

 This work is licensed under a
This work is licensed under a Abstract
Thyroid hormones have a central role in cardiovascular homeostasis. In myocardium, these hormones stimulate both diastolic myocardial relaxation and systolic myocardial contraction, have a pro-angiogenic effect and an important role in extracellular matrix maintenance. Thyroid hormones modulate cardiac mitochondrial function. Dysfunction of thyroid axis impairs myocardial bioenergetic status. Both overt and subclinical hypothyroidism are associated with a higher incidence of coronary events and an increased risk of heart failure progression. Endothelial function is also impaired in hypothyroid state, with decreased nitric oxide-mediated vascular relaxation. In heart disease, particularly in ischemic heart disease, abnormalities in thyroid hormone levels are common and are an important factor to be considered. In fact, low thyroid hormone levels should be interpreted as a cardiovascular risk factor. Regarding ischemic heart disease, during the late post-myocardial infarction period, thyroid hormones modulate left ventricular structure, function and geometry. Dysfunction of thyroid axis might even be more prevalent in the referred condition since there is an upregulation of type 3 deiodinase in myocardium, producing a state of local cardiac hypothyroidism. In this focused review, we summarize the central pathophysiological and clinical links between altered thyroid function and ischemic heart disease. Finally, we highlight the potential benefits of thyroid hormone supplementation as a therapeutic target in ischemic heart disease.
Keywords: thyroid hormones, hypothyroidism, hyperthyroidism, ischemic heart disease, ischemia/reperfusion injury, mitochondria
Introduction
Thyroid hormones (THs) play fundamental roles in cardiovascular homeostasis (1). Given that cardiovascular diseases are among the most prevalent illnesses worldwide, causing substantial mortality, morbidity and hospitalization, an understanding of the role of THs in the cardiovascular system is imperative (1). The main goal of this manuscript is to review the central pathophysiological and clinical links between altered thyroid function and cardiovascular diseases, particularly in ischemic heart disease.
Thyroid hormones
Nearly all organs have thyroid receptors and are in some way regulated by the thyroid axis (2). THs are produced by the thyroid gland, which is mainly regulated by thyroid-stimulating hormone (TSH). TSH is secreted by the pituitary gland and is regulated by thyrotropin-releasing hormone (TRH) secreted by the hypothalamus (2). Ninety percent of the TH secreted is thyroxine (T4) and the remaining 10% is triiodothyronine (T3) (3). T3 is 20 times more potent than T4, making T3 the biologically active hormone of the thyroid axis (3). Most T3 is generated peripherally from T4 conversion by deiodinases (Fig. 1). These enzymes are also responsible for converting THs into inactive isomers such as reverse T3 (rT3) and 3,3-diiodothyronine (T2). There are three deiodinases with different functions: (1) type 1 deiodinase (D1) is localized in the plasma membrane and is expressed in the liver, thyroid and kidney; this enzyme is mainly responsible for the peripheral conversion of T4 into T3; (2) type 2 deiodinase (D2) seems to be more efficient than D1; the major role of this enzyme is to regulate the intracellular concentration of T3, converting T4 into T3, especially in the brain, pituitary gland and skeletal muscle; and (3) type 3 deiodinase (D3) irreversibly inactivates THs generating T2 or rT3; thus, by lowering the levels of these hormones, D3 is considered an important regulator of the thyroid axis (4). Furthermore, THs are mainly active when not bound to transport proteins. Therefore, variations on binding protein levels can change the peripheral activity of THs (5). In order to perform their roles, THs must bind to thyroid hormone receptors. These receptors are intracellular DNA-binding proteins that bind as hormone-receptor complexes to thyroid hormone response elements (TREs) in the regulatory regions of target genes (6). Consequently, THs modulate essential functions in the growth, development and metabolism of a variety of tissues. There are different subtypes of receptors – TRα1, TRα2, TRβ1 and TRβ2 – which have different functions. TRα1 is the subtype most expressed in the myocardium, regulating important genes related to cell growth, contractile function and electrical activity (6, 7). In fact, inhibition of TRα1 was shown to markedly depress post-ischemic cardiac function in mice (8). Although TRβ1 is also expressed in the myocardium, it is expressed at a lower level (7). Though TRα2 does not bind T3, it is able to bind TRE, thereby exerting a substantial negative effect on gene expression (7).
Figure 1.
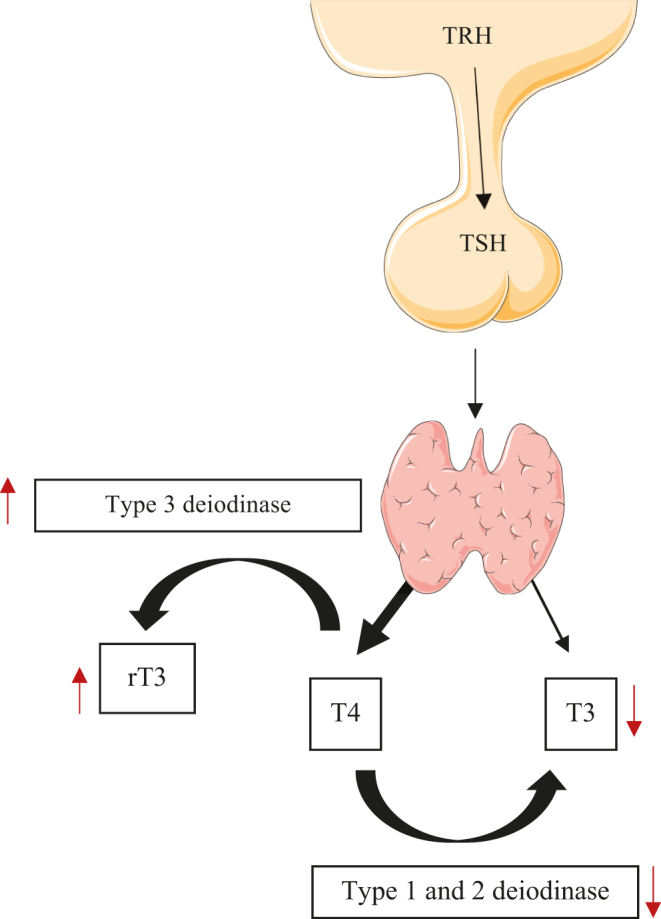
Thyroid axis and changes in nonthyroidal illness. Direction of arrows (↑, ↓) indicates increase or decrease, respectively. rT3, reverse triiodothyronine; T4, thyroxine; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
The effects of thyroid hormones on the cardiovascular system
THs exert a significant impact on the cardiovascular system via both genomic and non-genomic mechanisms. The major effects of THs on the myocardium are mediated by T3 (Table 1), which stimulates nearly all of the transporters and ion channels involved in calcium myocardial fluxes, upregulating sarcoplasmic reticulum calcium-activated ATPase 2 (SERCA2) and Na+/K+-ATPase and downregulating phospholamban (9). These changes enhance calcium uptake and release by the sarcoplasmatic reticulum, stimulating both diastolic myocardial relaxation and systolic myocardial contraction (9). The contractile apparatus of the cardiac myocyte has two subtypes of myosin heavy chains (MHCs): α-MHC and β-MHC – fast and slow myosin, respectively. T3 can upregulate α-MHC and downregulate β-MHC (10). Therefore, the myocardial hypothyroid state induces a so-called fetal gene reprogramming which increases the expression of β-MHC and decreases the expression of α-MHC and SERCA2 (11). This phenotype presents substantial implications on myocardial function and subsequent progression to heart failure (12). The fetal pattern is characterized by a preference for glucose over fatty acids as a substrate. Although such changes may lower oxygen demands, the yield of ATP per substrate also decreases, resulting in metabolic inefficiencies that decrease metabolic reserves – perhaps leading to cardiac pump dysfunction (12). Nevertheless, this phenotype seems to be reversible with the appropriate therapy (13). T3 is able to modify the activity of sodium, potassium and calcium channels, altering a variety of intracellular pathways in cardiac and vascular smooth-muscle cells. Consequently, THs can increase resting heart rate, cardiac contractility and venous tone almost immediately, increasing cardiac preload and cardiac output (14). T3 increases myocardial sensitivity to the adrenergic system by increasing the number of adrenergic membrane receptors. Additionally, T3 decreases systemic vascular resistance through vascular smooth-muscle relaxation, which in turn decreases renal perfusion and leads to renin-angiotensin-aldosterone axis activation (15, 16). The relaxation of vascular smooth muscle may lead to an increase in cardiac oxygenation. T3 increases metabolic and oxygen consumption, thereby enhancing the release of vasodilatory mediators (15). THs exhibited a dose-dependent direct effect in rat coronary arteries within a few seconds, indicating a non-genomic mechanism (17). Other effects of T3 that depend at least partially on non-genomic mechanisms include an increase in resting heart rate and left ventricular contractility (17). Non-genomic effects are poorly understood but are an important accessory mechanism in THs actions. These effects are receptor independent and regulate ion transporter activity in the plasma membrane (10). The non-genomic actions of THs in the myocardium involve different signaling cascades, such as cyclic adenosine monophosphate and protein kinases (18). A structural protein of the plasma membrane has been identified as a cell-surface receptor for THs. This receptor is an integrin αVβ3 that interacts with a variety of extracellular matrix proteins and induces serine-threonine kinase pathway (19). THs induce a rapid vascular relaxation that is mediated by NO produced by vascular smooth-muscle and endothelial cells. This is associated with the induction of phosphatidylinositol 3-kinase/protein (PI3K/Akt) signaling pathway. Oxidized low-density lipoprotein (LDL) may blunt the non-genomic action of THs and impair the NO production in endothelial cells (20). Furthermore, THs have a proangiogenic effect, stimulating arteriolar growth in the normal heart as well as after myocardial infarction (21). This proangiogenic effect is mediated by increased transcription of proangiogenic genes and expression of hypoxic inducible factor-1α (HIF-1α) and mediated by activation of integrin αVβ3 (19, 22). T3-induced angiogenesis has been observed in several experimental rat models of ischemia, hypertension and diabetic cardiomyopathy (23).
Table 1.
