

Abstract
Purpose:
The ganglioside fucosyl-GM1 (FucGM1) is a tumor-associated antigen expressed in a large percentage of human small cell lung cancer (SCLC) tumors, but absent in most normal adult tissues, making it a promising target in immuno-oncology. This study was undertaken to evaluate the preclinical efficacy of BMS-986012, a novel, nonfuco-sylated, fully human IgG1 antibody that binds specifically to FucGM1.
Experimental Design:
The antitumor activity of BMS-986012 was evaluated in in vitro assays using SCLC cells and in mouse xenograft and syngeneic tumor models, with and without chemotherapeutic agents and checkpoint inhibitors.
Results:
BMS-986012 showed a high binding affinity for FcγRIIIa (CD16), which resulted in enhanced antibody-dependent cellular cytotoxicity (ADCC) against FucGM1-expressing tumor cell lines. BMS-986012-mediated tumor cell killing was also observed in complement-dependent cytotoxicity (CDC) and antibody-dependent cellular phagocytosis (ADCP) assays. In several mouse SCLC models, BMS-986012 demonstrated efficacy and was well tolerated. In the DMS79 xenograft model, tumor regression was achieved with BMS-986012 doses of 0.3 mg/kg and greater; antitumor activity was enhanced when BMS-986012 was combined with standard-of-care cisplatin or etoposide. In a syngeneic model, tumors derived from a genetically engineered model of SCLC were treated with BMS-986012 or anti-FucGM1 with a mouse IgG2a Fc and their responses evaluated; when BMS-986012 was combined with anti–PD-1 or anti-CD137 antibody, therapeutic responses significantly improved.
Conclusions:
Single-agent BMS-986012 demonstrated robust antitumor activity, with the addition of chemotherapeutic or immunomodulatory agents further inhibiting SCLC growth in the same models. These preclinical data supported evaluation of BMS-986012 in a phase I clinical trial of patients with relapsed, refractory SCLC.
Introduction
Small cell lung cancer (SCLC) is an aggressive high-grade neuroendocrine neoplasia that accounts for about 15% of all lung cancers worldwide with approximately 30,000 new cases diagnosed in the United States each year (1, 2). Rapid proliferation, high growth fraction, and early, widespread metastases contribute to extremely poor prognosis (3, 4). Approximately 60% to 70% of the patients have clinically disseminated or extensive disease at presentation (5, 6). SCLC is highly sensitive to chemotherapy with an overall response rate of 50% to 80% obtained with standard-of-care (SOC) combination chemotherapy (7). However, almost all patients with disseminated disease relapse and have treatment-refractory tumors, resulting in a median overall survival of 8 to 13 months and a 5-year overall survival of 1% to 2% (1, 5, 6, 8, 9). New therapeutic approaches are clearly needed as existing treatments do not eradicate residual disease.
Gangliosides are cell surface glycosphingolipids that play important functional roles in cell-cell recognition, cell adhesion, and signal transduction (10). Although gangliosides are mostly expressed in neural tissue, abundant ganglioside expression has been observed in some tumors. For example, fucosylated monosialotetrahexosylganglioside (FucGM1) is normally expressed in a subset of peripheral sensory neurons and dorsal root ganglia, with occasional scattered expression on cells in the spleen, thymus, small intestine, and pancreas (11–16). However, a large percentage of SCLC tumors express very high levels of FucGM1 (11, 12, 16).
Therapeutic antibodies against gangliosides have been shown to inhibit tumor growth and metastasis, and to induce apoptosis of antigen-positive cells. Gangliosides such as GD2 and GD3 have been shown to function as effective targets for passive immunotherapy with monoclonal antibodies, as well as targets for active immunotherapy with vaccines (17, 18). Several studies have investigated FucGM1 as a tumor-associated antigen with therapeutic potential. FucGM1-specific antibodies have demonstrated complement activation and, in combination with cytostatic drugs, synergistic cytotoxic effects on FucGM1 expressing cells lines (19). In vivo, anti-FucGM1 mAbs have been reported to inhibit the engraftment of FucGM1-expressing tumor cells in nude mice (20, 21). In clinical studies, antibodies from patients with SCLC who were vaccinated with KLH-conjugated FucGM1 and developed antibody titers to the antigen (22, 23) demonstrated specific binding to tumor cells and complement-dependent cytotoxicity (CDC) against FucGM1-expressing cell lines in vitro (20, 21). However, the antibody response to FucGM1 was comprised predominantly of low-affinity IgM antibodies typical of theT-cell independent nature of immune responses to gangliosides (22, 23).
Fc receptor-dependent functional activity, such as antibody-dependent cellular cytotoxicity (ADCC), is an important mechanism for the clinical activity of many therapeutic antibodies (24–26). In addition, the structure of N-linked glycans linked to a conserved asparagine at amino acid position 297 (N297) of IgG1 heavy chains plays an important role in the conformation and stability of the Fc region (27, 28). It is well documented that absence of core fucose from the structure results in antibodies with higher binding affinity to FcγRIIIa on macrophages and natural killer (NK) cells, leading to increased ADCC effector function (29–36) and improved efficacy in vivo (37, 38).
Here, we report the results of preclinical studies of a novel nonfucosylated anti-FucGM1 antibody, BMS-986012. The antibody was profiled in vitro for binding affinity and effector functions, including ADCC, antibody-dependent cellular phagocytosis (ADCP), and CDC, and in vivo for antitumor activity using mouse xenograft and syngeneic tumor models. Mechanistically driven combinations with clinically actionable immuno-oncology antibodies targeting specific checkpoints involved in innate and adaptive immunity (CD137, PD-1) were also explored.
Materials and Methods
Cell lines
All human SCLC cell lines were obtained from ATCC. The mouse KP3 cell line was derived from a genetically engineered mouse model (39); these mice were from a mixed 129/Sv and C57BL/6 genetic background, allowing some allograft models to be expanded in F1 mice. All cell lines were cultured in RPMI1640 supplemented with 10% FBS and 2 mmol/L L-glutamine. All cell lines were authenticated and verified for absence of mouse and human pathogens (IDEXX BioResearch).
Antibodies and chemotherapy agents
BMS-986012 was generated from transgenic mice expressing human immunoglobulin genes immunized with FucGM1 conjugated to keyhole limpet hemocyanin (KLH) carrier protein (40). Heavy and light chain genes from hybridoma 7E4 were expressed recombinantly in CHO cells or in a fucosyltransferase-8 knockout (FUT8–/–) CHO cell line to produce a completely nonfucosylated antibody in-house. Human IgG4 Fc and mouse aglycosyl IgG1 (D265A) effector function-deficient Fc variants of BMS-986012 were produced by recombinant expression in wild-type CHO cells. Antibodies to mouse CD137, mouse PD-1, and isotype control antibodies were generated in-house and produced by recombinant expression in CHO cells or in FUT8–/– CHO cells for nonfucosylated antibodies. All other antibodies were purchased from commercial vendors. Chemotherapy agents, cisplatin (Sigma-Aldrich), and etoposide (Teva) were purchased from commercial vendors.
Antibody-binding assays
ELISA.
Purified FucGM1 was purchased from Matreya, LLC and solubilized in ethanol. Antigen solution was dispensed into PVC microtiter plates and air-dried onto the surface. GM1, the closely related ganglioside precursor, was similarly solubilized and coated on plates as a specificity control. Serial dilutions of BMS-986012 were applied to antigen-coated plates and bound antibody detected with an anti-human IgG secondary antibody conjugated with HRP.
Surface plasmon resonance.
