

Abstract
The optimal treatment of patients with cancer depends on establishing accurate diagnoses by using a complex combination of clinical and histopathological data. In some instances, this task is difficult or impossible because of atypical clinical presentation or histopathology. To determine whether the diagnosis of multiple common adult malignancies could be achieved purely by molecular classification, we subjected 218 tumor samples, spanning 14 common tumor types, and 90 normal tissue samples to oligonucleotide microarray gene expression analysis. The expression levels of 16,063 genes and expressed sequence tags were used to evaluate the accuracy of a multiclass classifier based on a support vector machine algorithm. Overall classification accuracy was 78%, far exceeding the accuracy of random classification (9%). Poorly differentiated cancers resulted in low-confidence predictions and could not be accurately classified according to their tissue of origin, indicating that they are molecularly distinct entities with dramatically different gene expression patterns compared with their well differentiated counterparts. Taken together, these results demonstrate the feasibility of accurate, multiclass molecular cancer classification and suggest a strategy for future clinical implementation of molecular cancer diagnostics.
Cancer classification relies on the subjective interpretation of both clinical and histopathological information with an eye toward placing tumors in currently accepted categories based on the tissue of origin of the tumor. However, clinical information can be incomplete or misleading. In addition, there is a wide spectrum in cancer morphology and many tumors are atypical or lack morphologic features that are useful for differential diagnosis (1). These difficulties can result in diagnostic confusion, prompting calls for mandatory second opinions in all surgical pathology cases (2). In the aggregate, these are significant limitations that may hinder patient care, add expense, and confound the results of clinical trials.
Molecular diagnostics offer the promise of precise, objective, and systematic human cancer classification, but these tests are not widely applied because characteristic molecular markers for most solid tumors have yet to be identified (3). Recently, DNA microarray-based tumor gene expression profiles have been used for cancer diagnosis. However, studies have been limited to few cancer types and have spanned multiple technology platforms complicating comparison among different datasets (4–10). The feasibility of cancer diagnosis across all of the common malignancies based on a single reference database has not been explored. In addition, comprehensive gene expression databases have yet to be developed, and there are no established analytical methods capable of solving complex, multiclass, gene expression-based classification problems.
To address these challenges, we created a gene expression database containing the expression profiles of 218 tumor samples representing 14 common human cancer classes. By using an innovative analytical method, we demonstrate that accurate multiclass cancer classification is indeed possible, suggesting the feasibility of molecular cancer diagnosis by means of comparison with a comprehensive and commonly accessible catalog of gene expression profiles.
Materials and Methods
Snap-frozen human tumor and normal tissue specimens, spanning 14 different tumor classes, were obtained from the National Cancer Institute/Cooperative Human Tissue Network, Massachusetts General Hospital Tumor Bank, Dana–Farber Cancer Institute, Brigham and Women's Hospital, Children's Hospital (all in Boston), and Memorial Sloan-Kettering Cancer Center (New York). Tissue was collected and studied under an anonymous discarded tissue protocol approved by the Dana–Farber Cancer Institute Institutional Review Board.
Initial diagnoses were made at university hospital referral centers by using all available clinical and histopathological information. Tissues underwent centralized clinical and pathology review at the Dana–Farber Cancer Institute and Brigham and Women's Hospital (by M.L.) or Memorial Sloan-Kettering Cancer Center (by E.L. and W.G.) to confirm initial diagnosis of site of origin and histological type. All tumors were biopsy specimens from primary sites (except where noted) obtained before any treatment and were enriched in malignant cells (>50%) but otherwise unselected. Normal tissue RNA (Biochain, Hayward, CA) was from snap-frozen autopsy specimens collected through the International Tissue Collection Network.
“Hybridization targets” were prepared with RNA from whole tumors by using published methods (4). Targets were hybridized sequentially to oligonucleotide microarrays [Hu6800 and Hu35KsubA GeneChips (Affymetrix, Santa Clara, CA)] containing a total of 16,063 probe sets representing 14,030 GenBank and 475 The Institute for Genomic Research (TIGR) accession nos., and arrays were scanned by using standard Affymetrix protocols and scanners. For subsequent analysis, each probe set was considered as a separate gene. Expression values for each gene were calculated by using Affymetrix genechip analysis software.
