-blocking Peptide HsTX1[R14A] for the treatment of autoimmune diseases. J. Pharm. Sci. 105, 650–656 (2016). [DOI] [PubMed] [Google Scholar]
- 113.Murray JK et al. Pharmaceutical optimization of peptide toxins for ion channel targets: Potent, selective, and long-lived antagonists of Kv1.3. J. Med. Chem. 58, 6784–6802 (2015). [DOI] [PubMed] [Google Scholar]
- 114.Edwards W et al. Targeting the ion channel Kv1.3 with scorpion venom peptides engineered for potency, selectivity, and half-life. J. Biol. Chem. 289, 22704–22714 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fung-Leung WP et al. T cell subset and stimulation strength-dependent modulation of T cell activation by Kv1.3 blockers. PLoS ONE 12, e0170102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chiang EY et al. Potassium channels Kv1.3 and KCa3.1 cooperatively and compensatorily regulate antigen-specific memory T cell functions. Nat. Commun. 8, 14644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Markgraf R et al. Mechanism and molecular basis for the sodium channel subtype specificity of micro-conopeptide CnIIIC. Br. J. Pharmacol. 167, 576–586 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Del Rio-Sancho S, Cros C, Coutaz B, Cuendet M & Kalia YN Cutaneous iontophoresis of μ-conotoxin CnIIIC. A potent Nav1.4 antagonist with analgesic, anaesthetic and myorelaxant properties. Int. J. Pharm. 518, 59–65 (2017). [DOI] [PubMed] [Google Scholar]
- 119.Balaev AN, Okhmanovich KA & Osipov v.N. A shortened, protecting group free, synthesis of the anti-wrinkle venom analogue Syn-Ake (R) exploiting an optimized Hofmann-type rearrangement. Tetrahedron Lett 55, 5745–5747 (2014). [Google Scholar]
- 120.Goliger JA & Paul DL Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Mol. Biol. Cell 6, 1491–1501 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grek CL et al. Topical administration of a connexin43-based peptide augments healing of chronic neuropathic diabetic foot ulcers: A multicenter, randomized trial. Wound Repair Regen. 23, 203–212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ghatnekar GS, Grek CL, Armstrong DG, Desai SC & Gourdie RG The effect of a connexin43-based peptide on the healing of chronic venous leg ulcers: a multicenter, randomized trial. J. Invest. Dermatol. 135, 289–298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Strohl WR Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs 29, 215–239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chandy KG & Norton RS Peptide blockers of Kv1.3 channels in T cells as therapeutics for autoimmune disease. Curr. Opin. Chem. Biol. 38, 97–107 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Kaspar AA & Reichert JM Future directions for peptide therapeutics development. Drug Discov. Today 18, 807–817 (2013). [DOI] [PubMed] [Google Scholar]
- 126.Bhardwaj G et al. Accurate de novo design of hyperstable constrained peptides. Nature 538, 329–335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Halai R et al. Effects of cyclization on stability, structure, and activity of α-conotoxin RgIA at the α9α10 nicotinic acetylcholine receptor and GABAB receptor. J. Med. Chem.54, 6984–6992 (2011) [DOI] [PubMed] [Google Scholar]
- 128.Kwon S et al. Efficient enzymatic cyclization of an inhibitory cystine knot-containing peptide. Biotechnol. Bioeng. 113, 2202–2212 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bergeron ZL & Bingham JP Scorpion toxins specific for potassium (K+) channels: a historical overview of peptide bioengineering. Toxins (Basel) 4, 1082–1119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Clark RJ et al. Engineering stable peptide toxins by means of backbone cyclization: stabilization of the α-conotoxin MII. Proc. Natl. Acad. Sci. USA 102, 13767–13772 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jensen JE et al. Cyclisation increases the stability of the sea anemone peptide APETx2 but decreases its activity at acid-sensing ion channel 3. Mar. Drugs 10, 1511–1527 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.van Lierop BJ et al. Dicarba α-conotoxin Vc1.1 analogues with differential selectivity for nicotinic acetylcholine and GABAB receptors. ACS Chem. Biol. 8, 1815–1821 (2013). [DOI] [PubMed] [Google Scholar]
