

Abstract
Atovaquone-proguanil (Malarone®) is used for malaria prophylaxis and treatment. While the cytochrome bc1-inhibitor atovaquone has potent activity, proguanil’s action is attributed to its cyclization-metabolite, cycloguanil. Evidence suggests that proguanil has limited intrinsic activity, associated with mitochondrial-function. Here we demonstrate that proguanil, and cyclization-blocked analogue tBuPG, have potent, but slow-acting, in vitro anti-plasmodial activity. Activity is folate-metabolism and isoprenoid biosynthesis-independent. In yeast dihydroorotate dehydrogenase-expressing parasites, proguanil and tBuPG slow-action remains, while bc1-inhibitor activity switches from comparatively fast to slow-acting. Like proguanil, tBuPG has activity against P. berghei liver-stage parasites. Both analogues act synergistically with bc1-inhibitors against blood-stages in vitro, however cycloguanil antagonizes activity. Together, these data suggest that proguanil is a potent slow-acting anti-plasmodial agent, that bc1 is essential to parasite survival independent of dihydroorotate dehydrogenase-activity, that Malarone® is a triple-drug combination that includes antagonistic partners and that a cyclization-blocked proguanil may be a superior combination partner for bc1-inhibitors in vivo.
Subject terms: Drug discovery, Diseases
Tina Skinner-Adams et al. show that proguanil and the cyclization-blocked analogue, tBuPG, have potent, anti-plasmodial activity in vitro. This study suggests that a cyclization-blocked proguanil may be a superior combination partner for bc1-inhibitors to suppress the survival of malarial parasites.
Introduction
The arylbiguanide proguanil (Paludrine®) was introduced for malaria treatment in the 1940’s, however resistance to the monotherapy was rapidly detected1. The potent fast-acting activity of proguanil is attributed to the dihydrofolate reductase inhibitor cycloguanil (the product of liver cytochrome P450 (CYP2C19) metabolism2) and resistance has been shown to be mediated by dihydrofolate reductase mutations3. To overcome clinical parasite resistance, and following the finding that proguanil can potentiate (increase>1000-fold) the activity of atovaquone (a cytochrome bc1 or mitochondrial electron transport chain complex III inhibitor4,5) the atovaquone-proguanil combination, Malarone®, was developed. This alleviated concerns regarding atovaquone resistance, which developed rapidly via the spread of cytochrome bc1 mutations6–8. Atovaquone-proguanil is used for the treatment of uncomplicated malaria in travellers or when first-line alternative treatments are not available or effective9. This drug is now recommended for malaria prophylaxis9, with each of the drugs in this combination having been shown to have activity against P. falciparum liver stage parasites alone10,11, and in combination12,13.
The synergistic activity of Malarone® is widely acknowledged to be associated with interactions between atovaquone and proguanil, not between atovaquone and cycloguanil5,14–16. However, there is evidence that proguanil itself also has intrinsic anti-parasitic activity. This includes evidence that proguanil has weak P. falciparum growth inhibition activity in vitro (IC50 2–71 μM; 42–72 h assays17–19), and thus independent of any metabolism, and that this activity is dihydrofolate reductase independent20,21. In vitro studies have also shown proguanil activity (IC50 3.2 µM) against P. yoelii sporozoites infecting HepG2-CD81 human hepatoma cells. These cells have impaired P450 activity22 and thus limited capacity to metabolise proguanil23. In addition, the idea that proguanil has intrinsic activity is supported by clinical observations of efficacy in regions with high levels of cycloguanil resistance24,25 and in people with impaired CYP2C19 activity (i.e., poor proguanil metabolizers)21,26. For example, in a study on the island of Malakula, Vanuatu, high antimalarial efficacy of proguanil monotherapy was observed in patients with CYP2C19-related poor proguanil metabolizer genotypes21. Together, these observations suggest that, in addition to factors such as the presence or absence of pre-existing resistance of infecting parasites to atovaquone and/or cycloguanil1,27,28, variations in how well individuals metabolise proguanil to cycloguanil21,26 may have an impact on the in vivo activity of Malarone®.
The intrinsic (i.e., in absence of metabolism) in vitro activity of proguanil against asexual blood stage parasites is not completely understood, however studies have shown that parasites with impaired mitochondrial electron transport chain function are hypersensitive to proguanil5,14,15,29. Plasmodium mitochondrial DNA (6 kb) encodes three mitochondrial electron transport chain proteins: cytochrome b and cytochrome c oxidase subunits I and III30–32. While canonical pathways central for carbon metabolism are maintained in Plasmodium mitochondrion33–36, parasite adenosine triphosphate (ATP) requirements are primarily met by cytosolic glycolysis35,37–40. An important role of mitochondria in Plasmodium asexual intraerythrocytic parasites is the provision of pyrimidine synthesis precursors. Central to this is a mitochondrion-located, essential dihydroorotate dehydrogenase enzyme, which requires ubiquinone turnover for activity14. However, it is believed that P. falciparum parasites expressing yeast dihydroorotate dehydrogenase, whose function is not linked to parasite mitochondria, must still maintain mitochondrial membrane potential to survive14. Mitochondrial membrane potential, maintained by the mitochondrial electron transport chain, is thought to be required for the function of various transporters that provide substrates for essential metabolic processes in the mitochondrion, including heme biosynthesis and iron-sulphur cluster biosynthesis14,41. Some, but not all, tricarboxylic acid cycle enzymes also appear to be essential in asexual intraerythrocytic Plasmodium parasites39. As parasites expressing yeast dihydroorotate dehydrogenase are resistant to cytochrome bc1 inhibitors, but sensitive to proguanil in combination with a cytochrome bc1 inhibitor5, it has been speculated that parasites have a secondary mechanism to maintain mitochondrial membrane potential that is only essential when the mitochondrial electron transport chain is inhibited14. It has been hypothesised14 that this secondary mechanism involves ATP synthase (complex V), that may operate in reverse to hydrolyse ATP and maintain membrane potential and that proguanil inhibits this process14. Nevertheless, while current data suggests that proguanil sensitises parasites to atovaquone-mediated mitochondrial membrane potential collapse, such a theory does not explain the intrinsic, antiplasmodial activity of proguanil reported in multiple in vitro assays17–19 and supported by clinical observations5,21,25,26. If ATP synthase is the target of proguanil it must be essential to parasites irrespective of mitochondrial electron transport chain inhibition. Additional studies are required to explain these observations and to understand the intrinsic activity of proguanil.
In this study, we explored the intrinsic17–20 in vitro action of proguanil against asexual intraerythrocytic stage parasites by profiling its temporal activity against different P. falciparum lines, demonstrating that this drug has potent, but slow-acting, activity. Furthermore, we distinguish the intrinsic anti-plasmodial activity of proguanil (1; Fig. 1) from the dihydrofolate reductase activity of cycloguanil (4; Fig. 1), by examining the activity of these compounds alongside a cyclization blocked proguanil analogue - tert-butyl proguanil (tBuPG; analogue 6; Fig. 1). tBuPG behaves similarly to proguanil, including having in vitro slow-acting activity, independent of folate, isoprenoid metabolism and pyrimidine synthesis, and synergistic activity with atovaquone and other cytochrome bc1 inhibitors. We also demonstrate antagonism between proguanil/tBuPG and cycloguanil, as well as between atovaquone and cycloguanil. Together these findings raise the possibility that a non-cyclizable proguanil analogue, similar to tBuPG, may be a better choice as a partner drug for atovaquone and other cytochrome bc1 inhibitors in vivo.
Fig. 1.

Schematic showing metabolism of proguanil to cycloguanil. a Proguanil (1) is metabolised to form the cyclization product cycloguanil (4) and the hydrolysis product 4-chlorophenylbiguanide (5). b Due to the lack of a methine proton in tBuPG (6), it is unable to undergo oxidative transformation into imine (3) and therefore cannot produce cycloguanil (4)
Results
Synthesis of a cyclization blocked proguanil analogue
The CYP2C19-mediated oxidation of proguanil (1; Fig. 1) involves homolytic C-H abstraction adjacent to the terminal nitrogen to give a radical (2; Fig. 1), which is oxidised (loss of a hydrogen atom) to give an imine intermediate (3; Fig. 1). This imine then either undergoes cyclisation of the adjacent nitrogen onto the imine to form cycloguanil (4; Fig. 1) or hydrolysis to form 4-chlorophenylbiguanide (5; Fig. 1). As the potent fast-action activity of proguanil (1; Fig. 1) is attributed to cycloguanil (4; Fig. 1) via the above in vivo metabolism mechanism, we synthesised the tert-butyl proguanil (tBuPG) analogue (6; Fig. 1b). In this analogue, the metabolism labile carbon-hydrogen bond is replaced with an inert carbon-carbon bond. As there is no methine proton in tBuPG (6; Fig. 1b), it is unable to undergo oxidative transformation into imine (3; Fig. 1b) and therefore cannot produce cycloguanil (4; Fig. 1). The cyclization blocked tert-butyl analogue of proguanil (6; Fig. 1b and Fig. 2) was synthesised from 3-(4-chlorophenyl)−1-cyanoguanidine (7; Fig. 2), as previously described42.
Fig. 2.
