

Abstract
Sequential enzymatic reactions on substrates tethered to carrier proteins (CPs) generate thiotemplated building blocks that are then delivered to nonribosomal peptide synthetases (NRPSs) to generate peptidic natural products. The underlying diversity of these thiotemplated building blocks is the principal driver of the chemical diversity of NRPS-derived natural products. Structural insights into recognition of CPs by tailoring enzymes that generate these building blocks are sparse. Here we present the crystal structure of a flavin-dependent prolyl oxidase that furnishes thiotemplated pyrrole as the product, in complex with its cognate CP in the holo and product-bound states. The thiotemplated pyrrole is an intermediate that is well-represented in natural product biosynthetic pathways. Our results delineate the interactions between the CP and the oxidase while also providing insights into the stereospecificity of the enzymatic oxidation of the prolyl heterocycle to the aromatic pyrrole. Biochemical validation of the interaction between the CP and the oxidase demonstrates that NRPSs recognize and bind to their CPs using interactions quite different from those of fatty acid and polyketide biosynthetic enzymes. Our results posit that structural diversity in natural product biosynthesis can be, and is, derived from subtle modifications of primary metabolic enzymes.
Graphical Abstract
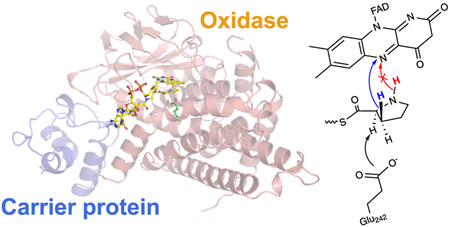
Nonribosomal peptide synthetases (NRPSs) deliver natural products of great utility as antibiotics and pharmaceuticals. A diverse set of proteinogenic and non-proteinogenic amino acid building blocks generates a tremendous chemical diversity of NRPS-derived peptidic natural products. Central to NRPS enzymology, and also polyketide and fatty acid biosynthesis, are small helical carrier proteins (CPs) that are posttranslationally modified with a coenzyme A (CoA)-derived phosphopantetheine appendage on a Ser side chain hydroxyl (Figure 1A). The phosphopantetheinyl thiol then acts as a transient shuttle for amino acids and for peptidyl chains by forming a high-energy thioester linkage with the carboxy termini. Type I NRPSs are large multimodular enzymes in which the CPs and NRPS catalytic enzymes are embedded as domains within a single polypeptide chain. Type I NRPS CPs access the active sites of adenylation domains that load the module specific amino acid onto the CP-phosphopantetheinyl thiol and of the condensation domains that accept the growing peptidyl chain on the upstream CP and condense it to the amino acid of the module specific CP via amide bond formation, in addition to other tailoring catalytic domains such as the terminal thioesterase domain (Figure 1B). On the other hand, type II NRPS CPs are not embedded within large multimodular enzymes and interact sequentially with stand-alone tailoring enzymes. In contrast to a wealth of crystal structures that are now available describing the modular architecture and interaction of type I NRPS CPs with the respective catalytic domains,1–4 our structural knowledge base for type II NRPSs is restricted to crystal structures of CPs alone and spectroscopic and mutational studies for interaction of type II NRPS CPs with their partner enzymes.5–7
Figure 1.

NRPS enzymology and construction of thiotemplated pyrroles. (A) The CoA-derived phosphopantetheine arm is appended to a serine chain hydroxyl to yield holo-CP that is abbreviated for the sake of clarity. (B) Representative type I NRPS module in which amino acids are specifically acylated to holo-CP by the adenyltransferase (A) domain, followed by ligation of the CP-acylated amino acid to the peptidyl chain acylated to the CP of the upstream NRPS module by the condensation (C) domain. (C) Biosynthesis of pyrrolyl-S-CPs as typified by the stand-alone type II NRPS adenyltransferase Bmp4 and prolyl-S-CP oxidase Bmp3 (boxed). Note that Bmp1 is a didomain protein for which only the N-terminal CP domain is shown here for the sake of clarity. In addition to Bmp3 and Bmp4, the flavin-dependent halogenase Bmp2 and the C-terminal thioesterase (TE) domain of Bmp1 participate in the biosynthesis of 2.
Several type II NRPS systems furnish thiotemplated initiator building blocks that are then delivered to downstream type I NRPSs, polyketide synthases, fatty acid synthases, and other modification systems. Derived from l-proline, thiotemplated pyrrole is one such building block. A widely represented enzymatic route for the construction of thiotemplated pyrroles involves the ATP-dependent acylation of l-proline to the phosphopantetheinyl thiol of a holo-CP followed by four-electron oxidation of the prolyl-S-CP to yield pyrrolyl-S-CP (Figure 1C).8–10 A downstream tailoring step common for thiotemplated pyrroles is halogenation, catalyzed by FAD-dependent halogenases, first characterized en route the biosynthesis of pyoluteorin [1 (Figure 1C)], and for several other halogenated pyrrole-containing natural products since then.11 Several steps along the biosynthetic route for thiotemplated pyrroles have been structurally interrogated. Burkart and co-workers have determined the holo and pyrrolyl-acylated structures of the pyoluteorin biosynthetic type II NRPS CP PltL and have queried the binding interaction between the CP and the l-proline adenyltransferase enzymes.6,7 However, the flavin-dependent prolyl-S-CP oxidase has evaded structural characterization and the manner of CP engagement by this enzyme is not clear at present; there are also mechanistic considerations that have not been resolved (Figure 1C, boxed).9,10
Flavin-dependent dehydrogenases and oxidases are ubiquitous in primary and secondary metabolism.12,13 The prolyl-S-CP oxidases are highly homologous to dehydrogenases from primary metabolism that participate in fatty acid catabolism to install the α,β-unsaturation in alkyl fatty acid chains that are thioesterified to CoA. The trans-Δ2-enoyl CoA products are then hydrated and oxidized to yield β-keto thioesters for thiolysis and release of acetyl-CoA. Crucially, fatty acid dehydrogenases employ substrates acylated to CoA, not CPs, and regenerate the oxidized flavin cofactor by delivering electrons to the respiratory chain. In contrast, the prolyl oxidases employ CP-acylated substrates and use molecular oxygen as the terminal electron acceptor, hence their designation as oxidases rather than dehydrogenases.14
The recently characterized biosynthetic scheme for the production of tetrabromopyrrole (2) and pentabromopseudilin (3) follows the thiotemplated pyrrole biosynthetic logic described above. Briefly, the adenyltransferase Bmp4 loads l-proline on the N-terminal CP domain of Bmp1, henceforth termed Bmp1(CP), followed by oxidation of the prolyl side chain by the FAD-dependent oxidase Bmp3.15 The halogenase Bmp2 and the C-terminal thioesterase (TE) domain of Bmp1 then furnish 2,16 which is subsequently debrominated and oxidatively coupled with 2,4-dibromophenol to yield 3.15, 17 The Bmp1–4 biosynthetic motif is widely distributed in oceanic metagenomes with multiple biological activities and ecological roles assigned to 2.18–21 Here, we report a 1.9 Å resolution crystal structure of the prolyl-S-Bmp1(CP) oxidase Bmp3 in complex with the product pyrrolyl-S-Bmp1(CP) and the cofactor FAD and a 2.7 Å resolution Bmp3/holo-Bmp1(CP) co-crystal structure. The complex crystal structures, obtained without any cross-linking, demonstrate the tetrameric quaternary structure of Bmp3 with four distinct binding sites for Bmp1(CP) leading to substrate engagement in a highly organized active site that is sterically tuned for binding the prolyl heterocycle. In addition to a finely tuned active site architecture and the manner of CP engagement in type II NRPS biosynthetic schemes, our results illuminate an overall structural and mechanistic conservation between a secondary metabolic enzyme participating in natural product biosynthesis and its close homologues from primary metabolism.