Surface plasmon resonance (SPR) spectroscopy studies of BMS-986012 binding to FucGM1 were conducted with a Biacore T-200 instrument. Approximately 150 RUs of BMS-986012 were captured on a Biacore CM5 chip surface coated with Protein G to a density of 320 RUs. FucGM1-conjugated BSA was used as target antigen and flowed over the chip at concentrations ranging from 12.5 to 100 nmol/L at 25°C and 37°C. GM1-conjugated BSA was used as an antibody specificity control, and isotype control antibody was used to account for nonspecific interactions. The Biacore chip was regenerated with 5 μL of 50 mmol/L sodium hydroxide solution at a flow rate of 100 μL/minute. Association rate constant (kon) and dissociation rate constant (koff) were computed from sensorgrams. FucGM1-conjugated BSA was produced by first treating FucGM1 with ozone to cleave the ceramide and generate an aldehyde. This modified FucGM1 was then conjugated to BSA by reductive amination.
Fc receptor binding.
BMS-986012 binding to human Fcγ receptors and FcRn was assessed by SPR using the Biacore 3000 system. BMS-986012 was immobilized onto the CM5 chip surface at 300 RU. Fcγ receptors and FcRn produced by recombinant expression in CHO cells was flowed over the chip at concentrations of 1.25, 3.125, 6.25, 12.5, and 25 nmol/L. Assessment of FcRn binding was carried out at pH 6.0 in 50 mmol/L 2-(N-morpholino) ethanesulfonic acid (MES) buffer. Association and dissociation times were set at 3 and 2 minutes, respectively. Chip regeneration was performed using 15 μL of 50 mmol/L sodium hydroxide followed by 15 μL of 25 mmol/L hydrochloric acid. Regeneration for FcRn binding was done using 250 mmol/L sodium hydroxide at a flow rate of 100 μL/minute.
Cell-binding assays.
BMS-986012 binding to cells was evaluated by flow cytometry. A panel of SCLC cell line cells were stained with serial dilutions of BMS-986012 from 100 mg/mL in flow cytometry buffer (PBS + 0.02% sodium azide, 2% FBS, and 20% Versene). Bound antibody was detected with a PE-labeled F(abʹ)2 fragment goat antihuman IgG (Jackson ImmunoResearch) and evaluation on a BD Flow cytometer (BD FACSCalibur or FAC-SArray). All flow cytometry data were analyzed using FlowJo software v8 (TreeStar).
Effector functions.
ADCC and ADCP assays were performed as described previously (41, 42). To evaluate CDC, SCLC cell lines (25,000 cells in 50 μL/well) were plated onto a 96-well plate in RPMI1640 supplemented with 0.5% BSA (Sigma-Aldrich). Cells were mixed with 25 μL/well of a 1:3 dilution of human complement (Quidel). Serial dilutions of anti-FucGM1 or isotype control antibodies were added and the plates incubated at 37° C for 4 hours. Cell viability was determined using CellTiter-Glo reagent (Promega). With signal proportional to viable cell number, percent viability was calculated relative to isotype control (100% viable). The resulting values were plotted against antibody concentration and EC50 values were calculated by nonlinear regression analysis, sigmoidal dose response using GraphPad Prism.
IHC
Unfixed cryostat sections at 5 μm were stored at –80°C. For staining, the slides were air-dried for 15 minutes and fixed with 4% PFA for 10 minutes and then acetone for 5 minutes at room temperature. They were washed twice with PBS and endogenous peroxidase activity was blocked by incubation with the Peroxidazed 1 (BioCare Medical) for 5 minutes (Dako). After rinsing with PBS, slides were incubated with Background Punisher (BioCare Medical). Primary FITC-labeled anti-FucGM1 antibody (Bristol-Myers Squibb, clone 19C8P3+) at 5 μg/mL or isotype control (FITC-DT-IgGnf) at 5 μg/mL was applied to tissue sections and incubated for 30 minutes under light-blocked condition. Following three washes with PBS, slides were incubated for 30 minutes with anti-FITC antibody (Invitrogen, Rabbit IgG). After three washes with PBS, slides were incubated for 30 minutes with the peroxidase-conjugated anti-rabbit IgG polymer (MACH2 Rabbit, BioCare Medical). Slides were washed three times with PBS and reacted with 3,3ʹ-diaminobenzidine (DAB) substrate-chromogen solution supplied in Betazoid DAB Chromogen kit (BioCare Medical) for 2 minutes, washed again with deionized water, and counterstained with CAT hematoxylin (BioCare Medical).
In vivo efficacy studies
Tumor cells (5 × 106 cells in 0.1 mL saline containing 50% Matrigel, Becton Dickinson) were implanted subcutaneously into the right flank of 7- to 9-week-old female C.B-17 SCID mice (Taconic) for human cell line xenograft models, or B6129SF1/J mice (The Jackson Laboratory) for the mouse syngeneic model. Patient-derived xenografts (PDX; Crown Bioscience) were engrafted in NOD/SCID mice. Tumor volumes, body weights, and clinical observations were recorded twice per week. Tumor caliper measurements were converted into tumor volumes using the formula: 1/2 × length × width × height. When tumors reached a mean size of 50 to 155 mm3, mice were randomized into treatment cohorts (typically, 8–12/group). Antibodies or SOC drugs were formulated in PBS and administered intraperitoneally twice weekly. Mice were euthanized when tumor volume exceeded 2,000 mm3 or bodyweight loss exceeded 20% of initial study weight. Tumor growth inhibition (TGI) at a specified day was calculated as [1-mean or median tumor volume of treatment group/tumor volume of vehicle control group] × 100. All mouse experimentation was carried out subsequent to proper review and approval by the Bristol-Myers Squibb Animal Care and Use Committee, accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All mice were maintained under specific pathogen-free conditions following the National Research Council’s “Guide for the Care and Use of Laboratory Animals,” 8th edition.
Statistical analysis
The EC50 values were calculated using nonlinear curve-fitting of the dose-response curves using a sigmoidal model (GraphPad Prism). Statistical comparisons of treatment groups were conducted using one-way ANOVA, followed by a Dunn or Tukey post hoc test (GraphPad Prism). Statistical significance was set at P values < 0.05.
Results
FucGM1 is expressed in SCLC tumors
Several studies have reported expression of FucGM1 in SCLC tumors with a prevalence ranging from 67% to over 90% (11–13). FucGM1 expression and prevalence in lung cancer tumors was independently confirmed by IHC using a well-characterized commercial mAb against FucGM1, F12 (Supplementary Table S1A; refs. 11–13). Using ≥10% tumor cell positive as a cutoff, FucGM1 was expressed in half (12/24) of the SCLC tumors in our panel. These data combined with data from previously published reports on FucGM1 expression in SCLC (106 total samples) showed 68% (76/106) as FucGM1+ (Supplementary Table S1B). FucGM1 expression was also observed in 20% (21/103) of NSCLC tumors tested (Supplementary Table S1A). In addition, FucGM1 expression levels on human SCLC tumor cell lines were compared with BMS-986012-mediated growth inhibition levels observed in xenograft studies (Supplementary Table S1C); high target antigen expression levels were generally, but not always, associated with high antibody efficacy.
BMS-986012 binds specifically to FucGM1 and shows enhanced binding to human FcγRIIIa
BMS-986012 was expressed in a FUT8–/– CHO cell line to produce a nonfucosylated IgG1 antibody. BMS-986012 binding specificity was evaluated by ELISA (Fig. 1A). Dose-dependent binding to purified FucGM1 was observed with BMS-986012, but no significant binding to the closely related precursor molecule, GM1, was detected with the antibody. BMS-986012 binding was next evaluated against a panel of SCLC cell lines by flow cytometry (Fig. 1B). Specific, dose-dependent binding of BMS-986012 to tumor cell lines was observed with an EC50 range from 6 to 132 nmol/L for cells representing high-level (H740) and intermediate-level (DMS79) FucGM1 expression, respectively, as assessed by maximum geometric mean fluorescence intensity (GMFI). NCI-H128 and NCI-H209 cells showed the lowest maximum GMFI. FucGM1 expression was confirmed on these cell lines grown as tumor xenografts in SCID mice by IHC (Fig. 1C). The rank order and relative expression level of FucGM1 by IHC was consistent with that observed by flow cytometry.
