

Summary
Chronic myeloid leukemia (CML) originates in a hematopoietic stem cell (HSC) transformed by the BCR-ABL oncogene and is effectively treated with tyrosine kinase inhibitors (TKIs). TKIs do not eliminate disease-propagating leukemic stem cells (LSC), suggesting a deeper understanding of niche-dependent regulation of CML LSC is required to eradicate disease. Cxcl12 is expressed in bone marrow niches and controls HSC maintenance, and here we show that targeted deletion of Cxcl12 from mesenchymal stromal cells (MSC) reduces normal HSC numbers but promotes LSC expansion by increasing self-renewing cell divisions, possibly through enhanced Ezh2 activity. In contrast, endothelial cell-specific Cxcl12 deletion decreases LSC proliferation, suggesting niche-specific effects. During CML development, abnormal clusters of colocalized MSC and LSC form, but disappear upon Cxcl12 deletion. Moreover, MSC-specific deletion of Cxcl12 increases LSC elimination by TKI treatment. These findings highlight a critical role of niche-specific effects of Cxcl12 expression in maintaining quiescence of TKI-resistant LSC populations.
Keywords: CXCL12, hematopoietic stem cells, chronic myelogenous leukemia, leukemic stem cells, bone marrow microenvironment, TKI, drug resistance, mesenchymal stromal cells
Graphical Abstract
Introduction
Chronic myelogenous leukemia (CML) is a clonal hematological disorder which results from the t (9: 22) (q34; q11) chromosomal translocation, leading to the formation of the BCR-ABL oncogene. The resulting protein demonstrates abnormal tyrosine kinase activity, and leads to hematopoietic stem cell (HSC) transformation and development of a myeloproliferative disorder, which can progress to a fatal blast crisis. BCR-ABL tyrosine kinase inhibitors (TKI) are highly effective in the treatment of CML, leading to deep remissions and prolonged survival in the majority of patients. However, TKI treatment is limited by treatment failure in some patients, and persistence of quiescent leukemic stem cells (LSC) in the broader patient population. Most patients require continued treatment to maintain remission, with associated risks of toxicities, non-compliance and high financial costs (Bhatia et al., 2003; Chu et al., 2011). A subset of patients achieving deep molecular remissions are able to discontinue TKI treatment without disease recurrence. However, persistence of BCR-ABL+ cells can be detected even in this select group (Chu et al., 2011).
Several studies indicate that LSC resistance to TKI-treatment occurs through kinase-independent mechanisms (Corbin et al., 2011; Hamilton et al., 2012; Perl and Carroll, 2011). These include alterations in intrinsic cell-regulatory mechanisms that persist despite inhibition of tyrosine kinase activity, as well as signals from the bone marrow (BM) microenvironment (BMM) that support LSC persistence (Agarwal et al., 2016; Zhang et al., 2016; Zhang et al., 2013). HSC in the BM are supported by complex niches, which are specific anatomic locations where HSC are found, and which regulate stem cell fate (Hoggatt et al., 2016; Morrison and Scadden, 2014). Niche components may include endothelial cells, mesenchymal stromal cells (Pinho et al., 2013), osteoblasts (Bowers et al., 2015), adipocytes and megakaryocytes (Birbrair and Frenette, 2016; Bruns et al., 2014). CML development induces significant alteration in the BMM, including markedly altered levels of inflammatory cytokines and chemokines (Zhang et al., 2012), that may enhance LSC and contribute to TKI resistance (Bhatia et al., 1995). Although niche-mediated leukemia maintenance has been investigated in myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPN), and acute myeloid leukemia (AML) models, the nature and regulatory role of BM niches for LSC remains poorly understood (Arranz et al., 2014; Balderman et al., 2016; Hanoun et al., 2014; Krause et al., 2013; Mead et al., 2017; Medyouf, 2017; Schneider et al., 2017).
C-X-C motif chemokine ligand 12 (CXCL12) is the major chemoattractant for HSC, and plays a major role in their localization to regulatory niches (Sugiyama et al., 2006). CXCL12-expressing cells include perivascular mesenchymal stem/stromal cells, also identified as CXCL12-abundant reticular cells (CAR cells), endothelial cells, osteoprogenitors, and mature osteoblasts (Asada et al., 2017). Studies using Cre-lox mediated targeting of the CXCL12 gene to evaluate the role of different BMM subpopulations in regulating normal HSC have shown an important role for mesenchymal progenitor cells, targeted by Prx1-Cre, and endothelial cells, targeted by Tek-Cre, for maintaining HSC (Ding and Morrison, 2013; Greenbaum et al., 2013). In contrast, cells targeted by Osx-Cre, which include osteoprogenitors, were required for B-lymphopoiesis, whereas cells targeted by Ocn-Cre, which include osteoblasts, did not have a significant effect. In previous studies, we have shown that CXCL12 levels are reduced in the CML BMM, and are only partially restored after TKI treatment (Zhang et al., 2012). In the present study, we used CXCL12-GFP mice (Tokoyoda et al., 2004) and CXCL12f/f mice crossed with Cre lines targeting specific CXCL12-expressing cells to investigate how CML development affected diverse CXCL12-expressing BMM populations, and the contribution of CXCL12-expressing populations to LSC regulation. In contrast to normal hematopoiesis, CXCL12 deletion from mesenchymal progenitors enhanced CML LSC numbers and function, and CXCL12 loss from endothelial cells reduced CML LSC numbers. Mechanisms associated with LSC quiescence were downregulated, and sensitivity to TKI treatment was enhanced following CXCL12 loss from mesenchymal progenitors. These results reveal that CXCL12-expressing BMM cells demonstrate distinct niche functions for CML LSC compared with normal HSC, and that targeting of interactions with CXCL12-expressing mesenchymal progenitors may enhance LSC elimination.
Results
CXCL12 produced from mesenchymal progenitor cells is important for normal murine LTHSC maintenance
To study the role of CXCL12-expressing BMM subpopulations in regulating LSC, we transplanted BM from a well characterized SCL-tTA/BCR-ABL transgenic inducible CML mouse model (Koschmieder et al., 2005) and control normal BM cells into mice with Cre-lox mediated targeting of the CXCL12 gene in different BMM cell types. The Cre-lines used included Tek-Cre, targeting endothelial cells, Prx1-Cre, targeting mesenchymal stromal cell progenitors (MSC) and CAR cell populations, Osx-Cre, targeting osteoprogenitor and CAR cell populations, and Ocn-Cre, targeting osteoblast and CAR cell populations. Since irradiation can damage BMM populations(Green and Rubin, 2014), we confirmed that endothelial, osteolineage and mesenchymal cell subpopulations had recovered by 4 to 8 weeks post-irradiation with either 400cGy or 800cGy, compared to non-irradiated mice (Figure S1A–E). We evaluated the function of mesenchymal cells (CD45-Ter119-CD31-) selected from the BM of irradiated compared to non-irradiated mice, 4 weeks after irradiation (Figure S1F). We did not observe a significant difference in CFU-F frequency (Figure S1G). RT-Q-PCR analysis showed that expression of differentiation markers and hematopoietic regulatory factors was not significantly changed in mesenchymal cells from irradiated mice, with the exception of increased expression of the adipocytic differentiation marker Adipoq, and reduced IL3 expression in mesenchymal cells from irradiated mice (Figure S1H). To test HSC supportive function, LTHSC isolated from CD45.1 mice were cocultured with BM mesenchymal cells for 3 days and transplanted into irradiated wild type CD45.2 mice (Figure S1I). We saw no significant difference in engraftment of WBC (Figure S1J) and neutrophil counts (Figure S1K), total donor (Figure S1L), myeloid (S1L), B and T cells (not shown) from LTHSC cultured with BM mesenchymal cells from irradiated and non-irradiated mice. Take together this data indicates that the phenotype and function of BM mesenchymal cells from previously irradiated mice is similar to non-irradiated mice. We observed increased engraftment of CML myeloid cells in mice irradiated at 800cGy compared to 400cGy (Fig S1N-O), and hence, used this dose for subsequent studies.
Lineage-specific targeting by Cre lines was confirmed by crossing Cre mice with Rosa26-CAG-tdTomatof/+ reporter mice. Evaluation of BM CD45-Ter119-cells revealed uniform expression of tdTomato in CD31+ cells in Tek-Cre+ mice, CD140α+ cells and CD140α+Sca-1+ (PαS) cells in Prx1-Cre+ mice, CD51+CD140α+ cells in Osx-Cre+ mice, and >70% of CD51+Sca-1-cells in Ocn-Cre+ mice (Figure S2A). RT-Q-PCR analysis for CXCL12 expression in populations sorted using these markers confirmed reduced CXCL12 expression in target populations in respective Cre-lines (Fig S2B–E).
Transplantation of normal BM into Prx1-Cre+CXCL12f/f mice (Figure 1A) resulted in significantly reduced WBC (Figure 1B), reduced donor myeloid and B cells, but not T cells (Figure 1C–E), and reduced BM cellularity (Figure 1F) compared to Cre-negative mice. Evaluation of BM stem and progenitor populations (Figure S2F,G) indicated significant reduction in long-term HSC (LTHSC) (Fig 1G), short-term HSC (STHSC) (Figure 1H), multipotent progenitors (MPP) (Figure 1I) and granulocyte-macrophage progenitors (GMP) (Figure 1J) compared to Cre-negative mice. In contrast, transplantation of normal BM into Tek-Cre+CXCL12f/f mice resulted in similar WBC counts as in Cre-negative mice (Figure 1B), and did not significantly reduce donor myeloid, B and T cells (not shown), BM cellularity (Figure 1F), LTHSC (Figure 1G) and MPP numbers (Figure 1H), but significantly reduced STHSC (Figure 1I) and GMP numbers (Figure 1J). Osx-Cre+CXCL12f/f and Ocn-Cre+CXCL12f/f mice transplanted with normal BM did not demonstrate alterations in WBC (Figure S2H), BM cellularity (Figure S2I), and stem and progenitor numbers compared to Cre-negative controls (Figure S2J–M). Transplantation of BM cells from Prx1-Cre+CXCL12f/f recipients into secondary recipients resulted in significantly reduced donor engraftment compared to BM cells from control mice (Figure 1K), BM cells from Tek-Cre+CXCL12f/f mice did not generate significantly different donor engraftment (Figure 1L), while BM cells from Osx− Cre+CXCL12f/f and Ocn-Cre+CXCL12f/f mice resulted in higher donor engraftment (Figure S2N, O) than BM from control mice. These results confirm the specific importance of CXCL12 expression by Prx1-Cre+ MSC, as opposed to other BMM populations, in maintenance of normal LTHSC, consistent with previous reports (Greenbaum et al., 2013).
Figure 1. CXCL12 produced from mesenchymal progenitor cells is important for normal murine LTHSC maintenance.

(A) Whole bone marrow cells (WBM) were obtained from wild-type normal mice and injected (n=7–11 mice/group) into age and sex matched CXCL12f/f (CXCL12f/f-XX-Cre-; Cre-negative), CXCL12f/f-Tek-Cre, CXCL12f/f-Prx1-Cre, (CXCL12f/f-XX-Cre+; XX representing either Tek or Prx1) 6–8 week old littermates irradiated at 8Gy. Blood draws were performed every 4 weeks to check donor engraftment in PB until 16 weeks after which mice were euthanized to analyze stem/progenitor populations. PB white blood cells (WBC) (B), donor-derived myeloid cells (C), -B Cells (D), -T Cells (E), BM total cellularity (F), LTHSC (G), STHSC (H), MPP (I), GMP (J), per 1 femur, 1 tibia (2 bones) are shown. WBM cells from primary transplanted mice were pooled and transplanted into secondary recipient normal mice (n=3–10 mice/group) irradiated at 8Gy. Serial analysis for donor engraftment every 4 weeks is shown in the PB of mice receiving BM from either No Cre or Tek-Cre (K), and No Cre or Prx1-Cre mice (L). Error bars represent mean ± sem. Significance values. ns (non-significant) P>0.05, *P< 0.05, **P< 0.01, ***P<0.001, ****P<0.0001. See also Figures S1 and S2.