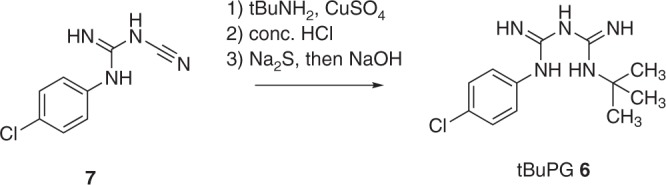
Reaction conditions for synthesis of a cyclization-blocked proguanil (tBuPG; 6)
Activity of proguanil and tBuPG against P. falciparum lines
The in vitro activity of proguanil and tBuPG in 48 h (corresponding to one asexual developmental cycle), 72 h and 96 h (corresponding to two asexual developmental cycles) assays (Fig. 3a, b) was compared to six clinically used antimalarial drugs (cycloguanil, chloroquine, pyrimethamine, atovaquone, clindamycin, and artemisinin; Fig. 3c–h). In vitro [3H]-hypoxanthine uptake assays were performed using synchronous ring-stage parasites. Comparative activity was assessed using five P. falciparum lines (3D7, Dd2, FCR3, K1 and C2B) with differing resistance profiles to control drugs (Fig. 3i, j; Supplementary Table 1). Lines were termed resistant or moderately resistant if the mean 48 h 50% inhibitory concentration (IC50) was > 10-fold or > 5-fold greater, respectively, than the drug-sensitive reference line 3D7 (Fig. 3i, j, red and white, respectively). Clindamycin had > 1000-fold better activity in 96 h (e.g., Pf3D7 IC50 0.004 µM) and 72 h (e.g., Pf3D7 IC50 0.013 µM) assays versus 48 h assays (e.g., Pf3D7 IC50 > 100 µM) (Fig. 3h; Supplementary Table 1), as expected43. Clindamycin is a protein synthesis inhibitor that causes a delayed death effect in Plasmodium with progeny inheriting a non-functional apicoplast organelle in the second cycle, following first cycle exposure43. The other control drugs generally showed a ten-fold or less change in IC50 in 48 h versus 72 h assays (Fig. 4; red circles) or 48 h versus 96 h assays (corresponding to one versus two generations of asexual blood stage intra-erythrocytic development; Fig. 4; blue squares). The IC50 for proguanil and tBuPG were lower at 72 h (e.g., Pf3D7 IC50 0.49 µM and 0.33 µM, respectively) and 96 h (e.g., Pf3D7 IC50 0.11 µM and 0.05 µM, respectively) versus 48 h (e.g., Pf3D7 IC50 46.23 µM and 7.58 µM, respectively) for all P. falciparum lines examined (P < 0.05; Supplementary Table 1; Fig. 3a, b, respectively). The IC50 fold-changes for proguanil and tBuPG were generally ≥ 20x fold higher for 48 h versus 72 h or 96 h assays (Fig. 4a, b). An exception was proguanil against FCR3 in 48 h (IC50 34.79 µM) versus 72 h assays (IC50 2.89 µM; Fig. 4a; ~12-fold IC50 change) and tBuPG against FCR3 in 48 h (IC50 16.76 µM) versus either 72 h (IC50 2.96 µM) or 96 h (IC50 1.42 µM) assays (Figure 4; ~6–12-fold IC50 change). None of the compounds examined (excluding clindamycin for which statistical analyses could not be carried as IC50s were not achieved) showed a difference in IC50s at 96 h versus 72 h (P > 0.01; Supplementary Table 1). Together these data indicate that proguanil and tBuPG have intrinsic slow-action activity that is not dependent on concurrent mitochondrial electron transport chain inhibition as previously hypothesised41.
Fig. 3.

In vitro IC50 of proguanil, tBuPG and control antimalarials at 48 h, 72 h and 96 h. P. falciparum synchronous ring-stage infected erythrocytes were assayed at 48 h (red circles), 72 h (blue squares) and 96 h (green triangles) in in vitro [3H]-hypoxanthine uptake growth inhibition assays with different antimalarial drugs (a–h). Mean ± SD IC50 values (Log10 scale) are shown for at least three independent experiments, each carried out in triplicate wells. Lines were termed resistant or moderately resistant if the mean 48 h IC50 was > 10-fold or > 5-fold greater, respectively, than the drug-sensitive reference line 3D7 (i and j; red and white, respectively). Statistical analysis (P values) of difference between 48 h, 72, and 96 h assays for each parasite line are shown in Supplementary Table 1
Fig. 4.

Fold-change in IC50 in 48 h versus 72 h or 96 h assays. P. falciparum synchronous ring stage infected erythrocytes were assayed in 48 h, 72 h and 96 h in in vitro [3H]-hypoxanthine uptake growth inhibition assays with different antimalarial drugs (a–h). Data are presented on a log-scale as fold change of mean IC50 values from Table1/Fig. 2 for 48 h/72 h (red) and 48 h/96 h (blue)
Proguanil and tBuPG activity is not rescued by isopentenyl pyrophosphate
Currently known slow-action antimalarial drugs including the antibiotic clindamycin, exhibit a delayed death phenotype. These compounds interfere with replication of the essential P. falciparum apicoplast organelle in the second cycle after exposure43,44. As the only essential function of the apicoplast during asexual blood stage growth is isoprenoid precursor biosynthesis, products of this pathway such as isopentenyl pyrophosphate can rescue the effects of apicoplast-associated delayed death agents in vitro45. To determine whether the slow-action activity observed for proguanil and tBuPG is due to apicoplast-associated delayed death, in vitro 96 h rescue assays were carried out in media with and without isopentenyl pyrophosphate supplementation. As expected, the activity of the control drug clindamycin was rescued by isopentenyl pyrophosphate (Fig. 5a). In contrast, as for the negative control compound cycloguanil (Fig. 5b), the activity of proguanil (Fig. 5c) and tBuPG (Fig. 5d) was not rescued by the addition of isopentenyl pyrophosphate. These data indicate that proguanil and tBuPG do not cause apicoplast-associated delayed death of the parasite.
Fig. 5.

In vitro activity of proguanil and tBuPG with and without isopentenyl pyrophosphate supplementation. Synchronous ring-stage P. falciparum 3D7 parasitized erythrocytes were assayed using proguanil (c), tBuPG (d) or the control drugs clindamycin (a) and cycloguanil (b) over 96 h in [3H]-hypoxanthine uptake growth inhibition assays. Assays were carried out in RPMI 1640 parasite culture media supplemented with ( + isopentenyl pyrophosphate; blue, closed circles) or without (-isopentenyl pyrophosphate; black, closed circles) 200 µM isopentenyl pyrophosphate. In each case, data are mean ( ± SD) % inhibition of two independent experiments, each carried out in triplicate wells
Proguanil and tBuPG action is not rescued by p-aminobenzoic acid-folic acid
To confirm that the slow-action phenotype of proguanil and tBuPG in P. falciparum is not related to inhibition of folate biosynthesis (mode of action of cycloguanil), rescue experiments were carried out using media supplemented with folic acid and the folate synthesis intermediate, p-aminobenzoic acid, versus media with low folic acid and p-aminobenzoic acid levels. As shown in Fig. 6, the 96 h activity of proguanil and tBuPG against P. falciparum 3D7 was not altered in media with low versus high levels of folic acid and p-aminobenzoic acid. In contrast, and as expected, the activity of cycloguanil, but not chloroquine, was rescued with folic acid and p-aminobenzoic acid.
Fig. 6.