T3-regulated genes.
| Positively regulated | Negatively regulated |
|---|---|
| α-MHC | β-MHC |
| SERCA2 | Phospholamban |
| Na+/K+ ATPase | p53 and JNK |
| β 1-adrenergic receptor | p38/MAPK |
| NRF1, NFR2 | |
| HIF-1α, mt-TFA, PPARC1α | |
| PGC-1α | |
| Bcl-2 (antiapoptotic) | |
| HSP27, HSP70 |
The therapeutic targets of cardioprotection should not be limited to cardiomyocytes, but should also include other cells such as fibroblasts and endothelial cells that play important roles in preserving myocardial function. THs can modulate metalloproteinases (MMP), increasing MMP 1 and 2 as well as collagen gene expression; consequently, they may have an important impact on the extracellular matrix of the heart (24). Tissue inhibitors of MMP are downregulated by THs (24). The antifibrotic effect of T3 is suggested by evidence that early T3 replacement after ischemia/reperfusion in rats is associated with a reduction in scar size (25). Moreover, hypothyroid status is marked by an increased susceptibility to collagen deposition and cardiac fibrosis (26).
Thyroid hormone as a cardiac mitochondrial regulator
THs modulate cardiac mitochondrial function by increasing mitochondrial mass, respiration, oxidative phosphorylation, enzyme activity and mitochondrial protein synthesis such as that of cytochrome as well as phospholipid and mtDNA content (27). Changes in the levels of circulating THs may impair myocardial bioenergetic status with consequences on cardiac function (28, 29). Mitochondrial dysfunction plays a central role in cardiac dysfunction and in the occurrence and progression of heart failure (30). The regulation of mitochondrial function and biogenesis by THs is an emerging mechanism in the therapeutics of cardioprotection. THs promote the upregulation of proteins that are functionally relevant to the rescue of mitochondrial function. Consequently, these hormones may reduce cardiomyocyte loss in the peri-infarct zone. Reversal of the post-ischemic decline of TH levels has been shown to downregulate tumor suppressor protein (p53) possibly via the upregulation of miRNA 30a (31). Additionally, premature activation of the c-Jun N-terminal kinase (JNK) cascade occurs minutes after myocardial infarction. JNK protein expression is associated with apoptosis in the infarction border zone, cardiac dilatation and pathological remodeling (32). Given that p53 can regulate the JNK pathway through a positive feedback loop, THs might reduce JNK levels through a p53-dependent mechanism (31). T3 treatment (14 ng/g body weight, dose given daily) for 3 days after acute myocardial infarction in rats reduced myocyte apoptosis in the border area, possibly via Akt signaling (33).
T3 administration in rats significantly increases the expression of transcription factors implicated in mitochondrial biogenesis, including nuclear regulatory factors – NRF-1 and NRF-2 which mediate the expression of HIF-1α, mitochondrial transcription factor A (mt-TFA) and peroxisome proliferator-activated receptor coactivator-1α (PPARc-1α), particularly in the peri-infarct zone (34). In fact, the overexpression of mt-TFA and HIF-1α can limit left ventricular remodeling and preserve cardiac performance after myocardial infarction (35, 36, 37). THs also enhance transcriptional coactivators such as peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α) which modulate the regulatory function of many factors (38). Additionally, PGC-1α also induces D2, which generates local THs and further enhances mitochondrial function (38). Downregulation of cytochrome c oxidase I (CcO-I) – a key enzyme of the mitochondrial respiratory chain – occurs in the border zone of infarcted tissue. THs increase mitochondrial proliferation, mitochondrial protein synthesis and cytochrome content. T3 treatment in rats attenuate the downregulation in CcO-I (27, 39). Ischemic/reperfusion injury (IRI) damages the mitochondrial outer membrane, and together with the activation of the proapoptotic proteins, enhances mitochondrial outer membrane permeabilization, releasing cytochrome c, activating caspases and increasing apoptosis (40). THs increase the expression of antiapoptotic protein Bcl-2, reducing the Bax:Bcl-2 ratio and improving cardiac function in infarcted rats (41). Furthermore, THs can prevent oxidative damage in the heart by limiting ROS levels (42). T3 also protects cardiomyocytes against oxidative stress-mediated cell death by opening the protective mitochondrial ATP-dependent K+ channel (mitoKATP) in rescued mitochondria (27). T3 replacement after myocardial infarction might offer a highly successful therapeutic strategy to improve myocardial mitochondrial function and cardiac cell metabolism, limiting infarct-scar size and preventing heart failure (34).
Cardiovascular effects of hyperthyroidism
Hyperthyroidism is characterized biochemically by low TSH levels and elevated free T4, free T3 or both. The prevalence of overt hyperthyroidism in the general population is 0.5% (43). Although the effects of THs might be beneficial for cardiovascular function, when exacerbated as they are in hyperthyroidism, they can be detrimental. THs increase resting heart rate, blood volume, myocardial contractility and ejection fraction (Table 2). The most common cardiovascular symptom in patients with hyperthyroidism is palpitation, with 20% exhibiting atrial fibrillation (44). The Rotterdam study, a cross-sectional analysis including 1149 women, showed that elderly women with high/normal thyroid function (low range of normal TSH levels) have an increased risk for atrial fibrillation (45). Systolic function is consistently increased – even at rest – as is the rate of ventricular relaxation and filling (46). At the same time, pulse pressure is wider because of the increased systolic arterial pressure and decreased diastolic arterial pressure due to vasodilatation (Table 2) (47). In patients with hyperthyroidism, cardiac output is notably elevated. Overt hyperthyroidism is associated with increased cardiovascular mortality and 16% increased risk of major cardiovascular events (48). High-output heart failure might be induced by hyperthyroidism even in patients without heart disease (48). Hyperthyroidism is also associated with pulmonary hypertension – albeit less frequently – and atrioventricular valve regurgitation, mainly of the tricuspid valve (49). Heart failure is the main cause of increased cardiovascular mortality in both overt hyperthyroidism and subclinical hyperthyroidism (50).
Table 2.
Effects of thyroid dysfunction in the cardiovascular system.
| Hypothyroidism: overt or subclinical | Hyperthyroidism: overt or subclinical |
|---|---|
| ↓ heart rate | ↑ heart rate |
| ↓ inotropism | ↑ myocardial contractility |
| ↑ peripheral vascular resistance | ↑ cardiac output |
| diastolic hypertension | Wider pulse pressure |
| sinus bradycardia | Vasodilatation |
| sinus bradycardia | ↑ blood volume |
| ↑ risk of atherosclerosis and dyslipidemia | ↓ mean arterial pressure |
| Pericarditis, pericardial effusion, cardiac tamponade | Atrial fibrillation, Pulmonary hypertension, atrioventricular valve regurgitation |
Subclinical hyperthyroidism is defined by normal free T4 and free T3 levels with a reduced TSH level. The prevalence of exogenous subclinical hyperthyroidism in patients prescribed with levothyroxine is 15–20% (51). Subclinical hyperthyroidism has been associated with increased all-cause mortality, coronary heart disease (CHD) events and mortality and atrial fibrillation (52, 53). CHD mortality and atrial fibrillation are correlated with TSH levels, being higher with TSH under 0.10 mIU/L when compared with levels between 0.10 and 0.44 mIU/L (54). In a meta-analysis of 55 cohort studies with 1,898,314 patients, subclinical and overt hypothyroidism were associated with higher risks of cardiac mortality and all-cause mortality (55).
Cardiovascular effects of hypothyroidism
Hypothyroidism is defined by high TSH levels with low THs levels. The prevalence of overt hypothyroidism in nonpregnant adults is 0.2–2.0% (43). Cardiac dysfunction observed in hypothyroidism is not solely explained by decreased inotropism and increased peripheral vascular resistance (Table 2). The hypothyroid state results in lower heart rate and decreased myocardial contraction and relaxation, with prolonged systolic and early diastolic time intervals, culminating in advanced stages of heart failure (12). Hypothyroidism is also associated with diastolic hypertension and sinus bradycardia due to sinus node dysfunction (56, 57). Diastolic dysfunction is one of the main cardiac anomalies found in patients with hypothyroidism, both at rest and with exertion. Left ventricular asynchrony is also present in hypothyroidism, which affects diastolic and systolic functions, exercise capacity, and quality of life, rendering potential heart failure patients more symptomatic and worsening their prognosis (58). CHD that occasionally coexists with hypothyroidism may be preexistent and aggravated by thyroid dysfunction. Although less common, pericarditis, pericardial effusion and cardiac tamponade are also potential manifestations of hypothyroidism (56, 57).
Subclinical hypothyroidism (SCH) is defined as serum THs within reference range in the presence of elevated serum TSH levels. SCH can be classified as grade 1 (TSH >4.0 or 4.5, but <10 mIU/L) or grade 2 (TSH >10 mIU/L) (10). The prevalence of SCH in the population varies from 4 to 20%, occurring more frequently in adults older than 65 years of age (59). SCH is also associated with a higher risk of heart failure, morbidity and mortality (50, 60). These patients are also at increased risk of atherosclerosis and coronary events (61). This increased risk of coronary events may be caused by increased cholesterol and homocysteine levels and decreased LDL receptors also seen in patients with overt hypothyroidism (50, 61). Patients with SCH have decreased nitric oxide (NO)-mediated vascular relaxation, which contributes to an increase in vascular resistance and consequently to increased left ventricular afterload (Fig. 2) (62). Several studies have shown that patients with hypothyroidism (both overt and subclinical) exhibit impaired endothelial function that improves with TH replacement therapy (61, 63). In the Rotterdam Study mentioned above, patients with SCH had higher prevalence of aortic calcification and myocardial infarction, especially those who were positive for thyroid autoantibodies (64). Patients with SCH also have a decreased left ventricular global longitudinal strain, especially in the lateral wall and interventricular septum (65). Along with these left ventricular disturbances, neovascularization in cardiac tissue after an ischemic event may be inhibited in SCH. Consequently, this may accelerate cardiac pathological remodeling contributing to the development of heart failure (66). In addition to hypercholesterolemia, increased homocysteine, systemic vascular resistance and oxidative stress described above, SCH might also increase peripheral insulin resistance and activate prothrombotic pathways and hypercoagulability (Fig. 2) (67). SCH may constitute a potentially reversible cardiovascular risk factor, a scenario which should be taken into account as it is present in almost 12% of patients with acute myocardial infarction (10, 67). The risk of cardiac events depends on the severity of hormonal dysfunction, tending to be higher when TSH is ≥7.0 mIU/L and even more evident when the value is ≥10 mIU/L (68).
Figure 2.