CXCL12 deletion from MSC and endothelial cells differentially regulates murine CML LSC maintenance and self-renewal
We next evaluated the effect of CXCL12 deletion on CML stem cells. LSC in the CML mouse model share the same phenotype as normal LTHSC (CD45+Lin-Sca-1+Kit+CD150+CD48-) (Zhang et al., 2012). In contrast to normal hematopoiesis, transplantation of CML BM into Prx1-Cre+CXCL12f/f mice (Figure 2A) resulted in reduced survival (Figure 2B), increased leukocytosis (Figure 2D), increased neutrophils and reduced lymphocytes (Figure S3A, B), increased donor CML cell engraftment (Figure S3C), increased BM cellularity (Figure 2E), and increased donor CML LSC (LTHSC), STHSC and MPP numbers (Figure 2F–H), compared to control mice. However, transplantation of CML BM into Tek-Cre+CXCL12f/f mice resulted in increased survival (Figure 2C), significantly reduced BM cellularity (Figure 2E), and reduced leukemic stem and progenitor cell numbers (Figure 2F–H), compared to control mice. Transplantation of CML cells into Osx-Cre+CXCL12f/f and Ocn-Cre+CXCL12f/f mice resulted in similar rate of survival (Figure S3D,E), minimal but significant increase in WBC with Osx-Cre and not Ocn-Cre mice (Figure S3F), no significant change in myeloid cells in blood (Figure S3G), and no change in BM or spleen cellularity (Figure S3H,I). Analysis of stem and progenitor cells numbers compared to control mice (Figure S3J), showed no significant change compared to control mice (Figure S3K–M). Splenic cellularity and stem cells and progenitor numbers in the spleens of all Cre-strains transplanted with CML cells were similar to control mice (data not shown). To exclude a confounding effect of irradiation, we also used a non-irradiation/non-cytotoxic regimen that combines anti-c-Kit antibody and anti-CD47 antibodies (Figure S4A). The anti-c-Kit antibody targets HSC in BM niches, and CD47 blockade enhances Fc-mediated antibody effector functions (Chhabra et al., 2016). Transplantation of CML c-Kit+ cells into non-irradiated mice resulted in similar patterns of engraftment as obtained using the irradiation-based regimen, with enhanced leukocytosis (not shown), total donor engraftment (Figure 2I, S4B), myeloid cell engraftment (not shown) and increased BM LTHSC (Figure 2J, S4C), STHSC (Figure 2K) and MPP (Figure 2L) in Prx1− Cre+CXCL12f/f mice compared to controls.
Figure 2. CXCL12 deletion from MSC and endothelial cells differentially regulates murine CML LSC maintenance and self-renewal.

(A) BCR-ABL expression was induced in SCL-tTA-BCR-ABL mice by tetracycline withdrawal. Whole BM cells were obtained 6–8 weeks after induction and injected into age and sex matched CXCL12f/f (CXCL12f/f-XX-Cre-; Cre-negative), CXCL12f/f-Tek-Cre, CXCL12f/f-Prx1-Cre, (CXCL12f/f-XX-Cre+; XX representing either Tek or Prx1) 6–8-week old littermates irradiated at 800cGy (n=7–11 mice/group) (B-H). Survival of Prx1-Cre and Tek-Cre mice is shown (B, C). Blood draws were performed every 2 weeks to check for leukemia development. Total white blood cells (WBC) are shown (D). Mice were euthanized at 10 weeks to analyze stem/progenitor populations. BM cellularity (E), LTHSC (F), STHSC (G), and MPP (H) per 1 femur, 1 tibia (2 bones) are shown. In another experiment, CML c-Kit+ enriched cells (2*106 cells/mouse) were injected into non-irradiated anti-c-Kit and anti-CD47 antibodies treated recipient Cre mice. Serial CML donor engraftment in the PB (I), donor LTHSC (J), STHSC (K), MPP (L) in the BM are shown after 16 weeks. After primary transplantation of CML WBM cells into respective WT and knock out mice from experiment A, LTHSC were FACS purified and transplanted into secondary healthy WT mice irradiated at 8Gy. Serial blood draw was performed every 4 weeks until 24 weeks. The total WBC (M), frequency of donor engraftment (N) and frequency of donor myeloid cells in the PB (O) from secondary transplant experiment are shown. Error bars represent mean ± sem. Significance values. ns (non-significant) P>0.05, *P< 0.05, **P< 0.01, ***P<0.001, ****P<0.0001. See also Figures S3 and S4.
Transplantation of CML BM cells from Prx1-Cre+CXCL12f/f mice into secondary recipients resulted in significantly enhanced donor engraftment, compared to BM cells from control mice (Figure S4D), whereas transplantation of BM cells from Tek-Cre+CXCL12f/f mice resulted in reduced short-term, but not long-term engraftment (Figure S4E). BM cells from Osx-Cre+CXCL12f/f and Ocn-Cre+CXCL12f/f mice resulted in similar donor engraftment as BM from control mice (Figure S5F,G). Since altered secondary repopulation could reflect changes in LSC frequency in BM, we also transplanted purified LSC from these mice into secondary recipients (Figure S4H). LSC from Prx1-Cre+CXCL12f/f mice generated higher WBC counts (Figure 2M), increased donor engraftment (Figure 2N) compared to LSC from control mice over 24 weeks, and increased donor myeloid cells (Figure 2O) at early time times. However, transplantation of LSC from Tek-Cre+CXCL12f/f mice resulted in similar engraftment as LSC from controls (Figure 2M–O). These results demonstrate that deletion of CXCL12 from Prx1− Cre+ MSC leads to expansion of LSC with increased self-renewal and repopulation capacity, in contrast to reduction in normal LTHSC numbers. On the other hand, deletion of CXCL12 from Tek-Cre+ endothelial cells reduces LSC numbers, without affecting normal LTHSC. We conclude that different CXCL12-expressing BM cells differ in their regulatory influences on CML LSC, and Prx1+ MSC also demonstrate differential regulatory effects on CML LSC compared to normal LTHSC.
CXCL12 deletion from MSC provides a competitive advantage to CML LSC
To evaluate whether CXCL12 deletion affected CML LSC homing to the BM, Lin-normal and CML BM cells were injected intravenous into Prx1-Cre+CXCL12f/f and Tek-Cre+CXCL12f/f transgenic mice, and BM was harvested 16 hours post-transplant (Figure S5A). The numbers of donor CML or normal LTHSC locating to the BM of Prx1-Cre+CXCL12f/f or Tek-Cre+CXCL12f/f mice were not significantly reduced compared to control mice (Figure S5B,C). These results may be explained by lower migration and adhesion of CML LSC in response to CXCL12(Peled et al., 2002), and indicate that altered growth of CML LTHSC in Prx1-Cre+CXCL12f/f or Tek-Cre+CXCL12f/f mice is not explained by altered homing to BM.
We next evaluated whether CXCL12 deletion affected competitive growth of CML LSC and normal LTHSC, by transplanting mixtures of equal numbers of normal and CML cells into Prx1-Cre+CXCL12f/f and Tek-Cre+CXCL12f/f mice (Figure S5D). CXCL12 deletion from Prx1-Cre targeted cells resulted in a significant competitive advantage for CML LSC and MPP over normal cells when compared to control mice, with increased repopulation of leukemic compared to normal stem and progenitor cells (Figure 3A–D). However, CXCL12 deletion from Tek-Cre targeted cells resulted in a significant advantage for normal over CML cells, and reduced repopulation of CML compared to normal total donor cells, LTHSC and MPP (Figure 3E–H). These results suggest that the differential effects of CXCL12 deletion from Prx1-Cre and Tek-Cre targeted cells on normal and CML LSC are related to altered growth regulation by these niches.
Figure 3. CXCL12 deletion from MSC results in increased CML LSC cycling and provides a competitive advantage to CML LTHSC.

Ratio of donor total CML cells over normal cells (A), LTHSC (B), STHSC (C), MPP (D), in BM of either Cre-negative or Prx1-Cre recipient mice. Ratio of donor total CML cells over normal cells (E), LTHSC (F), STHSC (G), MPP (H), in BM of either Cre-negative or Tek-Cre recipient mice. Pooled cell cycle analysis of CML LTHSC from EdU incorporation and DAPI assay (I), and Ki67 and DAPI labeling (J). (K) LTHSC were FACS sorted from normal or CML mice, and CD140α+ cells were FACS sorted from normal or Prx1-Cre+ mice. Normal (CD45.2+) or CML (CD45.1+/CD45.2+) LTHSC were co-cultured with either normal (NL) or Prx1-Cre+ CD140α+ cells for 3 days and cell numbers and immunophenotype (CD45, CD150, CD48) were analyzed. The absolute number of total CD45+ cells (L), LTHSC (M), MPP (N), are shown. Error bars represent mean ± sem. Significance values. ns (non-significant) P>0.05, *P< 0.05, **P< 0.01, ***P<0.001, ****P<0.0001. See also Figure S5.
CXCL12 deletion from MSC results in increased CML LSC cycling
We performed EdU labeling in vivo to evaluate cell cycling (Fig 4A). CXCL12 deletion from Prx1-Cre targeted cells caused significant increase in CML LSC cycling, with increased frequency of S/G2/M phase cells and reduced frequency of G0/G1 cells (Figure 3I, S5F). CXCL12 deletion from Prx1-Cre targeted cells also increased frequency of cycling normal LTHSC (data not shown), consistent with prior reports (Greenbaum et al., 2013). We further evaluated quiescence using Ki67 labeling. CXCL12 deletion from Prx1-Cre+ MSC reduced the frequency of quiescent CML LSC in G0 phase (Figure 3J, S5F). CXCL12 deletion from Tek-Cre+ endothelial cells did not alter cycling or quiescence of CML LSC.
Figure 4. CXCL12 deletion prevents formation of MSC clusters in CML BM with colocalization of leukemic progenitors.

Representative 3D confocal images of femoral BM volumes of Prx1-Cre+ Rosa26-tdTomatof/f (green) and Tek-Cre+ Rosa26-tdTomatof/f (red) mice transplanted with FACS sorted α-cat-GFP+ c-Kit+ cells from either normal (CTRL) or CML mice (CML) 8 weeks post-transplant. (A). Abnormal clusters of Prx1-Cre+ MSC appear in the BM during progression of CML (left panels: depth, 90 µm; size, 1429 µm*549 µm; scale bar, 200 µm; right panels: depth, 64 µm; size, 800 µm*400 µm; scale bar, 200 µm). CML progression has massive effects in BM microvascular structure compared to normal BM (right panels). A conspicuous increase in microvascular density, a loss of the typical sinusoidal morphology and prominent signs of vascular remodeling are apparent. (B) 3D images and tissue maps of typical MSC cluster and aggregation of c-Kit+ leukemic progenitors (magenta) inside and in the vicinity of these structures (delimited by dashed lines and highlighted) (top image: depth, 90.4 µm; size, 517 µm*526 µm; scale bar, 200 µm) (C) Representative images of MSC (green) in Prx1-Cre+ CXCL12+/+ (upper panel) and Prx1-Cre+ CXCL12fl/fl bones (lower panel) with CML, which shows normal homogeneous distribution of MSC and absence of clusters when CXCL12 is deleted from the mesenchymal Prx-1-Cre+ fraction (top left panel: depth, 107 µm; size, 800 µm*400 µm; scale bar, 200 µm; bottom left panel: depth, 64 µm; size, 800 µm*400 µm; scale bar, 200 µm). Right images show tissue density maps of MSC corresponding to BM volumes imaged in left panels. Automated microscopy images correspond to stitching of adjacent fields. See also Figure S6.
Short term in vitro co-culture of FACS sorted normal LTHSC with CXCL12− deleted CD140α+ cells (corresponding to cells targeted by Prx1-Cre; Figure 4K) resulted in significant reduction in LTHSC numbers and increase in MPP numbers compared to control stromal cells (Figure 4L–N). In contrast, CML LSC and MPP numbers were not affected by CXCL12 deletion, suggesting increased differentiation of normal compared to CML LSC upon CXCL12 deletion. These results suggest that CXCL12 deletion from Prx1-Cre target cells differentially affects self-renewal of cycling normal LTHSC and CML LSC. Similar results were obtained using CD140α+ stromal cells harvested from 8Gy-irradiated CXCL12-deleted and control mice (Figure S5G–I).