Activity of proguanil and tBuPG in low versus supplemented p-aminobenzoic acid and folic acid media. The sensitivity of synchronous ring-stage P. falciparum 3D7 parasitized erythrocytes to control drugs cycloguanil (a) and chloroquine (b) and test compounds proguanil (c) and tBuPG (d) was assessed in 96 h dose response [3H]-hypoxanthine growth inhibition assays. Assays were performed in paired plates, one containing media without supplemented p-aminobenzoic acid and folic acid (low p-aminobenzoic acid and folic acid; black solid line) and the other with supplemented p-aminobenzoic acid and folic acid (high p-aminobenzoic acid and folic acid; 1 mg /mL each; blue dashed line). Data are presented as mean ( ± SD) % inhibition of two independent experiments, each carried out in triplicate wells
Activity appears independent of pyrimidine synthesis
Data from previous studies show that parasites with impaired mitochondrial electron transport chain function are hypersensitive to proguanil5,14,15,29. A critical function of the Plasmodium mitochondrial electron transport chain is reoxidisation of dihydroubiquinone to ubiquinone, the electron acceptor for the dihydroorotate dehydrogenase enzyme that is critical to pyrimidine synthesis14,46. Here we show that proguanil and tBuPG activity appears independent of dihydroorotate dehydrogenase and pyrimidine biosynthesis by examining their activity against P. falciparum parasites episomally expressing yeast dihydroorotate dehydrogenase (3D7-yDHODH)47. As the activity of yeast dihydroorotate dehydrogenase is dependent on fumarate rather than ubiquinone, it is not linked to mitochondrial mediated ubiquinone turnover48. Activity against 3D7-yDHODH parasites was compared to wild type 3D7 parasites in 48 h and 96 h assays using proguanil, tBuPG and cytochrome bc1 inhibitors (atovaquone49, myxothiazole49, ELQ-30050, decoquinate51 and antimycin A49). As proguanil has been hypothsized to inhibit the mitochondrial electron transport chain, other eukaryote mitochondrial electron transport chain inhibitors (sodium azide52,53, oligomycin A53 and metformin54) were also examined for comparison purposes. However, as discussed below, the targets of these agents in P. falciparum are not known. The IC50 of proguanil was ≥ 50-fold lower for both 3D7 and 3D7-yeast dihydroorotate dehydrogenase parasites in 96 h (IC50 360 nM and 490 nM, respectively) versus 48 h (IC50 22.0 µM and 25.0 µM, respectively) assays (Fig. 7a, b; Supplementary Table 2; P < 0.01). A similar finding was seen for tBuPG for both 3D7 and 3D7-yeast dihydroorotate dehydrogenase parasites, with > 20-fold lower IC50 in 96 h (IC50 210 nM and 470 nM, respectively) versus 48 h (IC50 11.0 µM and 9.20 µM, respectively) assays (Fig. 7a, b; Supplementary Table 2; P < 0.01). This indicates that the slow-action activity of these compounds is retained in 3D7-yeast dihydroorotate dehydrogenase parasites. The activity of proguanil and tBuPG was also not different for 3D7 and 3D7-yeast dihydroorotate dehydrogenase parasites in 48 h or 96 h assays (3D7-yeast dihydroorotate dehydrogenase IC50/3D7 IC50 ratios of 1–2; Fig. 7b; Supplementary Table 2; P > 0.05). These data suggest that the slow-action activity of these compounds appears independent of pyrimidine biosynthesis, a critical pathway within P. falciparum mitochondria. Similar to proguanil and tBuPG, the activity of mitochondrial electron transport chain inhibitors metformin, sodium azide and oligomycin A was independent of pyrimidine synthesis, with the activity of each retained against 3D7-yeast dihydroorotate dehydrogenase parasites (3D7-yeast dihydroorotate dehydrogenase IC50/3D7 IC50 ratios of ~1–2; Fig. 7b; Supplementary Table 2; P > 0.05). However, in contrast to proguanil and tBuPG, metformin, sodium azide and oligomycin A did not display a slow-action phenotype (Fig. 7a; Supplementary Table 2; 3D7 96 h/48 h IC50 ≤ 5). As expected, the five complex III inhibitors were more potent against 3D7 than 3D7-yeast dihydroorotate dehydrogenase parasites in 48 h assays, with > 380-fold higher IC50s for 3D7-yeast dihydroorotate dehydrogenase versus 3D7 (excluding antimycin A, for which precise 3D7-yeast dihydroorotate dehydrogenase IC50 calculations were not possible; (Fig. 7a; Supplementary Table 2). This same effect was also seen for 96 h assays, with > 11-fold higher IC50s for 3D7-yeast dihydroorotate dehydrogenase versus 3D7 parasites (Fig. 7a; Supplementary Table 2; P < 0.05). Interestingly, in 96 h assays, the activity of the cytochrome bc1 inhibitors against 3D7-yeast dihydroorotate dehydrogenase parasites was >38–1000-fold greater than in 48 h assays, with the possible exception of antimycin A that was less potent than the other compounds, and for which a precise 48 h 3D7-yeast dihydroorotate dehydrogenase IC50 could not be calculated (Supplementary Table 2). Together these data suggest that in the absence of indirect activity due to effects on dihydroorotate dehydrogenase and pyrimidine biosynthesis, cytochrome bc1 inhibitors have a slow-action anti-plasmodial phenotype.
Fig. 7.

In vitro activity of compounds against P. falciparum 3D7 and 3D7- yeast dihydroorotate dehydrogenase parasites in 48 h and 96 h assays. a Compounds were tested in 48 h and 96 h assays against P. falciparum 3D7 and 3D7-yeast dihydroorotate dehydrogenase parasites (3D7-yDHODH), starting with synchronous ring-stage parasites. Mean IC50 ( ± SD) values are shown for 48 h (3D7 solid black bars; 3D7-yDHODH solid grey bars) versus 96 h (3D7 black striped bars; 3D7-yDHODH grey striped bars) assays for each line. Red star—IC50 not achieved (i.e., IC50 greater than value shown). b Fold change in IC50 of compounds against P. falciparum 3D7 and 3D7-yDHODH in 48 h (white bars) and 96 h (chequered bars) assays. Mean IC50 ( ± SD) values were used to compare activity against 3D7-yDHODH versus 3D7 for 48 h and 96 h assays. Solid and dashed horizontal lines show 1× and 10× fold change, respectively. In each case, 2–4 independent experiments were carried out, each in triplicate wells, before data were averaged and fold change calculated. Red star – IC50 not achieved: fold change calculated based on highest concentration tested (i.e., IC50 higher than this value). Selected eukaryote complex I and IV/V mitochondrial electron transport chain inhibitors (sodium azide52,53, oligomycin A53 and metformin54) were examined for comparison purposes
In vitro combination studies
The co-administration of compounds can result in effects that are less or greater than their predicted sum effects when used alone. These interactions can impact the activity of compounds in the clinical setting and can be assessed by examining the activity of compound combinations and then using these data to plot isobolograms. These plots indicate potential compound interactions, with concave, convex and linear isoboles being indicative of synergistic, antagonistic and additive interactions, respectively. Isobologram analyses were performed to examine the interactions of proguanil and tBuPG with cycloguanil and cyctochrome bc1 inhibitors atovaquone or ELQ-300. Assays were performed with 3D7 and 3D7-yeast dihydroorotate dehydrogenase parasites to examine compound interactions in the presence and absence of any indirect effect on pyrimidine synthesis. Proguanil and tBuPG behaved synergistically with atovaquone and ELQ-300 when assessed against 3D7 or 3D7-yeast dihydroorotate dehydrogenase parasites for 48 h or 96 h (Fig. 8), whereas combinations of cycloguanil with proguanil, tBuPG or atovaquone behaved antagonistically (Fig. 9). As 3D7-yeast dihydroorotate dehydrogenase parasites are resistant to cycloguanil, due to the presence of the human dihydrofolate reductase gene on the transfection plasmid, compound interactions with this drug against this parasite line were not assessed.
Fig. 8.

Isobolograms describing the interactions of progunail and tBuPG with cytochrome bc1 inhibitors ELQ-300 and atovaquone against P. falciparum 3D7 and 3D7-yeast dihydroorotate dehydrogenase (3D7-yDHODH) parasites in 48 h and 96 h assays. The interactions of antimalarial compounds was assessed using isobologram analysis. Combinations of compounds that resulted in 50% growth inhibition were determined and are presented as Fraction Inhibitory Concentrations (FICs), where a FIC = 1 represents the concentration of each compound required to inhibit parasite growth by 50% when used alone. Interaction (I) values were also determined4. Positive values of I indicate synergism, and negative values indicate antagonism; addition occurs when I equals 0. The significance of the difference of I from zero was assessed with Student’s t test
Fig. 9.

Isobolograms describing the interactions of proguanil or tBuPG with cycloguanil and cycloguanil with atovaquone against P. falciparum 3D7 parasites. The interactions of antimalarial compounds was assessed using isobologram analysis in 48 h assays. I values were calculated as previously described4. Positive values of I indicate synergism, and negative values indicate antagonism; addition occurs when I equals 0. The significance of the difference of I from zero was assessed with Student’s t test
Activity in a Plasmodium liver-cell infection model
The effect of proguanil and tBuPG on in vitro murine malaria parasite liver stage infection55 was assessed using P. berghei luciferase-expressing sporozoites and HepG2-A16-CD81EGFP cells (Fig. 10). The activity of tBuPG (IC50 2.94 µM) was approximately two-fold better than that of proguanil (IC50 6.02 µM). These values are consistent with the previously published activity of proguanil against P. yoelii infected HepG2-CD81 cells (IC50 3.2 µM)23.
Fig. 10.
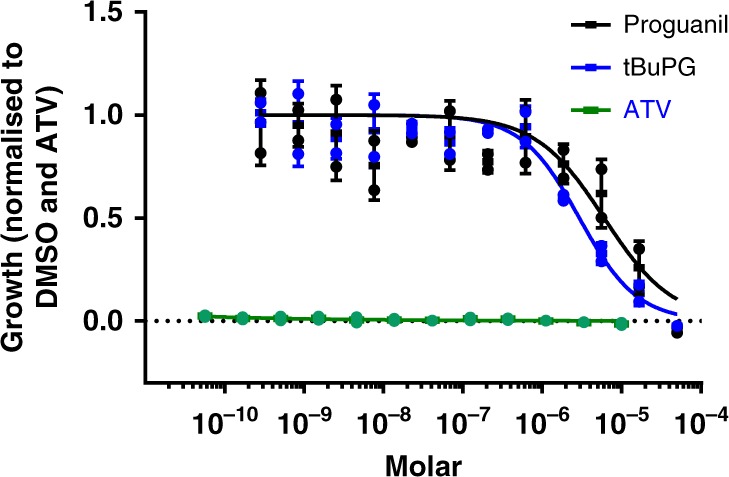
Proguanil and tBuPG show micromolar anti-P. berghei-Luciferase liver stage activity. Activity of proguanil and tBuPG was tested in HepG2-A16-CD81EGFP cells infected with P. berghei-Luciferase sporozoites. Data was normalised against atovaquone (ATV) and DMSO, and liver stage P. berghei-Luciferase IC50 values were determined using the average normalised bioluminescence intensity of 8 wells per concentration from two technical replicates of 1536- well plates, and a nonlinear variable slope four-parameter regression curve fitting model in Prism 6 (GraphPad Software Inc.). Proguanil (IC50 6.02 µM) and tBuPG (IC50 2.94 µM)
Discussion
With almost half the world’s population at risk of malaria and a global agenda focused on eradication56, there is an urgent need to develop new drugs with novel modes of action for malaria prevention and treatment57,58. To date, the development of malaria chemoprotection drugs has lagged behind the development of treatment drugs9,58. However, the shift towards achieving malaria eradication has highlighted the need for new and improved chemoprotection agents56–58. As malaria is eliminated from countries, infection dynamics will change, with people moving from being semi-immune to non-immune. Thus, drug combinations used to prevent disease and parasite transmission (chemoprotection) described by Target Product Profile 258, as opposed to those to treat malaria symptoms (treatment) described by Target Product Profile 158, will be needed for vulnerable populations and during outbreaks57. Chemoprotection strategies should ideally include drug combinations with causal liver and asexual blood stage58 activity (that does not need to be fast-onset57), a profile that has been modelled on atovaquone-proguanil (Malarone®)58. Few drugs are currently available to prevent malaria, with none currently suitable for wide-scale mass drug administration in malaria-endemic areas57,59. Malarone® is used for daily malaria prophylaxis, but also for treatment of uncomplicated malaria in travellers or when other first-line treatments are not available or effective9. While doxycycline and some other antibacterial agents can be used for short-term prevention (mainly for travellers)9, daily dosing is prescribed and there are contraindications for infants and pregnant women, the most at-risk individuals in malaria endemic regions. Mefloquine, in contrast, can cause neurological side-effects9. The development of new, well-tolerated and effective chemoprotection agents is vital to the eventual eradication of malaria57,58.