Main changes in cardiovascular system with low T3 levels in nonthyroidal illness. LDL, low-density lipoprotein; LV, left ventricle; MHC, myosin heavy chain; MMP-2, matrix metalloproteinase-2; NO, nitric oxide; SERCA2, sarcoplasmic/endoplasmic reticulum calcium ATPase 2.
Another subtype of TH dysfunction called nonthyroidal illness syndrome (NTIS) – also known as euthyroid sick syndrome or low-T3 syndrome – is characterized by a rapid decline in plasma-free THs levels (free T3 and free T4), with a marked elevation of rT3 in critically ill patients (69). TSH level is usually within the reference range, except in severe cases of this syndrome. Local and systemic bioavailability of THs is regulated by aberrant expression of deiodinases, SNPs and novel regulators of expression of deiodinases genes (70). Changes in deiodinases activity are more pronounced in conditions characterized by low tissue perfusion (71). The main mechanism seems to involve reduced activity of the deiodinases that convert T4 to T3 and an increase in deiodinase D3 that converts T4 to rT3 (72). D1 is the main pathway for rT3 clearance. The increase in rT3 is partially explained by a decrease in D1 activity (73). Peeters et al. evidenced that liver D1 activity was downregulated in critically ill patients and D3 activity in liver and skeletal muscle was induced. Skeletal muscle is an abundant tissue in humans, and it is likely that induction of D3 in this tissue also contributes to the pathophysiology of NTIS (71). As in hemangiomas that increase D3 expression and lead to ‘consumptive hypothyroidism’ (74). Recent studies show that expression of D3 is increased in some pathological contexts in a cell-specific manner, which are cancer, cardiac hypertrophy, myocardial infarction, chronic inflammation or critical illness (75). In NTIS, the decrease in T3-dependent gene expression is independent of circulating T3 concentration, demonstrating that after an ischemic event, there is potent and stable induction of D3 activity in cardiomyocytes, resulting in subsequent local cardiac hypothyroidism (10). This might be mediated by tissue catecholamines and inflammatory responses, via inflammatory cytokines such as tumor necrosis factor-α (TNF-α). In fact, D3 overexpression is a common mark of inflammatory response and is one of the leading events in NTIS, often seen in chronic inflammation (70). Experimental evidence showed that infusion of TNF-α, IFN-α, IL-1 and IL-6 in humans results in a decrease in serum T3, T4 and TSH levels and a rise in rT3 (76, 77, 78). In patients with acute myocardial infarction, the rise in IL-6 is closely linked with a decrease in T3 (79). In fact, IL-6 inhibits 5′-deiodinase activity, thereby reducing the conversion of T4 to T3 (80).
Hypoxia also contributes to decreased T3 levels, as it induces hypoxia-induced factor-1 (HIF-1α) that can activate D3 in the myocardium (81). In most conditions, NTIS seems to be an adaptive, compensatory and beneficial response, decreasing energy consumption in response to inflammation during various critical illnesses (82). Although T3 plays an important role in cardiac function, the impact of NTIS in patients with heart disease is unclear. Nevertheless, increased rT3 may be considered a predictor of both short- and long-term mortality in ischemic heart disease (10). NTIS is often associated with depressed myocardial function and is be a strong predictor of mortality in patients with heart disease, both in acute and chronic conditions (69, 83). Low T3 levels are also associated with dilated cardiomyopathy; indeed, 20–30% of patients with dilated cardiomyopathy have NTIS (11).
The impact of thyroid hormone dysfunction on myocardial ischemia
Myocardial ischemia is a major cause of mortality and morbidity worldwide (84). An understanding of the mechanisms of interaction between THs and their receptors is crucial to assess their impact in myocardial ischemia. TRα1 plays a key role during post-ischemic adaptation as it appears to present dual action and may be able to convert pathologic to physiologic growth depending on its ligand availability (85). In fact, TRα1 overexpression in the nucleus of cardiomyocytes in the absence of adequate THs as ligands may induce pathological hypertrophy and fetal phenotype, with predominant β-MHC expression. In contrast, higher levels of THs stimulate an α-MHC growth pattern, enhancing more physiological growth (86).
The precise prevalence of NTIS among patients with acute coronary syndrome has not been defined, but a prevalence of 5–35% has been reported in the literature (87). Several studies demonstrated a decrease in T3 and an increase in rT3 concentration in patients after an acute coronary event (88). Some factors may predict a more pronounced decline in T3 levels, such as worsening angina pectoris preceding acute myocardial infarction, known chronic heart failure or previous myocardial infarction and diabetes mellitus (89). Low T3 levels also induce oxidative stress and increase apoptotic rate, which may worsen ventricular dysfunction (90). Therefore, THs levels are an important factor modulating left ventricular structure, function and geometry during the late post-myocardial infarction period (91). Patients with ST-elevation myocardial infarction (STEMI) and alterations in thyroid function have almost a 3.5-fold increased risk of major adverse cardiac events, including cardiogenic shock and death, compared with patients with STEMI and no thyroid disorder (92). In fact, alterations in thyroid function seem to occur more frequently in STEMI than in NSTEMI (non-ST-elevation myocardial infarction), possibly because of poorer short-term prognosis and features of the occlusive coronary thrombus typical of STEMI (88, 92, 93).
Recent evidence indicates that circulating T3 levels are an independent determinant of the recovery of left ventricular ejection fraction 6 months after acute myocardial infarction in humans (94). Friberg et al. found a positive correlation between rT3 levels and 1-year mortality in patients with myocardial infarction, independent of other risk factors (95). In line with these results, a recent study with patients attending a cardiac rehabilitation program after an acute coronary syndrome also reported an association between lower T3 levels and all-cause mortality (96). In patients with myocardial injury, lower T3 levels have been correlated with increased serum levels of cardiac biomarkers such as troponin T and N-terminal pro-brain natriuretic peptide and with lower left ventricular ejection fraction (97). T3 levels may represent a predictor of the potential recovery of ventricular function (94).
One of the priorities in the treatment of myocardial ischemia is the reestablishment of coronary circulation. Early reperfusion has a great impact on short-term mortality after a myocardial ischemic event (98). Coronary revascularization by either coronary bypass surgery (CABG) or percutaneous coronary intervention (PCI) constitutes the primary option in the treatment of coronary artery disease. Despite its indisputable benefits, reperfusion after a myocardial ischemic event may contribute to adverse cardiac remodeling with possible evolution to heart failure. The pathophysiology of IRI is complex; however, recent evidence suggests that mitochondrial dysfunction may be one of the major mechanisms of IRI (99). The incidence of post-ischemic heart failure remains critical, increasing the risk of both cardiac and all-cause deaths (98). After reperfusion, extracellular washout of accumulated H+ ions creates a large gradient that increases the influx of sodium via the Na+/H+ exchanger. This stimulates the reverse action of the Na+/Ca2+ exchanger pump, increasing oxidative stress (98). THs improve the balance of proapoptotic and pro-survival signaling pathways which may limit IRI (100). T3 enhances the expression of HIF-1α, limiting the mitochondrial opening of permeability transition pores and thereby protecting the cardiomyocyte from reperfusion injury (101). Serum THs levels after CABG are often decreased (102). In fact, NTIS is reported in 50–75% of patients after cardiac surgery and some authors consider this as a poor prognostic factor and a predictor of mortality (28, 102). Pantos et al. were the first to observe that pretreatment with THs confers protection against IRI in isolated rat hearts in a pattern similar to ischemic preconditioning (1). The interest in the role of THs in cardioprotection is increasing. In fact, THs pretreatment may confer protection against subsequent IRI by inducing pharmacological preconditioning in cardiomyocytes, mainly by enhancing heat-shock protein 27 (HSP27) and heat-shock protein 70 (HSP70) and decreasing the activation of proapoptotic p38MAPK (1, 102, 103, 104). Recent studies using TH replacement therapy in animal models with regional or global myocardial ischemia followed by revascularization and/or reperfusion showed improved reversal of myocardial dysfunction compared with the absence of TH replacement therapy (69).
Thyroid hormones and cardiovascular risk factors
Overt and SCH are recognized as cardiovascular risk factors (59). In fact, thyroid disorders are associated with dyslipidemia, insulin resistance, hypertension, inflammation, atherosclerosis and other conditions (48). Hyperlipidemia seen in hypothyroidism is probably due to reduced cholesterol clearance, attributable to reduced hepatic LDL receptors and a decrease in 7α-hydroxylase levels in the liver (10). Elevated serum levels of total cholesterol, LDL cholesterol and apolipoprotein B are nearly reversed following THs therapy in patients with overt hypothyroidism (105). A recent meta-analysis found a statistically significant decrease of 9% in total cholesterol and 14% in LDL cholesterol in patients treated with levothyroxine (doses used vary from 67.5 μg/day to 85.5 μg/day) compared with the placebo group (59). While some studies show a decrease in triglyceride levels, other studies detected lower levels of apolipoprotein B-100 in the treated group (106). The role of THs in preventing cardiac ischemia has not been extensively investigated, mainly due to the possibility that THs may increase heart rate and cardiac work (107). However, the anti-ischemic and cardioprotective effects of THs suggest that supplementation may be more beneficial than harmful. Thus, the correction of overt and subclinical thyroid dysfunction may reduce the risk of future CHD and protect cardiomyocytes from future ischemic stress (107). TH replacement therapy may reverse atherosclerosis, lower peripheral vascular resistance and improve myocardial perfusion in patients with hypothyroidism (61). Treatment with levothyroxine in humans, mean dose 85.5 ± 4,3 μg/day, also has an impact on other predictors of cardiovascular events, such as carotid artery intima-media thickness (106).