- 133.Yu R et al. Less is More: Design of a highly stable disulfide-deleted mutant of analgesic cyclic α-conotoxin Vc1.1. Sci. Rep. 5, 13264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Platt RJ et al. Stapling mimics noncovalent interactions of γ-carboxyglutamates in conantokins, peptidic antagonists of N-methyl-D-aspartic acid receptors. J. Biol. Chem. 287, 20727–20736 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Harvey AJ, Gable RW & Baell JB A three-residue, continuous binding epitope peptidomimetic of ShK toxin as a Kv1.3 inhibitor. Bioorg. Med. Chem. Lett. 15, 3193–3196 (2005). [DOI] [PubMed] [Google Scholar]
- 136.Chen R et al. Peptidomimetic star polymers for targeting biological ion channels. PLoS ONE 11, e0152169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tranberg CE et al. ω-Conotoxin GVIA mimetics that bind and inhibit neuronal Cav2.2 ion channels. Mar. Drugs 10, 2349–2368 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mollica A et al. Design, synthesis and biological evaluation of two opioid agonist and Cav2.2 blocker multitarget ligands. Chem. Biol. Drug Des. 86, 156–162 (2015). [DOI] [PubMed] [Google Scholar]
- 139.Rusconi F et al. Peptidomimetic targeting of Cavβ2 overcomes dysregulation of the L-type calcium channel density and recovers cardiac function. Circulation 134, 534–546 (2016). [DOI] [PubMed] [Google Scholar]
- 140.Michne W et al. Novel inhibitors of potassium ion channels on human T lymphocytes. J. Med. Chem. 38, 1877–1883 (1995). [DOI] [PubMed] [Google Scholar]
- 141.Hill RJ et al. WIN 17317–3: novel nonpeptide antagonist of voltage-activated K+ channels in human T lymphocytes. Mol. Pharmacol. 48, 98–104 (1995). [PubMed] [Google Scholar]
- 142.Nguyen A et al. Novel nonpeptide agents potently block the C-type inactivated conformation of Kv1.3 and suppress T cell activation. Mol. Pharmacol. 50, 1672–1679 (1996). [PubMed] [Google Scholar]
- 143.Knaus HG et al. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry 33, 5819–5828 (1994). [DOI] [PubMed] [Google Scholar]
- 144.Carter PJ & Lazar GA Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 17, 197–223 (2018). [DOI] [PubMed] [Google Scholar]
- 145.Gilbert SM et al. A phase I clinical trial demonstrates that nfP2X7-targeted antibodies provide a novel, safe and tolerable topical therapy for basal cell carcinoma. Br. J. Dermatol. 177, 117–124 (2017). [DOI] [PubMed] [Google Scholar]
- 146.Kamath AV Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discov. Today Technol. 21-22, 75–83 (2016). [DOI] [PubMed] [Google Scholar]
- 147.Shcherbatko A et al. Modulation of P2X3 and P2X2/3 receptors by monoclonal antibodies. J. Biol. Chem. 291, 12254–12270 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vernino S Autoimmune and paraneoplastic channelopathies. Neurotherapeutics 4, 305–314 (2007). [DOI] [PubMed] [Google Scholar]
- 149.Sun H & Li M Antibody therapeutics targeting ion channels: are we there yet? Acta Pharmacol. Sin. 34, 199–204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gomez-Varela D et al. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 67, 7343–7349 (2007). [DOI] [PubMed] [Google Scholar]
- 151.Lin FF et al. Generation and characterization of fully human monoclonal antibodies against human Orai1 for autoimmune disease. J. Pharmacol. Exp. Ther. 345, 225–238 (2013). [DOI] [PubMed] [Google Scholar]
- 152.Cox JH et al. Antibody-mediated targeting of the Orai1 calcium channel inhibits T cell function. PLoS ONE 8, e82944 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sun H et al. A monoclonal antibody against KCNK9 K+ channel extracellular domain inhibits tumour growth and metastasis. Nat. Commun. 7, 10339 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.MacDonald L, Gao M, Morra M, Alessandri-Haber NM & LaCroix-Fralish ML, inventors; Regeneron Pharmaceuticals, Inc; (Tarrytown, NY: ) assignee. Anti-ASIC1 Antibodies and Uses Thereof. US patent US9371383 2016.