CXCL12 deletion prevents formation of MSC clusters in CML BM with colocalization of leukemic progenitors
We performed 3D in situ imaging to obtain further insight into the relative localization of leukemic progenitor cells to CXCl12+ niche elements (Gomariz et al., 2018; Nombela-Arrieta and Manz, 2017; Nombela-Arrieta et al., 2013). α-catulin-GFP expression identifies long-term repopulating primitive murine LSC (Acar et al., 2015). We crossed α-catulin-GFP mice with SCL-tTA-BCR-ABL mice to generate α-catulin-GFP-SCL-tTA-BCR-ABL mice. We confirmed that LSK+CD150+CD48− LTHSC were enriched within GFP+ cells, and that CML-α-catulin-GFP mice develop leukemia (Figure S6A). However, α-catulin-GFP+ HSC although clearly identified in thick BM slices from primary mice (data not shown), were not detected in BM from transplanted mice, suggesting that expression may be lost after transplantation. Prx1-Cre+ and Tek-Cre+ mice were crossed with Rosa26-tdTomatof/f mice and CXCL12f/f mice to allow simultaneous visualization of Prx1-Cre+ and Tek-Cre+ cells and evaluation of effects of CXCL12 deletion from these cells. We confirmed that CXCL12 deletion did not significantly affect Prx1-Cre+ MSC numbers in either CML or normal BM (Figure S6B).
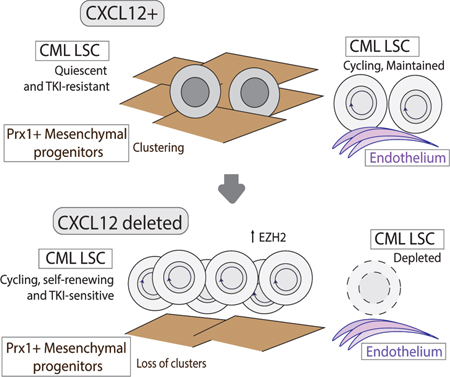
We transplanted CML and normal α-catulin-GFP+c-Kit+ cells into Prx1-Cre and Tek-Cre mice crossed with Rosa26-tdTomatof/f and CXCL12f/f or CXCL12+/+ mice (Figure S6C). The vast majority of c-Kit+ progenitors (>80%) in BM of transplanted mice are of donor origin. 3D imaging of BM volumes in Tek-Cre+ mice showed that CML development was associated with marked vascular disruption and remodeling, leading to fundamental alterations in BM sinusoidal vascular structure (Figure 4A, and Supplementary Videos 1 and 2). Similar vascular alterations were seen in CXCL12fl/fl mice, indicating that they are not dependent on CXCL12 expression on Tek+ cells (not shown). 3D imaging of BM volumes in Prx1-Cre+ mice showed that CML development resulted in formation of pathological tissue domains harboring an abnormally high density of spatially disorganized Prx1-Cre+ MSC (Figure 4A, S6D and Supplementary Videos 3–6) as well as c-Kit+ leukemia progenitors (Figure 4B). Quantitative analysis revealed increased density of Prx-1+ MSC within clusters, and a concomitant enhanced density of c-Kit+ cells around and inside these structures, compared to BM areas outside of clusters within the same bone (Figure S6E–F). CXCL12 deletion from Prx1+ MSC led to loss of MSC clustering, indicating that formation of these aberrant tissue aggregates is dependent on CXCL12 expression in Prx1+ MSC (Figure 4C).
CXCL12 deletion from MSC results in downregulation of gene signatures associated with quiescence and TKI resistance in CML LSC
RNA-Seq analysis revealed distinct patterns of gene expression of LSC from Prx1− Cre+CXCL12f/f and Tek-Cre+CXCL12f/f mice, compared to LSC from control mice. Gene set enrichment analysis (GSEA) revealed that LSC from Prx1-Cre+CXCL12f/f mice demonstrate positive enrichment of genes associated with cell cycling, consistent with our cell cycle analysis, and of MYC target genes (Figure 5A, Table S1). Polycomb Repressive Complex 2 (PRC2) target genes were significantly downregulated, indicating increased PRC2 activity (Figure 5B). LSC from Prx1-Cre+CXCL12f/f mice also demonstrated downregulation of genes related to TGF-β, and STAT3, important mechanisms for regulation of CML LSC quiescence and TKI resistance (Figure 5B, Table S1). TGFβ is a critical regulator of Akt activation and Foxo3a localization in CML LSC and plays a critical role in LSC maintenance (Naka et al., 2010). STAT3 and its targets have been shown to contribute to TKI resistance of CML LSC (Gleixner et al., 2017; Warsch et al., 2013). Therefore, CXCL12 expression on MSC modulates key regulatory mechanisms contributing to CML LSC quiescence, self-renewal and TKI resistance. In contrast, CML LSC from Tek-Cre+CXCL12f/f mice demonstrated enrichment of PRC2 target genes, TGF-β, and inflammatory signaling, and downregulation of genes related to cell cycle progression, LTHSC cycling, and MYC signaling, compared to LSC from control mice (Table S1). These results are consistent with the distinct regulatory effects of CXCL12 expressing endothelial cells on CML LSC.
Figure 5. CXCL12 deletion from MSC results in downregulation of gene signatures associated with quiescence and TKI resistance in CML LSC.

RNASeq analysis was performed on CML LTHSC FACS sorted from Cre-negative and Prx1-Cre mice. Gene-set Enrichment Analysis (GSEA) of differentially expressed genes showed that CML LTHSC from Prx1-Cre exhibited significantly increased expression of several gene sets related to cell cycling, hematopoietic stem cell proliferation, and MYC target genes when compared to LTHSC from Cre-negative mice. Representative plots for E2F target and MYC target genes are shown (A). CML LTHSC from Prx1-Cre showed decreased expression of gene sets related to TGF-β signaling, STAT3/5, and PRC2 target genes (B). (C) Expression of EZH1 and EZH2 among LTHSC from Prx1- Cre and Cre-negative mice from the RNA-Seq analysis. FACS sorted CML LSK cells from various groups of mice either untreated (D), or treated with vehicle or EZH2 inhibitor (GSK343) for 2 weeks and subjected to western blotting to analyze various protein levels (E) and densitometric analysis performed as shown. Effect of EZH2 inhibition on leukemia progression was assessed by analyzing total WBC levels (F), frequency of neutrophils in PB (G), BM cellularity (H), representative FACS plots showing CML LTHSC in BM (I), total no. of LTHSC (J), MPP (K), and GMP (L). Error bars represent mean ± sem. Significance values. ns (non-significant) P>0.05, *P< 0.05, **P< 0.01, ***P<0.001, ****P<0.0001. See also Table S1.
Consistent with increased PRC2 activity, LSC from Prx1-Cre+CXCL12f/f mice demonstrated increased EZH2 expression, but reduced expression of EZH1 (Figure 5C), a counter-balancing mechanism controlling resting cells (Hidalgo et al., 2012; Margueron et al., 2008). We confirmed that progenitor cells from Prx1-Cre mice demonstrated increased EZH2 protein expression and increased total H3K27 trimethylation (Figure 5D) compared to control and Tek-Cre mice. We further evaluated the role of enhanced EZH2 activity in expansion of LSC observed in Prx1− Cre+CXCL12f/f mice (Xie et al., 2016). Treatment with the EZH2 inhibitor, GSK 343(Scott et al., 2016), resulted in inhibition of H3K27 trimethylation (Figure 5E) and significant reduction in WBC and neutrophils (Figure 5F, G), BM cellularity (Figure 5H), LTHSC, MPP and GMP numbers (Figure 5I–L) in Prx1-Cre+CXCL12f/f mice, but did not significantly affect these parameters in Tek-Cre and Cre-negative control mice. These results support a role for enhanced EZH2 expression and activity in expansion of LSC and leukemic cells in Prx1-Cre+CXCL12f/f compared to control and Tek-Cre mice.
CXCL12 deletion from MSC sensitizes murine and human CML LSC to TKI treatment
Previous studies have shown that TKI treatment reduces peripheral blood WBC, spleen cellularity and splenic LSC numbers in this CML mouse model, but does not reduce BM cellularity or LSC numbers (Zhang et al., 2012). We evaluated the effect of the second-generation BCR-ABL TKI, Nilotinib, on CML cells engrafted in Prx1-Cre+CXCL12f/f and Tek-Cre+CXCL12f/f mice. Following establishment of leukemia, mice were treated for 2 weeks with vehicle or Nilotinib (50mg/kg) (Figure 6A). TKI treatment led to significantly reduced leukocytosis (Figure 6B) and neutrophils (Figure 6C), and increased lymphocytes (Figure S7A) in the PB of control and Prx1-Cre+CXCL12f/f mice, but did not alter neutrophils in Tek-Cre+CXCL12f/f mice compared to vehicle. Although TKI treatment significantly reduced spleen cellularity in all three groups (Figure S7B), BM cellularity was only reduced in Prx1-Cre+CXCL12f/f mice (Figure 6D). TKI treatment resulted in significantly increased survival in Prx1-Cre+CXCL12f/f mice (Figure 6E) but did not significantly impact survival of Tek-Cre+CXCL12f/f mice (Figure 6F) compared to control mice. Importantly, BM LSC and STHSC populations were significantly reduced following TKI treatment in Prx1-Cre+CXCL12f/f mice (Figure 6G–I) but were not reduced by TKI treatment of control or Tek-Cre+CXCL12f/f mice. Although TKI treatment reduced splenic stem and progenitor cell populations increased lymphocytes in PB in Tek-Cre+CXCL12f/f, Prx1-Cre+CXCL12f/f and control mice, this did not reach statistical significance (Figure S7C–E). The effect of TKI-treatment on CML LSC repopulation capacity was evaluated by transplanting BM from TKI-treated mice into secondary wild-type recipient mice (Figure S7F). Mice receiving BM from TKI-treated Prx1− Cre+CXCL12f/f mice exhibited significantly reduced WBC (Figure S7G,I), long-term donor engraftment (Figure S7J,L), donor BM cells (Figure S7M) and donor LTHSC (Figure S7N), compared to mice receiving BM from vehicle-treated mice. We next transplanted purified LSC from TKI or vehicle-treated mice into secondary wild-type recipients. Mice receiving LSC from Prx1+Cre+CXCL12f/f mice demonstrated increased WBC, total donor cell and myeloid donor cell engraftment compared to Cre-negative mice, whereas mice receiving LSC from TKI-treated Prx1-Cre+CXCL12f/f mice demonstrated significantly reduced WBC, total donor cell and myeloid donor cell engraftment compared to TKI-treated control Cre-negative mice (Figure 6J–L). These data confirm that CXCL12 deletion from Prx1-Cre+ MSC results in enhanced engraftment potential of individual LSC, as well as enhanced LSC sensitivity to TKI treatment with significant depletion of functional engraftment capacity of individual LSC. These results indicate that increased LSC cycling following CXCL12 deletion from Prx1+ MSC is associated with enhanced sensitivity to TKI treatment. Taken together this data shows that TKI treatment in mice with CXCL12 deletion from MSC results in significant depletion of LSC capacity. In contrast, recipients of BM from Tek− Cre+CXCL12f/f or control mice treated with TKI did not exhibit significant change in WBC, donor engraftment, or donor stem and progenitor cells in BM compared to mice receiving cells from vehicle-treated mice (Figure S7G–N, 6J–L).
Figure 6. CXCL12 deletion from MSC sensitizes murine and human CML LSC to TKI treatment.