Thyroid hormones and the prevention of heart failure after acute coronary events
In order to improve cardiovascular outcomes, cardioprotection represents the new purpose of therapeutic interventions to minimize infarct size and prevent the progression toward heart failure after an acute ischemic event (98). TH replacement therapy shortly after an infarction (or even when delayed) may improve cardiac function, mainly through TRα1 binding (90, 107). THs preserve mitochondrial function and have antifibrotic and proangiogenic effects that are crucial to the prevention of heart failure (98). As described above, after an ischemic event, THs increase MHC-α and the SERCA/PLB ratio which may improve contractile indices and energy expenditure (107, 108). The known effects of THs on MMP include decreased cardiac scar area and improved left ventricular relaxation (23). THs also reduce myocyte apoptosis in the myocardial infarction border area shortly after an ischemic event (23). Rajagopalan et al. observed an improved left ventricular ejection fraction without significant changes in heart rate in adult female rats treated with approximately 6 μg/kg/day of T3 after left coronary artery ligation (109). Moreover, atrial tachyarrhythmia was reduced by 88% in the T3-supplemented group (109). In an animal model of NTIS after an ischemic event, a subcutaneous infusion of 6 μg/kg/day of T3, a physiological or near-physiological dose, improved mitochondrial function, reducing cardiomyocyte loss in the peri-infarct zone (110). Another study in dogs with induced ischemia reported that plasma T3 levels decreased significantly during the ischemic period and continued to fall after reperfusion. The deterioration of left ventricular function was worse in untreated dogs compared with dogs treated with T3, in which hemodynamic function was maintained and improved to levels higher than those in the controls (111).
It is well recognized that chronic treatment with β-blockers improves left ventricular contractility and cardiac remodeling after myocardial infarction (101). A recent study by Zhang et al. aimed to compare T3 and metoprolol treatment in rats with myocardial infarction (101). The treatment with T3 (5 μg/kg/day) or metoprolol (100 mg/kg/day) was given in drinking water immediately after surgery for 8 weeks. Both treatments enhanced left ventricular contractility compared to the placebo and decreased the incidence of atrial tachyarrhythmia (101). Compared to the placebo, histological analysis indicated a significant reduction of 19% in the infarct area in the T3-treated group, which was similar to the group treated with metoprolol (101). Furthermore, the diameter of the left atrium was significantly enlarged after myocardial infarction. While this was ameliorated by T3, the metoprolol group showed results similar to the control group (101). The reversal of fetal genes expression induced by T3 treatment was not as complete upon treatment with metoprolol, suggesting additional improvement in physiological parameters with T3 treatment compared with metoprolol treatment (101).
Thyroid hormones: a future therapeutic option?
Given that the thyroid axis plays a key role in cardiovascular regulation, a thorough understanding its functions may be useful in preventing CHD, reducing ischemic lesions and improving the outcomes of acute coronary events. Although it is known that THs levels in plasma decrease after an ischemic event, the physiological relevance of this remains uncertain and the benefits of THs treatment is controversial. Regarding SCH, current guidelines recommend that it be treated during pregnancy in symptomatic patients according to individual factors (age, TSH levels, thyroid autoantibodies, the presence of atherosclerotic cardiovascular disease, heart failure and other associated risk factors) (112). A normalized thyroid axis may improve cardiovascular outcomes after an ischemic event. Thus, treatment with levothyroxine or T3 offers a potential therapeutic approach in coronary artery disease. The use of levothyroxine requires the preservation of peripheral deiodinase activity to convert T4 into the active hormone T3. Administration of T3 may be a better option in patients with impaired conversion, based on increased D3 activity and reduced D1 and D2 (28). Another therapeutic option not yet available for clinical practice is the genetic manipulation of deiodinases, increasing the local production of T3 or the expression of thyroid receptors and enhancing hormonal signaling (113). The first option involving THs analogues has been tested in some clinical trials, but the genetic approaches remain in the experimental animal stage (11). Recent observations highlighting the cardioprotective effects of the TRα1 receptor suggest that the development of selective agonists for this receptor may increase the efficacy and safety of TH replacement treatment by circumventing the potential side effects of excess of T3 administration (100). Future studies are necessary to assess which synthetic hormone (T4 or T3), dosage and timing of administration yields the best results with the least adverse effects. A TRβ1 agonist, GC-1, with no effect on cardiac rhythm has been shown to have a proangiogenic effect dependent on MAPK signaling (114). A chemical compound named CO23 has recently been synthesized as a TRα1 selective agonist in amphibian models. However, this compound loses its selectivity in rat models. Further investigation is required in order to understand the differences in thyroid receptor expression and development (115). Additional selective TRα1 agonists are now being synthesized (116). The available evidence indicates that TH replacement treatment should be administered at a physiological dose to obtain the maximal protective effect (117). The use of supra-physiological doses may increase heart rate and contractile function, which may enhance energy expenditure and thus aggravate ischemia, masking any potentially beneficial effects (42, 108). THs appears to achieve inotropic and antiapoptotic effects with acute infusion of T3 (0.8 μg/kg bolus followed by 0.113 μg/kg per hour) in humans (118). Supra-physiological doses may also be associated with increased risk of atrial and ventricular tachyarrhythmias, symptoms of hyperthyroidism and adverse extra-cardiac consequences, particularly the development or worsening of osteoporosis (119). Furthermore, the long-term effects of THs treatment on myocyte remodeling following myocardial infarction are still unknown and require further investigation. At the time of this review, clinical interventional studies involving THs supplementation are available in patients undergoing CABG surgery or with chronic heart failure (117). The randomized clinical trials known as the THIRST study (98) and ThyrAMI (120) are the first clinical trials investigating the use of THs treatment in acute myocardial infarction. The recently published THIRST study is a phase II, randomized, double-blind, placebo-controlled study involving the administration of T3 (maximum dosage 15 μg/m2/die) in patients with acute myocardial infarction treated with primary angioplasty with free T3 levels below the lower reference range (121). This study showed that T3 therapy is safe and improves regional dysfunction in patients with STEMI and NTIS. The results reveal the absence of major or minor side effects induced by T3 treatment such as arrhythmias and increased heart rate (121). In fact, T3-treated patients had a lower incidence of arrhythmias and slower heart rate at follow-up (98). Untreated group had significantly higher TSH compared to T3-treated group at discharge and after 1-month follow-up (121). Cardiac magnetic resonance showed a significant reduction in the global size of necrosis, while regional systolic function tended to improve (121). At follow-up, both groups experienced a significant reduction in wall motion score index, bur the difference value (discharge/follow-up) was significantly higher in treated group (121). Additionally, T3-treated group had a significantly increased stoke volume at follow-up and there was a tendency to higher increase in stroke volume compared to the untreated group (121). Given that the study imposed restrictive inclusion criteria that caused difficulties in the enrollment of patients, additional data are necessary to assess clinical targets and treatment doses using this approach (121). The results of ThyAMI and other studies will be essential to gain a better understanding of the effects of THs in ischemic heart disease and to further characterize the therapeutic potential of THs in this context.
The use of oral T3 as a pretreatment (125 µg/day for seven days prior to surgery) in patients undergoing CABG with LV dysfunction improved post-ischemic recovery of cardiac function and significantly lowered the mean inotropic requirements (122). In this study, the treated group exhibited no changes in blood pressure, heart rate, or cardiac rhythm (122). In fact, the low levels of T3 resulting from CABG can be reversed by oral T3 administration to patients with LV dysfunction. Although no differences between groups in the incidence of death and myocardial ischemia were reported, the length of stay in ICU was shorter in the treated group (122). Klemperer et al. showed that T3 treatment at reperfusion in patients undergoing CABG increased cardiac output and lowered systemic vascular resistance (14). In isolated dogs’ hearts, T3 treatment after reperfusion increased coronary blood flow by decreasing coronary arterial resistance (123). Additionally, T3 treatment at reperfusion in patients after CABG improved cardiac index decreased cardiac troponin I release (124). Other studies show the effect of THs supplementation on cardiac hemodynamics after acute myocardial infarction. Ojamaa et al. reported increased ejection fraction in rats after administration of 1.2 µg/day of T3 for 1 week after acute myocardial ischemia (125). Administration of 3.0 µg/day of T3 plus 12 µg/day of T4 for 2 weeks after acute myocardial ischemia in rats increases ejection fraction, decreases wall tension and improves left ventricular geometry (126). In isolated rat hearts, T3 was demonstrated to have cardioprotective properties during reperfusion after ischemia as evidenced by reduced LDH release and increased contractile recovery (127). In isolated dogs hearts, a bolus of T3 (0.2 µg/kg) after reperfusion increased coronary blood flow by decreasing coronary arterial resistance (123). The results of a double-blind, randomized placebo-controlled clinical trial suggested that 0.4 mg/kg taken orally once a day for 4 days before surgery may provide protection against myocardial IRI during cardiac surgery in children by increasing HSP70 and MHC-α expression, inducing pharmacological ischemic preconditioning (128). A study in humans that aimed to evaluate the effects of TH replacement therapy on systolic and diastolic function in SCH patients showed a statistically significantly increase in ejection fraction after five months of levothyroxine therapy (129). In this study the starting dose of levothyroxine was 25 µg, and the euthyroid state was achieved with a mean dose of 60.8 ± 19 µg (129). Another study aimed to compare the effects of long-term, low dose T3 (1.2 µg/100g/day) and T4 (4.8 µg/100g/day) hormone treatment to those provided by aerobic exercise training in rats post-myocardial infarction (130). The T3 and T4 hormone treatment enhanced cardiac function, with infarct size reduction and increased ejection fraction, without differences in heart rate, cardiac output and serum THs levels. Low-dose THs might offer a suitable treatment option after myocardial infarction in patients who are intolerant to aerobic exercise training (130). Evidence suggests that the hypothyroid tissue state may be present independent of normal circulating levels of THs. Therefore, it is important to identify a good biomarker of tissue hypothyroid-like state in order to treat patients effectively. The results are promising so far; experimental and clinical studies demonstrate that THs can limit ischemic injury, attenuate cardiac remodeling, and improve hemodynamics. However, the optimal timing for pretreatment and the treatment dose must be carefully evaluated to maximize the benefits.
Conclusions
It is now recognized that even subtle changes in TH levels can lead to adverse effects in the cardiovascular system. Experimental and clinical evidence suggests a close link between low TH levels and poor prognosis in ischemic heart disease. This condition should therefore be regarded as a cardiovascular risk factor. Accordingly, TH replacement therapy may yield improvements in lipid profiles, potentially reversing myocardial dysfunction and preventing the progression to heart failure. TH replacement treatment exhibits anti-ischemic and cardioprotective effects, acting as a promising target for ischemic heart disease. Moreover, subclinical hypothyroidism treatment and nonthyroidal illness syndrome constitute topics garnering increased interest; recent studies suggest that therapy with physiological doses of T3 are safe and provide beneficial effects on ischemic heart disease. Large clinical trials involving TH replacement treatment are necessary to evaluate the potential benefits on morbidity and mortality in patients with ischemic heart disease, as well as any potential long-term consequences.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Funding
This work was supported by the project DOCnet (Grant number NORTE-01-0145-FEDER-000003), supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF).