Of 314 tumor and 98 normal tissue samples processed, 218 tumor and 90 normal tissue samples passed quality control criteria and were used for subsequent data analysis. The remaining 104 samples either failed quality control measures of the amount and quality of RNA, as assessed by spectrophotometric measurement of OD and agarose gel electrophoresis, or yielded poor-quality scans. Scans were rejected if mean chip intensity exceeded 2 SDs from the average mean intensity for the entire scan set, if the proportion of “present” calls was less than 10%, or if microarray artifacts were visible. The resulting dataset contained ≈5 million gene expression values.
Clustering.
Gene expression data were subjected to a variation filter that excluded genes showing minimal variation across the samples as follows: genes were excluded if they exhibited less than 5-fold and 500 units absolute variation across the dataset after a threshold of 20 units and ceiling of 16,000 units was applied. Of 16,063 expression values considered, 11,322 passed this filter and were used for clustering. The dataset was normalized by standardizing each row (gene) to mean = 0 and variance = 1. Average-linkage hierarchical clustering was performed by using cluster and treeview software (11). Self-organizing map analysis was performed by using our genecluster analysis package (available at www-genome.wi.mit.edu/MPR) (12).
Support Vector Machine (SVM) Algorithm and One vs. All (OVA) Classification Scheme.
The SVM experiments described in this article were performed by using an implementation of svm-fu (available at www.ai.mit.edu/projects/cbcl). This linear SVM algorithm maximizes the distance between a hyperplane, w, and the closest samples to the hyperplane from two tumor classes, with the constraint that the samples from the two classes lie on separate sides of the hyperplane. This distance is calculated in 16,063-dimensional gene space, corresponding to the total number of expression values considered. This geometric property can be imposed by means of the following optimization problem: min½∥w∥2 subject to yi(w⋅xi + b) ≥ 1, for all i. An unknown test sample's position relative to the hyperplane determines its class, and the confidence of each SVM prediction is based on the distance of a test sample from the hyperplane. In going from binary to multiclass classification, we used an OVA approach (described in Results). Given m classes and m trained classifiers, a new sample takes the class of the classifier with the largest real valued output class = arg maxi=1...m fi, where fi is the real valued output of the ith classifier. A positive prediction strength corresponds to a test sample being assigned to a single class rather than to the “all other” class.
Recursive Feature Elimination.
This feature selection method recursively removes features based on the
absolute magnitude of each hyperplane element (13). Given microarray
data with n genes per sample, each OVA SVM classifier
outputs a hyperplane, w, that can be thought of as a vector
with n elements each corresponding to the expression of a
particular gene. Assuming that the expression values of each gene have
similar ranges, the absolute magnitude of each element in w
determines its importance in classifying a sample, because
f(x) =
∑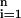 wixi +
b and the class label is sign[f(x)].
Each OVA SVM classifier is first trained with all genes, then genes
corresponding to |wi| in the bottom 10% are
removed, and each classifier is retrained with the smaller gene set.
This procedure is repeated iteratively to study prediction accuracy as
a function of gene number.
wixi +
b and the class label is sign[f(x)].
Each OVA SVM classifier is first trained with all genes, then genes
corresponding to |wi| in the bottom 10% are
removed, and each classifier is retrained with the smaller gene set.
This procedure is repeated iteratively to study prediction accuracy as
a function of gene number.
Statistical Analysis.
A class-proportional random predictor was used to determine the number of correct classifications that would be expected by chance for multiclass prediction. Associated P values were calculated based on the likelihood that the observed classification accuracy could be arrived at by chance, as described (14). Genes that correlate with each tumor class were identified by sorting all of the genes on the array according their signal to noise (S2N) values [(μ0 − μ1)/(σ0 + σ1)], where μ and σ represent the mean and SD of expression, respectively, for each class] as published (4). For the permutation tests, 1,000 permutations of the sample labels (tumor type) were performed on the dataset, and the S2N ratio was recalculated for each gene for each class label permutation. A gene is considered a statistically significant class-specific marker if the observed S2N exceeds the permuted S2N at least 99% of the time (P ≤ 0.01) (4).
Complete details regarding patient samples, pathology, molecular biology protocols, data analysis, raw gene expression data, and additional information are available at www-genome.wi.mit.edu/MPR/GCM.html.
Results
We determined the gene expression profiles of 144 primary tumors by using oligonucleotide microarrays containing 16,063 oligonucleotide probe sets. Tumor samples were primarily solid tumors of epithelial origin, spanning 14 common tumor classes that account for ≈80% of new cancer diagnoses in the U.S., as shown in Fig. 1.