Despite Malarone® being widely used for malaria chemoprotection, the antimalarial action of this drug combination is not completely understood. While many studies have suggested that the synergistic activity of Malarone® is mediated by atovaquone and proguanil, rather than proguanil’s active metabolite cycloguanil16,23, the anti-plasmodial action of proguanil has remained largely unexplored. Likewise, mechanisms to exploit the synergistic activity of atovaquone with proguanil, by preventing proguanil metabolism to cycloguanil, have not been investigated. Here we examined the possibility that a cyclization-blocked proguanil analogue may have the potential to replace proguanil through its beneficial properties as a synergistic partner drug for cytochrome bc1 inhibitors and by overcoming limitations associated with variability in how people metabolise proguanil to cycloguanil. This is important given that the activity of cyclogunail is compromised by widespread P. falciparum dihydrofolate reductase mutations that mediate drug resistance.
Published in vitro studies report that proguanil has weak and variable activity against asexual intraerythrocytic-stage P. falciparum parasites, with IC50s ranging from 2 to 71 μM in 42–72 h assays17–19. Adding to this data variability, in a high-throughput screen carried out by one of our groups several years ago, proguanil was found to have sub-µM activity in a 96 h assay (IC50 0.36 µM), but poor activity in a 48 h assay (IC50 2.87 μM)60. These data were deposited on PubChem61,62 as part of a larger dataset60, but the potential significance was not noted60. Indeed the dogma that proguanil has no or limited antiplasmodial activity prevails in the literature (e.g., recently reviewed in ref. 63), and as a result these findings remain essentially unrecognised. Here, we sought to clarify these data by investigating the intrinsic activity of proguanil and a cyclization-blocked analogue (tBuPG). Proguanil and tBuPG were first tested against five different P. falciparum lines in 48 h, 72 h and 96 h asexual stage, drug sensitivity assays. Our data demonstrate that proguanil and tBuPG have potent slow-action in vitro activity against P. falciparum parasites (Supplementary Table 1), at least an order of magnitude (10–420 fold) better than in 48 h fast-action assays. This slow-action activity is independent of folate metabolism (Fig. 6), and thus not associated with dihydrofolate reductase inhibition, cycloguanil’s primary mode of action. It is also independent of non-mevalonate isoprenoid biosynthesis (Fig. 5), the only essential function of the apicoplast organelle45 and the target of antibiotics with a delayed death phenotype64. These data suggest that slow-action activity of proguanil likely represents a different slow-action anti-plasmodial mechanism.
While proguanil’s mechanism of action is not completely understood, evidence suggests that it is linked to parasite mitochondria. In vitro combination studies have shown that proguanil lowers the concentration at which atovaquone collapses parasite mitochondrial membrane potential14,49. Investigations on the function of the mitochondrial ribosomal protein L13, involved in the translation of mitochondrial electron transport chain components, encoded by mitochondrial DNA, have shown that knock-down of this protein results in disruption of mitochondrial integrity, mitochondrial membrane potential collapse and hypersensitivity to proguanil29. The precise mechanism behind the mitochondrial membrane potential collapse mediated by proguanil is not known. However, proguanil’s poor activity in traditional fast-action assays has resulted in a hypothesis that this action is linked to an ATP synthase function that only becomes essential when the mitochondrial electron transport chain is inhibited14. It has been hypothesised14 that during mitochondrial electron transport chain inhibition, ATP synthase maintains mitochondrial membrane potential by operating in reverse and that proguanil inhibits this process14. This hypothesis fits well with functional observations in bovine mitochondria54, but does not account for reports that proguanil is active against parasites irrespective of mitochondrial electron transport chain inhibition, unless ATP synthase activity is essential to P. falciparum survival irrespective of mitochondrial electron transport chain inhibition. While the essentiality of this complex in P. falciparum remains to be determined, studies have shown ATP synthase is not essential in P. berghei asexual intraerythrocytic stage parasites37,40. It is possible that proguanil targets an alternative mechanism responsible for mitochondrial membrane potential maintenance. Alternatively, the slow-action activity of proguanil may be mediated by a mechanism different to that associated with its potentiation of mitochondrial membrane collapse.
One of the critical functions of mitochondria in malaria parasites is the re-oxidation of dihydroubiquinone to ubiquinone. Ubiquinone is the electron acceptor for several dehydrogenases including dihydroorotate dehydrogenase, a critical enzyme involved in pyrimidine synthesis14,46. We therefore further investigated the mode of action of proguanil by examining the effect of unlinking the pyrimidine pathway from the mitochondrial electron transport chain on proguanil and tBuPG activity. These assays involved a comparative assessment of the activity of proguanil and tBuPG with the activity of known mitochondrial electron transport chain inhibitors against wild-type P. falciparum parasites and parasites episomally expressing the ubiquinone-independent, fumarate-utilising, Saccharomyces cerevisiae (yeast) dihydroorotate dehydrogenase (Pf3D7-yeast dihydroorotate dehydrogenase)47. P. falciparum dihydroorotate dehydrogenase activity requires ubiquinone, however yeast dihydroorotate dehydrogenase uses fumarate, and is therefore not directly affected by cytochrome bc1 activity14,48. Our data demonstrate that the slow-action activity of proguanil and tBuPG is retained against Pf3D7-yeast dihydroorotate dehydrogenase parasites. This observation suggests that slow-action proguanil activity is independent of pyrimidine synthesis and more likely to be directly associated with mitochondrial electron potential, as previously hypothesised14. However, a particularly interesting observation from these data was that the activity of cytochrome bc1 inhibitors switched from comparatively fast-action when tested against 3D7 to slow-action when assessed against Pf3D7-yeast dihydroorotate dehydrogenase parasites. These data suggest that Pf3D7-yeast dihydroorotate dehydrogenase parasites remain sensitive to atovaquone, but that the activity of atovaquone changes in these parasites. While this may be explained by a separate atovaquone target, it is more likely due to mitochondrial electron transport chain inhibition given previous observations with similar parasite lines have shown that this inhibitory activity can be rescued with decylubiquinone65. This observation is important as it suggests the essentiality of additional ubiquinone dependent enzymes in these parasites that may warrant further investigation as drug targets. However, the strain specific nature of this observation as reported by Ke et al. requires further investigation. Interestingly, while assessments and parasites were different, Ke et al. reported yeast dihydroorotate dehydrogenase parasites derived from the 3D7 line to be resistant to slow-action atovoaquone activity65, we did not observe this resistance (Fig. 7 and Supplementary Table 2).
A mitochondrial mediated slow-action of atovaquone also fits well with the proposed slow-action phenotype and mechanism of action of proguanil and isobologram data that demonstrate that the interaction between atovaquone and proguanil remains synergistic against Pf3D7-yeast dihydroorotate dehydrogenase parasites (Fig. 8). Consistent synergy over time between atovaquone and proguanil or tBuPG against 3D7 and Pf3D7-yeast dihydroorotate dehydrogenase parasites also suggests that the slow-killing action of proguanil/tBuPG and atovaquone, when uncoupled from pyrimidine synthesis, are mechanistically related. While previous studies have demonstrated that proguanil and atovaquone act synergistically in vitro against wild-type4,5 and yeast dihydroorotate dehydrogenase-expressing parasites14, our study is the first, to our knowledge, to assess and compare synergy at early and late time points.
Our data on the activity of metformin, sodium azide and oligomycin A (included as eukaryotic control inhibitors of complex I, complex IV/V, and complex V, respectively) suggest that the anti-plasmodial activity of these compounds is independent of pyrimidine synthesis, but not slow-action. While these observations suggest that the slow-action mitochondrial electron transport chain inhibition phenotype may be limited to cytochrome bc1 inhibition, it is important to note that the efficacy of these classic eukaryotic mitochondrial electron transport chain inhibitors against Plasmodium mitochondrial electron transport chain complexes may be limited or may be complicated by other functions66,67. Metformin and sodium azide may have non-mitochondrial electron transport chain targets68–70 and have been shown to cause the generation of reactive oxygen species (ROS) following mitochondrial electron transport chain inhibition54,71. Thus, ROS toxicity could inhibit cells independently of mitochondrial electron transport chain dysfunction. Likewise, while oligomycin A has been shown to block the activity of ATP synthase (complex V)72, it has also been shown to interact with other ATPases/channels, such as Na + /K + -ATPase73. Interestingly, similar to ATP synthase, studies have shown that complex I (NADH:ubiquinone oxidoreductase (NDH2)) is not essential in asexual intraerythrocytic stage P. berghei parasites37,40. The essentiality of these complexes in P. falciparum remains to be determined.