Acknowledgments
All figures use templates from Servier Medical Art.
References
- 1.Pantos CI, Malliopoulou VA, Mourouzis IS, Karamanoli EP, Paizis IA, Steimberg N, Varonos DD, Cokkinos DV. Long-term thyroxine administration protects the heart in a pattern similar to ischemic preconditioning. Thyroid 2002. 12 325–329. ( 10.1089/10507250252949469) [DOI] [PubMed] [Google Scholar]
- 2.Klein I, Danzi S. Thyroid disease and the heart. Current Problems in Cardiology 2016. 41 65–92. ( 10.1016/j.cpcardiol.2015.04.002) [DOI] [PubMed] [Google Scholar]
- 3.Fliers E, Alkemade A, Wiersinga WM, Swaab DF. Hypothalamic thyroid hormone feedback in health and disease. Progress in Brain Research 2006. 153 189–207. ( 10.1016/S0079-6123(06)53011-0) [DOI] [PubMed] [Google Scholar]
- 4.Dentice M, Marsili A, Zavacki A, Larsen PR, Salvatore D. The deiodinases and the control of intracellular thyroid hormone signaling during cellular differentiation. Biochimica and Biophysica Acta 2013. 1830 3937–3945. ( 10.1016/j.bbagen.2012.05.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger HR, Creech MK, Hannoush Z, Watanabe Y, Kargi A, Weiss RE. A novel mutation causing complete thyroid binding globulin deficiency in a male with coexisting graves disease. AACE Clinical Case Reports 2017. 3 e134–e139. ( 10.4158/EP161421.CR) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin JZ, Sieglaff DH, Yuan C, Su J, Arumanayagam AS, Firouzbakht S, Cantu-Pompa JJ, Reynolds FD, Zhou X, Cvoro A, et al Gene specific actions of thyroid hormone receptor subtypes. PLOS ONE 2013. 8 e52407 ( 10.1371/journal.pone.0052407) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiersing WM. The role of thyroid hormone nuclear receptors in the heart: evidence from pharmacological approaches. Heart Failure Reviews 2010. 15 121–124. ( 10.1007/s10741-008-9131-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mourouzis I, Kostakou E, Galanopoulos G, Mantzouratou P, Pantos C. Inhibition of thyroid hormone receptor α1 impairs post-ischemic cardiac performance after myocardial infarction in mice. Molecular and Cellular Biochemistry 2013. 379 97–105. ( 10.1007/s11010-013-1631-9) [DOI] [PubMed] [Google Scholar]
- 9.Vargas-Uricoechea H, Sierra-Torres CH. Thyroid hormones and the heart. Hormone Molecular Biology and Clinical Investigation 2014. 18 15–26. ( 10.1515/hmbci-2013-0059) [DOI] [PubMed] [Google Scholar]
- 10.Razvi S, Jabbar A, Pingitore A, Danzi S, Biondi B, Klein I, Peeters R, Zaman A, Iervasi G. Thyroid hormones and cardiovascular function and diseases. Journal of the American College of Cardiology 2018. 71 1781–1796. ( 10.1016/j.jacc.2018.02.045) [DOI] [PubMed] [Google Scholar]
- 11.Gerdes AM, Lervasi G. Thyroid replacement therapy and heart failure. Circulation 2010. 122 385–393. ( 10.1161/CIRCULATIONAHA.109.917922) [DOI] [PubMed] [Google Scholar]
- 12.Villeneuve C, Guilbeau-Frugier C, Sicard P, Lairez O, Ordener C, Duparc T, De Paulis D, Couderc B, Spreux-Varoquaux O, Tortosa F, et al p53-PGC-1alpha pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxidants and Redox Signaling 2013. 18 5–18. ( 10.1089/ars.2011.4373) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mourouzis I, Forini F, Pantos C, Iervasi G. Thyroid hormone and cardiac disease: from basic concepts to clinical application. Journal of Thyroid Research 2011. 2011 958626 ( 10.4061/2011/958626) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klemperer JD, Klein I, Gomez M, Helm RE, Ojamaa K, Thomas SJ, Isom OW, Krieger K. Thyroid hormone treatment after coronary-artery bypass surgery. New England Journal of Medicine 1995. 333 1522–1527. ( 10.1056/NEJM199512073332302) [DOI] [PubMed] [Google Scholar]
- 15.Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. New England Journal of Medicine 2001. 344 501–509. ( 10.1056/NEJM200102153440707) [DOI] [PubMed] [Google Scholar]
- 16.Fukuyama K, Ichiki T, Takeda K, Tokunou T, Lino N, Masuda S, Ishibashi M, Egashira K, Shimokawa H, Hirano K, et al Downregulation of vascular angiotensin II type 1 receptor by thyroid hormone. Hypertension 2003. 41 598–603. ( 10.1161/01.HYP.0000056524.35294.80) [DOI] [PubMed] [Google Scholar]
- 17.Galli E, Pingitore A, Iervasi G. The role of thyroid hormone in the pathophysiology of heart failure: clinical evidence. Heart Failure Reviews 2010. 15 155–169. ( 10.1007/s10741-008-9126-6) [DOI] [PubMed] [Google Scholar]
- 18.Davis PJ, Leonard JL, Davis FB. Mechanisms of nongenomic actions of thyroid hormone. Frontiers in Neuroendocrinology 2008. 29 211–218. ( 10.1016/j.yfrne.2007.09.003) [DOI] [PubMed] [Google Scholar]
- 19.Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ. Integrin alphaVbeta3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology 2005. 146 2864–2871. ( 10.1210/en.2005-0102) [DOI] [PubMed] [Google Scholar]
- 20.Vicinanza R, Cappotelli G, Malacrino C, Nardo T, Buchetti B, Lenti L, Celi FS, Scarpa S. Oxidized low-density lipoproteins impair endothelial function by inhibiting non-genomic action of thyroid hormone-mediated nitric oxide production in human endothelial cells. Thyroid 2013. 23 231–238. ( 10.1089/thy.2011.0524) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis PJ, Davis FB, Mousa SA. Thyroid hormone-induced angiogenesis. Current Cardiology Reviews 2009. 5 12–16. ( 10.2174/157340309787048158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eckle T, Kohler D, Lehmann R, Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation 2008. 118 166–175. ( 10.1161/CIRCULATIONAHA.107.758516) [DOI] [PubMed] [Google Scholar]
- 23.Chen YF, Weltman NY, Li X, Youmans S, Krause D, Gerdes AM. Improvement of left ventricular remodeling after myocardial infarction with eight weeks L-thyroxine treatment in rats. Journal of Translational Medicine 2013. 11 40 ( 10.1186/1479-5876-11-40) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samuels HH, Tsai JS, Casanova J, Stanley F. Thyroid hormone action: in vitro characterization of solubilized nuclear receptors from rat liver and cultured GH1 cells. Journal of Clinical Investigation 1974. 54 853–865. ( 10.1172/JCI107825) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicolini G, Forini F, Kusmic C, Pitto L, Mariani L, Iervasi G. Early and short-term triiodothyronine supplementation prevents adverse post-ischemic cardiac remodeling: role of transforming growth factor-b1 and anti-fibrotic miRNA signaling. Molecular Medicine 2015. 21 900–911. ( 10.2119/molmed.2015.00140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinugawa K, Jeong MY, Bristow MR, Long CS. Thyroid Hormone Induces Cardiac Myocyte Hypertrophy in a thyroid hormone receptor α1-Specific Manner that Requires TAK1 and p38 mitogen-activated protein kinase. Molecular Endocrinology 2005. 19 1618–1628. ( 10.1210/me.2004-0503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marín-Garcia J. Thyroid hormone and myocardial mitochondrial biogenesis. Vascular Pharmacology 2010. 52 120–130. ( 10.1016/j.vph.2009.10.008) [DOI] [PubMed] [Google Scholar]
- 28.Jabbar A, Pingitore A, Pearce SH, Zaman A, Iervasi G, Razvi S. Thyroid hormones and cardiovascular disease. Nature Reviews Cardiology 2017. 14 39–55. ( 10.1038/nrcardio.2016.174) [DOI] [PubMed] [Google Scholar]
- 29.Goldenthal MJ, Ananthakrishnan R, Marín-García J. Nuclear-mitochondrial cross-talk in cardiomyocyte T3 signaling: a time-course analysis. Journal of Molecular and Cellular Cardiology 2005. 39 319–326. ( 10.1016/j.yjmcc.2005.03.016) [DOI] [PubMed] [Google Scholar]
- 30.Marín-García J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Failure Reviews 2008. 13 137–150. ( 10.1007/s10741-007-9079-1) [DOI] [PubMed] [Google Scholar]
- 31.Saha MN, Jiang H, Yang Y, Zhu X, Wang X, Schimmer AD, Qiu L, Chang H. Targeting p53 via JNK pathway: a novel role of RITA for apoptotic signaling in multiple myeloma. PLOS ONE 2012. 7 e30215 ( 10.1371/journal.pone.0030215) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida K, Yoshiyama M, Omura T, Nakamura Y, Kim S, Takeuchi K, Iwao H, Yoshikawa J. Activation of mitogen-activated protein kinases in the non-ischemic myocardium of an acute myocardial infarction in rats. Japanese Circulation Journal 2001. 65 808–814. ( 10.1253/jcj.65.808) [DOI] [PubMed] [Google Scholar]
- 33.Chen YF, Kobayashi S, Chen J, Redetzke RA, Said S, Liang Q, Gerdes AM. Short term triiodo-L-thyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. Journal of Molecular and Cellular Cardiology 2008. 44 180–187. ( 10.1016/j.yjmcc.2007.09.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forini F, Lionetti V, Ardehali H, Pucci A, Cecchetti F, Chanefar M, Nicolini G, Ichikawa Y, Nannipieri M, Recchia FA, et al Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. Journal of Cellular and Molecular Medicine 2011. 15 514–524. ( 10.1111/j.1582-4934.2010.01014.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y. Myocardial repair/remodelling following infarction: roles of local factors. Cardiovascular Research 2009. 81 482–490. ( 10.1093/cvr/cvn333) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garnier A, Fortin D, Deloménie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. Journal of Physiology 2003. 551 491–501. ( 10.1113/jphysiol.2003.045104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K, et al Overexpression of mitochondrial transcription factor A ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation 2005. 112 683–690. ( 10.1161/CIRCULATIONAHA.104.524835) [DOI] [PubMed] [Google Scholar]