MATERIALS AND METHODS
Methods describing cloning, site-directed mutagenesis to generate Bmp1(CP) and Bmp3 mutant strains, and protein purification are described in detail in the Supporting Information.
Sodium Dithionite Reduction and Reoxidation of Bmp3.
The ultraviolet–visible (UV–vis) absorbance of 25 μM Bmp3/Bmp1(CP) complex in buffer was recorded at room temperature using a sealed quartz cuvette in 2 nm wavelength increments in the range of 350–600 nm using the Thermo Fisher Scientific NanoDrop One spectrophotometer. Freshly prepared sodium dithionite at a final concentration of 4.5 mM was added to the protein sample in a cuvette, the solution mixed by inverting, and the UV–vis absorbance recorded. The protein solution was then exposed to air for 5 min and mixed by inverting, and the UV–vis absorbance recorded again.
Preparation of Bmp3/Holo-Bmp1(CP) and Bmp3/Pyrrolyl-S-Bmp1(CP).
Assays were conducted in a 2.5 mL total reaction volume, and the reactions were allowed to proceed overnight at room temperature. The assay for generating the Bmp3/holo-Bmp1(CP) complex contained 140 μM Bmp3/apo-Bmp1(CP), 1 mM CoA, 1 μM Sfp, 10 mM MgCl2, and 20 mM Tris-HCl (pH 8.0). Similarly, the assay for synthesis of pyrrolyl-S-Bmp1 contained 136 μM Bmp3/apo-Bmp1(CP), 1 mM CoA, 1 μM Sfp, 10 mM MgCl2, 2 mM ATP, 2 mM l-proline, 1 μM Bmp4, and 20 mM Tris-HCl (pH 8.0). Any observed precipitate was removed by centrifugation, and the supernatant was desalted using PD-10 gravity columns. The formation of both Bmp3/holo-Bmp1-(CP) and Bmp3/pyrrolyl-S-Bmp1(CP) was verified by mass spectrometry.
Rapid Reaction Kinetic Analyses.
For all spectroscopic studies, enzymes were prepared fresh. The molar ratio of FAD to Bmp3/Bmp1(CP) complex was determined by extracting FAD from the Bmp3/holo-Bmp1(CP) complex through heat denaturation at 100 °C for 10 min, centrifuging the protein precipitate, and estimating the total FAD concentration of the lysate using a molar extinction coefficient of 11300 M−1 cm−1 at 450 nm. From these measurements, the molar extinction coefficient of active, flavin-bound Bmp1-Bmp3 was determined to be around 11800 M−1 cm−1 at 440 nm. The purified Bmp3/holo-Bmp1(CP) stock was diluted to a concentration of 25 μM flavin-bound enzyme complex using 50 mM potassium phosphate buffer (pH 7.5) in the presence and absence of 150 μM Bmp4, 1 mM l-proline, 1 mM ATP, 5 mM MgCl2, and 1 mM sodium dithionite. The UV–vis absorption spectrum for each sample was recorded at 25 °C using a Cary 8454 UV–vis instrument (Agilent Technologies, Santa Clara, CA) equipped with a photo diode array to record spectra in the wavelength range of 200–1100 nm.
Rapid reaction mixing studies were performed using an Olis RSM1000 spectrometer equipped with a temperature-controlled stopped-flow handling unit. The spectrometer was configured to record 1000 absorption spectra per second over the wavelength range of 300–600 nm. The entire sample handling unit was made anaerobic by flushing the system with an oxygen scrubbing solution of 20 mM glucose and 10 units of glucose oxidase and permitted to incubate overnight to remove any trace oxygen. All experiments were performed at 25 °C by mixing an anaerobic solution of 150 μM Bmp4, 1 mM l-proline, 1 mM ATP, and 5 mM MgCl2 in one drive syringe against 25 μM Bmp3/holo-Bmp1(CP) in anaerobic buffer in the other drive syringe. To ensure complete anaerobiosis of reagents, all solutions were buffered in 25 mM potassium phosphate (pH 7.5) and stored for at least 16 h in an anaerobic chamber prior to addition of 20 mM glucose and 10 units of glucose oxidase. Bmp3/holo-Bmp1(CP) solutions were made anaerobic by repeated flushing and evacuation of a glass tonometer containing concentrated Bmp3/holo-Bmp1(CP) with argon gas before being transferred into an anaerobic chamber. Once in the anaerobic chamber, the Bmp3/holo-Bmp1(CP) solution was diluted to a concentration of 25 μM with an anaerobic buffer supplemented with glucose (substrate) and glucose oxidase, centrifuged at 10000g for 10 min, and returned to the glass tonometer. All experiments were carried out by mixing equal volumes of the solutions, and the reactions were monitored by observing the change in absorbance at 440 nm over 100 s. The resulting kinetic traces were best fit to eq 1 with KaleidaGraph (Synergy Software, Reading, PA)
| (1) |
where k1 is the apparent rate constant for flavin reduction, A is the absorbance at time t, A1 is the amplitude for k1, and C is the absorbance at the end of the reaction.
Size Exclusion Chromatography Analysis of the Bmp3/Bmp1(CP) Complex.
Size exclusion chromatography analysis of the Bmp3/Bmp1(CP) complex was conducted using a Superdex 75 gel filtration column (GE Healthcare). Twenty-four milligrams of the purified Bmp3/Bmp1(CP) protein complex at a concentration of 12 mg/mL was loaded onto a gel filtration column using an AKTA Prime liquid chromatography system. The sample was eluted using 20 mM HEPES (pH 7.5), 100 mM KCl buffer at a flow rate of 1 mL/min, and fractions corresponding to two protein peaks collected and aliquots of each fraction were analyzed by gel electrophoresis.
Crystallization and Structure Determination.
Bmp3/pyrrolyl-S-Bmp1(CP) and Bmp3/holo-Bmp1(CP) were crystallized using hanging drop vapor diffusion at 279 K against a reservoir solution of 15 mM magnesium acetate, 1.8 M ammonium sulfate, and 50 mM MES-Na (pH 6.0). Crystals appeared within 2 days and reached their maximum size in a week, which is when they were harvested in a cryoprotectant solution supplemented with 20% ethylene glycol and flash-frozen in liquid nitrogen. Diffraction data were collected at 100 K.
Diffraction data were indexed and scaled in space group P622 using HKL-2000.22 Though the diffraction intensities demonstrated the presence of a screw axis, no screw axis was assigned at this stage. An ensemble search model was generated using Protein Data Bank (PDB) entries 4IRN and 4ETW.23,24 Molecular replacement using Phaser, implemented within the Phenix suite, was used to find a preliminary solution, along with the final space group (P6422), that was extended using ARP/wARP followed by iterative cycles of manual building in Coot and refinement using REFMAC.25–29 Constraints for ligands were generated using the eLBOW implemented in the Phenix suite.30
RESULTS
Prolyl-S-CP Oxidase Activity of Bmp3.
The identification of the bmp gene cluster for the production of 2 and 3 in Marinomonas mediterranea MMB-1, among other marine proteobacteria, has been previously reported.15 Coexpression of M. mediterranea MMB-1 bmp1–4 genes in Escherichia coli in a medium supplemented with bromide led to the production of 2.16 Recombinant Bmp3 was recalcitrant to expression in the soluble form, as has been observed for other prolyl-S-CP oxidases.9 Our previous results demonstrated that Bmp3 could alternatively be expressed as a soluble complex with Bmp1-(CP).15 We thus purified the Bmp3/Bmp1(CP) complex by appending a His6 tag at the N-terminus of Bmp1(CP) while Bmp3 was untagged. The purified Bmp3/Bmp1(CP) complex was only transiently stable and dissociated during size exclusion chromatography leading to the precipitation of Bmp3 (Figure S1). While the inability to purify Bmp3 by itself precludes a detailed kinetic characterization of mutant enzymes described below, the coexpression of bmp1—4 allows us to monitor the effects of these mutations in the context of the entire biosynthetic scheme for the production of 2.