Figure 1.

Clustering of tumor gene expression data and identification of tumor-specific molecular markers. Hierarchical clustering (a) and a 5 × 5 self-organizing map (SOM) (b) were used to cluster 144 tumors spanning 14 tumor classes according to their gene expression patterns. (c) Gene expression values for class-specific OVA markers, as determined using the S2N metric, are shown. Columns represent 190 primary human tumor samples ordered by class. Rows represent 10 genes most highly correlated with each OVA distinction. Red indicates high relative level of expression, and blue represents low relative level of expression. The known cancer markers prostate-specific antigen (PSA), carcinoembryonic antigen (CEA), and estrogen receptor (ER) are identified. BR, breast adenocarcinoma; PR, prostate adenocarcinoma; LU, lung adenocarcinoma; CR, colorectal adenocarcinoma; LY, lymphoma; BL, bladder transitional cell carcinoma; ML, melanoma; UT, uterine adenocarcinoma; LE, leukemia; RE, renal cell carcinoma; PA, pancreatic adenocarcinoma; OV, ovarian adenocarcinoma; ME, pleural mesothelioma; CNS, central nervous system.
We explored two fundamentally different approaches to data analysis. The first, unsupervised learning, often referred to as clustering, allows the dominant structure in a dataset to dictate the separation of samples into clusters based on overall similarity in gene expression, without prior knowledge of sample identity. Fig. 1 shows the results of both hierarchical and self-organizing map clustering of this dataset. Although some tumor types [lymphoma, leukemia, and central nervous system (CNS)] formed relatively discrete clusters with both methods, others, in particular the epithelial tumors, were largely intermixed. This finding indicates that unsupervised learning does not adequately capture the tissue of origin distinctions among these molecularly complex tumors. This result possibly reflects the large degree of biological variability in gene expression data. In addition, because tumor specimens were unselected with regard to percentage of stromal infiltration or inflammation, these clustering results might reflect contributions from nonneoplastic cellular elements to gene expression signatures that confound tissue of origin distinctions. Alternatively, the hierarchical tree structure might reflect bona fide previously unrecognized relationships among tumors that transcend tissue of origin distinctions.
The second approach to this classification problem is to use a supervised learning method. This method involves “training” a classifier to recognize distinctions among the 14 clinically defined tumor classes based on gene expression patterns, and testing the accuracy of the classifier in a blinded manner. Supervised learning has been used to make pairwise distinctions with gene expression data [e.g., the distinction between acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML); ref. 4]. However, making multiclass distinctions can be a considerably more difficult challenge. For this purpose, we devised an analytical scheme, depicted in Fig. 2. First, we divide the multiclass problem into a series of 14 OVA pairwise comparisons. Each test sample is presented sequentially to these 14 pairwise classifiers, each of which either claims or rejects that sample as belonging to a single class. This method results in 14 separate OVA classifications per sample, each with an associated confidence. Each test sample is assigned to the class with the highest OVA classifier confidence.
Figure 2.

Multiclass classification scheme. The multiclass cancer classification problem is divided into a series of 14 OVA problems, and each OVA problem is addressed by a different class-specific classifier (e.g., “breast cancer” vs. “not breast cancer”). Each classifier uses the SVM algorithm to define a hyperplane that best separates training samples into two classes. In the example shown, a test sample is sequentially presented to each of 14 OVA classifiers and is predicted to be breast cancer, based on the breast OVA classifier having the highest confidence.
We evaluated several classification algorithms for these OVA pairwise classifiers including weighted voting (15), k-nearest neighbors (16), and SVM, all of which yielded significant prediction accuracy. Because the SVM algorithm consistently out-performed other algorithms, these results are described in detail (Figs. 3, 4, and 5). The SVM algorithm was used recently for pairwise gene expression-based classification (17, 18) and has a strong theoretical foundation (19, 20). This algorithm considers all profiled genes, to create descriptions of samples in this high-dimensional space, and then defines a hyperplane that best separates samples from two classes (Fig. 2). The position of an unknown sample relative to the hyperplane determines its membership in one or the other class (e.g., “breast cancer” vs. “not breast cancer”). Fourteen separate OVA classifiers classify each sample. The confidence of each OVA SVM prediction is based on the distance of the test sample to each hyperplane, with a value of 0 indicating that a sample falls directly on a hyperplane. The overall multiclass classifier assigns a sample to the class with the highest confidence among the 14 pairwise OVA analyses.