- 38.Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metabolism 2006. 3 333–341. ( 10.1016/j.cmet.2006.04.002) [DOI] [PubMed] [Google Scholar]
- 39.Paradise G, Ruggiero FM, Petrossil G, Quagliariello E. Enhanced cytochrome oxidase activity and modification of lipids in heart mitochondria from hyperthyroid rats. Biochimica et Biophysica Acta 1994. 1225 165–170. ( 10.1016/0925-4439(94)90074-4) [DOI] [PubMed] [Google Scholar]
- 40.Baines CP. The cardiac mitochondrion: nexus of stress. Annual Review of Physiology 2010. 72 61–80. ( 10.1146/annurev-physiol-021909-135929) [DOI] [PubMed] [Google Scholar]
- 41.de Castro AL, Fernandes RO, Ortiz VD, Campos C, Bonetto JH, Fernandes TR, Conzatti A, Siqueira R, Tavares AV, Schenkel PC, et al Thyroid hormones improve cardiac function and decrease expression of pro-apoptotic proteins in the heart of rats 14 days after infarction. Apoptosis 2016. 21 184–194. ( 10.1007/s10495-015-1204-3) [DOI] [PubMed] [Google Scholar]
- 42.de Castro AL, Tavares AV, Campos C, Fernandes RO, Siqueira R, Conzatti A, Bicca AM, Fernandes TRG, Sartorio CL, Schenkel PC, et al Cardioprotective effects of thyroid hormones in a rat model of myocardial infarction are associated with oxidative stress reduction. Molecular and Cellular Endocrinology 2014. 391 22–29. ( 10.1016/j.mce.2014.04.010) [DOI] [PubMed] [Google Scholar]
- 43.Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Archives of Internal Medicine 2000. 160 526–534. ( 10.1001/archinte.160.4.526) [DOI] [PubMed] [Google Scholar]
- 44.Biondi B, Palmieri EA, Lombardi G, Fazio S. Effects of thyroid hormone on cardiac function: the relative importance of heart rate, loading conditions, and myocardial contractility in the regulation of cardiac performance in human hyperthyroidism. Journal of Clinical Endocrinology and Metabolism 2002. 87 968–974. ( 10.1210/jcem.87.3.8302) [DOI] [PubMed] [Google Scholar]
- 45.Heeringa J, Hoogendoorn EH, van der Deure WM, Hofman A, Peeters RP, Hop WC, den Heijer M, Visser TJ, Witteman JC. High-normal thyroid function and risk of atrial fibrillation: the Rotterdam study. Archives of Internal Medicine 2008. 168 2219–2224. ( 10.1001/archinte.168.20.2219) [DOI] [PubMed] [Google Scholar]
- 46.Sahay RK. Thyrotoxicosis. Journal of the Association of Physicians of India 2011. 59 26–31. [PubMed] [Google Scholar]
- 47.Ertek S, Cicero AF. Hyperthyroidism and cardiovascular complications: a narrative review on the basis of pathophysiology. Archives of Medical Science 2013. 9 944–952. ( 10.5114/aoms.2013.38685) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Selmer C, Olesen JB, Hansen ML, von Kappelgaard LM, Madsen JC, Hansen PR, Pedersen OD, Faber J, Torp-Pedersen C, Gislason GH. Subclinical and overt thyroid dysfunction and risk of all-cause mortality and cardiovascular events: a large population study. Journal of Clinical Endocrinology and Metabolism 2014. 99 2372–2382. ( 10.1210/jc.2013-4184) [DOI] [PubMed] [Google Scholar]
- 49.Danzi S, Klein I. Thyroid disease and the cardiovascular system. Endocrinology and Metabolism Clinics of North America 2014. 43 517–528. ( 10.1016/j.ecl.2014.02.005) [DOI] [PubMed] [Google Scholar]
- 50.Vargas-Uricoechea H, Bonelo-Perdomo A, Sierra-Torres CH. Effects of thyroid hormones on the heart. Clinica e Investigacion en Arteriosclerosis 2014. 26 296–309. ( 10.1016/j.arteri.2014.07.003) [DOI] [PubMed] [Google Scholar]
- 51.Yeoh HL, Cheng AC, Cherry CL, Weir JM, Meikle PJ, Hoy JF, Crowe SM, Palmer CS. Immunometabolic and lipidomic markers associated with the frailty index and quality of life in aging HIV+ men on antiretroviral therapy. EBioMedicine 2017. 22 112–121. ( 10.1016/j.ebiom.2017.07.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tohidi M, Derakhshan A, Akbarpour S, Amouzegar A, Mehran L, Baghbani-Oskouei A, Azizi F, Hadaegh F. Thyroid dysfunction states and incident cardiovascular events: the Tehran thyroid study. Hormone and Metabolic Research 2018. 50 37–43. ( 10.1055/s-0043-121031) [DOI] [PubMed] [Google Scholar]
- 53.Sun J, Yao L, Fang Y, Yang R, Chen Y, Yang K, Tian L. Relationship between subclinical thyroid dysfunction and the risk of cardiovascular outcomes: a systematic review and meta-analysis of prospective cohort studies. International Journal of Endocrinology 2017. 2017 8130796 ( 10.1155/2017/8130796) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gencer B, Collet TH, Virgini V, Auer R, Rodondi N. Subclinical thyroid dysfunction and cardiovascular outcomes among prospective cohort studies. Endocrine, Metabolic and Immune Disorders Drug Targets 2013. 13 4–12. ( 10.2174/1871530311313010003) [DOI] [PubMed] [Google Scholar]
- 55.Ning Y, Cheng YJ, Liu LJ, Sara JD, Cao ZY, Zheng WP, Zhang TS, Han HJ, Yang ZY, Zhang Y, et al What is the association of hypothyroidism with risks of cardiovascular events and mortality? A meta-analysis of 55 cohort studies involving 1,898,314 participants. BMC Medicine 2017. 15 21 ( 10.1186/s12916-017-0777-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grais IM. Bedside skills: a 50-year personal retrospective. Texas Heart Institute Journal 2010. 37 629–632. [PMC free article] [PubMed] [Google Scholar]
- 57.Patil V, Patil HV, Agrawal V, Patil S. Cardiac tamponade in a patient with primary hypothyroidism. Indian Journal of Endocrinology and Metabolism 2011. 15 144 ( 10.4103/2230-8210.83358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ozturk S, Alcelik A, Ozyasar M, Dikbas O, Ayhan S, Ozlu F, Erdem A, Tosun M, Baltaci D, Yazici M. Evaluation of left ventricular systolic asynchrony in patients with subclinical hypothyroidism. Cardiology Journal 2012. 19 374–380. ( 10.5603/CJ.2012.0068) [DOI] [PubMed] [Google Scholar]
- 59.Abreu IM, Lau E, de Sousa Pinto B, Carvalho D. Subclinical hypothyroidism: to treat or not to treat, that is the question! A systematic review with meta-analysis on lipid profile. Endocrine Connections 2017. 6 188–199. ( 10.1530/EC-17-0028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pingitore A, Landi P, Taddei MC, Ripoli A, L’Abbate A, Lervasi G. Triiodothyronine levels for risk stratification of patients with chronic heart failure. American Journal of Medicine 2005. 118 132–136. ( 10.1016/j.amjmed.2004.07.052) [DOI] [PubMed] [Google Scholar]
- 61.Grais IM, Sowers JR. Thyroid and the heart. American Journal of Medicine 2014. 127 691–698. ( 10.1016/j.amjmed.2014.03.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alibaz Oner F, Yurdakul S, Oner E, Kubat Uzum A, Erguney M. Evaluation of the effect of L-thyroxin therapy on endothelial functions in patients with subclinical hypothyroidism. Endocrine 2011. 40 280–284. ( 10.1007/s12020-011-9465-2) [DOI] [PubMed] [Google Scholar]
- 63.Razvi S, Ingoe L, Keeka G, Oates C, McMillan C, Weaver JU. The beneficial effect of L-thyroxine on cardiovascular risk factors, endothelial function, and Quality of life in subclinical hypothyroidism: randomized, crossover trial. Journal of Clinical Endocrinology and Metabolism 2007. 92 1715–1723. ( 10.1210/jc.2006-1869) [DOI] [PubMed] [Google Scholar]
- 64.Hak AE, Pois HA, Visser TJ, Drexhage HA, Hofman A, Witteman JC. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: the Rotterdam Study. Annals of Internal Medicine 2000. 132 270–278. ( 10.7326/0003-4819-132-4-200002150-00004) [DOI] [PubMed] [Google Scholar]
- 65.Ilic S, Tadic M, Ivanovic B, Caparevic Z, Trbojevic B, Celic V. Left and right ventricular structure and function in subclinical hypothyroidism: the effects of one-year levothyroxine treatment. Medical Science Monitor 2013. 19 960–968. ( 10.12659/MSM.889621) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao N, Zhang W, Zhang YZ, Yang Q, Chen SH. Carotid intima-media thickness in patients with subclinical hypothyroidism: a meta-analysis. Atherosclerosis 2013. 227 18–25. ( 10.1016/j.atherosclerosis.2012.10.070) [DOI] [PubMed] [Google Scholar]
- 67.Suh S, Kim DK. Subclinical hypothyroidism and cardiovascular disease. Endocrinology and Metabolism 2015. 30 246–251. ( 10.3803/EnM.2015.30.3.246) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodondi N, den Elzen WP, Bauer DC, Cappola AR, Razvi S, Walsh JP, Asvold BO, Iervasi G, Imaizumi M, Collet TH, et al Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA 2010. 304 1365–1374. ( 10.1001/jama.2010.1361) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Novitzky D, Cooper DK. Thyroid hormone and the stunned myocardium. Journal of Endocrinology 2014. 223 R1–R8. ( 10.1530/JOE-14-0389) [DOI] [PubMed] [Google Scholar]