Figure 1.
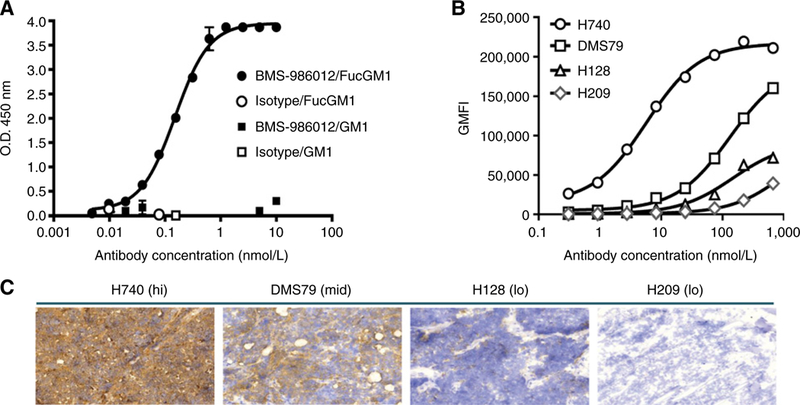
BMS-986012 binding to FucGM1 antigen. A, BMS-986012 binding to purified FucGM1 antigen by ELISA. BMS-986012 binds specifically to FucGM1 ( ) with an EC50 of 0.15 nmol/L, but not to the closely related precursor molecule GM1 (
) with an EC50 of 0.15 nmol/L, but not to the closely related precursor molecule GM1 ( ). No binding to FucGM1 (
). No binding to FucGM1 ( ) or GM1 (
) or GM1 ( ) was observed with isotype control antibody. B, BMS-986012 binding to FucGM1+ cell lines by flow cytometry. GMFI is plotted against antibody concentration. C, BMS-986012 binding to tumor Hs by IHC (×20 magnification). IHC was performed with BMS-986012 on tumor xenografts established in SCID mice with the same cell lines described in B.
) was observed with isotype control antibody. B, BMS-986012 binding to FucGM1+ cell lines by flow cytometry. GMFI is plotted against antibody concentration. C, BMS-986012 binding to tumor Hs by IHC (×20 magnification). IHC was performed with BMS-986012 on tumor xenografts established in SCID mice with the same cell lines described in B.
BMS-986012 binding to FucGM1 was further characterized by SPR. In aqueous solution, the glycolipid nature of FucGM1 results in micelle formation, making SPR analysis of specific binding a challenge; therefore, a FucGM1-BSA conjugate was used as the antigen. Equilibrium dissociation constants (KD) of 8.8 and 9.3 nmol/L were determined for BMS-986012 binding to FucGM1-BSA conjugate at 25°C and 37°C, respectively (Supplementary Table S2). No specific binding of BMS-986012 was observed using a structurally related GM1-BSA conjugate confirming specificity of the antibody for FucGM1.
SPR analysis was used to characterize binding of BMS-986012 to a panel of chip-immobilized human Fc receptors (Supplementary Table S3). AKD of 21 nmol/L for BMS-986012 binding to the FcγRIIIa F158 allele is in close agreement with KD values reported for other nonfucosylated antibodies and represents an approximate 100-fold increase in affinity over fucosylated antibody (43, 44). As expected, BMS-986012 binds less well to the FcγRIIIa V158 allele with a KD of 389 nmol/L. This value is slightly higher than the values reported for other nonfucosylated antibodies (43, 44).
BMS-986012 has enhanced ADCC effector function
Enhanced ADCC activity of nonfucosylated antibodies is well documented (29, 30, 35, 37, 38, 41, 42, 45, 46). Factors such as antigen density, epitope distance from the cell membrane, and binding affinity also contribute to antibody effector function (47, 48). To determine the effect of afucosylation on BMS-986012 FcγRIIIa binding and on antibody-mediated effector functions, FcγRIIIa-CHO transfectants were also analyzed by flow cytometry (Fig. 2A). Nonfucosylated BMS-986012 bound to FcγRIIIa-CHO transfectants with an EC50 of 3.5 nmol/L, a 39.4-fold increase in binding potency over the antibody in conventional fucosylated IgG1 format, 7E4 (EC50 = 137.8 nmol/L), and consistent with the enhanced binding observed by SPR.
Figure 2.
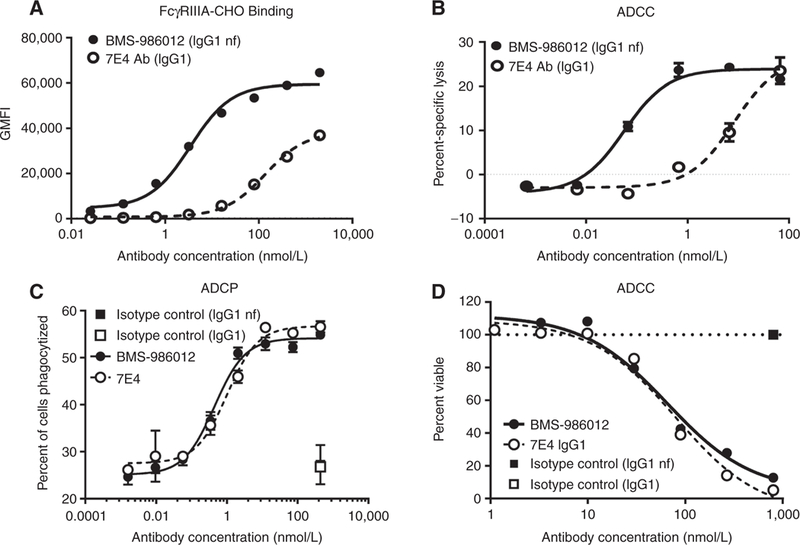
Comparison of FcγRIIIa binding and antibody-mediated effector functions of BMS-986012 and fucosylated parental antibody 7E4. Data are representative ofat least three independent experiments. A, FcγRIIIa receptor binding. BMS-986012demonstrates enhanced binding to CHOcells stablyexpressing human FcγRIIIa with binding EC5qs of 3.5 and 138 nmol/L for BMS-986012 () and 7E4 (), respectively. B, Enhanced BMS-986012-mediated FcγRIIIa-dependent ADCC activity in vitro. NK effector cells and DMS79 target cells were incubated with either BMS-986012 () or 7E4 (). Median percent-specific lysis ± SD is plotted against antibody concentration. C, BMS-986012-mediated ADCP activity in vitro. Macrophage effector cells and DMS79 target cells were incubated with BMS-986012 (), 7E4 (), or isotype control antibodies IgG1nf () and IgG1 (). Macrophage-phagocytosed tumor cells were identified by flow cytometry as PKH26+ CD14+ double-positive events. The median percent of double-positive events ± SD is plotted against antibody concentration. D, BMS-986012-mediated CDC activity in vitro. DMS79 target cells were incubated with BMS-986012 (), 7E4 (), or isotype control antibodies IgG1nf () and IgG1 () plus human complement. Cell viability was determined using CellTiter-Glo, and the percentage of viable cells is plotted against Ab concentration.