(A) BCR-ABL expression was induced in SCL-tTA-BCR-ABL mice by tetracycline withdrawal. Whole BM cells were obtained 6–8 weeks after induction and injected into age and sex matched CXCL12f/f (CXCL12f/f-XX-Cre-; Cre-negative), CXCL12f/f-Tek-Cre, CXCL12f/f-Prx1-Cre, (CXCL12f/f-XX-Cre+; XX representing either Tek or Prx1) 6–8-week old littermates irradiated at 8Gy (n=5–6 mice/group). 8 weeks post-transplantation, mice were treated with either vehicle (Veh) or nilotinib (50mg/kg; TKI) for 2 weeks once daily oral gavage after which mice were euthanized and PB, BM, spleen analyzed. Total white blood cells (WBC) (B) and frequency of neutrophils (C) in the PB are displayed. A cohort of mice were followed for their survival after stopping treatment among No Cre and Prx1-Cre mice (E), and No Cre and Tek-Cre mice (F). BM LTHSC (G), STHSC (H), MPP (I) per 1 femur, 1 tibia (2 bones) are shown. FACS sorted LTHSC from primary vehicle or TKI-treated mice (CD45.1/2+) were pooled and transplanted into secondary recipient (CD45.2+) healthy WT 6–8-week-old mice (1000 cells/mouse+250,000 helper WBM CD45.2+ cells) irradiated at 8Gy. Long-term donor engraftment was performed 16 weeks following transplantation. The total WBC levels (J), long-term CML donor engraftment (K), frequency of donor myeloid cells (L), in the PB of mice are shown. Human CML 34+ cells were stained with CFSE. CFSE+ primitive cells (34+ 38-) were FACS sorted and cultured in the presence or absence of FACS sorted tdTomato+ cells from CXCL12+/+-tdTomatof/+-Prx1-Cre+ and CXCL12f/f-tdTomatof/+-Prx1-Cre+ mice for 3 days and treated with or without Nil (1µM). Representative FACS plot (M), proliferation (N) and expansion (O) are shown. Error bars represent mean ± sem. ns (non-significant) P>0.05, *P< 0.05, **P< 0.01, ***P<0.001, ****P<0.0001. See also Figure S7.
We also evaluated the effect of CXCL12 deletion from MSC on human CML stem and progenitor cells. Human CML CD34+38− stem/primitive progenitor cells and CD34+CD38+ committed progenitor cells were labeled with CFSE and cocultured with media alone, control murine Prx1+ MSC (CXCL12+/+Prx1-Cre+), and CXCL12-deleted Prx1+ MSC (CXCL12f/fPrx1-Cre+), with or without exposure to Nilotinib (TKI). Consistent with results obtained with the murine CML model, CXCL12 deletion from MSC was associated with enhanced human CML stem cell division and cell expansion in culture and increased sensitivity to inhibition by Nilotinib (Figure 6M–O). In contrast, CXCL12 deletion from MSC did not alter cell division and expansion of human CML committed progenitors and their sensitivity to Nilotinib (not shown).
CML development leads to altered distribution of CXCL12-expressing cells in murine BM
We have reported that CXCL12 levels are significantly reduced in CML compared to normal BM (Zhang et al., 2012). Although CXCL12 expression increases upon TKI treatment, it remains significantly lower than normal BM samples. To investigate effects of CML development on CXCL12-expressing populations in BM, CXCL12-GFP knock-in mice (GFP inserted into the CXCL12 locus) (Omatsu et al., 2010; Tokoyoda et al., 2004), were crossed with SCL-tTA-BCR-ABL mice, and the effect of CML induction on CXCL12-expressing BMM cells analyzed (Figure 7A). CML BMM cells demonstrated significant reduction in CXCL12-expressing mesenchymal cells (Figure 7B,C), including those targeted by Prx1-Cre (Greenbaum et al., 2013) (Figure 7D), Osx-Cre (Figure 7E) and Ocn-Cre (Figure 7F), but significant increase in CXCL12-expressing endothelial cells (Figure 7G).
Figure 7. CML development leads to altered distribution of CXCL12-expressing cells in murine BM.

(A) Age and sex matched CXCL12-GFP knock-in mice (Normal) and CXCL12-GFP-SCL-tTA-BCR-ABL mice (CML) were subjected to tetracycline withdrawal. Mice were euthanized and FACS analysis for BM microenvironmental cells was performed every 5 weeks until 15 weeks (n=4–5 mice/group). FACS plots for CXCL12-expressing stromal (45-Ter119-CD31-) and endothelial cells (45-Ter119-CD31+) during CML development are shown (B). The number of CXCL12-expressing stromal cells (CD45-Ter119-CD31-GFP+) (C), CXCL12-expressing PDGFRα+ cells (CD45-Ter119-CD31-CD140α+GFP+) (D), CXCL12-expressing osteoprogenitors (CD45-Ter119-CD31-CD51+CD140α+GFP+) (E), CXCL12-expressing osteoblasts (CD45-Ter119-CD31-CD51+Sca-1-GFP+) (F), and CXCL12-expressing endothelial cells (CD45/Ter119-CD31+GFP+) (G) per femur are shown. Error bars represent mean ± sem. Significance values. ns (non-significant) P>0.05, *P< 0.05, **P< 0.01, ***P<0.001, ****P<0.0001. (H) Age and sex matched CXCL12-GFP knock-in mice (Normal) and CXCL12-GFP-SCL-tTA-BCR-ABL mice (CML) were subjected to tetracycline withdrawal for 8 weeks to induce leukemia. Then, a cohort of CML mice were placed back on tetracycline for further 9 weeks (CML+Tet). (I) Multilineage blood profile showing restoration of normal hematopoiesis upon tetracycline re-introduction. Mice were euthanized and FACS analysis was performed on BM (n=3–4 mice/group). The number of CXCL12-expressing stromal cells (CD4-Ter119-CD31-GFP+) (J), CXCL12-expressing PDGFRα+ cells (CD45-Ter119-CD31-CD140α+GFP+) (K), CXCL12-expressing osteoprogenitors (CD45-Ter119-CD31-CD51+CD140α+GFP+) (L), CXCL12-expressing osteoblasts (CD45-Ter119-CD31-CD51+Sca-1-GFP+) (M), and CXCL12-expressing endothelial cells (CD45-Ter119-CD31+GFP+) (N) per femur are shown. Error bars represent mean ± sem. Significance values. ns (non-significant) P>0.05, *P< 0.05, **P< 0.01, ***P<0.001, ****P<0.0001.
The SCL promoter is active and expresses BCR-ABL in endothelial cells. To determine whether alterations in CXCL12 expressing non-hematopoietic cells in CML BM were mediated by leukemic cells, we transplanted CML BM cells into CXCL12-GFP mice. CXCL12-GFP reporter mice irradiated at 8Gy and rescued with FACS sorted CD45+/Ter119+CD31− BM cells had similar numbers of CXCL12+ mesenchymal or endothelial cells in BM as non-irradiated mice (not shown). CML development was confirmed 12 weeks after transplant. CXCL12-expressing mesenchymal cell populations were reduced and CXCL12-expressing endothelial cells were increased in mice transplanted with CML compared to normal BM cells, confirming that changes in BMM populations were indeed leukemia-induced (not shown).
We also evaluated whether changes in CXCL12-expressing BMM cells were reversed following suppression of BCR-ABL expression. Leukemia was induced in CML CXCL12-GFP mice by tetracycline withdrawal for 8 weeks, and tetracycline reintroduced to suppress BCR-ABL expression (Figure 7H). Reversal of leukemia was confirmed after 9 weeks (Figure 7I). CXCL12-expressing mesenchymal populations remained significantly lower than normal controls (Figure 7J–M) following inhibition of BCR-ABL expression, whereas CXCL12-expressing endothelial cell numbers returned to normal levels (Figure 7N). Therefore, leukemia-induced alterations in CXCL12-expressing endothelial cells are corrected, but changes in BM mesenchymal cells are not fully reversed in leukemia remission.
Discussion
Our studies show that CXCL12 deletion from BM Prx1+MSC, but not other CXCL12-expressing BM microenvironment populations, results in enhanced leukemia development and reduced survival, related to increased cell cycling and expansion of CML LSC, and increased LSC inhibition by TKI treatment. These results therefore reveal an important role of CXCL12-expressing Prx1+MSC as specific regulatory niches that maintains quiescent, treatment-resistant LSC in the BM.
We show that BM Prx1+MSC differentially regulate CML LSC compared to normal HSC. In contrast to the observed expansion of CML LSC, CXCL12 deletion from Prx1+MSC result in depletion of normal HSC, which is consistent with prior reports also indicating an important role for this population in normal HSC maintenance. Indeed, deletion of CXCL12 from Prx1+MSC results in a competitive advantage for CML LSC over normal HSC. Both HSC and LSC demonstrate increased cell cycling, indicating that CXCL12-expressing Prx1+MSC are required to maintain stem cell quiescence in both populations. In contrast to loss of normal HSC, increased cycling was associated with expansion of CML LSC with maintenance of long-term engraftment capacity, suggesting that CML LSC, unlike normal HSC, do not require CXCL12-dependent MSC interactions to maintain self-renewal.
Previous reports suggest indicate that CML LSC may show reduced migration and adhesion in response to CXCL12. Our imaging studies revealed the presence of abnormal clusters of Prx1-Cre+ MSC with colocalization of leukemic progenitor cells in CML but not normal BM. Cluster formation was dependent on CXCL12 expression and was lost on CXCL12 deletion from Prx1+ MSC. We speculate that these MSC clusters may retain quiescent LSC, and that disrupting their formation by CXCL12 deletion may allow increased LSC cycling and sensitivity to TKIs.
In contrast to MSC, loss of CXCL12 expression from endothelial cells resulted in reduction of CML LSC numbers and extended survival and resulted in a competitive disadvantage for CML LSC compared to normal HSC, indicating that role for CXC12-expression on endothelial cells contributed to LSC maintenance. CXCL12-expressing MSC and endothelial niches thus have markedly different regulatory effects on LSC. Imaging studies revealed profound vascular disruption and remodeling in CML BM leading to altered BM sinusoidal vasculature structure. These observations are consistent with histological studies of BM biopsies from CML patients which show a significant increase in microvascular density (Kvasnicka and Thiele, 2004), altered vasculature structure with irregularity of shape and tortuosity, and a complex branching network of irregular shaped sinuses on 3-dimensional reconstruction. The mechanisms underlying leukemia-induced alterations in the BM vasculature are unclear, but were not corrected by CXCL12 deletion from endothelial cells, but may be related to elevated levels of angiogenic cytokines in CML BM.
Our studies support a potential role for enhanced PRC2 activity in increased LSC cycling and self-renewal following CXCL12 deletion from Prx1+MSC (Koschmieder and Vetrie, 2017). PRC2 targets genomic regions for silencing through EZH2 H3K27 histone methyltransferase activity. EZH2 overexpression prevents HSC exhaustion under replicative stress (Kamminga et al., 2006), and enhances leukemogenesis by maintaining stem cell properties in cycling cells (Lund et al., 2014; Xie et al., 2016). Increased PRC2 activity in LSC following CXCL12 deletion from Prx1+MSC could be related to increased MYC activation, which upregulates transcription of the core PRC2 components (Neri et al., 2012). EZH2 is reported to be dysregulated in CML LSC, with extensive reprogramming of H3K27me3 targets, and to be important for CML LSC maintenance (Xie et al., 2016), (Scott et al., 2016). CXCL12 deletion from Prx1+MSC further increased EZH2 expression and activity in CML progenitors, and an EZH2 inhibitor reduced leukemia development and leukemic stem and progenitor cell expansion in Prx1-Cre+CXCL12f/f mice. The role of specific niche interactions in regulating MYC and EZH2 activity in LSC requires further investigation.
CML development resulted in reduction in CXCL12-expressing BM mesenchymal cells and increase in CXCL12-expressing endothelial cells, both of which alterations may provide a competitive advantage to LSC over HSC. These alterations are reproduced by transplantation of leukemic hematopoietic cells, and could potentially be related to altered inflammatory cytokine and growth factor expression (Reynaud et al., 2011; Zhang et al., 2012) or extracellular vesicles production by leukemic cells (Kumar et al., 2017). Leukemia-induced alterations in CXCL12-expressing endothelial cells were reversed whereas mesenchymal cells remained significantly altered even after leukemia remission. Retention of niche alterations could continue to provide a hospitable environment to LSC during CML remission, and warrants further investigation.