The purified Bmp3/Bmp1(CP) complex demonstrated an UV–vis absorbance profile characteristic of a bound flavin cofactor in the oxidized state. Reduction with sodium dithionite quenches the flavin absorbance that is recovered upon exposure to air (Figure S2). We next treated the Bmp3/Bmp1(CP) complex with the Bacillus subtilis phosphopante-theinyl transferase Sfp and CoA to generate Bmp3/holo-Bmp1(CP), as monitored by the mass spectrometric phosphopantetheine ejection assay.31,32 Aerobic treatment of the Bmp3/holo-Bmp1(CP) complex with Bmp4 in the presence of ATP/Mg2+ and l-proline led to stoichiometric production of Bmp3/pyrrolyl-S-Bmp1(CP) as determined by the characteristic pyrrolyl-S-(cyclo)pantetheine MS2 product ion (Figure S3). The transient nature of the Bmp3/Bmp1(CP) complex is reflected in the observation that Sfp and Bmp4 could access Bmp1(CP) while bound to Bmp3. Pyrrolyl product formation was abolished in the absence of oxygen. The rate constant associated with hydride transfer leading to flavin reduction was determined by the anaerobic treatment of the Bmp3/holo-Bmp1(CP) complex in the presence of l-proline, ATP/Mg2+, and a 6-fold molar excess of Bmp4 relative to the Bmp3/holo-Bmp1(CP) complex (Figure 1C). The reaction was monitored by observing the temporal change in the flavin absorption spectra using a rapid reaction stopped-flow spectrophotometer (Figure 2A). These data were analyzed to reveal kinetic traces corresponding to the decrease in the absorbance at 440 nm (Figure 2B). These traces were best fit to a single-order rate equation to yield a flavin reduction rate constant (kred) of 0.05 ± 0.01 s−1. The kred of Bmp3 is in agreement with the kcat reported for the prolyl-S-CP oxidase AnaB (1 min−1), which yields an (S)-1-pyrroline-S-CP rather than a pyrrolyl-S-CP product.14 Agreement between the kred and kcat values for Bmp3 and AnaB, respectively, exists despite the difference in experimental techniques employed, an anaerobic spectrophotometric assay for Bmp3 as opposed to chromatographic separation of the substrate and product for AnaB.14 This perhaps alludes to hydride transfer being the rate-determining step in prolyl oxidation by AnaB and Bmp3, as has been proposed for FAD-dependent acyl-CoA dehydrogenases.13
Figure 2.

Bmp3/Bmp1(CP) complex. (A) Rapid reaction kinetic analysis of flavin reduction upon treatment of 25 μM Bmp3/holo-Bmp1(CP) with 150 μM Bmp4, 1 mM l-proline, 1 mM ATP, and 5 mM MgCl2 under anaerobic conditions after (—) 0.11, (-—-) 5.0, (–––) 10, (---) 30, (…) 50, and (…) 60 s. The flavin absorbance maximum at 440 nm is denoted by an arrow. (B) Single-turnover kinetic trace of prolyl oxidation monitored by the decrease in the absorbance at 440 nm. Single-turnover stopped-flow kinetic experiments were performed in triplicate, and the kinetic curve shown represents an average of three separate experiments. (C) Crystallographic asymmetric unit organization for the Bmp3/pyrrolyl-S-Bmp1(CP) complex. Bound FAD and pyrrolyl-S-phosphopantetheine are shown in ball-and-stick representation with carbon atoms colored yellow. (D) The biological tetramer is reconstituted by crystallographic symmetry. One Bmp1(CP)/Bmp3 monomeric unit is shown in surface representation with the other three Bmp1(CP)/Bmp3 units shown in cartoon representation. Difference Fourier electron density map contoured at (E) 3.0σ in blue and (F) 9.0σ in red calculated with ∣Fobs∣ – ∣Fcalc∣ coefficients and phases from the final refined model with the coordinates of FAD and pyrrolyl-S-phosphopantetheine (abbreviated as pyrrolyl-S-Ppant) deleted prior to one round of refinement. Pyrrolyl-S-phosphopantetheine is covalently acylated to the Bmp1(CP)-Ser35 side chain hydroxyl. The pantetheine sulfur atom, differentiated on the basis of the ∣Fobs∣ – ∣Fcalc∣ map contoured at 9.0σ, is marked by an arrow.
Overall Structure of the Bmp3/Bmp1(CP) Complex.
The Bmp3/holo-Bmp1(CP) and Bmp3/pyrrolyl-S-Bmp1(CP) complexes were desalted and directly used for crystallization without further purification. X-ray diffraction data were scaled to the preliminary P622 space group with the screw axis (P6422) determined during phasing by molecular replacement using PDB entries 4IRN (AnaB) and 4ETW (CP polypeptide only) as search models (Table 1).23,24 The Bmp3/Bmp1(CP) crystallographic asymmetric unit constitutes one Bmp3 monomer in complex with one Bmp1(CP) unit (Figure 2C). Apart from eight continuous solvent-exposed residues (Ser24–Asn31), the entirety of the Bmp3 peptide chain could be reliably built into the observed electron density maps (Figure 2C). Consistent with sequence homology, Bmp3 exhibits an overall tertiary structure similar to that of FAD-dependent dehydrogenases and oxidases (Table S1 and Figure S4). The Bmp3 monomer consists of an N-terminal α-helical domain followed by a β-sheet domain in the middle and finally a second C-terminal helical domain (Figure S5). The Bmp1-(CP) is folded into a helical bundle characteristic of CPs (Table S1). Hydrophobic amino acid side chains generate a compact solvent-occluded Bmp1(CP) core with polar amino acid side chains decorating the surface. The catalytic serine residue, Ser35, is positioned at the N-terminus of Bmp1(CP) helix II. Even though only a monomeric Bmp3/Bmp1(CP) complex is present in the asymmetric unit, crystallographic symmetry reveals that the quaternary structure comprises a Bmp3/Bmp1(CP) tetramer (Figure 2D). The tetrameric assembly for Bmp3 is supported by size exclusion chromatography and nondenaturing polyacrylamide gel electrophoresis (Figure S1). Each Bmp3 monomer contacts two other Bmp3 monomers via multiple interactions between Arg-Asp/Glu side chains while burying 3300 Å2 of surface area, as determined by the PDBePISA server.33 A similar tetrameric quaternary structure has been observed for acyl-CoA dehydrogenases, as well.34 The Bmp3 tetramer neatly organizes four distinct binding sites for Bmp1(CP). Unlike the Bmp3 monomers, the individual Bmp1(CP) units do not contact each other (Figure 2D).
Table 1.