FcγRIIIa is the primary receptor for IgG expressed by human NK cells, and therefore, BMS-986012 and parental wild-type IgG1 (7E4) were evaluated for NK cell-mediated ADCC activity against the FucGM1+ SCLC cell line, DMS79 (Fig. 2B). BMS-986012-mediated ADCC (EC50 = 0.06 nmol/L) was enhanced 138-fold compared with the activity observed with fucosylated 7E4 (EC50 = 8 nmol/L).
Macrophages can also participate in direct tumor cell killing by Fc receptor-mediated mechanisms including ADCP. Fcγ receptor expression on macrophages is more complex than on NK cells. In addition to FcγRIIIa, macrophages express FcγRI and FcγRIIa. BMS-986012 was tested for ADCP activity against DMS79 tumor cells (Fig. 2C). Macrophages effectively phagocytosed DMS79 cells in the presence of BMS-986012 (EC50 = 0.5 nmol/L). No difference in macrophage phagocytosis of DMS79 cells was observed between BMS-986012 and 7E4 (EC50 = 1.0 nmol/L). The lack of enhanced ADCP with nonfucosylated antibody is likely the result of differences in expression of specific Fcγ receptors between macrophages and NK cells, as well as the minimal effect that afucosylation has on antibody binding affinity to FcγRI and FcγRIIa (43, 44). Nevertheless, BMS-986012 is capable of mediating ADCP of FucGM1+ cells.
Finally, BMS-986012 was evaluated for CDC activity (Fig. 2D). BMS-986012 elicited dose-dependent CDC against DMS79 cells (EC50 = 59 nmol/L). Similar to ADCP data, no difference in CDC activity was observed between BMS-986012 and 7E4 (EC50 = 67 nmol/L). This is consistent with studies reporting that core fucose has no effect on antibody C1q binding or CDC activity (35, 45).
BMS-986012 demonstrates antitumor activity in human SCLC tumor xenograft and PDX models
Pharmacokinetic analysis of BMS-986012 following single-dose administration to mice was conducted in support of in vivo efficacy studies. As a biochemically defined ganglioside, FucGM1 antigen is identical across species; thus, BMS-986012 recognized FucGM1 in mice. Methods and results are provided in Supplementary Table S4.
Efficacy of BMS-986012 was evaluated in the DMS79 model at antibody concentrations ranging from 0.01 to 3 mg/kg. BMS-986012 monotherapy induced dose-dependent TGI (Supplementary Fig. S1A). Statistically significant TGI was observed at 0.1 mg/kg (78% TGI, day 36, P < 0.05 vs. vehicle) with maximal tumor stasis occurring at doses above 0.3 mg/kg (>95%–98%TGI, day 36, P < 0.001). As expected, the parental 7E4 antibody and nonfucosylated BMS-986012 were similarly efficacious in vivo (Supplementary Fig. S1B), as no significant increase in binding affinity to mouse FcγRs is observed with nonfucosylated human IgG1 (37). No antitumor efficacy was observed with the nonfucosylated IgG1 isotype control antibody at 3 mg/kg. BMS-986012 was also efficacious in a number of other xenograft models (Supplementary Fig. S2). All doses of BMS-986012 were well tolerated in these studies.
Unlike the uniform expression of FucGM1 on the DMS79 tumor cell line, expression of FucGM1 on established DMS79 xenografts was more heterogeneous (Supplementary Fig. S3) and similar to the range of FucGM1 expression observed on primary SCLC tumors. To support and extend the tumor xenograft efficacy results and to investigate the effect of FucGM1 expression level and heterogeneity on antitumor activity, BMS-986012 was evaluated in a small set of limited passage SCLC PDX models (Fig. 3). H-scores, an IHC assessment of antigen expression capturing both staining intensity and percentage of cells stained, were used as a semiquantitative measure of FucGM1 expression. In the models tested, TGI roughly tracks with target expression. BMS-986012 treatment of model LU5202 (H-score = 290) resulted in complete tumor regression (Fig. 3, right), while TGI of model LU5170 (H-score = 265) was less robust (75% TGI at 22 days after initial dosing; Fig. 3, middle). No TGI was observed with model LU5249 (H-score = 65; Fig. 3, left). Further studies are needed, however, to confirm these findings and more precisely characterize the relationship between the level of FucGM1 expression and efficacy.
Figure 3.
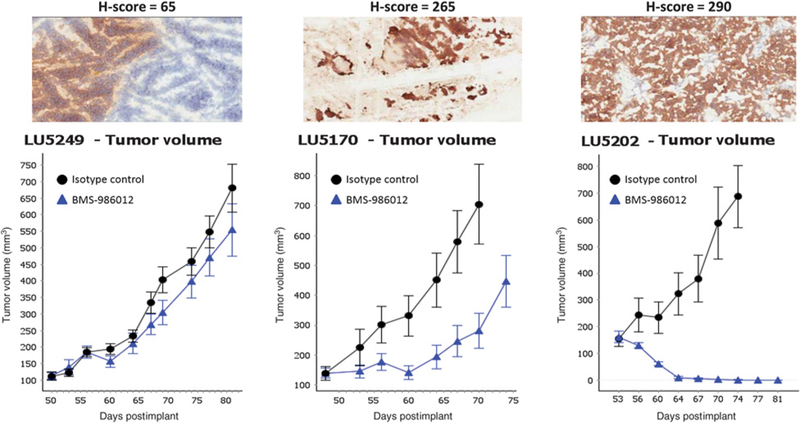
Effects of BMS-986012 on the growth of SCLC PDX models. Three SCLC PDX models with different levels of FucGM1 expression were selected for study. Mice were dosed with 10 mg/kg BMS-986012 or isotype control three times per week for 3 weeks.
BMS-986012 efficacy in SCLC xenograft models is enhanced by chemotherapeutics
Chemotherapy is an important component of SCLC clinical management. Efficacy of BMS-986012 was evaluated in the DMS79 xenograft model in combination with SOC chemotherapy agents, etoposide, or cisplatin (Fig. 4). For the BMS-986012/ etoposide combination, mice were dosed with either vehicle (PBS), isotype control antibody (3 mg/kg), or BMS-986012 (3 mg/kg), alone or in combination with etoposide (15 mg/kg). BMS-986012 showed good TGI, while no efficacy was observed with isotype control antibody (Fig. 4A). At day 35 after tumor implantation, the 3 mg/kg BMS-986012 group had a mean TGI of 61% (P < 0.05) compared with the isotype control group. The etoposide/isotype control antibody combination showed mean TGI of 51% (P < 0.05). Compared with single-treatment arms, the BMS-986012/etoposide combination had dramatically better efficacy, with a mean TGI of 94% (P < 0.05).
Figure 4.