The majority of CML patients achieve remission with TKI treatment, but require life-long therapy to prevent leukemia regrowth and relapse. The insensitivity of quiescent, primitive LSC to TKI treatment is a major barrier to a cure in CML. This work demonstrates the critical role for CXCL12-expressing MSC niches for maintaining LSC in a quiescent, TKI-resistant state, and indicates that strategies to target these niche interactions could enhance LSC elimination. Although CXCL12 deletion significantly enhances TKI-mediated targeting of the LSC pool from Prx1+MSC, it also results in expansion of leukemic cells in the absence of TKI, and may be best used after achievement of disease control with TKI. CXCL12 expression in the microenvironment plays an important role in several cancers (Abe-Suzuki et al., 2014; Azab et al., 2009; Balandran et al., 2016; Flores-Figueroa et al., 2012; Pandey et al., 2017; Pitt et al., 2015), and antagonists of CXCR4, the receptor for CXCL12, have been (Beider et al., 2014; Dillmann et al., 2009; Weisberg et al., 2012), and are currently being tested in clinical trials (Abraham et al., 2017; Cho et al., 2017; Liu et al., 2015; Nervi et al., 2009). Given the diverse functions of different CXCL12-expressing cells, targeting of specific LSC interactions with CXCL12-expressing mesenchymal progenitor niches, or downstream effectors of these interactions, may be preferable (Holtz et al., 2007; Jorgensen et al., 2006).
STAR METHODS
CONTACT FOR REAGENT OR RESOURCE SHARING
Further information and requests for resources and reagents may be directed to and will be fulfilled by the Lead Contact, Dr. Ravi Bhatia (rbhatia@uabmc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Samples
Samples were obtained from CML patients in chronic phase (CP) without prior imatinib treatment seen at the University of Alabama at Birmingham Hospital. Samples were processed for CD34+ cell selection with CliniMACS (Miltenyi Biotech, Teterow, Germany). Mononuclear cells were isolated by Ficoll-Hypaque (Sigma Diagnostics, St. Louis, MO) separation. CD34+ cells were isolated by using immunomagnetic beads (Miltenyi Biotech, Auburn, CA). CD34+ CD38− cells were obtained by flow cytometry sorting. Equal numbers of male and female subjects were used. Although numbers are insufficient for statistical analysis, review of the data did not show a significant impact of sex on the results obtained. All subjects signed an informed consent form. Sample acquisition was approved by the Institutional Review Board at the University of Alabama at Birmingham Hospital, in accordance with an assurance filed with and approved by the Department of Health and Human Services, and met all requirements of the Declaration of Helsinki.
Mice
SCL-tTA-BCR-ABL mice (CD45.2+; immune competent, C57BL/6J) were crossed with B6-Ly5.1/Cr mice (CD45.1+; immune competent, C57BL/6J; Charles River, Frederick, MD) to generate mice on the CD45.1/CD45.2 C57BL/6 background. CXCL12-GFP knock-in mice (exon 2 of CXCL12 gene replaced by GFP expression cassette; CD45.2+; immune competent, C57BL/6J) were provided by Dr. Takashi Nagasawa (Kyoto University, Japan), and crossed with SCL-tTA-BCR-ABL mice to generate CXCL12-GFP-SCL-tTA-BCR-ABL mice. Ai9 (B6.Cg-Gt (Rosa)26Sortm9(CAG-tdTomato)Hze/J) (immune competent, C57BL/6J, stock no. 007909), Tek-Cre (immune competent, C57BL/6J, stock no. 008863), Prx1-Cre (immune competent, C57BL/6J, stock no. 005584), Osx1-GFP-Cre (immune competent, C57BL/6J, stock no. 006361), Ocn-Cre (immune competent, C57BL/6J, stock no. 019509), B6(FVB)-Cxcl12tm1.1Link/J (immune competent, back-crossed to C57BL/6J for >20 generations, stock no. 021773), and Ctnnal1tm1.1Sjm/J (α-catulin-GFP, immune competent, C57BL/6J stock no. 028342) mice were from Jackson Laboratory (Bar Harbor, ME), and C57BL/6 mice (CD45.2+; immune competent, C57BL/6J, stock no. 556) from Charles River. CXCL12f/f-XX-Cre+ males (XX representing Tek, Prx1, Osx, or Ocn) were crossed with corresponding CXCL12f/f-XX-Cre-females to generate CXCL12f/f-XX-Cre+ experimental mice. α-catulin-GFP (CD45.2+) were crossed with SCL-tTA-BCR-ABL mice (CD45.2+) to generate α-catulin-GFP-SCL-tTA-BCR-ABL CML mice. All CML mice of various genotypes were maintained on tetracycline by feeding them doxycycline-containing green colored food (cat no. TD.09761, Envigo, New Jersey). Unless otherwise mentioned, all experiments were performed with 6–10 weeks old mice of both sexes. Experimental mice were separated by sex and housed with 5 mice per cage. All the mice were drug or test naïve and not involved in previous procedures. Mice of similar age were randomly divided into experimental groups. Investigators were blinded to the mice genotype during treatments or monitoring survival. All experimental mice received autoclaved water and clean rodent chow diet. Mice were subjected to 12-hour light/dark cycles, and controlled ambient room temperature (20–22°C), and air humidity (30%−70%). All mice were maintained in an AAALAC-accredited specific-pathogen-free animal care facilities, and all procedures were carried out in accordance with federal guidelines and protocols approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. All the various animal models utilized in this study are summarized in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD3 | eBioscience | Cat#13–0031-85; RRID:AB_466320 |
| Anti-mouse CD4 | eBioscience | Cat#13–0041-85; RRID:AB_466326 |
| Anti-mouse CD8 | eBioscience | Cat#13–0083-85; RRID:AB_657763 |
| Anti-mouse B220 | eBioscience | Cat#13–0452-85; RRID:AB_466450 |
| Anti-mouse CD19 | eBioscience | Cat#13–0193-85; RRID:AB_657658 |
| Anti-mouse IgM | eBioscience | Cat#13–5790-85; RRID:AB_466676 |
| Anti-mouse Gr-1 | eBioscience | Cat#13–5931-85; RRID:AB_466801 |
| Anti-mouse CD11b | eBioscience | Cat#13–0112-85; RRID:AB_466360 |
| Anti-mouse NK1.1 | eBioscience | Cat#13–5941-85; RRID:AB_466805 |
| Anti-mouse Ter119 | eBioscience | Cat#13–5921-85; RRID:AB_466798 |
| Anti-mouse CD117 | eBioscience | Cat#47–1172-82; RRID:AB_1582226 |
| Anti-mouse CD11b | eBioscience | Cat#47–0112-82; RRID:AB_1603193 |
| Anti-mouse CD16/32 | eBioscience | Cat#56–0161-82; RRID:AB_493994 |
| Anti-mouse CD45 | eBioscience | Cat#56–0451-82; RRID:AB_891454 |
| Anti-mouse B220 | eBioscience | Cat#56–0452-82; RRID:AB_891458 |
| Anti-mouse CD135 | eBioscience | Cat#15–1351-82; RRID:AB_494219 |
| Anti-mouse Sca-1 | eBioscience | Cat#12–5981-82; RRID:AB_466086 |
| Anti-mouse CD48 | eBioscience | Cat#17–0481-82; RRID:AB_469408 |
| Anti-mouse CD34 | eBioscience | Cat#50–0341-82; RRID:AB_10596826 |
| Anti-mouse CD45.1 | eBioscience | Cat#25–0453-82; RRID:AB_469629 |
| Anti-mouse CD45.2 | eBioscience | Cat#11–0454-85; RRID:AB_465062 |
| Anti-mouse CD31 | eBioscience | Cat#48–0311-82; RRID:AB_10598807 |
| Anti-mouse CD51 | eBioscience | Cat#13–0512-85; RRID:AB_466478 |
| Anti-mouse CD140α | eBioscience | Cat#25–1401-82; RRID:AB_2573400 |
| Anti-mouse Gr-1 | eBioscience | Cat#12–5931-83; RRID:AB_466046 |
| Anti-mouse CD19 | eBioscience | Cat#17–0193-82; RRID:AB_1659676 |
| Anti-mouse Ter119 | BioLegend | Cat#116220; RRID:AB_528963 |
| Anti-mouse Sca-1 | BioLegend | Cat#108126; RRID:AB_10645327 |
| Anti-mouse CD3 | BioLegend | Cat#100218; RRID:AB_1595492 |
| Anti-mouse CD150 | BioLegend | Cat#115922; RRID:AB_2303663 |
| Streptavidin | BioLegend | Cat#405229 |
| Goat Anti-Mouse Scf r / c-kit | R&D Systems | Cat#AF1356; RRID:AB_354750 |
| Endomucin (V.7C7) | Santa Cruz Biotechnology | Cat#sc-65495; RRID:AB_2100037 |
| Anti-GFP | Clontech Laboratories | Cat#632592; RRID:AB_2336883 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-21208; RRID:AB_141709 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Thermo Fisher Scientific | Cat#A-21084; RRID:AB_141494 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-21209; RRID:AB_2535795 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 546 | Thermo Fisher Scientific | Cat#A-11056; RRID:AB_142628 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Thermo Fisher Scientific | Cat#A10043; RRID:AB_2534018 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-21206; RRID:AB_2535792 |
| Anti-ACK2 | BioLegend | Cat#135131; RRID:AB_2571992 |
| Anti-CD47 | BioXCell | Cat#BE0283; RRID:AB_2687806 |
| Anti-Ki67 | Thermo Fisher Scientific | Cat#11–5698-80; RRID:AB_11151689 |
| Anti-EZH1 | Abcam | Cat#ab176115 |
| Anti-EZH2 | Active Motif | Cat#39933 |
| Anti-Phospho-Erk1/2 | Cell Signaling Technology | Cat#4370S |
| Anti-Phospho-AKT | Cell Signaling Technology | Cat#4060S |
| Anti-H3 | Abcam | Cat#ab1791 |
| Anti-H3K27me3 | Abcam | Cat#ab6002 |
| Anti-Actin | Sigma-Aldrich | Cat#A5441 |
| Biological Samples | ||
| Peripheral blood or bone marrow CML samples | University of Alabama at Birmingham Hospital | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline Rodent Diet | Envigo | Cat#TD.09761 |
| Sulfatrim-based diet (5053/.025%Tri/.1242%Sulf ½ IRR) | TestDiet | Cat#5W8F |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#472301 |
| Nilotinib | Novartis Pharmaceuticals | N/A |
| Collagenase II | Sigma-Aldrich | Cat# C1764 |
| Dispase II | Sigma-Aldrich | Cat#4942078001 |
| DNase I | Sigma-Aldrich | Cat#D4263 |
| BSA | Sigma-Aldrich | Cat#A9418 |
| DAPI | Thermo Fisher Scientific | Cat#D1306; RRID:AB_2629482 |
| EZH2 Inhibitor (GSK343) | MedChem Express | Cat#HY-13500 |
| Critical Commercial Assays | ||
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| CellTrace CFSE Cell Proliferation Kit | Thermo Fisher Scientific | Cat#C34554 |
| CD34 Microbead Kit | Miltenyi Biotech | Cat#130–046-702 |
| Click-iT Plus EdU Alexa Fluor 488 Assay Kit | Thermo Fisher Scientific | Cat#C10633 |
| Super Signal West Pico Kit | Thermo Fisher Scientific | Cat#34577 |
| Super Signal West Femto Kit | Thermo Fisher Scientific | Cat#34095 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat#FC-131–1096 |
| SMART-Seq v4 Ultra Low Input RNA Kit | Takara Bio | Cat#634891 |
| Deposited Data | ||
| RNA Seq data | UAB Bioinformatics Core | GEO: GSE124125 |
| Experimental Models: Organisms/Strains | ||
| Mouse: SCL-tTA-BCR-ABL | Ravi Bhatia Lab | N/A |
| Mouse: CXCL12-GFP | Takashi Nagasawa Lab | N/A |
| Mouse: B6.SJL-PtprcaPepcb/BoyCrCrl | Charles River Laboratory | Cat#564 |
| Mouse: C57BL/6NCr | Charles River Laboratory | Cat#556 |
| Mouse: Ai9 (B6.Cg-Gt (Rosa)26Sortm9(CAG-tdTomato)Hze/J) | Jackson Laboratory | Stock No: 007909 |
| Mouse: B6.Cg-Tg(Tek-cre)1Ywa/J | Jackson Laboratory | Stock No: 008863 |
| Mouse: B6.Cg-Tg(Prrx1-cre)1Cjt/J | Jackson Laboratory | Stock No: 005584 |
| Mouse: B6.Cg-Tg(Sp7-tTA,tetO-EGFP/cre)1Amc/J | Jackson Laboratory | Stock No: 006361 |