Similar to previous studies with proguanil and atovaquone5, our data demonstrate that tBuPG behaves synergistically with cytochrome bc1 inhibitors (Fig. 8). Proguanil and tBuPG also interacted synergistically with ELQ-300, a cyctochrome bc1 inhibitor recently suggested as a partner drug for atovaquone74. ELQ-300 is an attractive combination partner for atovaquone as it binds the quinone reductase site instead of the quinol oxidase site of cytochrome bc175, rendering atovaquone-resistant parasites sensitive to ELQ-30076. Recent data also suggest that atovaquone and ELQ-300 interact synergistically74. In contrast to these synergistic combinations our results show that combinations of the dihydrofolate reductase inhibitor cycloguanil with atovaquone are antagonistic (Interaction values < −5.0 in 48 h 3D7 assays; Fig. 9). While this observation has been supported by others16, additive activity for this combination has also been described15. The inconsistencies in these reports are difficult to reconcile, however, antagonistic interaction between atovaquone and WR99210, a compound with structural similarities to cycloguanil, have also been reported15. The mechanism of the reported antagonistic activity between atovaquone and cycloguanil remains to be determined, but may be associated with cellular entry mechanisms rather than direct mode of action. Combinations of cycloguanil with proguanil or tBuPG also behaved antagonistically in these studies (Fig. 9). These findings have potentially important ramifications for the activity of Malarone® in vivo, as they suggest that the synergistic activity of this combination may vary with proguanil metabolism and may be sub-optimal when proguanil is efficiently metabolised to cycloguanil. The rapid metabolism of proguanil to cycloguanil in a setting of high cycloguanil resistance and antagonistic activity may also impact the development of resistance to atovaquone or other cytochrome bc1 inhibitors partnered with proguanil, a factor that would be particularly problematic in instances where the partner drug has a long elimination half-life. While additional factors, including the pharmacokinetics of individual partner drugs, would need to be considered and further in vivo studies performed, these data also suggest that the highly synergistic nature of combinations of cytochrome bc1 inhibitors with proguanil can be maintained with compounds unable to be converted to cycloguanil, and that these combinations may be better than combinations with proguanil. As the cyctochrome bc1 inhibitor combination of atovaquone and ELQ-300 has recently been shown to be synergistic in P. yoelii infected mice74, it is also possible that a cyclization-blocked proguanil analogue may be a useful addition to dual cytochrome bc1 inhibitor combinations. While additional liver stage investigations, including P. vivax hypnozoite assays77, will be important to expand our findings, data showing that tBuPG (IC50 2.94 µM; Fig. 10) is active against P. yoelii infected HepG2-CD81 cells provides additional support for a cyclization-blocked proguanil analogue and cyctochrome bc1 inhibitor combination as a chemoprevention strategy23.
In summary, we report that proguanil and tBuPG have potent slow-acting activity that is independent of folate metabolism, pyrimidine synthesis and isoprenoid biosynthesis. Our data support the hypothesis that these compounds function within malaria parasite mitochondria and that this activity is responsible for the synergistic activity of Malarone® but in contrast to previous hypotheses, suggest that this action is effective in the absence of concurrent mitochondrial electron transport chain inhibition. Although the slow-action of these compounds means that they may not be ideal for clinical disease use, these compounds may still be well-suited for use as prophylactics. As we have previously hypothesised18, our data also raise the question as to whether Malarone® is actually acting as a triple combination, dependent on where and how it is used (i.e., for chemoprotection or treatment, whether pre-existing resistance to combination components is present and whether patients are high or low proguanil metabolisers). Taking into account the pharmacokinetics of atovaquone-proguanil (proguanil t1/2 12–21 h; atovaquone t1/2 1–6 days78,79), together with the slow-action pharmacodynamics of proguanil reported here, there may be an intrinsic benefit to the use of Malarone® as a prophylactic agent. When administered for treatment Malarone® is taken for three days versus a minimum of nine days as a prophylactic80. The metabolism of proguanil to cycloguanil may also result in a sub-optimal combination therapy strategy given the synergy of Malarone® is driven by proguanil not its cyclized metabolite cycloguanil. Thus, an alternative, and perhaps more effective strategy, may be the incorporation of a cyclization blocked proguanil such as tBuPG or a suitable analogue. Such a strategy would also overcome variations in activity associated with differences in proguanil metabolism.
Methods
Parasites and culture
P. falciparum parasites were maintained in vitro with human O+ erythrocytes in RPMI 1640 (cat # R8758; Sigma) containing 10% heat inactivated human serum and 5 mg/L gentamycin at 37oC in 5% O2 and 5% CO2 in N2, essentially as previously described81. The following P. falciparum lines were used: 3D7, Dd2, FCR3, K1 and C2B. P. falciparum 3D7 parasites overexpressing the yeast dihydroorotate dehydrogenase gene (3D7-yeast dihydroorotate dehydrogenase) were kindly provided by Dr Ben Dickerman, Burnet Institute, Melbourne Australia.
Compounds
Pyrimethamine, decoquinate, myxothiazol, sodium azide, atovaquone, clindamycin hydrochloride, artemisinin, antimycin A, oligomycin A, metformin hydrochloride and chloroquine diphosphate salt were all purchased from Sigma Aldrich, USA. Cycloguanil hydrochloride was purchased from Santa Cruz Biotechnology Inc, USA. ELQ-300 was provided by the Medicines for Malaria Venture. Proguanil was purchased from Sigma Aldrich, USA or synthesised alongside cyclization blocked proguanil as described below. Isopentenyl pyrophosphate was prepared according to a literature procedure82 and the NMR data is in accordance with that previously reported for isopentenyl pyrophosphate82,83. 1H NMR (400 MHz, D2O/ND4OD): δ 4.86 (d, J = 8.8 Hz, 2 H), 4.08 (q, J = 6.8 Hz, J = 13.6 Hz, 2 H), 2.41 (t, J = 6.8 Hz, 2 H) and 1.78 (s, 3 H); 13C NMR (100 MHz, D2O/ND4OD): δ 143.9, 111.4, 64.0, 37.8 and 21.6; 31P NMR (161 MHz, D2O/ND4OD): δ −6.37 (d, J = 22.5 Hz, 1 P) and −10. 43 (d, J = 22.5 Hz, 1 P). All compounds were prepared as 10–20 mM stock solutions in 100% DMSO or in the case of chloroquine PBS.
Proguanil and tBuPG (; 1-(4-Chlorophenyl)−5-tert-butyldiguanide) were synthesised from 3-(4-chlorophenyl)−1-cyanoguanidine. 3-(4-chlorophenyl)−1-cyanoguanidine was prepared according to a previously published method84. Briefly, a solution of p-chloroaniline (17.0 g, 0.133 mmol) in water (63 mL) and concentrated HCl (11.1 mL) was added over 1 h to a 50 °C solution of sodium dicyanamide (23.7 g, 0.266 mmol) in water (203 mL). The reaction mixture was heated to 80 °C for 24 h. After cooling to ambient temperature, saturated NaHCO3 solution (150 mL) was added. After stirring for 15 min the precipitated solid was collected by filtration, washed with water and air-dried to give 3-(4-chlorophenyl)−1-cyanoguanidine as a cream-coloured solid (20.2 g, 78%). 1H NMR (d6-DMSO) 400 MHz δ 9.15 (br.s, 1 H) and 7.40–7.33 (m, 4 H); 7.07 (br.s, 2 H).
Proguanil was prepared from 3-(4-chlorophenyl)−1-cyanoguanidine according to the method of Curd and Rose (1946)42. 3-(4-chlorophenyl)−1-cyanoguanidine (1.0 g, 5.14 mmol) was dissolved in ethanol (7.6 mL) and water (3.0 mL) and copper sulfate were added, followed by isopropylamine (0.92 g, 15.6 mmol) and the mixture heated to reflux for 20 h. After cooling to ambient temperature, water (23 mL) was added, followed by a solution of concentrated HCl (2.6 mL) in water (15.5 mL). After stirring for 30 min a solution of sodium sulfide nonahydrate (2.06 g) in water (10 mL) was added and the mixture stirred for 30 min. The mixture was filtered to remove copper sulfide, and the filtered solid washed with hot water (50 mL). The filtrate was cooled to 10 °C and a solution of sodium hydroxide (1.16 g in water 8.3 mL) was added slowly. The precipitated solid was collected by filtration and dried. Recrystallisation from toluene gave proguanil as a colourless solid (825 mg, 63%). 1H NMR (d6-DMSO) 400 MHz δ 7.40 (br. s, 1 H), 7.22–7.17 (m, 2 H), 6.85–6.76 (m, 2 H), 4.89 (br. s, 2 H), 3.85–3.80 (m, 1 H) and 1.08 (d, J = 1.8 Hz, 6 H). HRMS (): calcd. for C11H17ClN5 [M]+ 254.1170, found 254.1167.