Figure 3.

Multiclass classification results. (a) Results of multiclass classification by using cross-validation on a training set (144 primary tumors) and independent testing with 2 test sets: Test (54 tumors; 46 primary and 8 metastatic) and PD (20 poorly differentiated tumors; 14 primary and 6 metastatic). (b) Scatter plot showing SVM OVA classifier confidence as a function of correct calls (blue) or errors (red) for Training, Test, and PD samples. A, accuracy of prediction; %, percentage of total sample number.
Figure 4.

Multiclass classification error analysis. Matrices delineate distribution of actual compared with predicted class membership for multiclass prediction on training (crossvalidation) and test sets.
Figure 5.

Multiclass classification as a function of gene number. Training and test datasets were combined (190 tumors; 14 classes), then were randomly split into 100 training and test sets of 144 and 46 samples (all primary tumors) in a class-proportional manner. SVM OVA prediction was performed, and mean classification accuracy for the 100 splits was plotted as a function of number of genes used by each of the 14 OVA classifiers, showing decreasing prediction accuracy with decreasing gene number. Results using other algorithms (k-NN, k-nearest neighbors; WV, weighted voting) and classification schemes (AP, all-pairs) are also shown.
The accuracy of this multiclass SVM-based classifier in cancer diagnosis was first evaluated by cross-validation in a set of 144 training samples. This method involves randomly withholding 1 of the 144 primary tumor samples, building a predictor based only on the remaining samples, then predicting the class of the withheld sample. The process is repeated for each sample, and the cumulative error rate is calculated. As shown in Fig. 3, the majority (80%) of the 144 calls was high confidence (defined as confidence >0) and these had an accuracy of 90%, using the patient's clinical diagnosis as the “gold standard.” The remaining 20% of the tumors had low confidence calls (confidence ≤0), and these predictions had an accuracy of 28%. Overall, the multiclass prediction corresponded to the correct assignment for 78% of the tumors. For half of the errors, the correct classification corresponded to the second- or third-most confident OVA prediction.
We confirmed these results by training the multiclass SVM classifier on the entire set of 144 samples and applying this classifier without further modification to an independent test set of 54 tumor samples. Overall prediction accuracy on this test set was 78%, a result similar to cross-validation accuracy and highly statistically significant when compared with class-proportional random prediction (P ≤ 10−16). The majority of these 54 predictions (78%) were high confidence, with an accuracy of 83%, whereas low-confidence calls were made on the remaining 22% of tumors with an accuracy of 58%. Again, for one-half of the errors, the correct classification corresponded to the second- or third-best prediction. Of note, classification of 100 random splits of a combined training and test dataset gave similar results, confirming the stability of prediction for this collection of samples (Fig. 5).
Among these 54 test samples, were 8 metastatic samples, 6 of which were correctly classified despite the classifier having been trained solely with gene expression data derived from primary tumors (P = 0.005 vs. random multiclass assignment). This finding implies that prediction is being driven by cancer-intrinsic gene expression patterns rather than by gene expression signatures derived from contaminating nonmalignant tissue elements. These results further indicate that many cancers retain their tissue of origin identity throughout metastatic evolution, suggesting that gene expression-based approaches to the diagnosis of clinically problematic metastases of unknown primary origin (21) may be feasible.
We next investigated the number of genes contributing to the high accuracy of the SVM OVA classifier. The SVM algorithm considers all 16,063 input genes and naturally utilizes all genes that contain information for each OVA distinction. Genes are assigned weights based on their relative contribution to the determination of each hyperplane, and genes that do not contribute to a distinction are weighted at zero. Virtually all genes on the array were assigned weakly positive and negative weights in each OVA classifier (data not shown), indicating that thousands of genes potentially carry information relevant for the 14 OVA class distinctions. To determine whether the inclusion of this large number of genes was actually required for the observed high-accuracy predictions, we examined the relationship between classification accuracy and gene number by using recursive feature elimination. As shown in Fig. 5, maximal classification accuracy is achieved when the predictor utilizes all genes for each OVA distinction. Nevertheless, significant prediction can still be achieved by using smaller gene numbers. Alternate feature selection methods with different properties, such as S2N (4), radius-margin scaling (22), and gene shaving (23), also resulted in reduced classification accuracy (data not shown).