| Mouse: B6N.FVB-Tg(BGLAP-cre)1Clem/J | Jackson Laboratory | Stock No: 19509 |
| Mouse: B6(FVB)-Cxcl12tm1.1Link/J | Jackson Laboratory | Stock No: 021773 |
| Mouse: Ctnnal1tm1.1Sjm/J | Jackson Laboratory | Stock No: 028342 |
| Mouse: Ctnnal1tm1.1Sjm/J-SCL-tTA-BCR-ABL | This paper | N/A |
| Oligonucleotides | ||
| mCxcl12 qPCR probe | Thermo Fisher Scientific | Cat# Mm00445553_m1 |
| mActb qPCR probe | Thermo Fisher Scientific | Cat#4352933E |
| mIl1a qPCR probe | Thermo Fisher Scientific | Cat#Mm00439620_m1 |
| mIl1b qPCR probe | Thermo Fisher Scientific | Cat#Mm00434228_m1 |
| mIl3 qPCR probe | Thermo Fisher Scientific | Cat#Mm00439631_m1 |
| mIl6 qPCR probe | Thermo Fisher Scientific | Cat#Mm00446190_m1 |
| mTpo qPCR probe | Thermo Fisher Scientific | Cat#Mm00456355_m1 |
| mKitl qPCR probe | Thermo Fisher Scientific | Cat#Mm00442972_m1 |
| mAngpt1 qPCR probe | Thermo Fisher Scientific | Cat#Mm00456503_m1 |
| mFlt3l qPCR probe | Thermo Fisher Scientific | Cat#Mm00442801_m1 |
| mTgfb1 qPCR probe | Thermo Fisher Scientific | Cat#Mm01178820_m1 |
| mBglap qPCR probe | Thermo Fisher Scientific | Cat#Mm03413826_mH |
| mSp7 qPCR probe | Thermo Fisher Scientific | Cat#Mm04209856_m1 |
| mAdipoq qPCR probe | Thermo Fisher Scientific | Cat#Mm00456425_m1 |
| mAcan qPCR probe | Thermo Fisher Scientific | Cat#Mm00545794_m1 |
| Software and Algorithms | ||
| FlowJo software (version 8.5.2) | FlowJo | RRID:SCR_008520 |
| BD FACSDiva | BD Biosciences | RRID:SCR_001456 |
| GraphPad Prism 7 | GraphPad | RRID:SCR_002798 |
| ImageJ (version 1.51) | Imagej.nih.gov | RRID:SCR_003073 |
| Imaris software | Bitplane | RRID:SCR_007370 |
| ModFit LT | Verity | RRID:SCR_016106 |
METHOD DETAILS
Flow cytometry and cell sorting
PB, BM and spleen cells were labeled with respective antibodies as summarized in the Key Resources Table, and analyzed using a BD Fortessa X-20 flow cytometer (BD Biosciences, San Jose, CA). Mature populations were identified as follows: myeloid cells (Gr-1+Mac-1+), B cells (B220+CD19+), T cells (CD3+). For analysis of hematopoietic stem/progenitor cells, spleen, femurs and tibias were gently crushed and filtered through a 70-µm strainer (BD Biosciences). Filtered cells were analyzed as follows: granulocyte-macrophage progenitors (GMP; Lin−Sca-1−c-Kit+CD34+FcγR+), megakaryocyte-erythroid progenitors (MEP; Lin−Sca-1−c-Kit+CD34-FcγR−), long-term hematopoietic stem cells (LTHSC; Lin−Sca-1+c-Kit+Flt3-CD150+CD48−), short-term hematopoietic stem cells (STHSC; Lin−Sca-1+c-Kit+Flt3-CD150−CD48−), multipotent progenitors (MPP; Lin−Sca-1+c-Kit+Flt3-CD48+). For analysis of BMM cells, residual BM chips were digested with digestion buffer containing collagenase II (1 mg/mL; Cat: C1764; Sigma-Aldrich), dispase II (5 mg/mL; Cat: 4942078001; Sigma-Aldrich), DNase I (0.1 mg/mL; Cat: D4263; Sigma-Aldrich), BSA (1 mg/mL; Cat: A9418; Sigma-Aldrich) and HEPES, for 60 minutes at 37°C, and then combined with the marrow suspension. Filtered cells were analyzed as follows: non-hematopoietic non-endothelial stromal cells (CD45/Ter119/CD31-), endothelial cells (CD45/Ter119-CD31+), perivascular cells (CD45/Ter119/CD31-CD140α+), PαS cells (CD45/Ter119/CD31-CD140α+Sca-1+), osteoprogenitors (CD45/Ter119/CD31-CD51+CD140α+), and mature osteoblasts (CD45/Ter119/CD31-CD51+Sca-1-).
Primary cell co-culture
FACS sorted CD45/Ter119/CD31-CD140α+ stromal cells from non-irradiated or irradiated CXCL12f/f-Prx1-Cre- or CXCL12f/f-Prx1-Cre+ mice were plated in gelatin-coated 96-well plates for 24hrs in Stemspan serum-free enhanced medium (SFEM; StemCell Technologies). LTHSC from either normal (CD45.2+) or CML mice (CD45.1+/CD45.2+) were FACS sorted and co-cultured in 1:1 ratio with CD140α+ stromal cells in SFEM for 3 days at 37°C, 5% CO2. Cells were analyzed by FACS for total CD45+ cells, LTHSC and MPP subsets. To assess the functional capacity of BMM cells upon IRR, mice were either non-irradiated (Non-IRR) or irradiated at 800cGy (800cGy-IRR), and rescued with FACS sorted CD45/Ter119+CD31-non-stromal hematopoietic cells (1*105 cells/mouse). 4 weeks post-transplant, FACS sorted CD45/Ter119/CD31-non-hematopoietic BM stromal cells were co-cultured with normal FACS sorted LTHSC (CD45.1+) for 3 days. Normal donor CD45.1+ (300 cells/mouse) were transplanted into lethally irradiated recipient mice (CD45.2+) and long-term engraftment capacity determined. Proliferation of human CML cells was assessed by labeling with 5-(and 6-) carboxy-fluorescein diacetate succinimidyl ester (CFSE; Cat: C34554, Molecular Probes, Eugene, OR), FACS sorting CFSEmax, followed by culture for 3 days either alone (no MSC), with FACS sorted murine tdTomato+ cells from CXCL12+/+-tdTomatof/+-Prx1-Cre+ or CXCL12f/f-tdTomatof/+-Prx1-Cre+ mice, and treated with vehicle or TKI. At the end of the culture period, CFSE levels were analyzed by flow cytometry. ModFit software (Verity, Topsham, ME) was used to fit the data, determine the percentage of cells in each generation, and generate a proliferation index. The position of the parent generation was set on the basis of an aliquot of cells treated with 4% paraformaldehyde immediately after sorting.
Bone marrow transplantation
In all the transplantation experiments, recipient mice were placed on sulfatrim-based food (5053/.025%Tri/.1242%Sulf ½ IRR; Cat No: 5W8F; TestDiet, Richmond, IN) for 2–4 weeks post-transplantation to avoid any infection/toxicity-associated with irradiation. To analyze effects of irradiation on BMM cells, 6–8-week-old adult mice (CD45.2+) were irradiated at 400cGy or 800cGy and evaluated after 4 and 8 weeks. Mice receiving 800cGy radiation also received rescue BM cells (0.5*106 per mouse) from CD45.1 WT mice. For primary transplants, Cre-expressing recipient mice (CD45.2) irradiated at 800 cGy received BM cells (2*106 cells/mouse) from WT (CD45.1+) or CML (CD45.1+/CD45.2+) mice. Engraftment in PB was monitored every 2–4 weeks until 16 weeks in mice receiving WT cells, whereas mice receiving CML cells were sacrificed at 10 weeks because of disease-related morbidity. A cohort of mice was followed for survival. For secondary repopulation, BM cells from primary recipients were pooled and transplanted into WT secondary recipients (2*106 cells/mouse) irradiated at 800 cGy. Secondary transplants were also performed using LTHSC selected from primary recipient mice by FACS sorting (500 CD45.1/2+ LTHSC/mice, with 0.5*106 supporting WBM CD45.2+ cells). For competitive repopulation experiments, BM cells from normal (CD45.1+ cells) and CML (CD45.1+/CD45.2+ cells) mice were mixed in a 1:1 ratio and transplanted (2*106 cells/mouse) into CD45.2+ Cre-transgenic recipient mice irradiated at 800 cGy. Normal and CML donor multi-lineage engraftment was monitored in PB every 4 weeks until 16 weeks, when PB, BM, and spleen cells were analyzed. For non-irradiation regimen transplantation experiments, recipient mice (No Cre, Tek Cre, Prx1− Cre) were treated with anti-ACK2 antibody (cat no. 135131; Biolegend) (ACK2; 0.5mg/mouse on day 0, I.P.), anti-CD47 (cat no. BE0283, BioXCell) (MIAP410; 0.5mg/mouse daily for five days, I.P.), and then transplanted with CML c-Kit-enriched stem/progenitor cells (2*106 cells/mouse I.V.) on day 6. CML donor engraftment and leukemia progression were assessed beginning 4 weeks post-transplantation until 16 weeks. Secondary transplants were performed using LTHSC selected from primary recipient mice treated with either vehicle or TKI by FACS sorting (1000 CD45.1/2+ LTHSC/mice, with 0.25*106 supporting WBM CD45.2+ cells). For experiments with normal- and CML-α-catulin-GFP mice, CML was induced in the latter cohort of mice by doxycycline withdrawal and analyzing PB and BM after 9 weeks. Several mice were pooled to FACS sort either normal- or CML-α-catulin-GFP+c-Kit+ cells for transplantation into tdTomato+Prx1-Cre or tdTomato+Tek-Cre mice for BM imaging.
BM Homing Assay
CML or normal BM cells were lineage depleted (Direct lineage cell depletion kit, mouse, Miltenyi Biotech, catalog no. 130–110-470), and transplanted (0.5*106 per mouse for CML, 1*106 per mouse for normal BM) via tail vein into recipient mice irradiated at 8Gy. Mice were euthanized 16hrs post-transplantation, and stem/progenitor populations that homed to BM were analyzed.
Cell Cycle Analysis
Mice were injected intraperitoneally (I.P.) with EdU (Invitrogen, 1 mg/ mouse), euthanized 6 hours later, and EdU incorporation analyzed using the Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit (ThermoFisher Scientific, catalog no. C10633). Cell cycling was also analyzed by labeling with anti-Ki-67 (eBiosciences, catalog no. 11–5698-80) and DAPI.
Inhibitors
Nilotinib (TKI) supplied by Novartis Pharmaceuticals, and EZH2 inhibitor (GSK343) purchased from MedChem Express (cat no. HY-13500, Deer Park, NJ) were stored in 10mM dimethylsulfoxide (DMSO) at −20°C.
In vivo drug treatment
CML cells were transplanted into WT or Cre transgenic lines and development of neutrophilic leukocytosis confirmed after 8 weeks (data not shown). Mice were treated with vehicle, Nilotinib (50mg/kg once daily), EZH2i (GSK343 10mg/kg once daily) or combination by oral gavage for 2 weeks and PB, BM, and spleen cells analyzed. Secondary repopulation was analyzed by transplanting pooled BM cells from treated mice (2*106 cells/mouse) into WT mice irradiated at 800 cGy. Engraftment (CD45.1+/CD45.2+ cells) was monitored every 4 weeks until 16 weeks.
3D confocal imaging of BM
3D Immunohistochemistry of mouse femoral bones was performed as previously described (Gomariz et al., 2018; Nombela-Arrieta et al., 2013). Briefly, femoral bones were isolated, cleaned and immersed in 2% paraformaldehyde fixing solution for 6h at 4°C. Subsequent dehydration in 30% sucrose solution for 72h at 4°C was followed by O.C.T. embedding and snap freezing in liquid nitrogen. Frozen, dehydrated bone sections were then iteratively sectioned on two opposing sides along the longitudinal axis until full exposure of the marrow was confirmed by visual inspection. Residual O.C.T. was removed with three PBS washing steps. To avoid unspecific binding, exposed and cleaned marrow slices were then incubated in blocking solution (0.2% Triton X-100, 1% bovine serum albumin, 10% donkey serum, in PBS) O/N at 4°C. Primary and secondary immunostainings were performed in blocking solution for 3–4 days at 4°C, which was followed by an O/N washing s tep in PBS. Immunostained femoral slices were immersed in RapiClear 1.52 medium for 6–12 hours. Samples were imaged using 10x (HCX PL FLUOTAR), 20x (HC PL APO CS2) and 63x (HCX PL APO CS2) mounted on SP8 Leica confocal microscopes equipped with hybrid detectors. Antibodies related to immunostainings are provided in the Key Resources Table.