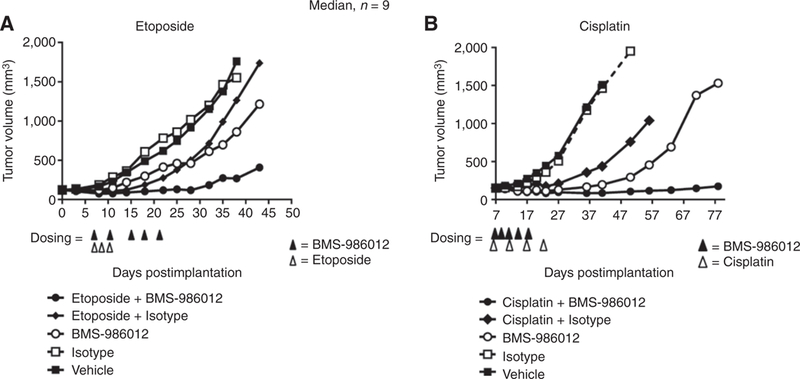
Anti-FucGM1 antibody combined with chemotherapy results in greater tumor growth inhibition. BMS-986012 and SOC chemotherapy were evaluated in the DMS79 xenograft tumor model. A, Mice were dosed intraperitoneally on days 7,11,15,18, and 21 after tumor implantation, with either vehicle (PBS), 3 mg/kg of isotype control antibody, or 3 mg/kg BMS-986012. Etoposide (15 mg/kg) was administered intraperitoneally on days 7,9, and 11. At day 35 after tumor implantation, BMS-986012 single-agent cohort had a mean TGI of 61% (P < 0.05) compared with isotype control group. The etoposide/isotype control antibody combination showed mean TGI of 51% (P < 0.05). Compared with single treatment arms etoposide/BMS-986012 combination showed better efficacy with a mean TGI of 94% (P < 0.05). B, Cisplatin (3 mg/kg) was dosed intraperitoneally on day 7,14,21, and 28 days after tumor implantation in combination with either 3 mg/kg isotype control antibody or 0.3 mg/kg of BMS-986012 on days 7,10,13,17, and 21 after tumor implantation. On day 36 after tumor implantation, the BMS-986012 single-agent cohort had a mean TGI of 84% (P < 0.05) compared with the isotype control group. TGI in the cisplatin/isotype control cohort was 64% (P < 0.05). Combining BMS-986012 with cisplatin significantly improved efficacy with mean TGI of 91% (P < 0.05). Data are representative of two independent experiments.
A similar study examined BMS-986012 combined with cisplatin (3 mg/kg; Fig. 4B). On day 36 after tumor implantation, the BMS-986012 0.3 mg/kg group had a mean TGI of 84% (P < 0.05) compared with the isotype control group. A mean TGI of 64% (P < 0.05) was observed for the cisplatin/isotype control cohort. As seen with the etoposide, combining BMS-986012 with cisplatin significantly improved efficacy, with a mean TGI of 91% (P < 0.05).
Both combinations were well tolerated with no increased body weight loss compared with the chemotherapy and isotype control antibody groups.
BMS-986012 efficacy in SCLC xenograft model is enhanced by anti-CD137 agonist antibody
NK cells are well-recognized mediators of antitumor ADCC (49). Cross-linking of FcγRIIIa on NKcells by antibody-opsonized tumor cells triggers NK-cell activation, including upregulation of CD137. Anti-CD137 agonist antibody has been shown to synergize with tumor antigen-specific antibodies in vivo (50–52). To determine whether BMS-986012 can synergize with an anti-CD137 agonist antibody, NK cells from healthy donors were cocultured with FucGM1-expressing DMS79 cells and antibody (Fig. 5A, left). After 24 hours, a dose-dependent increase in the number of CD137+ NK cells was observed with 36.5% of NK cells expressing CD137 at 10 mg/mL BMS-986012, the highest concentration tested. In contrast, cocultures with isotype control antibody added at the same concentration showed that only 4.5% of NK cells were CD137+. Upregulation of CD137 was also coincident with other markers of NK-cell activation, including cell surface expression of CD107a, a marker of cellular degranulation (Fig. 5A, middle), and a dose-dependent release of IFNγ into the coculture supernatant (Fig. 5A, right).
Figure 5.
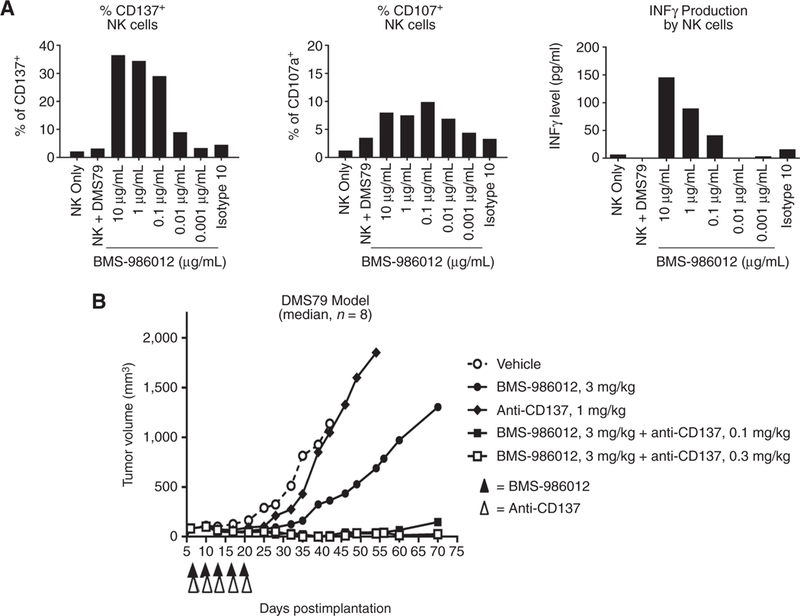
BMS-986012 efficacy is enhanced by anti-CD137 antibody. A, BMS-986012 induces NK-cell activation in the presence of DMS79 target cells. Negatively selected human NK cells and DMS79 target cells were cocultured 1:1 for 24 hours in RPMI1640 plus 10% FBS without IL2 with serially diluted BMS-986012 or isotype control antibody. Expression of CD137 (left) and CD107a (middle) was determined by flow cytometry. IFNγ secreted into culture supernatants was determined by ELISA (right). Data are representative of three independent experiments. B, Mice were dosed every 3 to 4 days with 3 mg/kg BMS-986012 beginning on day 7 postimplantation or 1 mg/kg anti-mouse CD137 beginning day 8 posttransplantation to assess single-agent efficacy. To evaluate combined activity, mice were dosed with 3 mg/kg BMS-986012 every 3 to 4 days intraperitoneally beginning on day 7 after tumor implantation and either 0.1 or 0.3 mg/kg anti-mouse CD137 intraperitoneally 1 day after each dose of BMS-986012 beginning on day 8 post implantation. Data are representative of two independent experiments.
In vivo efficacy of BMS-986012 plus anti-mouse CD137 agonist antibody was evaluated in the DMS79 tumor xenograft model (Fig. 5B). Considering the expected time course for CD137 upregulation on NK cells following FcγRllla cross-linking, anti-CD137 antibody was administered 24 hours after BMS-986012. Anti-CD137 antibody treatment alone (1 mg/kg) demonstrated minimal efficacy with 53% TGI on day 35 (P = n.s.). Treatment with BMS-986012 (3 mg/kg), however, resulted in an 89% (P < 0.01) inhibition of tumor growth with 1/8 mice tumor free. When combined, BMS-986012 and lower doses of anti-CD137 antibody demonstrated greater efficacy than either antibody administered as a single agent (antibody combination TGI > 100% vs. BMS-986012 only TGI = 89%, P < 0.05). Significant tumor regressions and curative responses (CR) were observed in the combined treatment arms at both anti-CD137 antibody dose levels tested (3/8 with 0.1 mg/kg and 5/8 with 0.3 mg/kg).
BMS-986012 efficacy in an SCLC syngeneic model is enhanced by immunomodulating antibodies
To evaluate efficacy of BMS-986012 in the context of a fully competent immune system, as well as in combination with immunomodulating antibodies, cell lines established from a genetically engineered mouse model of SCLC (39) were screened for FucGM1 expression. By flow cytometry, KP3 cells were found to have homogeneous expression of FucGM1 (Fig. 6A. top) at a level approximately 5-fold lower than DMS79 cells. Expression of FucGM1 on KP3 tumors grown in immunocompetent mice (B6129S1/J F1) was evaluated by IHC (Fig. 6A, bottom). Compared with flow cytometry, FucGM1 expression in tumors by IHC was more heterogeneous but similar to that observed for DMS79 xenograft tumors (Fig. 1C). Further characterization of the KP3 tumors by IHC showed limited tumor-infiltrating lymphocytes with sparse numbers of CD45+ cells located primarily around the tumor periphery (Supplementary Fig. S4), a finding similar to that reported for another genetically modified mouse SCLC model (53).