Our gene expression dataset is also useful for biological discovery. For example, the genes most highly correlated with each of the 14 tumor classes are displayed in Fig. 1, and a complete list of marker genes is available at www-genome.wi.mit.edu/MPR/GCM.html. Many genes already in routine clinical use for cancer diagnosis were identified, including prostate-specific antigen (prostate cancer), carcinoembryonic antigen (colon cancer), CD20 (lymphoid cancers), S100 (melanoma), and estrogen receptor (uterine cancer). In addition, many previously unrecognized markers were discovered, the vast majority of which are tissue-specific genes, reflecting a recurring onco-developmental connection that has been described for many cancers (24). For example, a search for colorectal adenocarcinoma-specific markers revealed 27 that were statistically significant (P < 0.01) based on permutation testing. These genes include intestine-specific transcription factors, cytoskeletal and adhesion molecules, signaling molecules, and membrane-bound tumor markers. Notably, the two transcription factors, Cdx-1 and Bteb-2, are both downstream targets of the Wnt-1/β-Catenin signaling pathway, which is mutated in most colorectal cancers (25–27). The other statistically significant colon adenocarcinoma marker genes are thus also candidates for being under Wnt-1/β-Catenin control. This observation suggests that the gene expression database described here may be useful not only for cancer diagnosis, but also for the generation of new biological hypotheses into the pathogenesis of cancer.
The significant degree of shared gene expression between tumors and their normal tissue counterparts prompted us to ask whether supervised learning could be used to distinguish 210 primary tumors considered as a single class from a collection of 90 normal tissues. By using the S2N metric, we were unable to identify single gene markers that are uniformly expressed only in cancer and not normal tissue. Nevertheless, using the SVM algorithm in cross-validation, we were able to make this pairwise distinction with high accuracy (92%), indicating the presence of a cancer-specific gene expression fingerprint common to all tumors.
We next considered the 28 samples that yielded low-confidence predictions in cross-validation, as these samples are generally misclassified by the multiclass predictor. We found that a large number (17 of 28) were moderately or poorly differentiated (high-grade) carcinomas. It can be difficult to classify such tumors with traditional methods because they often lack the characteristic morphological hallmarks of the organ from which they arise. It has been assumed that these tumors are nonetheless fundamentally molecularly similar to their better-differentiated counterparts, apart from a few differences that might account for their clinically aggressive nature. We directly tested this hypothesis by applying our multiclass classifier, trained on the original 144-tumor dataset, to an independent set of poorly differentiated tumors.
Gene expression data were collected from 20 poorly differentiated adenocarcinomas (14 primary and 6 metastatic), representing 5 tumor types: breast, lung, colon, ovary, and uterus. The technical quality of this dataset was indistinguishable from the other samples in the study. However, these tumors could not be accurately classified according to their tissues of origin, compared with the high overall accuracy seen with lower-grade tumors. Overall, only 6/20 samples (30%) were correctly classified, which is statistically no better than what one would expect by chance alone (P = 0.38) (Fig. 3). Because the classifier relies on the expression of thousands of similarly weighted tissue-specific molecular markers to determine the class of a tumor, these findings indicate that poorly differentiated tumors do not simply lack a few key markers of differentiation, but rather have fundamentally distinct gene expression patterns. This result has significant implications for the future management of patients with these cancers.
Discussion
We report here the creation of a gene expression database from 308 common human cancers and normal tissues by using oligonucleotide microarrays and demonstrate that multiclass cancer diagnosis is feasible by means of comparison of an unknown sample to this reference database. Notably, molecularly complex solid tumors can be distinguished with this method despite the presence of varying proportions of nonneoplastic elements in clinical specimens. These findings suggest a new strategy for the future uniform and comprehensive molecular classification of primary and metastatic tumors.
The multiclass classifier that we describe is highly accurate, but is not perfect. That errors were evenly distributed throughout most solid tumor classes and that half of the errors were “close calls” imply that improved accuracy might be possible by increasing the number of samples from these classes in the training set, beyond the modest number used in this study.
Our findings also imply that information useful for multiclass tumor classification is encoded in complex gene expression patterns not adequately captured by a small number of genes. Although pairwise distinctions can be made between select tumor classes using fewer genes, multiclass distinctions among highly related tumor types (i.e., adenocarcinomas) are intrinsically more difficult. The effects of biological and measurement noise, contaminating nonmalignant tumor components, and inclusion of genetically heterogeneous samples within clinically defined tumor classes may all effectively decrease predictive power in the multiclass setting. Increased gene number likely allows for highly accurate prediction despite these factors. A greater variety and large number of tumors with detailed clinico-pathological characterization will be required to fully explore the true limitations of gene expression-based multiclass classification. In addition, the SVM-based classification strategy used here may not be the optimal method for every type of multiclass problem. Other classification schemes, classification algorithms, or novel marker selection methods might also be useful for making multiclass distinctions.