Data visualization and quantitative image analysis
3D volumes were rendered and imaging data visualized using Imaris software (Bitplane). Gamma correction was applied in some cases exclusively for visualization purposes. To represent distribution of cells in the form of BM tissue density maps we made use of the empirical probability density, which was calculated with a custom algorithm that adapted the kernel-based density estimation method to account for the different boundaries of the BM as reported33. For density estimation of Prx1-Cre+ tdTomato+ cells, annotation was done using the Imaris spot segmentation tool using a predefined radius of 3.75µm. Accuracy of the segmentation was confirmed by careful visual inspection of the segmented cells. For optimal visualization of Prx1-Cre+ tdTomato+ clusters, regions of interest were created and masks were manually defined using Imaris surface drawing tool. When large regions were imaged, the different tiles were stitched with the Leica Application Suite (LAS) X software (Leica microsystems). Distinct colors were applied to highlight Prx1-Cre+ tdTomato+ cells within or outside of the boundaries of cellular clusters. c-Kit+ cells were semi-automatically detected with the Surfaces module of Imaris v9.2 (Bitplane AG), which classifies voxels according to their intensities. Due to local inhomogeneities in the c-kit signal intensity this method required manual correction of automatic segmentation, including deletion of false positives and addition of undetected false negatives. The 2D density maps were generated with a custom MATLAB tool based on the one proposed in Gomariz et al (Gomariz et al., 2018). This tool represents annotated cells as 3D Gaussian spheres centered on their coordinates. The functions representing each cell are added, and then averaged along the z axis of the image. The resulting file is transferred to Imaris for visualization.
Quantitative reverse transcription PCR
BM cells were pooled from 2–3 mice for each replicate, and BMM subpopulations sorted directly into RLT lysis buffer. RNA was extracted using RNAeasy micro kit (Qiagen, Valencia, CA), and cDNA synthesized using the Superscript III First-Strand Kit (Invitrogen, Grand Island, NY). QPCR was performed using TaqMan gene expression assays for mouse CXCL12 (Mm00445553_m1) and mouse ACTB (cat no. 4352933E).
Colony forming Unit-Fibroblast (CFU-F) Assay
FACS sorted CD45/Ter119/CD31- non-hematopoietic BM stromal cells from either non-irradiated (Non-IRR) or irradiated at 800cGy (800cGy-IRR) mice were cultured in 96- well plate for 2 weeks. Cells were rinsed with PBS, and visible colonies scored by staining with 0.5% crystal violet in methanol, and clusters with >50 cells were counted as a colony.
RNA Sequencing
LTHSC were sorted from BM of Cre-negative, Tek-Cre, and Prx1-Cre mice directly into RLT buffer and RNA prepared, with four biological replicates from each group. Sequencing libraries were prepared with the SMARTer Ultra Low Input RNA Kit for Sequencing (v4, TaKaRa Clontech, catalog no. 634891) and Nextera XT DNA Library Preparation Kit (96 samples, Illumina, cat no. FC-131–1096). Sequencing was performed using the HiSeq 2500 platform with the HiSeq SBS Kit V4 (Illumina). STAR (version 2.5.3a) was used to align raw RNA-Seq FASTQ reads to the mouse reference genome (Gencode Release M11)(Dobin et al., 2013), and number of reads mapping to each gene enumerated using HTSeq-count(Anders et al., 2015). Normalization and differential expression was calculated using DESeq2(Love et al., 2014). Pathway analysis was performed using gene set enrichment analysis (GSEA).
Western Blotting
FACS sorted CML LSK cells were treated as described in the results or directly lysed in buffer containing 0.5% Nonidet P-40 (Sigma Diagnostics) and 0.5% sodium deoxycholate supplemented with phenylmethylsulfonyl fluoride (1 mM/L), protease inhibitor mixture, and phosphatase inhibitors (50 mM/L sodium fluoride, 1 mM/L sodium vanadate; all from Sigma Diagnostics). Proteins were resolved on 4% to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to nitrocellulose membrane. Primary antibodies used were anti–EZH1 (cat: ab176115; Abcam, Cambridge, MA), anti–EZH2 (cat: 39933; Active Motif, Carlsbad, CA), anti– H3K27me3 (cat: ab6002; Abcam, Cambridge, MA), anti-actin (clone: AC15; cat: A5441; Sigma Aldrich, St. Louis, MO), anti-H3 (cat: ab1791; Abcam, Cambridge, MA), anti-P-ERK1/2 (Thr202/Tyr204) (cat: 4370S; Cell Signaling, Danvers, MA), anti-P-AKT (Ser473) (cat: 4060S; Cell Signaling, Danvers, MA), and Horseradish peroxidase– conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). Antibody detection was performed by using the SuperPico and SuperFemto kits (Pierce Biotechnology, Rockford, IL).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise specified, all results obtained from independent experiments are reported as means ± standard errors of means(SEM) of multiple replicates, and statistical analyses were performed using unpaired, nonparametric Student’s t test or 2-way analysis of variance (ANOVA), adjusting for multiple comparisons (GraphPad Prism version 6.0, La Jolla, CA). Data was examined for normality by evaluating skewness and for equivalence of variance by comparing variance between groups. Survival probabilities were estimated using Kaplan-Meier analysis and significance calculated using the log-rank (Mantel-Cox) test. p values < 0.05 were considered statistically significant. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; ns, not significant. The sample size was estimated based on assumptions of one-sided Type I error of 0.05 and estimates of fold change and variance from preliminary data to have >80% power to detect differences, if present, in our groups. All data and subjects were included in the analysis. The “n” in the figures and results represents the number of samples or animals utilized in the indicated experiments and values are reported in respective Figure Legends.
DATA AND SOFTWARE AVAILABILITY
The RNA sequencing data discussed in this publication has been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number: GSE124125.
Supplementary Material
Supplementary Video 1: Structure of the normal BM vascular compartment labeled in the diaphyseal region of Tek-Cre+Rosa26-tdTomato mice (CTRL) as in Fig 4a. Wide, labyrinth like sinusoids and arteries are labeled in red (Related to Figure 4).
Supplementary Video 2: 3D image of the BM diaphyseal vasculature in Tek-Cre+Rosa26-tdTomato mice (CML) as in Fig 4a. Disease progression results in conspicuous remodeling of the BM vasculature, loss of typical sinusoidal morphology and an overall significant increase in vascular density (Related to Figure 4).
Supplementary Video 3: 3D rendered volume of the femoral diaphysis of non-leukemic control Prx1-Cre+Rosa26-tdTomato mice (CTRL) as in Fig 4b, showing normal distribution of mesenchymal stromal Prx-1-Cre+ cells (green) and c-kit+ hematopoietic progenitor cells (magenta) (Related to Figure 4).
Supplementary Video 4: 3D rendered volume of the femoral diaphysis of Prx1-Cre+Rosa26-tdTomato mice (CML) as in Fig 4b. Representative image of the emergence of abnormal clusters of Prx1+ mesenchymal stromal cells (green), in which increased density of c-kit+ hematopoietic cells (magenta) is apparent. A high-resolution image of a CML-induced cluster is also shown in Supplementary Video 5. (Related to Figure 4 and Video 5).
Supplementary Video 5: High-resolution 3D render of confocal image stacks of the femoral diaphysis of mice after CML induction. Prx1+ mesenchymal stromal cells (green) organize in pathological high-density clusters. c-kit+ leukemic progenitor cells accumulate within or in close proximity to these abnormal niches. During the course of the video, mesenchymal and hematopoietic cells within clusters are highlighted by masking the cellular content out of these abnormal niches (Related to Figures 4 and S6).
Supplementary Video 6: 3D rendering of a large volume of the femoral diaphysis after CML induction. The video sequentially shows the quantitative cellular analysis of MSC and the co-localization of c-Kit+ cells in the vicinity and inside CML-induced MSC clusters. Prrx1-Cre+ tdTomato cells are shown in green and segmented cells in blue, while staining for c-Kit is depicted in magenta and segmentation of c-Kit+ cells are shown in multiple colors. The quantification of the densities of both subsets in regions inside and outside clusters of this image is shown in Figure S6 (Related to Figures 4 and S6).
Acknowledgements
This work was supported by the National Institutes of Health grants (R01 CA172447 and P30CA033572 to R.B.). We thank Dr. Robert S. Welner and Dr. Marcus Jaras for helpful scientific discussions and comments; HudsonAlpha Institute for Biotechnology Sequencing Facility, Huntsville, Alabama for performing RNA sequencing; the UAB Comprehensive Flow Cytometry Core and Animal Resource Center for providing help with FACS sorting and maintaining mice colonies, respectively; Dr. David Crossman at the Heflin Center for Genomic Science Core Laboratories at University of Alabama Birmingham for analysis of the RNA-seq data; and Andrew Zhang for help with the analysis of RNA seq data. Confocal imaging was performed at the Center for Microscopy and Image Analysis of the University of Zurich.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing financial interests.