- 155.Stortelers C, Pinto-Espinoza C, Van Hoorick D & Koch-Nolte F Modulating ion channel function with antibodies and nanobodies. Curr. Opin. Immunol. 52, 18–26 (2018). [DOI] [PubMed] [Google Scholar]
- 156.Buell G et al. Blockade of human P2X7 receptor function with a monoclonal antibody. Blood 92, 3521–3528 (1998). [PubMed] [Google Scholar]
- 157.Mettler Izquierdo S et al. High-efficiency antibody discovery achieved with multiplexed microscopy. Microscopy (Oxf) 65, 341–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bednenko J et al. A multiplatform strategy for the discovery of conventional monoclonal antibodies that inhibit the voltage-gated potassium channel Kv1.3. MAbs 10, 636–650 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Qiang M et al. Selection of an ASIC1a-blocking combinatorial antibody that protects cells from ischemic death. Proc. Natl. Acad. Sci. USA 115, E7469–E7477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hamers-Casterman C et al. Naturally occurring antibodies devoid of light chains. Nature 363, 446–448 (1993). [DOI] [PubMed] [Google Scholar]
- 161.Muyldermans S Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem.82, 775–797 (2013). [DOI] [PubMed] [Google Scholar]
- 162.Ingram JR, Schmidt FI & Ploegh HL Exploiting nanobodies’ singular traits. Ann. Rev. Immunol. 36, 695–715 (2018). [DOI] [PubMed] [Google Scholar]
- 163.Vazquez-Lombardi R et al. Challenges and opportunities for non-antibody scaffold drugs. Drug Discov. Today 20, 1271–1283 (2015). [DOI] [PubMed] [Google Scholar]
- 164.Stockbridge RB, Koide A, Miller C & Koide S Proof of dual-topology architecture of Fluc F- channels with monobody blockers. Nat. Commun. 5, 5120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sennhauser G, Amstutz P, Briand C, Storchenegger O & Grutter MG Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol 5, e7(2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gilbert SM et al. ATP in the tumour microenvironment drives expression of nfP2X7, a key mediator of cancer cell survival. Oncogene (2018). Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Danquah W et al. Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Sci. Transl. Med. 8, 366ra162 (2016). [DOI] [PubMed] [Google Scholar]
- 168.Matheu MP et al. Imaging of effector memory T cells during a delayed-type hypersensitivity reaction and suppression by Kv1.3 channel block. Immunity 29, 602–614(2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yang XF et al. The antibody targeting the E314 peptide of human Kv1.3 pore region serves as a novel, potent and specific channel blocker. PLoS ONE 7, e36379 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fan C et al. A novel PADRE-Ky1.3 vaccine effectively induces therapeutic antibodies and ameliorates experimental autoimmune encephalomyelitis in rats. Clin. Immunol.193, 98–109 (2018). [DOI] [PubMed] [Google Scholar]
- 171.Vellatt AK KnotBodiesTM: creating ion channel blocking antibodies by fusing Knottins into peripheral CDR loops. Precision Medicine and Ion Channel Retreat 2017; 2017. [Google Scholar]
- 172.Lee JH et al. A monoclonal antibody that targets a Nay1.7 channel voltage sensor for pain and itch relief. Cell 157, 1393–1404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 173.Dong L et al. Evaluation of recombinant monoclonal antibody Symab1 binding to Nay1.7 target sequences and block of human Nay1.7 currents. F1000Res 5, 2764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bang S et al. Differential inhibition of Nay1.7 and neuropathic pain by hybridoma-produced and recombinant monoclonal antibodies that target Nay1.7: Differential activities of Nay1.7-targeting monoclonal antibodies. Neurosci. Bull. 34, 22–41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Biswas K et al. Engineering antibody reactivity for efficient derivatization to generate Nay1.7 inhibitory GpTx-1 peptide-antibody conjugates. ACS Chem. Biol. 12, 2427–2435 (2017). [DOI] [PubMed] [Google Scholar]