Data Collection, Phasing, and Refinement Statistics
| Bmp3/pyrrolyl-S- Bmp1(CP) |
Bmp3/holo- Bmp1(CP) |
|
|---|---|---|
| PDB entry | 6CXT | 6CY8 |
| Data Collectiona | ||
| space group | P6422 | P6422 |
| a, b, c (Å) | 104.4, 104.4, 234.1 | 104.4, 104.4, 235.7 |
| α, β, γ (deg) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 |
| resolution (Å) | 1.90 (1.93–1.90) | 2.70 (2.75–2.70) |
| total no. of reflections | 60078 | 19969 |
| Rmerge | 0.102 (0.660) | 0.171 (0.444) |
| Rpim | 0.021 (0.139) | 0.045 (0.126) |
| I/σ(I) | 45.4 (4.1) | 12.5 (6.9) |
| completeness | 100.0 (97.0) | 95.9 (97.5) |
| redundancy | 23.1 (22.2) | 11.5 (10.4) |
| Refinement | ||
| resolution (Å) | 1.90 (1.95–1.90) | 2.73 (2.80–2.73) |
| no. of reflections | 56340 | 18816 |
| Rwork/Rfreeb | 0.181/0.210 | 0.169/0.233 |
| no. of atoms | ||
| protein | 3438 | 3422 |
| solvent | 345 | 159 |
| heteroatoms | 89 | 74 |
| B factor | ||
| protein | 28.9 | 43.3 |
| solvent | 39.7 | 38.0 |
| FAD | 22.4 | 31.9 |
| pyrrolyl-S-phosphopantetheine | 25.3 | – |
| phosphopantetheine | – | 41.1 |
| Ramachandran analysis (%) | ||
| favored | 96.1 | 95.4 |
| allowed | 3.6 | 3.9 |
| outliers | 0.3 | 0.7 |
| root-mean-square deviation | ||
| bond lengths (Å) | 0.02 | 0.02 |
| bond angles (deg) | 1.90 | 1.94 |
Values for the highest-resolution shell are shown in parentheses.
, and Rfree is the R value for a test set of reflections consisting of a random 5% of the diffraction data not used in refinement.
The observed difference electron density allowed us to assign all atoms for the cofactor FAD and the phosphopantetheine appendage acylated to the Bmp1(CP) Ser35 side chain hydroxyl with full occupancy (Figure 2E,F). The functional relevance of the Bmp3 tetramer is illustrated by the interactions contributing to cofactor engagement. FAD is bound at the interface of two Bmp3 monomers (Figure S6). Interactions provided by neighboring Bmp3 monomers include π-stacking of the FAD adenine ring against the Phe278 side chain and ionic interactions of the FAD pyrophosphate with an Arg268 side chain guanidino moiety. The redox active isoalloxazine ring is largely bound within a single Bmp3 subunit and is positioned by hydrogen bonding between the isoalloxazine N5 atom and the Bmp3 Thr158 side chain hydroxyl and by interactions of the isoalloxazine C2 and C4 carbonyls with Bmp3 main chain amides.
Curiously, the phosphopantetheine phosphoryl moiety resides in a solvent-filled cavity and does not contact any of the Bmp3 or Bmp1(CP) side chains directly; only water-mediated interactions with the Bmp3 Tyr178, Arg243, and Arg376 side chains can be discerned (Figure S7). The side chain of Arg243 also contacts the two pantetheinyl amide carbonyls. Several hydrophobic amino acid side chains guide the pantetheine chain to appropriately position the thioesterified substrate in the Bmp3 active site. The organization of the pantetheine chain relative to Bmp3 is similar to that of the pantetheine chain of CoA as observed in the crystal structures of acyl-CoA dehydrogenases. However, the positioning of the phosphopantetheine phosphoryl moiety relative to Bmp3 and that of the CoA-derived pyrophosphoryl group in acyl-CoA dehydrogenases are markedly different (Figure S8).35–38
Active Site Organization.
The difference electron density map demonstrates that the pantetheine chain terminates with a planar moiety bound to the thiol (Figure S9). In a manner convergent with the mass spectrometry data that demonstrated Bmp3-catalyzed conversion of l-proline to pyrrole (Figure S3), we have modeled pyrrolyl-S-phosphopantetheine, the product state, to be acylated to the Bmp1(CP) Ser35 side chain hydroxyl. The modeled pyrrole ring is in good agreement with the observed difference electron density (Figure 2E,F). The pyrrolyl 2-carbonyl oxygen is within binding distance of one of the FAD ribityl hydroxyls (interatomic distance of 2.6 Å) and the Ser363 main chain amide nitrogen (2.9 Å) that constitute the oxyanion hole to stabilize the enolate intermediate formed after proton abstraction by the catalytic base (vide infra). The pyrrolyl nitrogen is within hydrogen bonding distance of the Asn123 side chain amide (3.3 Å). At the si face, the pyrrole ring is coplanar with the flavin isoalloxazine ring. At the re face, side chains of Bmp3 residues Phe85, Ala89, Ala93, Met239, Leu246, and Phe362 generate a desolvated compact cavity (Figure 3A). Of these, Bmp3 residues Phe85, Ala89, and Ala93 are placed on adjoining turns of helix E of the Bmp3 N-terminal helical domain (Figure S5). The Bmp3 residue Glu242 is positioned in the middle of residues Met239 and Leu246 on adjoining turns of helix G of the Bmp3 C-terminal helical domain. The Glu242 side chain carboxylate is within binding distance of the Ser363 side chain hydroxyl and is positioned 3 Å from the pyrrolyl C2 position. All Bmp3 residues described above that constitute the pyrrole binding pocket are strictly conserved in prolyl-S-CP oxidases (Figure S5). The crystal structure of Bmp3/holo-Bmp1(CP) reveals that the Bmp3 active site is preorganized for substrate binding. No changes in the position of the cofactor or the active site amino acid side chains could be discerned between the holo and pyrrolyl states.
Figure 3.

Bmp3 active site. (A) The cofactor FAD and the pyrrolyl-S-phosphopantetheine are shown in ball-and-stick representation with carbon atoms colored yellow. Side chains of Bmp3 active residues discussed in the text are also shown in ball-and-stick representation with the pyrrolyl binding pocket shown as a partially transparent gray surface. The side chain of the catalytic base Glu242 is shown with carbon atoms colored green. A majority of Bmp3 residues, the side chains of which constitute the active site, are placed on Bmp3 helices E and G. (B) Area under the extracted ion chromatogram curve for the ion at m/z 381.75, the most abundant [M – H]− isotopic mass corresponding to 2, for coexpression of bmp1, bmp2, and bmp4 with bmp3 and bmp3 mutants after induction of protein expression for 5 h. The mean and standard deviation from three independent measurements are reported. (C) Comparison of the active sites of Bmp3 and fatty acid acyl-CoA dehydrogenase (PDB entry 3B96).38 Carbon atoms for the acyl-CoA dehydrogenase along with the FAD cofactor and the myristoyl-CoA substrate are colored cyan. Bmp3 atoms are colored as in panel A. For the sake of clarity, Bmp3 FAD is not shown. Side chains of Bmp3 amino acids Phe85 and Leu246 obstruct the cavity through which the myristoyl-CoA acyl chain is threaded. Replacement of the acyl-CoA dehydrogenase branched Ile144 side chain with Bmp3 Ala93 expands the active site to allow for pyrrole ring binding. Note that the myristoyl-CoA Cα and Cβ atoms align with the pyrrolyl C2 and C3 atoms. The pantetheinyl sulfur atoms are marked by an arrow.