- 70.Dentice M, Salvatore D. Deiodinases: the balance of thyroid hormone: local impact of thyroid hormone inactivation. Journal of Endocrinology 2011. 209 273–282. ( 10.1530/JOE-11-0002) [DOI] [PubMed] [Google Scholar]
- 71.Peeters RP, Wouters PJ, Kaptein E, van Toor H, Visser TJ, Van den Berghe G. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients. Journal of Clinical Endocrinology and Metabolism 2003. 88 3202–3211. ( 10.1210/jc.2002-022013) [DOI] [PubMed] [Google Scholar]
- 72.Warner MH, Beckett GJ. Mechanisms behind the non-thyroidal illness syndrome: an update. Journal of Endocrinology 2010. 205 1–13. ( 10.1677/JOE-09-0412) [DOI] [PubMed] [Google Scholar]
- 73.Kaptein EM, Feinstein EI, Nicoloff JT, Massry SG. Serum reverse triiodothyronine and thyroxine kinetics in patients with chronic renal failure. Journal of Clinical Endocrinology and Metabolism 1983. 57 181–189. ( 10.1210/jcem-57-1-181) [DOI] [PubMed] [Google Scholar]
- 74.Huang SA, Tu HM, Harney JW, Venihaki M, Butte AJ, Kozakewich HP, Fishman SJ, Larsen PR. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. New England Journal of Medicine 2000. 343 185–189. ( 10.1056/NEJM200007203430305) [DOI] [PubMed] [Google Scholar]
- 75.Gereben B, Zeöld A, Dentice M, Salvatore D, Bianco AC. Activation and inactivation of thyroid hormone by deiodinases: local action with general consequences. Cellular and Molecular Life Sciences 2008. 65 570–590. ( 10.1007/s00018-007-7396-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van der Poll T, Romijn JA, Wiersinga WM, Sauerwein HP. Tumor necrosis factor: a putative mediator of the sick euthyroid syndrome in man. Journal of Clinical Endocrinology and Metabolism 1990. 71 1567–1572. ( 10.1210/jcem-71-6-1567) [DOI] [PubMed] [Google Scholar]
- 77.Hermus RM, Sweep CG, van der Meer MJ, Ross HA, Smals AG, Benraad TJ, Kloppenborg PW. Continuous infusion of interleukin-1 beta induces a nonthyroidal illness syndrome in the rat. Endocrinology 1992. 131 2139–2146. ( 10.1210/endo.131.5.1425414) [DOI] [PubMed] [Google Scholar]
- 78.Bartalena L, Brogioni S, Grasso L, Velluzzi F, Martino E. Relationship of the increased serum interleukin-6 concentration to changes of thyroid function in nonthyroidal illness. Journal of Endocrinological Investigation 1994. 17 269–274. ( 10.1007/BF03348974) [DOI] [PubMed] [Google Scholar]
- 79.Kimura T, Kanda T, Kotajima N, Kuwabara A, Fukumura Y, Kobayashi I. Involvement of circulating interleukin-6 and its receptor in the development of euthyroid sick syndrome in patients with acute myocardial infarction. European Journal of Endocrinology 2000. 143 179–184. ( 10.1530/eje.0.1430179) [DOI] [PubMed] [Google Scholar]
- 80.Boelen A, Maas MA, Lowik CW, Platvoet MC, Wiersinga WM. Induced illness in interleukin-6 (IL-6) knock- out mice: a causal role of IL-6 in the development of the low 3,5,3’-triiodothyronine syndrome. Endocrinology 1996. 137 5250–5254. ( 10.1210/endo.137.12.8940342) [DOI] [PubMed] [Google Scholar]
- 81.Zhang B, Peng W, Wang C, Li W, Xu YA. A low fT3 level as a prognostic marker in patients with acute myocardial infarctions. Internal Medicine 2012. 51 3009–3015. ( 10.2169/internalmedicine.51.7902) [DOI] [PubMed] [Google Scholar]
- 82.Chopra IJ. Clinical review 86: Euthyroid sick syndrome: is it a misnomer? Journal of Clinical Endocrinology and Metabolism 1997. 82 329–334. ( 10.1210/jcem.82.2.3745) [DOI] [PubMed] [Google Scholar]
- 83.Lervasi G, Pingitore A, Landi P, Raciti M, Ripoli A, Scarlattini M, L’Abbate A, Donato L. Low-T3 syndrome: a strong prognostic predictor of death in patients with heart disease. Circulation 2003. 107 708–713. ( 10.1161/01.CIR.0000048124.64204.3F) [DOI] [PubMed] [Google Scholar]
- 84.Shimokawa H, Yasuda S. Myocardial ischemia: current concepts and future perspectives. Journal of Cardiology 2008. 52 67–78. ( 10.1016/j.jjcc.2008.07.016) [DOI] [PubMed] [Google Scholar]
- 85.Mai W, Janier MF, Allioli N, Quignodon L, Chuzel T, Flamant F, Samarut J. Thyroid hormone receptor alpha is a molecular switch of cardiac function between fetal and postnatal life. PNAS 2004. 101 10332–10337. ( 10.1073/pnas.0401843101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pantos C, Xinaris C, Mourouzis I, Perimenis P, Politi E, Spanou D, Cokkinos DV. Thyroid hormone receptor alpha 1: a switch to cardiac cell “metamorphosis”? Journal of Physiology and Pharmacology 2008. 59 253–269. [PubMed] [Google Scholar]
- 87.Lamprou V, Varvarousis D, Polytarchou K, Varvarousi G, Xanthos T. The role of thyroid hormones in acute coronary syndromes: prognostic value of alterations in thyroid hormones. Clinical Cardiology 2017. 40 528–533. ( 10.1002/clc.22689) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Franklyn JA, Gammage MD, Ramsden DB, Sheppard MC. Thyroid status in patients after acute myocardial infarction. Clinical Science 1984. 67 585–590. ( 10.1042/cs0670585) [DOI] [PubMed] [Google Scholar]
- 89.Friberg L, Werner S, Eggertsen G, Ahnve S. Rapid down-regulation of thyroid hormones in acute myocardial infarction: is it cardioprotective in patients with angina? Archives of Internal Medicine 2002. 162 1388–1394. ( 10.1001/archinte.162.12.1388) [DOI] [PubMed] [Google Scholar]
- 90.Pantos C, Mourouzis I. The emerging role of TRalpha1 in cardiac repair: potential therapeutic implications. Oxidative Medicine and Cellular Longevity 2014. 2014 481482 ( 10.1155/2014/481482) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jankauskiene E, Orda P, Barauskiene G, Mickuviene N, Brozaitiene J, Vaskelyte JJ, Bunevicius R. Relationship between left ventricular mechanics and low free triiodothyronine levels after myocardial infarction: a prospective study. Internal and Emergency Medicine 2016. 11 391–398. ( 10.1007/s11739-015-1370-x) [DOI] [PubMed] [Google Scholar]
- 92.Ozcan KS, Osmonov D, Toprak E, Gungor B, Tatlisu A, Ekmekci A, Kaya A, Tayyareci G, Erdinler İ. Sick euthyroid syndrome is associated with poor prognosis in patients with ST segment elevation myocardial infarction undergoing primary percutaneous intervention. Cardiology Journal 2014. 21 238–244. ( 10.5603/CJ.a2013.0108) [DOI] [PubMed] [Google Scholar]
- 93.O’Gara PT, Kushner FG, Ascheim DD, Casey DE, Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, et al 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Journal of the American College of Cardiology 2013. 61 e78–e140. ( 10.1016/j.jacc.2012.11.019) [DOI] [PubMed] [Google Scholar]
- 94.Lymvaios I, Mourouzis I, Cokkinos DV, Dimopoulos MA, Toumanidis ST, Pantos C. Thyroid hormone and recovery of cardiac function in patients with acute myocardial infarction: a strong association? European Journal of Endocrinology 2011. 165 107–114. ( 10.1530/EJE-11-0062) [DOI] [PubMed] [Google Scholar]
- 95.Friberg L, Drvota V, Bjelak AH, Eggertsen G, Ahnve S. Association between increased levels of reverse triiodothyronine and mortality after acute myocardial infarction. American Journal of Medicine 2001. 111 699–703. ( 10.1016/S0002-9343(01)00980-9) [DOI] [PubMed] [Google Scholar]
- 96.Brozaitiene J, Mickuviene N, Podlipskyte A, Burkauskas J, Bunevicius R. Relationship and prognostic importance of thyroid hormone and N-terminal pro-B-Type natriuretic peptide for patients after acute coronary syndromes: a longitudinal observational study. BMC Cardiovascular Disorders 2016. 16 45 ( 10.1186/s12872-016-0226-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang WY, Tang YD, Yang M, Cui C, Mu M, Qian J, Yang YJ. Free triiodothyronine level indicates the degree of myocardial injury in patients with acute ST-elevation myocardial infarction. Chinese Medical Journal 2013. 126 3926–3929. ( 10.20431/2455-7153.0301003) [DOI] [PubMed] [Google Scholar]
- 98.Pingitore A, Nicolini G, Kusmic C, Iervasi G, Grigolini P, Forini F. Cardioprotection and thyroid hormones. Heart Failure Reviews 2016. 21 391–399. ( 10.1007/s10741-016-9545-8) [DOI] [PubMed] [Google Scholar]
- 99.Forini F, Nicolini G, Iervasi G. Mitochondria as key targets of cardioprotection in cardiac ischemic disease: role of thyroid hormone triiodothyronine. International Journal of Molecular Sciences 2015. 16 6312–6336. ( 10.3390/ijms16036312) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pantos C, Mourouzis I. Translating thyroid hormone effects into clinical practice: the relevance of thyroid hormone receptor alpha1 in cardiac repair. Heart Failure Reviews 2015. 20 273–282. ( 10.1007/s10741-014-9465-4) [DOI] [PubMed] [Google Scholar]
- 101.Zhang K, Tang YD, Zhang Y, Ojamaa K, Li Y, Saini AS, Carrillo-Sepulveda MA, Rajagopalan V, Gerdes AM. Comparison of therapeutic triiodothyronine versus metoprolol in treatment of myocardial infarction in rats. Thyroid 2018. 28 799–810. ( 10.1089/thy.2017.0544) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang GZ, Xue FS, Liu YY, Li HX, Liu Q, Liao X. Effects of enteral different-dose levothyroxine- sodium pretreatment on serum thyroid hormone levels and myocardial ischemia- reperfusion injury. Perfusion 2018. 33 584–592. ( 10.1177/0267659118769228) [DOI] [PubMed] [Google Scholar]