Interestingly, the poorly differentiated tumors analyzed in this study could not be classified according to their tissues of origin, despite the classifier's use of thousands of tissue-specific molecular markers. We had expected that these tumors would have fundamentally similar gene expression patterns compared with their well differentiated counterparts, with only minor differences. To the contrary, our data indicate that poorly differentiated tumors have a very different gene expression program. On a fundamental level, this finding raises the possibilities that poorly differentiated tumors arise from distinct cellular precursors, have different molecular mechanisms of transformation, or have unique natural histories in some other respect. This finding also has important clinical implications in that it suggests that these tumors should be classified distinctly, rather than lumped with well differentiated tumors arising from the same organ. Given the clinically aggressive nature of poorly differentiated cancers, some markers of poorly differentiated tumors might prove generally useful for predicting poorer clinical outcome.
Expression-based multiclass cancer classification is not a substitute for traditional diagnostics, but it represents a potentially important adjunct. Molecular characteristics of a tumor sample may remain intact despite atypical clinical or histological features. Classification occurs through an algorithmic rather than subjective approach in which classification confidence is quantified. In addition, all samples are evaluated by a uniform method that can be standardized throughout the medical community. Currently, diagnostic advances are disseminated into clinical practice in a slow and uneven fashion. By contrast, a centralized classification database may allow classification accuracy to rapidly improve as the classification algorithm “learns” from an ever-growing database. As robust molecular correlates of stage, natural history, and treatment response in multiple tumor classes are discovered (5, 28, 29), computational methods for making multiclass distinctions using gene expression or proteomic data will take on increasing importance.
Clinical trials will be required to determine how best to integrate genomics-based diagnostics into standard patient care. This study provides insight into the form such molecular diagnosis might take. A future challenge is to directly apply this approach to the diagnosis of clinically ambiguous tumors. In addition, many have assumed that DNA microarrays will be useful for the high-throughput discovery of tumor-specific marker genes, but that clinical implementation will use routine immunohistochemistry or other traditional methods. Indeed, some of the markers that we describe may prove useful in this realm. However, our results indicate that optimal multiclass molecular classification may require gene numbers that are beyond the scope of traditional molecular diagnostics such as immunohistochemistry. This finding suggests that the successful clinical deployment of comprehensive molecular-based classification may require the introduction of highly parallel platforms such as DNA microarrays into the clinical setting.
Acknowledgments
We thank Scott Pomeroy, Margaret Shipp, Raphael Bueno, Kevin Loughlin, and Phil Febbo for contributing tumor samples to this study. We thank David Waltregny for initial review of pathology, Christine Huard and Michelle Gaasenbeek for expert technical assistance, and Leslie Gaffney for insightful editorial review. We are also indebted to members of the Cancer Genomics Group (Whitehead/Massachusetts Institute of Technology Center for Genome Research) and the Golub Laboratory (Dana–Farber Cancer Institute) for many valuable discussions. This work was supported in part by a Harvard/National Institutes of Health training grant in Molecular Hematology (S.R.) and by grants from Affymetrix, Millennium Pharmaceuticals (Cambridge, MA), and Bristol-Myers Squibb (E.S.L.).
Abbreviations
- SVM
support vector machine
- OVA
one vs. all
- S2N
signal to noise
Note Added in Proof.
Recently, Su et al. (30) also reported using human tumor gene expression profiles to distinguish a number of carcinoma classes.