Figure 6.
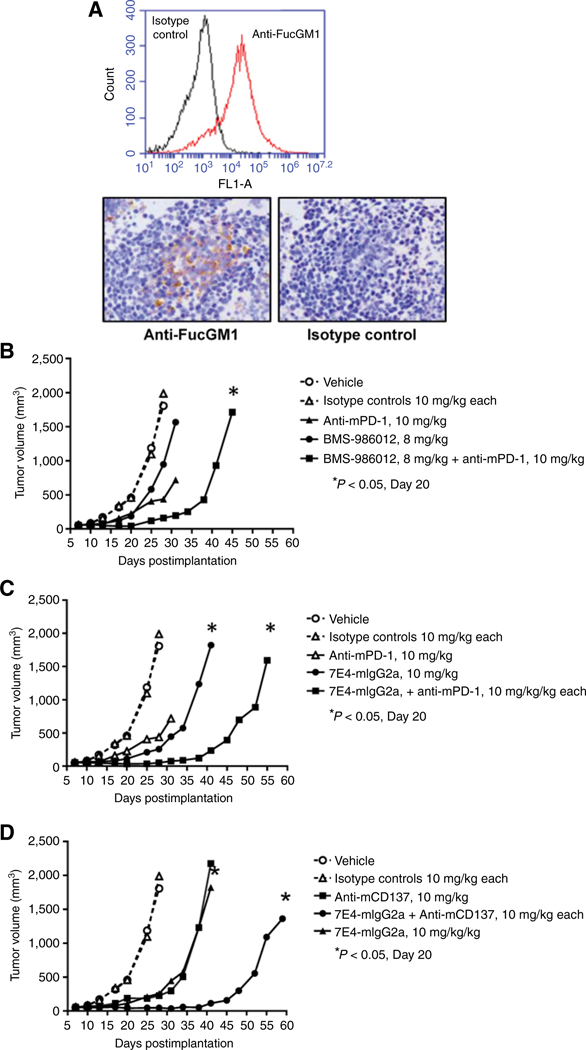
BMS-986012 efficacy in a syngeneic model. A, Top, in vitro cultured KP3 cells were stained with anti-FucGM1 (red line) or isotype control (black line) antibody and assessed by flow cytometry. Bottom, tumors from B6129S1/J F1 mice implanted with KP3 cells were evaluated for FucGM1 expression by IHC (×20 magnification). Left, F12 anti-FucGM1 antibody; right, isotype control antibody. B, Mice were dosed intraperitoneally on days 7,11,14,17, 20, and 25 after tumor implantation with either vehicle (PBS), isotype control antibody, or BMS-986012 ± anti-mPD-1 antibody intraperitoneally on the same schedule. TGI observed on day 20 with BMS-986012 and anti-mPD-1 was not significant. Significant TGI was observed on day 20 with combined BMS-986012 and anti-mPD-1 antibodies. C, Mice were dosed intraperitoneally on days 7, 11, 14,17, 20, and 25 after tumor implantation with either vehicle (PBS), isotype control antibody, or anti-FucGM1 7E4-mIgG2a chimeric antibody ± anti-mPD-1 antibody on the same schedule. Significant TGI was observed on day 20 with 7E4-mIgG2a and with combined 7E4-mIgG2a plus anti-mPD-1 antibody. D, Mice were dosed intraperitoneally on days 7, 11, 14, 17, 20, and 25 after tumor implantationwith either vehicle (PBS), isotype control antibody, or anti-FucGM1 7E4-mIgG2a chimeric antibody ± anti-mCD137 antibody intraperitoneally on days 7,12,14,18,21 and 26 after tumor implantation. Significant TGI was observed on day 20 with 7E4-mIgG2a and with combined 7E4-mIgG2a plus anti- mCD137 antibody.
Compared with the efficacy observed in the DMS79 model, BMS-986012 was less effective as a single agent in the KP3 model with TGI of 47% at day 20 (Fig. 6B). Similar TGI (53%) was observed with anti–PD-1 alone. TGI was improved significantly when the two antibodies were combined (TGI 89% vs. isotype, P < 0.05).
To better model BMS-986012 Fc receptor–dependent effector functions in the mouse system, the antibody was reengineered as a mouse IgG2a Fc chimeric, which more closely recapitulates species-specific Fcγ receptor interactions and effector functions in the relevant immune cell types in mice. When tested as a single agent, efficacy of the IgG2a chimeric anti-FucGM1 antibody dosed at 10 mg/kg was improved compared with BMS-986012 (47% and 81% TGI, respectively, at day 20, P < 0.05; Fig. 6B). The improved efficacy was also observed when the antibody was combined with anti–PD-1 (97% TGI at day 20, P < 0.05). Finally, efficacy in combination with anti-CD137 was reexamined using the mouse IgG2a Fc chimeric antibody (Fig. 6D). Significant growth inhibition was observed with 7E4-IgG2a chimeric combined with anti-mCD137 (100% TGI at day 20, P < 0.05).
Discussion
Clinical management of SCLC remains a challenge. Existing treatments do not eradicate residual disease, and essentially, all patients with extensive disease quickly relapse. With the clear need for new therapeutic approaches, there has been considerable interest in targeting tumor-associated gangliosides. Several gangliosides have been identified as potential immunotherapy targets for SCLC including GD2, GD3, and FucGM1. Therapeutic antibodies against gangliosides GD2 and GD3 have been pursued in multiple indications, with the antibody to GD2 (dinutuximab) approved for pediatric high-risk glioblastoma (50).
Of the gangliosides expressed in SCLC, FucGM1 shows the most restricted expression on normal tissues. Targeting FucGM1 has been approached utilizing a FucGM1-KLH vaccine conjugate to stimulate an immune response against FucGM1+ SCLC tumors (22, 23). Although anti-FucGM1 antibody responses were observed in a number of subjects, serum titers were generally low and comprised mainly of low-affinity IgM antibodies. Moreover, such low titer and low-affinity responses are typical of carbohydrate antigens, which are poorly immunogenic and predominately T-cell independent. Vaccine conjugates produced with synthetic FucGM1 rather than purified from natural sources were particularly ineffective at generating antibody responses that recognized FucGM1+ tumor cells (23). BMS-986012 addresses many of the issues associated with FucGM1 vaccine approach, including antibody-binding affinity and effector functions. The data presented here demonstrate that BMS-986012 specifically binds to FucGM1 with high affinity, and as an IgG1 mAb, the need for T-cell–dependent class switching is avoided. Indeed, by producing the antibody in a nonfucosylated format, antibody effector function has been enhanced.
BMS-986012 efficacy was profiled in a number of SCLC models. Expression of FucGM1 on DMS79 SCLC cells established as xenografts was heterogeneous, similar to the expression observed in clinical disease. In this model, BMS-986012 demonstrated pronounced activity with tumor stasis occurring at low doses. Evaluation in a number of xenograft models, as well as in a limited set of SCLC PDX models suggests efficacy of BMS-986012 is roughly correlated with the level of target expression (Supplementary Table S1C). However, complement receptor expression (CD46, CD55, CD59) and antiapoptotic pathways can mitigate the response. Because combination regimens with SOC chemotherapy comprise an important part of SCLC treatment, we explored chemotherapeutic combinations with BMS-986012. In the DMS79 model, combination treatment with BMS-986012 plus cisplatin or etoposide resulted in greater efficacy than with either treatment alone, and in the absence of any observable adverse effects.