1-(4-Chlorophenyl)−5-tert-butyldiguanide (tBuPG) was prepared from 3-(4-chlorophenyl)−1-cyanoguanidine as follows. 1-(4-Chlorophenyl)−2-cyanoguanidine (1.0 g, 5.14 mmol) was dissolved in ethanol (7.6 mL) and water (3.0 mL) and copper sulfate (650 mg, 2.60 mmol) was added, followed by tert-butylamine (0.92 g, 15.6 mmol) and the mixture heated to reflux for 24 h. After cooling to ambient temperature water (23 mL) was added, followed by 2 M HCl (15.5 mL). After stirring for 30 min a solution of sodium sulfide nonahydrate (2.06 g) in water (10 mL) was added and the mixture stirred for another 30 min. The mixture was filtered to remove copper sulfide, and the solid washed with hot water (50 mL). The combined filtrate was cooled to 10 °C and a solution of sodium hydroxide (1.16 g) in water (8.3 mL) was added slowly. A cream-coloured solid precipitated and was collected by filtration. Recrystallisation from toluene gave 1-(4-chlorophenyl)−5-tert-butyldiguanide (436 mg, 32%) as a colourless solid. 1H NMR (d6-DMSO) 400 MHz δ 9.98 (br. s, 1 H), 7.76 (br. s, 1 H), 7.42–7.32 (m, 4 H), 7.22–7.00 (m, 3 H) and 1.24 (s, 9 H). HRMS: calcd. for C12H19ClN5 268.1329, found 268.1327.
In vitro P. falciparum growth inhibition assays
Fast-action (48 h) in vitro growth inhibitory activity of compounds was tested against synchronous early ring cultures (final 1% parasitemia and 1% haematocrit) of P. falciparum parasitized erythrocytes using [3H]-hypoxanthine incorporation (0.5 µCi/well; 96 well plates), essentially as previously described85. Following incubation at 37oC for 48 h (5% CO2; 5% O2; 90% N2) [3H]-hypoxanthine incorporation was determined by harvesting cultures onto glass fibre filter mats and counting using a Perkin Elmer/Wallac Trilux 1450 MicroBeta scintillation counter. Percentage inhibition of growth compared to matched vehicle controls was determined and IC50 values calculated using log-linear interpolation of inhibition curves86. Data are presented as mean ( ± SD) of at least three independent assays, each carried out in triplicate. The slow-action in vitro growth inhibitory activity of compounds was also determined using the [3H]-hypoxanthine incorporation against synchronous early ring cultures. However parasites were added to drug dilutions at a final parasitemia and haematocrit of 0.1% and 2% respectively. Assay plates were incubated at 37oC for 72 h (5% CO2; 5% O2; 90% N2) before media refreshment (100 µL) and the addition of [3H]-hypoxanthine (0.5 µCi/well). After a further 24 h incubation (total 96 h), [3H]-hypoxanthine incorporation was assessed. Growth inhibition and IC50 values were then determined as described for 48 h assays.
Rescue assays
Isopentenyl pyrophosphate and folate + p-aminobenzoic acid rescue experiments were carried in 96-well plates over 96 h essentially as previously described for in vitro growth inhibition assays. However, for each comparison, two identical 96 h assays were performed simultaneously. For isopentenyl pyrophosphate rescue experiments one plate was supplemented with 200 µM isopentenyl pyrophosphate45 whereas the other was not. Clindamycin and cycloguanil were included as positive and negative controls respectively. For folate + p-aminobenzoic acid rescue experiments all assays were performed in custom RPMI 1640 containing no folate or p-aminobenzoic acid (Life Technologies; USA), one plate was then supplemented with folate 1 mg/mL and 1 mg/mL p-aminobenzoic acid, while the remaining plate was left without supplementation. Cycloguanil and chloroquine served as positive and negative controls respectively in these assays. In each case two independent assays were performed, each in triplicate wells.
Isobologram assays
Antimalarial drug combinations were assessed against P. falciparum 3D7 or 3D7-yeast dihydroorotate dehydrogenase. Assays were performed in 96-well microtiter plates essentially as previously described87. However parasites were prepared at 0.1% parasitemia and 2% hematocrit for 96 h assays. Each combination was assessed at least twice in triplicate and IC50 values were determined by log-linear interpolation86. Isobolograms were constructed from normalised fractional-inhibitory concentration (FIC) values and Isoboles were fitted to define the interaction parameter, I, as previously described4,87. Positive values of I indicate synergism, and negative values indicate antagonism; addition occurs when I equals 0. The significance of I for each compound combination was tested using a Student’s t test87.
In vitro P. berghei-Luciferase liver stage assays
Human hepatoma, HepG2-A16-CD81EGFP, cells were cultured at 37 °C in 5% CO2 in DMEM media (DMEM (Life Technology, CA) supplemented with 10% FBS and 1x Pen-Strep-Glutamine (Gibco, CA). For both P. berghei-Luciferase and HepG2 cytotoxicity assays, 20–26 h prior to sporozoite infection, HepG2-A16-CD81EGFP20 cells (6 × 105 cells/ml in assay medium; DMEM without Phenol Red (Life Technologies, CA), 5% FBS, and 5x Pen-Strep-Glutamine (Gibco, CA)) were seeded into white solid bottom 1536-well plates (custom GNF mold ref# 789173-F, Greiner Bio-One) at 5 μL per well. Compounds (50 nL in DMSO) were transferred to wells using an Acoustic Transfer System (Biosero). Atovaquone and Puromycin were used as positive controls for P. berghei-Luciferase liver stage assays and HepG2 cytotoxicity assays, respectively, and tested starting at a highest concentration of 10 μM, with 1:3 serial dilutions. DMSO (0.5% DMSO final concentration) was used as negative control for both assays. P. berghei-ANKA-GFP-Luc-SMCON (P. berghei-Luciferase)88 sporozoites were freshly dissected from infected Anophelese stephensi mosquitoes received from the Insectary Core Facility at New York University (NYU). Sporozoites were then filtered twice through a 20 μm nylon net filter (Steriflip, Millipore) and 200 sporozoites/1 μL seeded into assay media. HepG2-A16-CD81EGFP cells were infected with 1 × 103 sporozoites per well (5 μL) using a single tip Bottle Valve liquid handler (GNF). Plates were centrifuged at 24 °C for 3 min at 330×g on normal acceleration and brake settings (Eppendorf 5810 R centrifuge). HepG2-A16-CD81EGFP cells for toxicity assessment were left uninfected, with 5 µL assay media added to each well to match test plates. Plates were then incubated at 37 °C for 48 h in 5% CO2 with high humidity chamber. After the incubation, P. berghei-Luciferase exoerythrocytic form growth and HepG2-A16-CD81EGFP cell viability were assessed by a bioluminescence measurement by spinning the inverted plates at 150xg for 30 s and adding 2 μL per well of Bright-Glo (Promega) for quantification of P. berghei-Luciferase exoerythrocytic forms or CellTiter-Glo (Promega) (diluted 1:2 with deionized water) for quantification of HepG2-A16-CD81EGFP cell viability. Immediately after addition of the luminescence reagent, the luminescence was measured by the Envision Multilabel Reader (PerkinElmer). For IC50 determination, the background for the P. berghei-Luciferase growth inhibition was defined as average bioluminescence of the 16 wells with atovaquone (10⌠M), and the background for the HepG2 Cytotoxicity was determined as the average of the 12 wells with puromycin at single concentration (10⌠M). The baseline was defined as average bioluminescence of the 150 wells with 0.5% DMSO. IC50 values were determined using the average normalised bioluminescence intensity of eight wells per concentration from two technical replicates of the 1536- well plates, and a nonlinear variable slope four-parameter regression curve fitting model in Prism 6 (GraphPad Software Inc.).
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
Thanks to Ben Dickerman, Burnet Institute, Melbourne Australia for the supply of 3D7-yeast dihydroorotate dehydrogenase and the Australian Red Cross Blood Service for the provision of human blood and sera for culture of Plasmodium parasites. This work was supported by the Medicines for Malaria Venture (Geneva, Switzerland) and an Australian National Health and Medical Research Council Project grant (APP1098848) to K.T.A., T.S.A., O.H., D.A.F. and J.H.R. E.A.W. and D.A.F. gratefully acknowledge funding support for their work from the NIH (R01 AI103058) and the Medicines for Malaria Venture (Geneva, Switzerland).
Author contributions
T.S.-A., A.G.R., O.H., J.B, J.R. and K.T.A. contributed to the conception and design of the project. T.S.-A., G.M.F., A.G.R., K.E.J., T.W., P.C., Y.A.-K., S.M., M.C. and K.T.A. contributed to experimental work. All authors (T.S.S.-A., G.M.F., A.G.R.,O.H., K.E.J., T.W., M.v.I., P.C., Y.A.-K., S.M., E.A.W., M.C., D.A.F., J.B., J.H.R. and K.T.A.) contributed to data analysis/interpretation. T.S.-A. and K.T.A. wrote the first draft of the manuscript. All authors contributed to writing and editing sections of the manuscript. T.S.-A. and K.T.A. contributed equally to this work.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information, or are available from the authors upon request. The source data underlying the plots in figures are provided in Supplementary Data 1 (Excel file).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Tina S. Skinner-Adams, John H. Ryan, Katherine T. Andrews.
Contributor Information
Tina S. Skinner-Adams, Email: t.skinner-adams@griffith.edu.au
John H. Ryan, Email: jack.ryan@csiro.au
Katherine T. Andrews, Email: k.andrews@griffith.edu.au
Supplementary information
Supplementary information accompanies this paper at 10.1038/s42003-019-0397-3.