References
- 1.Ramaswamy S, Osteen R T, Shulman L N. In: Clinical Oncology. Lenhard R E, Osteen R T, Gansler T, editors. Atlanta: Am. Cancer Soc.; 2001. pp. 711–719. [Google Scholar]
- 2.Tomaszewski J E, LiVolsi V A. Cancer. 1999;86:2198–2200. doi: 10.1002/(sici)1097-0142(19991201)86:11<2198::aid-cncr4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 3.Connolly J L, Schnitt S J, Wang H H, Dvorak A M, Dvorak H F. In: Cancer Medicine. Holland J F, Frei E, Bast R C, Kufe D W, Morton D L, Weichselbaum R R, editors. Baltimore: Williams & Wilkins; 1997. pp. 533–555. [Google Scholar]
- 4.Golub T R, Slonim D K, Tamayo P, Huard C, Gaasenbeek M, Mesirov J P, Coller H, Loh M L, Downing J R, Caligiuri M A, et al. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 5.Alizadeh A A, Eisen M B, Davis R E, Ma C, Lossos I S, Rosenwald A, Boldrick J C, Sabet H, Tran T, Yu X, et al. Nature (London) 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 6.Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, et al. Nature (London) 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 7.Perou C M, Sorlie T, Eisen M B, van de Rijn M, Jeffrey S S, Rees C A, Pollack J R, Ross D T, Johnsen H, Akslen L A, et al. Nature (London) 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 8.Hedenfalk I, Duggan D, Chen Y, Radmacher M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi O P, Wilfond B, et al. N Engl J Med. 2001;344:539–548. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- 9.Khan J, Wei J S, Ringner M, Saal L H, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu C R, Peterson C, et al. Nat Med. 2001;7:673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhanasekaran S M, Barrette T R, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta K J, Rubin M A, Chinnaiyan A M. Nature. 2001;412:822–826. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 11.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E S, Golub T R. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guyon, I., Weston, J., Barnhill, S. & Vapnik, V. (2002) Mach. Learn., in press.
- 14.Hair J F, Anderson R E, Tatham R L, Black W C. Multivariate Data Analysis. Englewood Cliffs, NJ: Prentice–Hall; 1998. [Google Scholar]
- 15.Slonim D K. Proceedings of the Fourth Annual International Conference on Computational Molecular Biology. Tokyo: Universal Acad. Press; 2000. pp. 263–272. [Google Scholar]
- 16.Dasarathy V B. NN Pattern Classification Techniques. Los Alamitos, CA: IEEE Comp. Soc. Press; 1991. [Google Scholar]
- 17.Brown M P, Grundy W N, Lin D, Christianini N, Sugnet C W, Furey T S, Ares M, Haussler D. Proc Natl Acad Sci USA. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furey T, Christianini N, Duffy N, Bednarski D W, Schummer M, Haussler D. Bioinformatics. 2000;16:906–914. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- 19.Vapnik V N. Statistical Learning Theory. New York: Wiley; 1998. [Google Scholar]
- 20.Evgeniou T, Pontil M, Poggio T. Adv Comput Math. 2000;13:1–50. [Google Scholar]
- 21.Hainsworth J D, Greco F A. N Engl J Med. 1993;329:257–263. doi: 10.1056/NEJM199307223290407. [DOI] [PubMed] [Google Scholar]
- 22.Chapelle, O., Vapnik, V., Bousquet, O. & Mukherjee, S. (2002) Mach. Learn., in press.
- 23.Hastie, T., Tibshirani, R., Eisen, M. B., Alizadeh, A., Levy, R., Staudt, L., Chan, W. C., Botstein, D. & Brown, P. (2000) Genome Biol.1, RESEARCH003. [DOI] [PMC free article] [PubMed]
- 24.Taipale J, Beachy P A. Nature (London) 2001;411:349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 25.Lickert H, Domon C, Huls G, Wehrle C, Duluc I, Clevers H, Meyer B I, Freund J N, Kemler R. Development (Cambridge, UK) 2000;127:3805–3813. doi: 10.1242/dev.127.17.3805. [DOI] [PubMed] [Google Scholar]
- 26.Ziemer L T, Pennica D, Levine A J. Mol Cell Biol. 2001;21:562–574. doi: 10.1128/MCB.21.2.562-574.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bienz M, Clevers H. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 28.Scherf U, Ross D T, Waltham M, Smith L H, Lee J K, Tanabe L, Kohn K W, Reinhold W C, Myers T G, Andrews D T, et al. Nat Genet. 2000;24:236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- 29.Staunton J E, Slonim D K, Coller H A, Tamayo P, Angelo M J, Park J, Scherf U, Lee J K, Reinhold W O, Weinstein J N, et al. Proc Natl Acad Sci USA. 2001;98:10787–10792. doi: 10.1073/pnas.191368598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su A I, Welsh J B, Sapinoso L M, Kern S G, Dimitrov P, Lapp H, Schultz P G, Powell S M, Moskaluk C A, Frierson H F, Jr, Hampton G M. Cancer Res. 2001;61:7388–7393. [PubMed] [Google Scholar]