Several studies have demonstrated that anti-CD137 agonist antibodies can synergize with antitumor antibodies, such as rituximab, trastuzumab, and cetuximab to enhance NK-cell activity (51, 52, 54). It has also been suggested that NK-cell activation through anti-CD137 would enhance ADCC (55). Thus, we investigated BMS-986012 in combination with anti-CD137 antibody and showed that human NK cells upregulate CD137 after exposure to FucGM1 mAb and tumor cells in vitro, enhancing mAb-dependent cytotoxicity against tumor cells. On the basis of these studies, sequential administration of FucGM1 and anti-CD137 mAb was preferred over concurrent administration. Similar to published reports on the combination of anti-CD137 with trastuzumab, rituximab, or cetuximab, BMS-986012 with anti- CD137 was synergistic, suggesting that these prior observations could be expanded to antibodies targeting cell surface gangliosides (51, 52, 54). Although NK cells are the most likely target of the anti-CD137 agonist antibody in vivo, particularly in the SCID xenograft models, CD137 is expressed by other immune cells as well as in some tumors. Further studies are needed to confirm NK cells as the primary mediator of anti-CD137 agonist antibodies in vivo.
The enhanced effector functions of the nonfucosylated IgG1 described thus far act primarily through NK and myeloid cells that are present in SCID mice. To evaluate T-cell–dependent combinations with checkpoint inhibitor anti–PD-1 in mice with fully intact immune systems, we used a syngeneic mouse tumor cell line derived from a genetically engineered SCLC mouse model. BMS-986012 combined with anti–PD-1 showed improved efficacy over either antibody alone. Indeed, many clinical studies combining anti–PD-1 with antibodies directed to other antigens are underway.
The functional role of FucGM1 in mammalian tissues is unclear, and the potential liabilities of targeting FucGM1 in humans remain unknown. FucGM1 is known to associate with membrane lipids and cluster into lipid rafts with scaffolding proteins that enable cross-talk of intracellular proteins involved in neuronal signaling. Although FucGM1 is developmentally regulated, its expression in spinal ganglia, along with sparse, occasional distribution in other tissues, has been reported and needs further assessment (11–16). Although tissue or cell-specific expression may differ, the biochemical nature of FucGM1 as an antigen is the same across species, and we have shown that BMS-986012 binds to FucGM1 in mouse tissues. Encouragingly, we did not observe adverse effects (i.e., bodyweight loss or atypical findings in tissue necropsies) in our mouse tumor model studies. An additional inference, based on the fairly long mouse plasma half-life (T1/2) of BMS-986012, is that tissue-mediated drug disposition may not play a significant role in the circulating clearance of the antibody.
In conclusion, nonfucosylated anti-FucGM1 mAb BMS-986012 shows robust tumor growth inhibition as a monotherapy, and the addition of SOC chemotherapy agents or immunomodulatory antibody further inhibits tumor growth. The preclinical data described here provide support for the clinical evaluation of BMS-986012.
Supplementary Material
Translational Relevance.
BMS-986012 is a first-in-class fully human mAb to fucosyl-GM1 that maybe an effective therapeutic antibody for treating fucosyl-GM1–positive tumors. With the capacity to mediate enhanced ADCC, CDC, and ADCP, monotherapy with BMS-986012 shows significant efficacy in small cell lung cancer (SCLC) xenograft, PDX, and syngeneic tumor models. Combining BMS-986012 with standard-of-care chemotherapy agent cisplatin or etoposide elicits additive therapeutic responses. BMS-986012 combined with anti-CD137 or anti–PD-1 immunomodulatory antibodies results in synergistic efficacy. Intervention with novel immunotherapeutics such as BMS-986012 may succeed in overcoming drug resistance typically seen in SCLC. Collectively, the data reported in this article provide preclinical support for evaluation of BMS-986012 as a therapeutic agent for the treatment of antigen-positive SCLC either alone or in combination with chemotherapeutic or immunomodulatory agents.
Acknowledgments
The authors thank Xingjie Chen, Yu Ching Chang, Yichong Wang, and Remie Mandawe for their contributions. The authors are grateful to Olufemi Adelakun and Linlin Luo for their IHC analyses of human lung cancer tissues. The authors also thank Catherine A. Bolger for her help in preparing this manuscript.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
P. Ponath is an employee of and has ownership interests (including patents) at Bristol-Myers Squibb. D. Menezes has ownership interests (including patents) at Bristol-Myers Squibb, Celgene, Novartis, and Roche, and a provisional published patent disclosure related to the therapeutic agent described in the publication. M. Oyasu has ownership interests (including patents) at Bristol-Myers Squibb. H. LeBlanc has ownership interests (including patents) at Bristol-Myers Squibb. V.S. Rangan has ownership interests at Bristol-Myers Squibb. J. Sage reports receiving commercial research support from AbbVie. P.M. Cardarelli has ownership interests at Bristol-Myers Squibb. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008;359:1367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011;61:212–36. [DOI] [PubMed] [Google Scholar]
- 3.SabariJK LokBH, Laird JH Poirier JT, RudinCM. Unravelling the biology of SCLC: implications for therapy. Nat Rev Clin Oncol 2017;14:549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koinis F, Kotsakis A, Georgoulias V. Small cell lung cancer (SCLC): no treatment advances in recent years. Transl Lung Cancer Res 2016;5: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan BA, Coward JI. Chemotherapy advances in small-cell lung cancer. J Thorac Dis 2013;Suppl 5:S565–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandler AB. Chemotherapy for small cell lung cancer. Semin Oncol 2003;30:9–25. [DOI] [PubMed] [Google Scholar]
- 7.Ettinger DS. New drugs for chemotherapy-naive patients with extensive-disease small cell lung cancer. Semin Oncol 2001;28(Suppl. 4):27–9. [PubMed] [Google Scholar]
- 8.Lally BE, Urbanic JJ, Blackstock AW, Miller AA, Perry MC. Small cell lung cancer: have we made any progress over the last 25 years? Oncologist 2007;12:1096–104. [DOI] [PubMed] [Google Scholar]
- 9.Hurwitz JL, McCoy F, Scullin P, Fennell DA. New advances in the second-line treatment of small cell lung cancer. Oncologist 2009;14:986–94. [DOI] [PubMed] [Google Scholar]
- 10.Sonnino S, Mauri L, Chigorno V, Prinetti A. Gangliosides as components of lipid membrane domains. Glycobiology 2007;17:1R–13R. [DOI] [PubMed] [Google Scholar]
- 11.Brezicka FT, Olling S, Nilsson O, Bergh J, Holmgren J, Sorenson S, et al. Immunohistological detection of fucosyl-GM1 ganglioside in human lung cancer and normal tissues with monoclonal antibodies. Cancer Res 1989;49:1300–5. [PubMed] [Google Scholar]
- 12.Brezicka FT, Olling S, Bergman B, Berggren H, Engstroom CP, Holmgren J, et al. Immunohistochemical detection of two small cell lung carcinoma- associated antigens defined by MAbs F12 and 123C3 in bronchoscopy biopsy tissues. APMIS 1991;99:797–802. [DOI] [PubMed] [Google Scholar]
- 13.Brezicka T, Bergman B, Olling S, Fredman P. Reactivity of monoclonal antibodies with ganglioside antigens in human small cell lung cancer tissues. Lung Cancer 2000;28:29–36. [DOI] [PubMed] [Google Scholar]
- 14.Fredman P, Brezicka T, Holmgren J, Lindholm L, Nilsson O, Svennerholm L. Binding specificity of monoclonal antibodies to ganglioside, Fuc-GM1. Biochim Biophys Acta 1986;875:316–23. [DOI] [PubMed] [Google Scholar]