We next used the coexpression of bmp1–4 genes in E. coli for the production of 2 to evaluate the catalytic roles of residues described above. Upon induction of recombinant protein expression, a time-dependent production of 2 was observed, reaching a maximum 5 h post-induction after which degradative dehalogenation products were observed (Figures S10 and S11). Dehalogenation of in situ-produced 2 in E. coli has been observed previously.16,17 Mutation of Glu242 to Ala or Asp completely abolished the production of 2, corroborating its role as the catalytic base (Figure 3B). A decrease in the level of production of 2 was observed for the Asn123Ala mutation, while the production of 2 was unchanged with the Ser363Ala mutation. Though Bmp3 Ser363 is strictly conserved among prolyl-S-CP oxidases (Figure S5), it is noteworthy that the isovaleryl-CoA dehydrogenase, for which the catalytic Glu is positioned analogously to Bmp3, bears an Ala residue at this position (Figure S12).35
Comparison of Bmp3 with acyl-CoA dehydrogenases demonstrates the primacy of Bmp3 helices E and G in sculpting the Bmp3 active site (Figure 3A). Two major changes are apparent. First, helix E and helix G for Bmp3 move closer to each other to constrict the cavity through which the fatty acid acyl chains are threaded. The side chains of Bmp3 Phe85 and Leu246 also obstruct this cavity precluding the binding of long acyl chains (Figure 3C). Second, to accommodate the five-membered substrate heterocycle in the Bmp3 active site, a branched amino acid in the acyl-CoA dehydrogenase active site, Ile144 in human very long chain acyl-CoA dehydrogenase (VLCAD, PDB entry 3B96), is replaced with Ala93. This change allows for room to accommodate the C4, C5, and N1 atoms of the Bmp3 substrate heterocycle (Figure 3C).38 A similar expansion of the Bmp3 substrate binding pocket is observed when compared to the active sites of medium chain acyl CoA dehydrogenase (Leu103 in PDB entry 3MDE replaced with Bmp3 Ala93), isovaleryl-CoA dehydrogenase (Leu103 in PDB entry 1IVH replaced with Bmp3 Ala93), isobutyryl-CoA dehydrogenase (Ile103 in PDB entry 1RX0 replaced with Bmp3 Ala89), and glutaryl-CoA dehydrogenase (Val99 and Ile103 in PDB entry 1SIR replaced with Bmp3 Ala89 and Bmp3 Ala93, respectively).35–37,39 In each of the acyl-CoA dehydrogenase structures mentioned above, the site analogous to Bmp3 Asn123 is occupied by a hydrophobic amino acid that does not interact with the bound substrate. It is likely that subtle changes in active sites allow for the expansion of the catalytic repertoire of primary metabolic enzymes to allow them to participate in natural product biosynthesis (Figure S13).
Bmp3/Bmp1(CP) Binding Interactions.
Bmp1(CP) is bound at the interface of two Bmp3 monomers. The N-terminus of Bmp1(CP) helix II is inserted into a funnel-shaped surface cavity that is generated by Bmp3 helices G and K and the interconnecting loops of the Bmp3 β-sheet domain (Figure 4A). Surface complementarity between Bmp1(CP) and Bmp3 positions the Bmp1(CP) Ser35 side chain appropriately to direct the phosphopantetheine arm toward the Bmp3 active site. In the vicinity of Bmp1(CP) Ser35, the Bmp3 Asn375 side chain is hydrogen bonded to the Bmp1(CP) Leu36 main chain amide (interatomic distance of 3.0 Å). The Bmp1(CP) Leu36 side chain is inserted into a small hydrophobic cavity defined by the side chains of Bmp3 Ile368 and Leu372 that also houses the pantetheinyl gemdimethyl moiety (Figure 4A).
Figure 4.

Bmp3/Bmp1(CP) binding interactions. (A) Bmp1(CP) (blue) binds at the interface of two Bmp3 monomers (pink and brown). Residues involved in binding interactions are shown in ball-and-stick representation. The pyrrolyl-S-phosphopantetheine, acylated to the Bmp1(CP) Ser35 side chain, is shown with carbon atoms colored yellow. Note that the Arg277 side chain interacts with the terminal main chain carboxylate (residue Leu380) of the adjoining Bmp3 neighbor. The cofactor FAD has been omitted for the sake of clarity. (B) Effect of Bmp1(CP) and Bmp3 mutations on the in vivo production of 2. Relative abundances of 2 are plotted. The mean and the standard deviation from three independent measurements are reported. (C) Solution structure of type II NRPS CP PltL with the pyrrolyl-N-phosphopantetheine (in ball-and-stick representation with carbon atoms colored yellow) in the “retracted” conformation (PDB entry 2N5I).7 The CP residues implicated in interacting with the l-proline adenyltransferase are shown in ball-and-stick representation with carbon atoms colored pink.6 (D) Crystal structure of Bmp1(CP) with the pyrrolyl-S-phosphopantetheine in the extended conformation. Residues at homologous positions for Bmp1(CP) that constitute the pyrrolyl binding pocket in PltL in the retracted state are involved in hydrophobic contacts with Bmp3 residues Tyr178 and Leu179.
As for cofactor binding, cooperativity between Bmp3 monomers is observed for binding to Bmp1(CP). The Arg277 side chain from an adjoining Bmp3 monomer is hydrogen bonded to the Bmp1(CP) Leu13 main chain amide carbonyl (2.8 Å). This Arg277 side chain also makes a salt bridge with the terminal main chain carboxylate of its neighboring Bmp3 monomer [2.9 Å (Figure 4A)]. Mutation of Arg277 leads to a significant reduction in the level of production of 2, further underscoring the functional relevance of the Bmp3 quaternary structure (Figure 4B). In the vicinity of Arg277, the Bmp1(CP) Glu15 side chain carboxylate is hydrogen bonded to the adjoining Bmp3 Thr274 main chain amide nitrogen (2.8 Å). The interactions mentioned above are all positioned at the C-terminus of Bmp1(CP) helix I (Figure 4A).
Bmp1(CP) helices II and III also make extensive contact with Bmp3. The side chains of Bmp1(CP) residues Leu28, Met38, Ile58, Pro60, and Phe63 generate a large concave hydrophobic patch on the Bmp1(CP) surface that recruits the Bmp3 Tyr178 and Leu179 side chains for binding (Figure 4A). The Bmp3 Tyr178 side chain phenoxyl is also hydrogen bonded to the guanidine side chain of Bmp3 Arg233 (2.9 Å), which additionally coordinates to the Bmp1(CP) Glu55 side chain carboxylate in a bidentate manner (2.7 and 2.8 Å). Mutation of either Bmp3 Arg233 or Bmp1(CP) Glu55 to Ala leads to an only slight reduction in the level of production of 2 (Figure 4B). However, disrupting the hydrophobic interaction by the Bmp3 Tyr178Ala/Leu179Ala double mutation leads to complete elimination of the production of 2. Relative contributions of the hydrogen bonding/salt bridge interactions to the hydrophobic interactions between Bmp1(CP) and Bmp3 are provided by the Bmp3 Arg233Ala/Arg277Ala double mutant in the absence or presence of the Bmp1(CP) Glu55Ala mutation. Either set of mutations does not abolish the production of 2 as observed for the Bmp3 Tyr178Ala/Leu179Ala double mutant. Furthermore, sequence alignment reveals a greater conservation of the Tyr178/Leu179 pair than of Arg233 or Arg277 among prolyl-S-CP oxidases (Figure S5). Taken together, these results indicate hydrophobic interactions are the primary determinants for Bmp1(CP) by Bmp3 with a smaller contribution of hydrogen bonding and salt bridge interactions. Curiously, the Bmp3 Arg233 and Bmp1(CP) Glu55 side chains present the solitary instance of salt bridge interactions between Bmp1(CP) and Bmp3. At Bmp1(CP) helix II, the Asp34 and Asp46 side chains are completely bereft of interactions with Bmp3.