- 103.Pantos C, Malliopoulou V, Paizis I, Moraitis P, Mourouzis I, Tzeis S, Karamanoli E, Cokkinos DD, Carageorgiou H, Varonos D, et al Thyroid hormone and cardioprotection: study of p38 MAPK and JNKs during ischaemia and at reperfusion in isolated rat heart. Molecular and Cellular Biochemistry 2003. 242 173–180. ( 10.1023/A:1021162417490) [DOI] [PubMed] [Google Scholar]
- 104.Pantos C, Malliopoulou V, Mourouzis I, Karamanoli E, Moraitis P, Tzeis S, Paizis I, Cokkinos AD, Carageorgiou H, Varonos DD, et al Thyroxine pretreatment increases basal myocardial heat-shock protein 27 expression and accelerates translocation and phosphorylation of this protein upon ischaemia. European Journal of Pharmacology 2003. 478 53–60. ( 10.1016/j.ejphar.2003.08.030) [DOI] [PubMed] [Google Scholar]
- 105.Liu XL, He S, Zhang SF, Wang J, Sun XF, Gong CM, Zheng SJ, Zhou JC, Xu J. Alteration of lipid profile in subclinical hypothyroidism: a meta-analysis. Medical Science Monitor 2014. 20 1432–1441. ( 10.12659/MSM.891163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Meier C, Staub JJ, Roth CB, Guglielmetti M, Kunz M, Miserez AR, Drewe J, Huber P, Herzog R, Muller B. TSH-controlled L-thyroxine therapy reduces cholesterol levels and clinical symptoms in subclinical hypothyroidism: a double blind, placebo-controlled trial (Basel Thyroid Study). Journal of Clinical Endocrinology and Metabolism 2001. 86 4860–4866. ( 10.1210/jcem.86.10.7973) [DOI] [PubMed] [Google Scholar]
- 107.Pantos C, Mourouzis I, Cokkinos DV. New insights into the role of thyroid hormone in cardiac remodeling: time to reconsider? Heart Failure Reviews 2011. 16 79–96. ( 10.1007/s10741-010-9185-3) [DOI] [PubMed] [Google Scholar]
- 108.Mourouzis I, Mantzouratou P, Galanopoulos G, Kostakou E, Roukounakis N, Kokkinos AD, Cokkinos DV, Pantos C. Dose-dependent effects of thyroid hormone on post-ischemic cardiac performance: potential involvement of Akt and ERK signalings. Molecular and Cellular Biochemistry 2012. 363 235–243. ( 10.1007/s11010-011-1175-9) [DOI] [PubMed] [Google Scholar]
- 109.Rajagopalan V, Zhang Y, Ojamaa K, Chen YF, Pingitore A, Pol CJ, Saunders D, Balasubramanian K, Towner RA, Gerdes AM. Safe oral triiodo-L-thyronine therapy protects from post-infarct cardiac dysfunction and arrhythmias without cardiovascular adverse effects. PLoS ONE 2016. 11 e0151413 ( 10.1371/journal.pone.0151413) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Forini F, Kusmic C, Nicolini G, Mariani L, Zucchi R, Matteucci M, Iervasi G, Pitto L. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology 2014. 155 4581–4590. ( 10.1210/en.2014-1106) [DOI] [PubMed] [Google Scholar]
- 111.Novitzky D, Matthews N, Shawley D, Cooper DK, Zuhdi N. Triiodothyronine in the recovery of stunned myocardium in dogs. Annals of Thoracic Surgery 1991. 51 10–16; discussion 16. ( 10.1016/0003-4975(91)90438-V) [DOI] [PubMed] [Google Scholar]
- 112.Garber JR, Cobin RH, Gharib H, Hennessey JV, Klein I, Mechanick JI, Pessah-Pollack R, Singer PA, Woeber KA. & American Association of Clinical Endocrinologists and American Thyroid Association Taskforce on Hypothyroidism in Adults. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocrine Practice 2012. 18 988–1028. ( 10.4158/EP12280.GL) [DOI] [PubMed] [Google Scholar]
- 113.Iervasi G, Nicolini G. Thyroid hormone and cardiovascular system: from basic concepts to clinical application. Internal and Emergency Medicine 2013. 8 S71–S74. ( 10.1007/s11739-013-0911-4) [DOI] [PubMed] [Google Scholar]
- 114.Mousa SA, O’Connor LJ, Bergh JJ, Davis FB, Scanlan TS, Davis PJ. The proangiogenic action of thyroid hormone analogue GC-1 is initiated at an integrin. Journal of Cardiovascular Pharmacology 2005. 46 356–360. ( 10.1097/01.fjc.0000175438.94906.a0) [DOI] [PubMed] [Google Scholar]
- 115.Grijota-Martinez C, Samarut E, Scanlan TS, Morte B, Bernal J. In vivo activity of the thyroid hormone receptor beta- and α-selective agonists GC-24 and CO23 on rat liver, heart, and brain. Endocrinology 2011. 152 1136–1142. ( 10.1210/en.2010-0813) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ocasio CA, Scanlan TS. Design and characterization of a thyroid hormone receptor alpha (TRalpha)-specific agonist. ACS Chemical Biology 2006. 1 585–593. ( 10.1021/cb600311v) [DOI] [PubMed] [Google Scholar]
- 117.Pingitore A, Chen Y, Gerdes AM, Iervasi G. Acute myocardial infarction and thyroid function: new pathophysiological and therapeutic perspectives. Annals of Medicine 2012. 44 745–757. ( 10.3109/07853890.2011.573501) [DOI] [PubMed] [Google Scholar]
- 118.Kaptein EM, Sanchez A, Beale E, Chan LS. Clinical review: thyroid hormone therapy for postoperative nonthyroidal illnesses: a systematic review and synthesis. Journal of Clinical Endocrinology and Metabolism 2010. 95 4526–4534. ( 10.1210/jc.2010-1052) [DOI] [PubMed] [Google Scholar]
- 119.Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, Rivkees SA, Samuels M, Sosa JA, Stan MN, et al 2016 American Thyroid Association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid 2016. 26 1343–1421. ( 10.1089/thy.2016.0229) [DOI] [PubMed] [Google Scholar]
- 120.Jabbar A, Ingoe L, Pearce S, Zaman A, Razvi S. Thyroxine in acute myocardial infarction (ThyrAMI) - levothyroxine in subclinical hypothyroidism post-acute myocardial infarction: study protocol for a randomised controlled trial. Trials 2015. 16 115 ( 10.1186/s13063-015-0621-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pingitore A, Mastorci F, Piaggi P, Aquaoro GD, Molinaro S, Ravani M, Caterina A, Trianni G, Ndreu R, Berti S, et al Usefulness of triiodothyronine replacement therapy in patients with ST elevation myocardial infarction and borderline/reduced triiodothyronine levels (from the THIRST study). American Journal of Cardiology 2019. 123 905–912. ( 10.1016/j.amjcard.2018.12.020) [DOI] [PubMed] [Google Scholar]
- 122.Sirlak M, Yazicioglu L, Inan MB, Eryilmaz S, Tasoz R, Aral A, Ozyurda U. Oral thyroid hormone pretreatment in left ventricular dysfunction. European Journal of Cardio-Thoracic Surgery 2004. 26 720–725. ( 10.1016/j.ejcts.2004.07.003) [DOI] [PubMed] [Google Scholar]
- 123.Klemperer JD, Zelando J, Helm RE, Berman K, Ojamaa K, Klein I, Isom OW, Krieger K. Triiodothyronine improves left ventricular function without oxygen wasting effects after global hypothermic ischemia. Journal of Thoracic and Cardiovascular Surgery 1995. 109 457–465. ( 10.1016/S0022-5223(95)70276-8) [DOI] [PubMed] [Google Scholar]
- 124.Ranasinghe AM, Quinn DW, Pagano D, Edwards N, Faroqui M, Graham TR, Keogh BE, Mascaro J, Riddington DW, Rooney SJ, et al Glucose-insulin-potassium and tri-iodothyronine individually improve hemodynamic performance and are associated with reduced troponin I release after on-pump coronary artery bypass grafting. Circulation 2006. 114 I245–I250. ( 10.1161/CIRCULATIONAHA.105.000786) [DOI] [PubMed] [Google Scholar]
- 125.Ojamaa K, Kenessey A, Shenoy R, Klein I. Thyroid hormone metabolism and cardiac gene expression after acute myocardial infarction in the rat. American Journal of Physiology. Endocrinology and Metabolism 2000. 279 E1319–E1324. ( 10.1152/ajpendo.2000.279.6.E1319) [DOI] [PubMed] [Google Scholar]
- 126.Iliopoulou I, Mourouzis I, Lambrou GI, Iliopoulou D, Koutsouris DD, Pantos C. Time‐dependent and independent effects of thyroid hormone administration following myocardial infarction in rats. Molecular Medicine Reports 2018. 18 864–876. ( 10.3892/mmr.2018.9008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pantos C, Mourouzis I, Saranteas T, Brozou V, Galanopoulos G, Kostopanagiotou G, Cokkinos DV. Acute T3 treatment protects the heart against ischemia-reperfusion injury via TRα1 receptor. Molecular and Cellular Biochemistry 2011. 353 235–241. ( 10.1007/s11010-011-0791-8) [DOI] [PubMed] [Google Scholar]
- 128.Zhang JQ, Yang QY, Xue FS, Zhang W, Yang GZ, Liao X, Meng FM. Preoperative oral thyroid hormones to prevent euthyroid sick syndrome and attenuate myocardial ischemia-reperfusion injury after cardiac surgery with cardiopulmonary bypass in children: A randomized, double-blind, placebo-controlled trial. Medicine 2018. 97 e12100 ( 10.1097/MD.0000000000012100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nakova VV, Krstevska B, Kostovska ES, Vaskova O, Ismail LG. The effect of levothyroxine treatment on left ventricular function in subclinical hypothyroidism. Archives of Endocrinology and Metabolism 2018. 62 392–398. ( 10.20945/2359-3997000000052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Teixeira RB, Zimmer A, de Castro AL, de Lima-seolin BG, Turck P, Siqueira R, Belló-Klein A, Singal PK, da Rosa Araujo AS. Long-term T3 and T4 treatment as an alternative to aerobic exercise training in improving cardiac function post-myocardial infarction. Biomedicine and Pharmacotherapy 2017. 95 965–973. ( 10.1016/j.biopha.2017.09.021) [DOI] [PubMed] [Google Scholar]