- 15.Zhang S, Cordon-Cardo C, Zhang HS, Reuter VE, Adluri S, Hamilton WB, et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int J Cancer 1997;73: 42–9. [DOI] [PubMed] [Google Scholar]
- 16.Nilsson O, Brezicka FT, Holmgren J, Sörenson S, Svennerholm L, Yngvason F, et al. Detection of a ganglioside antigen associated with small cell lung carcinoma using monoclonal antibodies directed against fucosyl-GM1. Cancer Res 1986;46:1403–7. [PubMed] [Google Scholar]
- 17.Houghton AN, Mintzer D, Cordon-Cardo C, Welt S, Fliegel B, Vadhan S, et al. Mouse monoclonal IgG3 antibody detecting GD3 ganglioside: a phase I trial in patients with malignant melanoma. Proc Natl Acad Sci USA 1985;82:1242–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irie RF, Morton DL. Regression of cutaneous metastatic melanoma by intralesional injection with human monoclonal antibody to ganglioside GD2. Proc Natl Acad Sci USA 1986;83:8694–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brezicka T, Einbeigi Z. Supra-additive cytotoxic effects of a combination of cytostatic drugs and antibody-induced complement activation on tumor cells in vitro. Tumour Biol 2001;22:97–103. [DOI] [PubMed] [Google Scholar]
- 20.Brezicka FT, Holmgren J, Kalies I, Lindholm L. Tumor-cell killing by MAbs against fucosyl GM1, a ganglioside antigen associated with small-cell lung carcinoma. Int J Cancer 1991;49:911–8. [DOI] [PubMed] [Google Scholar]
- 21.Brezicka T, Einbeigi Z, Bergman B. Functional assessment in vitro of human- complement-dependent antibody-induced cytotoxicity of neoplastic cells. Cancer Immunol Immunother 2000;49:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dickler MN, Ragupathi G, Liu NX, Musselli C, Martino DJ, Miller VA, et al. Immunogenicity of a fucosyl-GM1 keyhole limpet hemocyanin conjugate vaccine in patients with small cell lung cancer. Clin Cancer Res 1999;5:2773–9. [PubMed] [Google Scholar]
- 23.Krug LM, Ragupathi R, Hood C, Kris MG, Miller VA, Allen JR, et al. Vaccination of patients with small-cell lung cancer with synthetic Fucosyl-GM1 conjugated to keyhole limpet hemocyanin. Clin Cancer Res 2004;10:6094–100. [DOI] [PubMed] [Google Scholar]
- 24.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol 2006;6:343–57. [DOI] [PubMed] [Google Scholar]
- 25.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med 2000; 6:443–6. [DOI] [PubMed] [Google Scholar]
- 26.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRlla gene. Blood 2002;99:754–8. [DOI] [PubMed] [Google Scholar]
- 27.Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs 2011;3:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang J, Richardson J, Du Z, Zhang Z. Effect of Fc-Glycan structure on the conformational stability of IgG revealed by hydrogen/deuterium exchange and limited proteolysis. Biochemistry 2016;55:860–8. [DOI] [PubMed] [Google Scholar]
- 29.Scallon B, McCarthy S, Radewonuk J, Cai A, Naso M, Raju TS, et al. Quantitative in vivo comparisons of the Fc gamma receptor-dependent agonist activities of different fucosylation variants of an immunoglobulin G antibody. Int Immunopharmacol 2007;7:761–72. [DOI] [PubMed] [Google Scholar]
- 30.Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res 2007;67:8882–90. [DOI] [PubMed] [Google Scholar]
- 31.Presta LG, Shields RL, Namenuk AK, Hong K, Meng YG. Engineering therapeutic antibodies for improved function. Biochem Soc Trans 2002; 30:487–90. [DOI] [PubMed] [Google Scholar]
- 32.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem 2002;277:26733–40. [DOI] [PubMed] [Google Scholar]
- 33.Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose orbisecting N-acetylglucosamineofhuman IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem 2003;278:3466–73. [DOI] [PubMed] [Google Scholar]
- 34.Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody dependent cellular cytotoxic activity. Nat Biotechnol 1999;17:176–80. [DOI] [PubMed] [Google Scholar]
- 35.Niwa R, Natsume A, Uehara A, Wakitani M, Iida S, Uchida K, et al. IgG subclass-independent improvement of antibody-dependent cellular cytotoxicity by fucose removal from Asn297-linked oligosaccharides. J Immunol Methods 2005;306:151–60. [DOI] [PubMed] [Google Scholar]
- 36.Ferrara C, Stuart F, Sondermann P, Brünker P, Umana P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to nonfucosylated IgG glycoforms. J Biol Chem 2006;281:5032–6. [DOI] [PubMed] [Google Scholar]
- 37.Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, Theriault J, et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2- amplified breast cancer. Cancer Res 2010;70:4481–9. [DOI] [PubMed] [Google Scholar]
- 38.Gerdes CA, Nicolini VG, Heiter S, van Puijenbroek E, Lang S, Roemmele M, et al. GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin CancerRes 2013;19:1126–38. [DOI] [PubMed] [Google Scholar]
- 39.Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med 2011;17:1504–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Capello S, Liu NX, Musselli C, Brezicka FT, Livingston PO, Ragupathi G. Immunization of mice with fucosyl-GM1 conjugated with keyhole limpet hemocyanin results in antibodies against human small-cell lung cancer cells. Cancer Immunol Tmmunother 1999;48:483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cardarelli PM, Moldovan-Loomis MC, Preston B, Black A, Passmore D, Chen TH, et al. in vitro and in vivo characterization of MDX-1401 fortherapy of malignant lymphoma. Clin Cancer Res 2009;15:3376–83. [DOI] [PubMed] [Google Scholar]
- 42.Cardarelli PM, Rao-Naik C, Chen S, Huang H, Pham A, Moldovan-Loomis MC, et al. A nonfucosylated human antibody to CD19 with potent B-cell depletive activity for therapy of B-cell malignancies. Cancer Immunol Immunother 2010;59:257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibodybinding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther 2008;7:2517–27. [DOI] [PubMed] [Google Scholar]
- 44.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009;113: 3716–25. [DOI] [PubMed] [Google Scholar]
- 45.Natsume A, Takamura H, Nakagawa T, Shimizu Y, Kitajima K, Wakitani M,. et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res 2008;68:3863–72. [DOI] [PubMed] [Google Scholar]
- 46.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping ofthe binding site on human IgG1 forFcgamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 2001;276:6591–604. [DOI] [PubMed] [Google Scholar]
- 47.Velders MP, van Rhijn CM, Oskam E, Fleuren GJ, Warnaar SO, Litvinov SV. The impact of antigen density and antibody affinity on antibody- dependent cellular cytotoxicity: relevance for immunotherapy of carcinomas. Br J Cancer 1998;78:478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cleary KLS, Chan HTC, James S, Glennie MJ, Cragg MS. Antibody distance from the cell membrane regulates antibody effector mechanisms. J Immunol 2017;198:3999–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zamai L, Ponti C, Mirandola P, Gobbi G, Papa S, Galeotti L, et al. NK cells and cancer. J Immunol 2007;178:4011–6. [DOI] [PubMed] [Google Scholar]
- 50.Chiavenna SM, Jaworski JP, Vendrell A. State of the art in anti-cancer mAbs. J Biomed Sci 2017;4:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest 2014;124:2668–82. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood 2011;117:2423–32. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung cancer subtypes generate unique immune responses. J Immunol 2016;197:4493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kohrt HE, Houot R, Weiskopf K, Goldstein MJ, Scheeren F, Czerwinski D, et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J Clin Invest 2012;122:1066–75. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Yonezawa A, Dutt S, Chester C, Kim J, Kohrt HE. Boosting cancer immunotherapy with anti-CD137 antibody therapy. Clin Cancer Res 2015;21: 3113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.