DISCUSSION
CPs are ubiquitous participants in fatty acid biosynthesis and in polyketide synthase and NRPS-derived natural product biosynthetic schemes. CPs directionally shuttle carboxylic acid intermediates through enzyme active sites by covalently tethering them to the phosphopantetheinyl thiol via a high-energy thioester bond. In addition to substrate channeling, the flexibility of the phosphopantetheine arm allows CPs to sequester their thioesterified payloads within their internal hydrophobic core to prevent deleterious hydrolysis of the thioester bond and other off-pathway reactions. It is thus instructive to contrast the conformations of the pyrrolyl-S-phosphopantetheine arm in the “extended” state, which is described in this study, and the “retracted” state, as was reported for the highly homologous type II NRPS CP-PltL that participates in the biosynthesis of 1 (Figure 4C,D, root-mean-square deviation of 1.6 Å over all atoms of 71 aligned residues).7 In a correlation with catalysis, the extended state allows delivery of the substrate into the enzyme active sites while the retracted state likely involves acylated substrate/product protection. Both, Bmp1(CP) and PltL have similar tetrahelical structures with an interrupted helix III [by Pro60 in Bmp1(CP) and Gly67 in PltL]. The solution structure of PltL pyrrolyl-N-phosphopantetheine (the thioester bond was replaced with an amide in this study) demonstrates that the aromatic pyrrole ring is sequestered by a surface hydrophobic patch on the CP constituted by the side chains of PltL residues Leu35, Ile45, Ile65, and Phe70. The corresponding positions for Bmp1(CP) are all occupied by hydrophobic amino acid side chains, as well, namely, Leu28, Met38, Ile58, and Phe63. This analogous pyrrolyl sequestering hydrophobic patch on the Bmp1(CP) surface is the site of hydrophobic interaction with Bmp3, as mediated by the side chains of Bmp3 residues Tyr178 and Leu179 that are critical for catalysis (Figure 4). Thus, dual roles for the pyrrolyl-CP surface hydrophobic patch emerge, those being recruitment of the prolyl-S-CP dehydrogenase and shielding of the aromatic pyrrole ring prior to delivery to the downstream pyrrolyl-S-CP halogenase. Notably, product sequestration by CPs involved in fatty acid biosynthesis seems to be much tighter than for PltL. Only one edge of the pyrrolyl heterocycle contacts the PltL, while the cysteamine moiety is completely bereft of interactions with PltL, akin to other observations for NRPS CPs.7,40,41 Using NMR titrations and mutational analyses, Burkart and co-workers have mapped out the interaction surface between the CP PltL and the l-proline adenyltransferase PltF.6 Crucially, residues implicated in interaction of PltL with PltF are distinct from the Bmp3/Bmp1(CP) interaction surface (Figure 4C). Hence, using a combination of these results, a synergistic view emerges in which type II NRPS CPs likely employ distinct binding interfaces to interact with different catalytic partners. A detailed understanding of CP/tailoring enzyme interactions is undoubtedly critical for the engineering of biosynthetic pathways.
The primacy of salt bridges between Asp/Glu and Arg/Lys side chains in mediating interactions between CPs and their partner enzymes in fatty acid and polyketide synthesis is well-established.23,42–48 Typically, salt bridges are predicated upon CP helix II termini where they serve to position the phosphopantetheine-bearing serine residue, which is at the N-terminus of CP helix II, in a catalytically proficient manner relative to the partner enzyme. Crystal structures of excised type I NRPS domains in complex with CPs and of multidomain NRPS modules reveal a reduced reliance on salt bridges with hydrophobic interactions and hydrogen bonds being the primary contributors to CP binding.49–56 The Bmp3/Bmp1(CP) structure reveals that this differential mode of binding is likely shared by type II NRPS tailoring enzymes, as has also been observed for the CP-tethered amino acid hydroxylating cytochrome P450 enzyme P450sky, which remarkably recognizes three different CPs in the skyllamycin biosynthetic scheme.57,58 Still, the complete disengagement of Bmp1(CP) helix II from the binding surface is striking, as is the contribution of two Bmp3 monomers in constructing the Bmp1(CP) binding interface. A third Bmp3 monomer contributes the Arg268 and Phe278 side chains for binding the FAD cofactor, illustrating a highly cooperative Bmp3 tetrameric core to enable catalysis (Figure 4A and Figure S6). While the functional relevance of this unexpected mode of CP binding is unclear, hydrophobic interactions between Bmp1-(CP) and Bmp3 perhaps explain the insolubility of recombinant Bmp3 in the absence of the Bmp1(CP) interaction. The fact that Bmp3/apo-Bmp1(CP) can be modified by Sfp and the fact that Bmp3/holo-Bmp1(CP) can then be acylated by Bmp4 point toward significant conformational flexibility in the binding interactions that allow Bmp1(CP) to access different catalytic sites while still engaged to Bmp3.
Despite extensive attempts, we were unable to capture the prolyl-S-Bmp1(CP) in complex with an inactive mutant form of Bmp3, frustrating our efforts to describe the stereochemistry of the substrate l-prolyl heterocycle relative to the catalytic base Glu242 side chain carboxylate. However, the observed arrangement of the product pyrrole ring in the enzyme active site helps rationalize that the Glu242 side chain is indeed present in a catalytically proficient conformation. Assuming that the prolyl heterocyclic amine maintains the binding interaction with the Asn123 side chain amide, the C2–H for the l-prolyl heterocycle will be directed toward the Glu242 side chain carboxylate for abstraction as a proton, while the prolyl heterocycle is puckered up toward the FAD isoalloxazine ring [Figure 5, substrate binding catalytic state I (CS-I)]. Desolvation of the enzyme active site by hydrophobic amino acid side chains surrounding Glu242 and by the cofactor isoalloxazine ring modulates the pKa of the substrate prolyl C2–H and of the catalytic base to enable proton abstraction.13 The oxyanion hole generated by the FAD ribityl hydroxyl and the Bmp3 Ser363 main chain amide stabilize the enolate intermediate (CS-II). Following proton abstraction from C2, there are two mechanistic possibilities, which have been proposed previously.9,10 Tautomerization of the enolate can result in the transfer of the pro-R hydrogen as the hydride from the C3 position leading to the formation of the 2,3-dehydroprolyl-S-Bmp1(CP) intermediate (Figure 5A, CS-III). Alternatively, enamine–imine tautomerization followed by hydride transfer from the prolyl N1 to FAD N5 will result in the formation of the C2=NH+ imine (Figure 5B). Which of the two, C2–C3 or the C2–N1 oxidative routes, does Bmp3 follow? On the basis of the positioning of the pyrrolyl C3 proximal to FAD N5 (interatomic distance of 3.3 Å), we posit that the C2–C3 oxidative scheme is likely operative. The pyrrolyl N1 is too distant (4.7 Å) from the flavin redox center, and the N1–H will not have been positioned appropriately for hydride transfer. Furthermore, the pyrrolyl C3 aligns precisely with fatty acid Cβ from where the hydride is transferred to FAD in the acyl-CoA dehydrogenase active site to generate the Cα–Cβ oxidized product (Figure 3C). An alternate arrangement in which the product pyrrolyl ring is flipped 180°, thus swapping the positions of the C3 and N1 atoms, is crystallographically indistinguishable from the model of the product described above (Figure S14). However, the alternate flipped conformation of the pyrrole ring will lead the l-prolyl C2–H to point toward the isoalloxazine ring and away from the catalytic base, which would not be catalytically proficient.
Figure 5.

C2–N1 and C2–C3 oxidative routes. The catalytic base Glu242 side chain abstracts the C2 proton, leading to the formation of the enolate intermediate that can tautomerize with the prolyl (A) C3-pro-R-H or (B) N1–H being transferred as hydride to the FAD. The prochiral assignments for the prolyl C3–C5 hydrogen atoms are shown in the inset. Different catalytic states (CS-I–VI) are described in the text. (C) Slight difference in the positioning of the catalytic base side chains between AnaB and Bmp3. The alignment was generated by overlapping the FAD isoalloxazine rings. The cofactor for AnaB has been omitted for the sake of clarity. The C4 and C5 positions are labeled.
Progressing from the 2,3-dehydroprolyl-S-Bmp1(CP) intermediate (Figure 5, CS-III), enamine–imine tautomerization could furnish the 1,2-dehydroprolyl-S-Bmp1(CP) intermediate (CS-IV) that serves to modulate the acidity of the C5 pro-R proton for abstraction by the catalytic base. Stereospecificity for proton abstraction at C5 has indeed been experimentally demonstrated for the homologous cyanobacterial prolyl-S-CP oxidase AnaB using isotopically labeled substrates.14 Subsequent pro-R proton abstraction from C4 of the 1,5-dehydroprolyl-S-Bmp1(CP) intermediate (CS-V) furnishes the 4,5-dehydroprolyl-S-Bmp1(CP) intermediate (CS-VI) followed by a second round of proton abstraction from C2 and hydride transfer from C3 to furnish the pyrrolyl-S-Bmp1(CP) product.