References
- Abe-Suzuki S, Kurata M, Abe S, Onishi I, Kirimura S, Nashimoto M, Murayama T, Hidaka M, and Kitagawa M (2014). CXCL12+ stromal cells as bone marrow niche for CD34+ hematopoietic cells and their association with disease progression in myelodysplastic syndromes. Laboratory investigation; a journal of technical methods and pathology 94, 1212–1223. [DOI] [PubMed] [Google Scholar]
- Abraham M, Klein S, Bulvik B, Wald H, Weiss ID, Olam D, Weiss L, Beider K, Eizenberg O, Wald O, et al. (2017). The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16–1 expression. Leukemia [DOI] [PubMed]
- Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, Jaiyeola C, Zhao Z, Luby-Phelps K, and Morrison SJ (2015). Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 526, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P, Zhang B, Ho Y, Cook A, Li L, Mikhail FM, Wang Y, McLaughlin ME, and Bhatia R (2016). Enhanced targeting of CML stem and progenitor cells by inhibition of porcupine acyltransferase in combination with TKI. Blood [DOI] [PMC free article] [PubMed]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arranz L, Sanchez-Aguilera A, Martin-Perez D, Isern J, Langa X, Tzankov A, Lundberg P, Muntion S, Tzeng YS, Lai DM, et al. (2014). Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 512, 78–81. [DOI] [PubMed] [Google Scholar]
- Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, Ma’ayan A, and Frenette PS (2017). Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nature cell biology [DOI] [PMC free article] [PubMed]
- Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X, Jia X, Wright R, Ospina B, Carlson AL, et al. (2009). CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 113, 4341–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balandran JC, Purizaca J, Enciso J, Dozal D, Sandoval A, Jimenez-Hernandez E, Aleman-Lazarini L, Perez-Koldenkova V, Quintela-Nunez Del Prado H, Rios de Los Rios J, et al. (2016). Pro-inflammatory-Related Loss of CXCL12 Niche Promotes Acute Lymphoblastic Leukemic Progression at the Expense of Normal Lymphopoiesis. Front Immunol 7, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balderman SR, Li AJ, Hoffman CM, Frisch BJ, Goodman AN, LaMere MW, Georger MA, Evans AG, Liesveld JL, Becker MW, et al. (2016). Targeting of the bone marrow microenvironment improves outcome in a murine model of myelodysplastic syndrome. Blood 127, 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beider K, Darash-Yahana M, Blaier O, Koren-Michowitz M, Abraham M, Wald H, Wald O, Galun E, Eizenberg O, Peled A, et al. (2014). Combination of imatinib with CXCR4 antagonist BKT140 overcomes the protective effect of stroma and targets CML in vitro and in vivo. Molecular cancer therapeutics 13, 1155–1169. [DOI] [PubMed] [Google Scholar]
- Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL, Arber DA, Slovak ML, and Forman SJ (2003). Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 101, 4701–4707. [DOI] [PubMed] [Google Scholar]
- Bhatia R, McGlave PB, Dewald GW, Blazar BR, and Verfaillie CM (1995). Abnormal function of the bone marrow microenvironment in chronic myelogenous leukemia: role of malignant stromal macrophages. Blood 85, 3636–3645. [PubMed] [Google Scholar]
- Birbrair A, and Frenette PS (2016). Niche heterogeneity in the bone marrow. Annals of the New York Academy of Sciences 1370, 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers M, Zhang B, Ho Y, Agarwal P, Chen CC, and Bhatia R (2015). Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 125, 2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. (2014). Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nature medicine 20, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra A, Ring AM, Weiskopf K, Schnorr PJ, Gordon S, Le AC, Kwon HS, Ring NG, Volkmer J, Ho PY, et al. (2016). Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Science translational medicine 8, 351ra105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho BS, Kim HJ, and Konopleva M (2017). Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: from bench to bedside. Korean J Intern Med 32, 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, McDonald T, Lin A, Chakraborty S, Huang Q, Snyder DS, and Bhatia R (2011). Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood 118, 5565–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, and Druker BJ (2011). Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. The Journal of clinical investigation 121, 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillmann F, Veldwijk MR, Laufs S, Sperandio M, Calandra G, Wenz F, Zeller J, and Fruehauf S (2009). Plerixafor inhibits chemotaxis toward SDF-1 and CXCR4-mediated stroma contact in a dose-dependent manner resulting in increased susceptibility of BCR-ABL+ cell to Imatinib and Nilotinib. Leukemia & lymphoma 50, 1676–1686. [DOI] [PubMed] [Google Scholar]
- Ding L, and Morrison SJ (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Figueroa E, Varma S, Montgomery K, Greenberg PL, and Gratzinger D (2012). Distinctive contact between CD34+ hematopoietic progenitors and CXCL12+ CD271+ mesenchymal stromal cells in benign and myelodysplastic bone marrow. Laboratory investigation; a journal of technical methods and pathology 92, 1330–1341. [DOI] [PubMed] [Google Scholar]
- Gleixner KV, Schneeweiss M, Eisenwort G, Berger D, Herrmann H, Blatt K, Greiner G, Byrgazov K, Hoermann G, Konopleva M, et al. (2017). Combined targeting of STAT3 and STAT5: a novel approach to overcome drug resistance in chronic myeloid leukemia. Haematologica 102, 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomariz A, Helbling PM, Isringhausen S, Suessbier U, Becker A, Boss A, Nagasawa T, Paul G, Goksel O, Szekely G, et al. (2018). Quantitative spatial analysis of haematopoiesis-regulating stromal cells in the bone marrow microenvironment by 3D microscopy. Nat Commun 9, 2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DE, and Rubin CT (2014). Consequences of irradiation on bone and marrow phenotypes, and its relation to disruption of hematopoietic precursors. Bone 63, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, and Link DC (2013). CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, Nicolini FE, Muller-Tidow C, Bhatia R, Brunton VG, et al. (2012). Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 119, 1501–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, Lacombe J, Armstrong SA, Duhrsen U, and Frenette PS (2014). Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell stem cell 15, 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo I, Herrera-Merchan A, Ligos JM, Carramolino L, Nunez J, Martinez F, Dominguez O, Torres M, and Gonzalez S (2012). Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell stem cell 11, 649–662. [DOI] [PubMed] [Google Scholar]
- Hoggatt J, Kfoury Y, and Scadden DT (2016). Hematopoietic Stem Cell Niche in Health and Disease. Annu Rev Pathol 11, 555–581. [DOI] [PubMed] [Google Scholar]
- Holtz M, Forman SJ, and Bhatia R (2007). Growth factor stimulation reduces residual quiescent chronic myelogenous leukemia progenitors remaining after imatinib treatment. Cancer research 67, 1113–1120. [DOI] [PubMed] [Google Scholar]
- Jorgensen HG, Copland M, Allan EK, Jiang X, Eaves A, Eaves C, and Holyoake TL (2006). Intermittent exposure of primitive quiescent chronic myeloid leukemia cells to granulocyte-colony stimulating factor in vitro promotes their elimination by imatinib mesylate. Clinical cancer research : an official journal of the American Association for Cancer Research 12, 626–633. [DOI] [PubMed] [Google Scholar]
- Kamminga LM, Bystrykh LV, de Boer A, Houwer S, Douma J, Weersing E, Dontje B, and de Haan G (2006). The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood 107, 2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschmieder S, Gottgens B, Zhang P, Iwasaki-Arai J, Akashi K, Kutok JL, Dayaram T, Geary K, Green AR, Tenen DG, et al. (2005). Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood 105, 324–334. [DOI] [PubMed] [Google Scholar]
- Koschmieder S, and Vetrie D (2017). Epigenetic dysregulation in chronic myeloid leukaemia: A myriad of mechanisms and therapeutic options. Semin Cancer Biol [DOI] [PubMed]
- Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, Hurley MP, Lezeau S, Attar E, Wu JY, Lin HY, et al. (2013). Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nature medicine 19, 1513–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, McDonald T, Lin A, Kumar AR, DiGiusto DL, et al. (2017). Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia [DOI] [PMC free article] [PubMed]
- Kvasnicka HM, and Thiele J (2004). Bone marrow angiogenesis: methods of quantification and changes evolving in chronic myeloproliferative disorders. Histol Histopathol 19, 1245–1260. [DOI] [PubMed] [Google Scholar]
- Liu T, Li X, You S, Bhuyan SS, and Dong L (2015). Effectiveness of AMD3100 in treatment of leukemia and solid tumors: from original discovery to use in current clinical practice. Exp Hematol Oncol 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund K, Adams PD, and Copland M (2014). EZH2 in normal and malignant hematopoiesis. Leukemia 28, 44–49. [DOI] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, and Reinberg D (2008). Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32, 503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead AJ, Neo WH, Barkas N, Matsuoka S, Giustacchini A, Facchini R, Thongjuea S, Jamieson L, Booth CAG, Fordham N, et al. (2017). Niche-mediated depletion of the normal hematopoietic stem cell reservoir by Flt3-ITD-induced myeloproliferation. The Journal of experimental medicine 214, 2005–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medyouf H (2017). The microenvironment in human myeloid malignancies: emerging concepts and therapeutic implications. Blood 129, 1617–1626. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, and Scadden DT (2014). The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N, and Hirao A (2010). TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 463, 676–680. [DOI] [PubMed] [Google Scholar]
- Neri F, Zippo A, Krepelova A, Cherubini A, Rocchigiani M, and Oliviero S (2012). Myc regulates the transcription of the PRC2 gene to control the expression of developmental genes in embryonic stem cells. Mol Cell Biol 32, 840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, Prior JL, Piwnica-Worms D, Bridger G, Ley TJ, et al. (2009). Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 113, 6206–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nombela-Arrieta C, and Manz MG (2017). Quantification and three-dimensional microanatomical organization of the bone marrow. Blood Adv 1, 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, Park SY, Lu J, Protopopov A, and Silberstein LE (2013). Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nature cell biology 15, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, and Nagasawa T (2010). The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33, 387–399. [DOI] [PubMed] [Google Scholar]
- Pandey S, Mourcin F, Marchand T, Nayar S, Guirriec M, Pangault C, Monvoisin C, Ame-Thomas P, Guilloton F, Dulong J, et al. (2017). IL-4/CXCL12 loop is a key regulator of lymphoid stroma function in follicular lymphoma. Blood [DOI] [PubMed]
- Peled A, Hardan I, Trakhtenbrot L, Gur E, Magid M, Darash-Yahana M, Cohen N, Grabovsky V, Franitza S, Kollet O, et al. (2002). Immature leukemic CD34+CXCR4+ cells from CML patients have lower integrin-dependent migration and adhesion in response to the chemokine SDF-1. Stem cells 20, 259–266. [DOI] [PubMed] [Google Scholar]
- Perl A, and Carroll M (2011). BCR-ABL kinase is dead; long live the CML stem cell. The Journal of clinical investigation 121, 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, and Frenette PS (2013). PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. The Journal of experimental medicine 210, 1351–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA, et al. (2015). CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer cell 27, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, Sala-Torra O, Radich JP, and Passegue E (2011). IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer cell 20, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RK, Mullally A, Dugourd A, Peisker F, Hoogenboezem R, Van Strien PMH, Bindels EM, Heckl D, Busche G, Fleck D, et al. (2017). Gli1+ Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell stem cell 20, 785–800 e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MT, Korfi K, Saffrey P, Hopcroft LE, Kinstrie R, Pellicano F, Guenther C, Gallipoli P, Cruz M, Dunn K, et al. (2016). Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov 6, 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Kohara H, Noda M, and Nagasawa T (2006). Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988. [DOI] [PubMed] [Google Scholar]
- Tokoyoda K, Egawa T, Sugiyama T, Choi BI, and Nagasawa T (2004). Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 20, 707–718. [DOI] [PubMed] [Google Scholar]
- Warsch W, Walz C, and Sexl V (2013). JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1+ chronic myeloid leukemia. Blood 122, 2167–2175. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Azab AK, Manley PW, Kung AL, Christie AL, Bronson R, Ghobrial IM, and Griffin JD (2012). Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 26, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC, Fujiwara Y, Qi J, Cheloni G, Das PP, et al. (2016). Chronic Myelogenous Leukemia-Initiating Cells Require Polycomb Group Protein EZH2. Cancer Discov 6, 1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chu S, Agarwal P, Campbell VL, Hopcroft L, Jorgensen HG, Lin A, Gaal K, Holyoake TL, and Bhatia R (2016). Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood 128, 2671–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee SU, Hair A, Holyoake TL, Huettner C, and Bhatia R (2012). Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer cell 21, 577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, Shultz L, and Bhatia R (2013). Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 121, 1824–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1: Structure of the normal BM vascular compartment labeled in the diaphyseal region of Tek-Cre+Rosa26-tdTomato mice (CTRL) as in Fig 4a. Wide, labyrinth like sinusoids and arteries are labeled in red (Related to Figure 4).
Supplementary Video 2: 3D image of the BM diaphyseal vasculature in Tek-Cre+Rosa26-tdTomato mice (CML) as in Fig 4a. Disease progression results in conspicuous remodeling of the BM vasculature, loss of typical sinusoidal morphology and an overall significant increase in vascular density (Related to Figure 4).
Supplementary Video 3: 3D rendered volume of the femoral diaphysis of non-leukemic control Prx1-Cre+Rosa26-tdTomato mice (CTRL) as in Fig 4b, showing normal distribution of mesenchymal stromal Prx-1-Cre+ cells (green) and c-kit+ hematopoietic progenitor cells (magenta) (Related to Figure 4).
Supplementary Video 4: 3D rendered volume of the femoral diaphysis of Prx1-Cre+Rosa26-tdTomato mice (CML) as in Fig 4b. Representative image of the emergence of abnormal clusters of Prx1+ mesenchymal stromal cells (green), in which increased density of c-kit+ hematopoietic cells (magenta) is apparent. A high-resolution image of a CML-induced cluster is also shown in Supplementary Video 5. (Related to Figure 4 and Video 5).
Supplementary Video 5: High-resolution 3D render of confocal image stacks of the femoral diaphysis of mice after CML induction. Prx1+ mesenchymal stromal cells (green) organize in pathological high-density clusters. c-kit+ leukemic progenitor cells accumulate within or in close proximity to these abnormal niches. During the course of the video, mesenchymal and hematopoietic cells within clusters are highlighted by masking the cellular content out of these abnormal niches (Related to Figures 4 and S6).
Supplementary Video 6: 3D rendering of a large volume of the femoral diaphysis after CML induction. The video sequentially shows the quantitative cellular analysis of MSC and the co-localization of c-Kit+ cells in the vicinity and inside CML-induced MSC clusters. Prrx1-Cre+ tdTomato cells are shown in green and segmented cells in blue, while staining for c-Kit is depicted in magenta and segmentation of c-Kit+ cells are shown in multiple colors. The quantification of the densities of both subsets in regions inside and outside clusters of this image is shown in Figure S6 (Related to Figures 4 and S6).
Data Availability Statement
The RNA sequencing data discussed in this publication has been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number: GSE124125.