References
- 1.Blasco B, Leroy D, Fidock DA. Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017;23:917–928. doi: 10.1038/nm.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward SA, et al. The activation of the biguanide antimalarial proguanil co-segregates with the mephenytoin oxidation polymorphism--a panel study. Br. J. Clin. Pharmacol. 1991;31:689–692. doi: 10.1111/j.1365-2125.1991.tb05594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peterson DS, Milhous WK, Wellems TE. Molecular basis of differential resistance to cycloguanil and pyrimethamine in Plasmodium falciparum malaria. Proc. Natl Acad. Sci. USA. 1990;87:3018–3022. doi: 10.1073/pnas.87.8.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canfield CJ, Pudney M, Gutteridge WE. Interactions of atovaquone with other antimalarial drugs against Plasmodium falciparum in vitro. Exp. Parasitol. 1995;80:373–381. doi: 10.1006/expr.1995.1049. [DOI] [PubMed] [Google Scholar]
- 5.Srivastava IK, Vaidya AB. A mechanism for the synergistic antimalarial action of atovaquone and proguanil. Antimicrob. Agents Chemother. 1999;43:1334–1339. doi: 10.1128/AAC.43.6.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Looareesuwan S, et al. Efficacy and safety of atovaquone/proguanil compared with mefloquine for treatment of acute Plasmodium falciparum malaria in Thailand. Am. J. Trop. Med. Hyg. 1999;60:526–532. doi: 10.4269/ajtmh.1999.60.526. [DOI] [PubMed] [Google Scholar]
- 7.Looareesuwan S, et al. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am. J. Trop. Med. Hyg. 1996;54:62–66. doi: 10.4269/ajtmh.1996.54.62. [DOI] [PubMed] [Google Scholar]
- 8.Chiodini PL, et al. Evaluation of atovaquone in the treatment of patients with uncomplicated Plasmodium falciparum malaria. J. Antimicrob. Chemother. 1995;36:1073–1078. doi: 10.1093/jac/36.6.1073. [DOI] [PubMed] [Google Scholar]
- 9.World Health Organization. Guidelines for the treatment of malaria. 3rd edition. (2015). [PubMed]
- 10.Fairley NH. Researches on paludrine (M.4888) in malaria; an experimental investigation undertaken by the L.H.Q. Medical Research Unit (A.I.F.) Cairns, Australia. Trans. R. Soc. Trop. Med. Hyg. 1946;40:105–162. doi: 10.1016/0035-9203(46)90052-1. [DOI] [PubMed] [Google Scholar]
- 11.Shapiro TA, Ranasinha CD, Kumar N, Barditch-Crovo P. Prophylactic activity of atovaquone against Plasmodium falciparum in humans. Am. J. Trop. Med. Hyg. 1999;60:831–836. doi: 10.4269/ajtmh.1999.60.831. [DOI] [PubMed] [Google Scholar]
- 12.Shanks GD, et al. Atovaquone and proguanil hydrochloride for prophylaxis of malaria. J. Travel. Med. 1999;6:S21–S27. [PubMed] [Google Scholar]
- 13.Berman JD, et al. Causal prophylactic efficacy of atovaquone-proguanil (Malarone (TM)) in a human challenge model. Trans. R. Soc. Trop. Med. Hygiene. 2001;95:429–432. doi: 10.1016/S0035-9203(01)90206-8. [DOI] [PubMed] [Google Scholar]
- 14.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 15.Jones K, Ward SA. Biguanide-atovaquone synergy against Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 2002;46:2700–2703. doi: 10.1128/AAC.46.8.2700-2703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thapar MM, Gupta S, Spindler C, Wernsdorfer WH, Bjorkman A. Pharmacodynamic interactions among atovaquone, proguanil and cycloguanil against Plasmodium falciparum in vitro. Trans. R. Soc. Trop. Med. Hyg. 2003;97:331–337. doi: 10.1016/S0035-9203(03)90162-3. [DOI] [PubMed] [Google Scholar]
- 17.Canfield CJ, et al. PS-15: a potent, orally active antimalarial from a new class of folic acid antagonists. Am. J. Trop. Med. Hyg. 1993;49:121–126. doi: 10.4269/ajtmh.1993.49.121. [DOI] [PubMed] [Google Scholar]
- 18.Fidock DA, Nomura T, Wellems TE. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Mol. Pharmacol. 1998;54:1140–1147. doi: 10.1124/mol.54.6.1140. [DOI] [PubMed] [Google Scholar]
- 19.Watkins WM, Sixsmith DG, Chulay JD. The activity of proguanil and its metabolites, cycloguanil and p-chlorophenylbiguanide, against Plasmodium falciparum in vitro. Ann. Trop. Med. Parasitol. 1984;78:273–278. doi: 10.1080/00034983.1984.11811816. [DOI] [PubMed] [Google Scholar]
- 20.Fidock DA, Wellems TE. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc. Natl Acad. Sci. USA. 1997;94:10931–10936. doi: 10.1073/pnas.94.20.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaneko A, et al. Intrinsic efficacy of proguanil against falciparum and vivax malaria independent of the metabolite cycloguanil. J. Infect. Dis. 1999;179:974–979. doi: 10.1086/314683. [DOI] [PubMed] [Google Scholar]
- 22.Donato MT, Lahoz A, Castell JV, Gomez-Lechon MJ. Cell lines: a tool for in vitro drug metabolism studies. Curr. Drug. Metab. 2008;9:1–11. doi: 10.2174/138920008783331086. [DOI] [PubMed] [Google Scholar]
- 23.Barata L, et al. In Vitro analysis of the interaction between atovaquone and proguanil against liver stage malaria parasites. Antimicrob. Agents Chemother. 2016;60:4333–4335. doi: 10.1128/AAC.01685-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lell B, Luckner D, Ndjave M, Scott T, Kremsner PG. Randomised placebo-controlled study of atovaquone plus proguanil for malaria prophylaxis in children. Lancet. 1998;351:709–713. doi: 10.1016/S0140-6736(97)09222-2. [DOI] [PubMed] [Google Scholar]
- 25.Blanchard TJ, et al. Multiresistant falciparum malaria cured using atovaquone and proguanil. Trans. R. Soc. Trop. Med. Hyg. 1994;88:693. doi: 10.1016/0035-9203(94)90233-X. [DOI] [PubMed] [Google Scholar]
- 26.Mberu EK, Wansor T, Sato H, Nishikawa Y, Watkins WM. Japanese poor metabolizers of proguanil do not have an increased risk of malaria chemoprophylaxis breakthrough. Trans. R. Soc. Trop. Med. Hyg. 1995;89:658–659. doi: 10.1016/0035-9203(95)90434-4. [DOI] [PubMed] [Google Scholar]
- 27.Krudsood S, et al. Efficacy of atovaquone-proguanil for treatment of acute multidrug-resistant Plasmodium falciparum malaria in Thailand. Am. J. Trop. Med. Hyg. 2007;76:655–658. doi: 10.4269/ajtmh.2007.76.655. [DOI] [PubMed] [Google Scholar]
- 28.Kuhn S, Gill MJ, Kain KC. Emergence of atovaquone-proguanil resistance during treatment of Plasmodium falciparum malaria acquired by a non-immune north American traveller to west Africa. Am. J. Trop. Med. Hyg. 2005;72:407–409. doi: 10.4269/ajtmh.2005.72.407. [DOI] [PubMed] [Google Scholar]
- 29.Ke, H., Dass, S., Morrisey, J. M., Mather, M. W. & Vaidya, A. B. The mitochondrial ribosomal protein L13 is critical for the structural and functional integrity of the mitochondrion in Plasmodium falciparum. J. Biol. Chem. 293, 8128–8137 (2018). [DOI] [PMC free article] [PubMed]
- 30.Vaidya AB, Akella R, Suplick K. Sequences similar to genes for two mitochondrial proteins and portions of ribosomal RNA in tandemly arrayed 6-kilobase-pair DNA of a malarial parasite. Mol. Biochem. Parasitol. 1989;35:97–107. doi: 10.1016/0166-6851(89)90112-6. [DOI] [PubMed] [Google Scholar]
- 31.Suplick K, Akella R, Saul A, Vaidya AB. Molecular cloning and partial sequence of a 5.8 kilobase pair repetitive DNA from Plasmodium falciparum. Mol. Biochem. Parasitol. 1988;30:289–290. doi: 10.1016/0166-6851(88)90098-9. [DOI] [PubMed] [Google Scholar]
- 32.Feagin JE. The 6-kb element of Plasmodium falciparum encodes mitochondrial cytochrome genes. Mol. Biochem. Parasitol. 1992;52:145–148. doi: 10.1016/0166-6851(92)90046-M. [DOI] [PubMed] [Google Scholar]
- 33.Vaidya AB, Mather MW. Mitochondrial evolution and functions in malaria parasites. Annu. Rev. Microbiol. 2009;63:249–267. doi: 10.1146/annurev.micro.091208.073424. [DOI] [PubMed] [Google Scholar]
- 34.Jacot D, Waller RF, Soldati-Favre D, MacPherson DA, MacRae JI. Apicomplexan energy metabolism: carbon source promiscuity and the quiescence hyperbole. Trends. Parasitol. 2016;32:56–70. doi: 10.1016/j.pt.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 35.MacRae JI, et al. Mitochondrial metabolism of sexual and asexual blood stages of the malaria parasite Plasmodium falciparum. BMC Biol. 2013;11:67. doi: 10.1186/1741-7007-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheiner L, Vaidya AB, McFadden GI. The metabolic roles of the endosymbiotic organelles of Toxoplasma and Plasmodium spp. Curr. Opin. Microbiol. 2013;16:452–458. doi: 10.1016/j.mib.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boysen KE, Matuschewski K. Arrested oocyst maturation in Plasmodium parasites lacking type II NADH:ubiquinone dehydrogenase. J. Biol. Chem. 2011;286:32661–32671. doi: 10.1074/jbc.M111.269399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hino A, et al. Critical roles of the mitochondrial complex II in oocyst formation of rodent malaria parasite Plasmodium berghei. J. Biochem. 2012;152:259–268. doi: 10.1093/jb/mvs058. [DOI] [PubMed] [Google Scholar]
- 39.Ke H, et al. Genetic investigation of tricarboxylic acid metabolism during the Plasmodium falciparum life cycle. Cell Rep. 2015;11:164–174. doi: 10.1016/j.celrep.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sturm A, et al. synthase is dispensable in blood-stage Plasmodium berghei rodent malaria but essential in the mosquito phase. Proc. Natl Acad. Sci. USA. 2015;112:10216–10223. doi: 10.1073/pnas.1423959112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Painter HJ, Morrisey JM, Vaidya AB. Mitochondrial electron transport inhibition and viability of intraerythrocytic Plasmodium falciparum. Antimicrob. Agents Chemother. 2010;54:5281–5287. doi: 10.1128/AAC.00937-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curd, F. H. & Rose, F. L. Synthetic antimalarials; some aryl-diguanide (“-biguanide”) derivatives. J. Chem. Soc., 729–737 (1946). [PubMed]