The active site of Bmp3 reveals that C2, C4 pro-R, and C5 pro-R protons are all accessible to the Bmp3 Glu242 side chain. Accessibility to the C5 site justifies that both Bmp3 and AnaB can generate pyrrolyl-S-CP products starting from 3,4-dehydroprolyl-S-CP by isomerizing this substrate to 4,5-dehydroprolyl-S-CP (CS-VI) via proton abstraction from C5 and subsequent oxidation of the C2–C3 bond.9,14 It is noteworthy that the catalytic scheme for AnaB stalls at the 1,5-dehydroprolyl-S-CP state (CS-V), likely due to inaccessibility of the C4 site to the AnaB catalytic base, which would disfavor the formation of the 4,5-dehydroprolyl-S-CP intermediate for AnaB, as has been proposed previously (Figure 5).14 A comparison of the Bmp3 and AnaB active sites reveals differential positioning of the respective catalytic base Glu side chains (Glu242 for Bmp3 and Glu244 for AnaB). An alignment of the AnaB and Bmp3 active sites demonstrates that the AnaB Glu244 side chain likely cannot access the pyrrolyl-C4 site while the Bmp3 Glu242 side chain can (Figure 5C). The Bmp3 Glu242 side chain conformation is conserved between the Bmp3/holo-Bmp1(CP) and Bmp3/pyrrolyl-S-Bmp1(CP) structures and is thus not an artifact introduced by the presence of the pyrrolyl ring in the Bmp3 active site as compared to the crystal structure of AnaB, which does not possess the phosphopantetheinyl substrate or the product in the active site.24 Reactivation of the reduced flavin cofactor by molecular oxygen is unlikely to dictate the differential activities of AnaB and Bmp3. AnaB is readily oxidized by exposure to air upon dithionite reduction.14 No additional basic amino acid side chains, which have been implicated in activation of molecular oxygen for the oxidation of flavin hydroquinone, can be detected in or in the vicinity of the Bmp3 active site.59
Biosynthetic schemes for NRPS- and polyketide-derived natural products bearing pyrrolyl moieties typically use l-proline-derived thiotemplated pyrrole as the initiator unit that is subsequently handed off for modular elongation. Why has nature not evolved, rather, why have we not yet detected type I NRPS modules that employ pyrroles as intermediate, rather than initiator, building blocks in assembly line biosynthetic schemes? Possible rationales against the conversion of freestanding l-proline to pyrrole-2-carboxylic acid, which could then be incorporated into a growing peptide chain by an appropriately selective adenylation domain, have been presented previously.10 While type I NRPS l-proline adenyltransferase domains do indeed exist, the homotetrameric structure of the prolyl-S-CP oxidase would perhaps posit an overly large steric constraint on the organization of the type I NRPS module in which each of the other of domains will need to be present in a quadrimolar stoichiometry as well to generate thiotemplated pyrrole-2-carboxylate in situ. It is thus tantalizing to propose that the quaternary structure of the prolyl-S-CP oxidase has restricted the generation of thiotemplated pyrroles to type II NRPS systems only where the CP, the l-proline adenyltransferase, and the prolyl-S-CP oxidase are stand-alone enzymes and not physically organized into a singular megasynthase.
CONCLUSIONS
Pyrrole as a biosynthetic synthon is widely represented in bioactive natural products where it is derived from enzymatic oxidation of the l-proline side chain with the amino acid ligated to the phosphopantetheinyl thiol of a carrier protein. The crystal structure of the prolyl oxidase in complex with the carrier protein reveals binding interactions that are then validated using an in vivo assay monitoring the production of a marine natural product, tetrabromopyrrole. The binding interactions identified thus are distinct from the ionic interactions that typically operate in fatty acid and polyketide biosynthetic schemes to appropriately position the carrier proteins relative to the respective tailoring enzymes. The active site of the oxidase, as described in the product-bound form here, is highly similar to that of fatty acid catabolic oxidases with subtle changes that allow for the diversification of the enzymatic activity for pyrrole biosynthesis.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge Dr. Joseph P. Noel (The Salk Institute for Biological Studies, La Jolla, CA) for access to synchrotron beamtime, Dr. Amit R. Reddi and Claudia Montllor Albalate (Georgia Institute of Technology) for access to the anaerobic chamber and assistance with native polyacrylamide gel electrophoresis, Dr. Wilfred A. van der Donk (University of Illinois Urbana-Champaign, Urbana, IL) for insightful discussions, and the Biopolymer Characterization core facility at the Parker H. Petit Institute for Bioengineering and Bioscience (Georgia Institute of Technology) for the use of their shared equipment, services, and expertise.
Funding
This work was supported by the National Institutes of Health (Grant P01-ES030316 to B.S.M. and Grant R00-ES026620 to V.A.), the National Science Foundation (Grant OCE-1837116 to B.S.M.), and a Sloan Foundation research fellowship (to V.A.).
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b01177.
A description of methods employed for in vivo production of 2 and for protein purifications, Table S1, and Figures S1–S14 (PDF)
Accession Codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank as entries 6CXT [Bmp3/pyrrolyl-S-Bmp1(CP)] and 6CY8 [Bmp3/holo-Bmp1(CP)]. Bmp3, UniProtKB entry F2K077; Bmp1, UniProtKB entry F2K074; Bmp4, UniProtKB entry F2K078.