- 176.Douthwaite JA, Finch DK, Mustelin T & Wilkinson TC Development of therapeutic antibodies to G protein-coupled receptors and ion channels: Opportunities, challenges and their therapeutic potential in respiratory diseases. Pharmacol. Ther. 169, 113–123 (2017). [DOI] [PubMed] [Google Scholar]
- 177.Ayoub MA et al. Antibodies targeting G protein-coupled receptors: Recent advances and therapeutic challenges. MAbs 9, 735–741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Magnani F et al. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 11, 1554–1571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Serrano-yega MJ, Magnani F, Shibata Y & Tate CG Conformational thermostabilization of the p1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. USA 105, 877–882 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Starkie DO, Compson JE, Rapecki S & Lightwood DJ Generation of recombinant monoclonal antibodies from immunised mice and rabbits via flow cytometry and sorting of antigen-specific IgG+ memory B cells. PLoS ONE 11, e0152282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rouet R, Jackson KJL, Langley DB & Christ D Next-generation sequencing of antibody display repertoires. Front. Immunol. 9, 118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yang W et al. Next-generation sequencing enables the discovery of more diverse positive clones from a phage-displayed antibody library. Exp. Mol. Med. 49, e308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Earl LA, Falconieri, y., Milne, J.L. & Subramaniam, S. Cryo-EM: beyond the microscope. Curr. Opin. Struct. Biol. 46, 71–78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang RY et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife 5, e17219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shen H et al. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 362, 6412 (2018). [DOI] [PubMed] [Google Scholar]
- 186.Gonzalez-Sapienza G, Rossotti MA & Tabares-da Rosa S Single-domain antibodies as versatile affinity reagents for analytical and diagnostic applications. Front. Immunol. 8, 977 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vincke C & Muyldermans S Introduction to heavy chain-only antibodies and derived nanobodies In: Saerens D & Muyldermans S (eds). Single Domain Antibodies, vol. 911 Humana: Totowa, NJ, 2012, pp 15–26. [DOI] [PubMed] [Google Scholar]
- 188.Sela-Culang I, Kunik V & Ofran Y The structural basis of antibody-antigen recognition. Front. Immunol. 4, 302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Inoue A, Sawata SY, Taira K & Wadhwa R Loss-of-function screening by randomized intracellular antibodies: identification of hnRNP-K as a potential target for metastasis. Proc. Natl. Acad. Sci. USA 104, 8983–8988 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Schmidt FI et al. Phenotypic lentivirus screens to identify functional single domain antibodies. Nat. Microbiol. 1, 16080 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Caussinus E, Kanca O & Affolter M Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 19, 117–121 (2011). [DOI] [PubMed] [Google Scholar]
- 192.Kuo C-L, Oyler GA & Shoemaker CB Accelerated neuronal cell recovery from Botulinum neurotoxin intoxication by targeted ubiquitination. PLoS ONE 6, e20352 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rasmussen SGF et al. Structure of a nanobody-stabilized active state of the β2-adrenoceptor. Nature 469, 175–180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kruse AC et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Heijman J & Dobrev D Ion channels as part of macromolecular multiprotein complexes: Clinical significance. Herzschrittmacherther. Elektrophysiol. 29, 30–35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Long SB, Tao X, Campbell EB & MacKinnon R Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382 (2007). [DOI] [PubMed] [Google Scholar]
- 197.Shen H et al. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, eaal4326 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.