- 43.Dahl EL, Rosenthal PJ. Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 2007;51:3485–3490. doi: 10.1128/AAC.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goodman CD, Pasaje CFA, Kennedy K, McFadden GI, Ralph SA. Targeting protein translation in organelles of the Apicomplexa. Trends. Parasitol. 2016;32:953–965. doi: 10.1016/j.pt.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 45.Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011;9:e1001138. doi: 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fry M, Beesley JE. Mitochondria of mammalian Plasmodium spp. Parasitology. 1991;102:17–26. doi: 10.1017/S0031182000060297. [DOI] [PubMed] [Google Scholar]
- 47.Dickerman BK, et al. Identification of inhibitors that dually target the new permeability pathway and dihydroorotate dehydrogenase in the blood stage of Plasmodium falciparum. Sci. Rep. 2016;6:37502. doi: 10.1038/srep37502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagy M, Lacroute F, Thomas D. Divergent evolution of pyrimidine biosynthesis between anaerobic and aerobic yeasts. Proc. Natl Acad. Sci. USA. 1992;89:8966–8970. doi: 10.1073/pnas.89.19.8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srivastava IK, Rottenberg H, Vaidya AB. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J. Biol. Chem. 1997;272:3961–3966. doi: 10.1074/jbc.272.7.3961. [DOI] [PubMed] [Google Scholar]
- 50.Nilsen A, et al. Discovery, synthesis, and optimization of antimalarial 4(1H)-quinolone-3-diarylethers. J. Med. Chem. 2014;57:3818–3834. doi: 10.1021/jm500147k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nam TG, et al. A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem. Biol. 2011;6:1214–1222. doi: 10.1021/cb200105d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haldar K, de Amorim AF, Cross GA. Transport of fluorescent phospholipid analogues from the erythrocyte membrane to the parasite in Plasmodium falciparum-infected cells. J. Cell. Biol. 1989;108:2183–2192. doi: 10.1083/jcb.108.6.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uyemura SA, Luo S, Moreno SN, Docampo R. Oxidative phosphorylation, Ca(2+) transport, and fatty acid-induced uncoupling in malaria parasites mitochondria. J. Biol. Chem. 2000;275:9709–9715. doi: 10.1074/jbc.275.13.9709. [DOI] [PubMed] [Google Scholar]
- 54.Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014;462:475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baragana B, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522:315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.World Health Organization. Global technical strategy for malaria 2016–2030. (2015).
- 57.Burrows JN, et al. Antimalarial drug discovery - the path towards eradication. Parasitology. 2014;141:128–139. doi: 10.1017/S0031182013000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burrows JN, et al. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017;16:26. doi: 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.World Health Organization. World Malaria Report. (2017).
- 60.Ekland EH, Schneider J, Fidock DA. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J. 2011;25:3583–3593. doi: 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim S, et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016;44:D1202–D1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.National Center for Biotechnology Information, PubChem BioAssay Database; AID=504834,.
- 63.Staines Henry M, Burrow Rebekah, Teo Beatrix Huei-Yi, Chis Ster Irina, Kremsner Peter G, Krishna Sanjeev. Clinical implications of Plasmodium resistance to atovaquone/proguanil: a systematic review and meta-analysis. Journal of Antimicrobial Chemotherapy. 2017;73(3):581–595. doi: 10.1093/jac/dkx431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McFadden GI, Yeh E. The apicoplast: now you see it, now you don’t. Int. J. Parasitol. 2017;47:137–144. doi: 10.1016/j.ijpara.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ke H, et al. Variation among Plasmodium falciparum strains in their reliance on mitochondrial electron transport chain function. Eukaryot. Cell. 2011;10:1053–1061. doi: 10.1128/EC.05049-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balabaskaran Nina P, et al. ATP synthase complex of Plasmodium falciparum: dimeric assembly in mitochondrial membranes and resistance to genetic disruption. J. Biol. Chem. 2011;286:41312–41322. doi: 10.1074/jbc.M111.290973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mather MW, Morrisey JM, Vaidya AB. Hemozoin-free Plasmodium falciparum mitochondria for physiological and drug susceptibility studies. Mol. Biochem. Parasitol. 2010;174:150–153. doi: 10.1016/j.molbiopara.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou J, Massey S, Li L. Metformin: an old drug with new applications. Int. J. Mol. Sci. 2018;19:E2863. doi: 10.3390/ijms19102863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell. Metab. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 70.Ju R, Mao Y, Glick MJ, Muller MT, Snyder RD. Catalytic inhibition of DNA topoisomerase IIalpha by sodium azide. Toxicol. Lett. 2001;121:119–126. doi: 10.1016/S0378-4274(01)00328-9. [DOI] [PubMed] [Google Scholar]
- 71.Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J. Biol. Chem. 1998;273:11619–11624. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- 72.Lardy HA, Johnson D, Mcmurray WC. Antibiotics as tools for metabolic studies .1. A survey of toxic antibiotics in respiratory, phosphorylative and glycolytic systems. Arch. Biochem. Biophys. 1958;78:587–597. doi: 10.1016/0003-9861(58)90383-7. [DOI] [PubMed] [Google Scholar]
- 73.Glynn IM, Karlish SJ. The sodium pump. Annu. Rev. Physiol. 1975;37:13–55. doi: 10.1146/annurev.ph.37.030175.000305. [DOI] [PubMed] [Google Scholar]
- 74.Stickles AM, et al. Atovaquone and ELQ-300 combination therapy as a novel dual-site cytochrome bc1 inhibition strategy for malaria. Antimicrob. Agents Chemother. 2016;60:4853–4859. doi: 10.1128/AAC.00791-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stickles AM, et al. Subtle changes in endochin-like quinolone structure alter the site of inhibition within the cytochrome bc1 complex of Plasmodium falciparum. Antimicrob. Agents Chemother. 2015;59:1977–1982. doi: 10.1128/AAC.04149-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nilsen, A. et al. Quinolone-3-diarylethers: A new class of antimalarial drug. Sci. Transl. Med.5, ARTN 177ra3710.1126/scitranslmed.3005029 (2013). [DOI] [PMC free article] [PubMed]
- 77.Armistead JS, Adams JH. Advancing research models and technologies to overcome biological barriers to Plasmodium vivax Control. Trends. Parasitol. 2018;34:114–126. doi: 10.1016/j.pt.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boggild AK, Parise ME, Lewis LS, Kain KC. Atovaquone-proguanil: report from the CDC expert meeting on malaria chemoprophylaxis (II) Am. J. Trop. Med. Hyg. 2007;76:208–223. doi: 10.4269/ajtmh.2007.76.208. [DOI] [PubMed] [Google Scholar]
- 79.Edstein MD, et al. Lengthy antimalarial activity of atovaquone in human plasma following atovaquone-proguanil administration. Antimicrob. Agents Chemother. 2005;49:4421–4422. doi: 10.1128/AAC.49.10.4421-4422.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Freedman DO, Chen LH, Kozarsky PE. Medical considerations before international travel. N. Engl. J. Med. 2016;375:247–260. doi: 10.1056/NEJMra1508815. [DOI] [PubMed] [Google Scholar]
- 81.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 82.Davisson VJ, Woodside AB, Poulter CD. Synthesis of allylic and homoallylic isoprenoid pyrophosphates. Methods Enzymol. 1985;110:130–144. doi: 10.1016/S0076-6879(85)10068-6. [DOI] [PubMed] [Google Scholar]
- 83.Davisson VJ, et al. Phosphorylation of isoprenoid alcohols. J. Org. Chem. 1986;51:4768–4779. doi: 10.1021/jo00375a005. [DOI] [Google Scholar]
- 84.Curd FH, et al. Synthetic antimalarials; an alternative route to N1-aryl-N5-alkyldiguanides. J. Chem. Soc. 1948;3:1630–1636. doi: 10.1039/jr9480001630. [DOI] [PubMed] [Google Scholar]
- 85.Andrews KT, et al. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob. Agents Chemother. 2008;52:1454–1461. doi: 10.1128/AAC.00757-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huber W, Koella JC. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993;55:257–261. doi: 10.1016/0001-706X(93)90083-N. [DOI] [PubMed] [Google Scholar]
- 87.Skinner-Adams TS, Andrews KT, Melville L, McCarthy J, Gardiner DL. Synergistic interactions of the antiretroviral protease inhibitors saquinavir and ritonavir with chloroquine and mefloquine against Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 2007;51:759–762. doi: 10.1128/AAC.00840-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Franke-Fayard B, et al. Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc. Natl Acad. Sci. USA. 2005;102:11468–11473. doi: 10.1073/pnas.0503386102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information, or are available from the authors upon request. The source data underlying the plots in figures are provided in Supplementary Data 1 (Excel file).