REFERENCES
- (1).Miller BR, and Gulick AM (2016) Structural biology of nonribosomal peptide synthetases. Methods Mol. Biol. 1401, 3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Walsh CT (2016) Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep. 33, 127–135. [DOI] [PubMed] [Google Scholar]
- (3).Marahiel MA (2016) A structural model for multimodular NRPS assembly lines. Nat. Prod. Rep. 33, 136–140. [DOI] [PubMed] [Google Scholar]
- (4).Kittila T, Mollo A, Charkoudian LK, and Cryle MJ (2016) New structural data reveal the motion of carrier proteins in nonribosomal peptide synthesis. Angew. Chem., Int. Ed. 55, 9834–9840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lohman JR, Ma M, Cuff ME, Bigelow L, Bearden J, Babnigg G, Joachimiak A, Phillips GN Jr., and Shen B (2014) The crystal structure of BlmI as a model for nonribosomal peptide synthetase peptidyl carrier proteins. Proteins: Struct., Funct., Genet 82, 1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jaremko MJ, Lee DJ, Patel A, Winslow V, Opella SJ, McCammon JA, and Burkart MD (2017) Manipulating protein-protein interactions in nonribosomal peptide synthetase type II peptidyl carrier proteins. Biochemistry 56, 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jaremko MJ, Lee DJ, Opella SJ, and Burkart MD (2015) Structure and substrate sequestration in the pyoluteorin type II peptidyl carrier protein PltL. J. Am. Chem. Soc. 137, 11546–11549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Thomas MG, Burkart MD, and Walsh CT (2002) Conversion of l-proline to pyrrolyl-2-carboxyl-S-PCP during undecylprodigiosin and pyoluteorin biosynthesis. Chem. Biol. 9, 171–184. [DOI] [PubMed] [Google Scholar]
- (9).Garneau S, Dorrestein PC, Kelleher NL, and Walsh CT (2005) Characterization of the formation of the pyrrole moiety during clorobiocin and coumermycin A1 biosynthesis. Biochemistry 44, 2770–2780. [DOI] [PubMed] [Google Scholar]
- (10).Walsh CT, Garneau-Tsodikova S, and Howard-Jones AR (2006) Biological formation of pyrroles: nature’s logic and enzymatic machinery. Nat. Prod. Rep. 23, 517–531. [DOI] [PubMed] [Google Scholar]
- (11).Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, and Walsh CT (2005) Dichlorination of a pyrrolyl-S-carrier protein by FADH2-dependent halogenase PltA during pyoluteorin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 102, 13843–13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Walsh CT, and Wencewicz TA (2013) Flavoenzymes: versatile catalysts in biosynthetic pathways. Nat. Prod. Rep. 30, 175–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mansoorabadi SO, Thibodeaux CJ, and Liu HW (2007) The diverse roles of flavin coenzymes–nature’s most versatile thespians. J. Org. Chem. 72, 6329–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mann S, Lombard B, Loew D, Mejean A, and Ploux O (2011) Insights into the reaction mechanism of the prolyl-acyl carrier protein oxidase involved in anatoxin-a and homoanatoxin-a biosynthesis. Biochemistry 50, 7184–7197. [DOI] [PubMed] [Google Scholar]
- (15).Agarwal V, El Gamal AA, Yamanaka K, Poth D, Kersten RD, Schorn M, Allen EE, and Moore BS (2014) Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat. Chem. Biol. 10, 640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).El Gamal A, Agarwal V, Diethelm S, Rahman I, Schorn MA, Sneed JM, Louie GV, Whalen KE, Mincer TJ, Noel JP, Paul VJ, and Moore BS (2016) Biosynthesis of coral settlement cue tetrabromopyrrole in marine bacteria by a uniquely adapted brominase-thioesterase enzyme pair. Proc. Natl. Acad. Sci. U. S. A. 113, 3797–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).El Gamal A, Agarwal V, Rahman I, and Moore BS (2016) Enzymatic reductive dehalogenation controls the biosynthesis of marine bacterial pyrroles. J. Am. Chem. Soc. 138, 13167–13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Whalen KE, Kirby C, Nicholson RM, O’Reilly M, Moore BS, and Harvey EL (2018) The chemical cue tetrabromopyrrole induces rapid cellular stress and mortality in phytoplankton. Sci. Rep. 8, 15498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zheng J, McKinnie SMK, El Gamal A, Feng W, Dong Y, Agarwal V, Fenical W, Kumar A, Cao Z, Moore BS, and Pessah IN (2018) Organohalogens naturally biosynthesized in marine environments and produced as disinfection byproducts alter sarco/endoplasmic reticulum Ca(2+) dynamics. Environ. Sci. Technol. 52, 5469–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sneed JM, Sharp KH, Ritchie KB, and Paul VJ (2014) The chemical cue tetrabromopyrrole from a biofilm bacterium induces settlement of multiple Caribbean corals. Proc. R. Soc. London, Ser. B 281, 20133086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tebben J, Tapiolas DM, Motti CA, Abrego D, Negri AP, Blackall LL, Steinberg PD, and Harder T (2011) Induction of larval metamorphosis of the coral Acropora millepora by tetrabromopyrrole isolated from a Pseudoalteromonas bacterium. PLoS One 6, e19082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Otwinowski Z, Borek D, Majewski W, and Minor W (2003) Multiparametric scaling of diffraction intensities. Acta Crystallogr., Sect. A: Found. Crystallogr. 59, 228–234. [DOI] [PubMed] [Google Scholar]
- (23).Agarwal V, Lin S, Lukk T, Nair SK, and Cronan JE (2012) Structure of the enzyme-acyl carrier protein (ACP) substrate gatekeeper complex required for biotin synthesis. Proc. Natl. Acad. Sci. U. S. A. 109, 17406–17411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Moncoq K, Regad L, Mann S, Mejean A, and Ploux O (2013) Structure of the prolyl-acyl carrier protein oxidase involved in the biosynthesis of the cyanotoxin anatoxin-a. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 2340–2352. [DOI] [PubMed] [Google Scholar]
- (25).Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, and Adams PD (2008) Automated structure solution with the PHENIX suite. Methods Mol. Biol. 426, 419–435. [DOI] [PubMed] [Google Scholar]
- (26).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (27).Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (28).Perrakis A, Sixma TK, Wilson KS, and Lamzin VS (1997) wARP: improvement and extension of crystallographic phases by weighted averaging of multiple-refined dummy atomic models. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 448–455. [DOI] [PubMed] [Google Scholar]
- (29).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Moriarty NW, Grosse-Kunstleve RW, and Adams PD (2009) electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr., Sect. D: Biol. Crystallogr. 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, and Kelleher NL (2006) Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopante-theinyl elimination reactions during tandem mass spectrometry. Biochemistry 45, 12756–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, and Walsh CT (1998) Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 37, 1585–1595. [DOI] [PubMed] [Google Scholar]
- (33).Krissinel E, and Henrick K (2007) Inference of macro-molecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. [DOI] [PubMed] [Google Scholar]
- (34).Battaile KP, Molin-Case J, Paschke R, Wang M, Bennett D, Vockley J, and Kim JJ (2002) Crystal structure of rat short chain acyl-CoA dehydrogenase complexed with acetoacetyl-CoA: comparison with other acyl-CoA dehydrogenases. J. Biol. Chem. 277, 12200–12207. [DOI] [PubMed] [Google Scholar]
- (35).Tiffany KA, Roberts DL, Wang M, Paschke R, Mohsen AW, Vockley J, and Kim JJ (1997) Structure of human isovaleryl-CoA dehydrogenase at 2.6 Å resolution: structural basis for substrate specificity. Biochemistry 36, 8455–8464. [DOI] [PubMed] [Google Scholar]
- (36).Battaile KP, Nguyen TV, Vockley J, and Kim JJ (2004) Structures of isobutyryl-CoA dehydrogenase and enzyme-product complex: comparison with isovaleryl- and short-chain acyl-CoA dehydrogenases. J. Biol. Chem. 279, 16526–16534. [DOI] [PubMed] [Google Scholar]
- (37).Fu Z, Wang M, Paschke R, Rao KS, Frerman FE, and Kim JJ (2004) Crystal structures of human glutaryl-CoA dehydrogenase with and without an alternate substrate: structural bases of dehydrogenation and decarboxylation reactions. Biochemistry 43, 9674–9684. [DOI] [PubMed] [Google Scholar]
- (38).McAndrew RP, Wang Y, Mohsen AW, He M, Vockley J, and Kim JJ (2008) Structural basis for substrate fatty acyl chain specificity: crystal structure of human very-long-chain acyl-CoA dehydrogenase. J. Biol. Chem. 283, 9435–9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kim JJ, Wang M, and Paschke R (1993) Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc. Natl. Acad. Sci. U. S. A. 90, 7523–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Roujeinikova A, Baldock C, Simon WJ, Gilroy J, Baker PJ, Stuitje AR, Rice DW, Slabas AR, and Rafferty JB (2002) X-ray crystallographic studies on butyryl-ACP reveal flexibility of the structure around a putative acyl chain binding site. Structure 10, 825–835. [DOI] [PubMed] [Google Scholar]
- (41).Goodrich AC, Harden BJ, and Frueh DP (2015) Solution structure of a nonribosomal peptide synthetase carrier protein loaded with its substrate reveals transient, well-defined contacts. J. Am. Chem. Soc. 137, 12100–12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Miyanaga A, Iwasawa S, Shinohara Y, Kudo F, and Eguchi T (2016) Structure-based analysis of the molecular interactions between acyltransferase and acyl carrier protein in vicenistatin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 113, 1802–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Maloney FP, Gerwick L, Gerwick WH, Sherman DH, and Smith JL (2016) Anatomy of the beta-branching enzyme of polyketide biosynthesis and its interaction with an acyl-ACP substrate. Proc. Natl. Acad. Sci. U. S. A. 113, 10316–10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Cryle MJ, and Schlichting I (2008) Structural insights from a P450 Carrier Protein complex reveal how specificity is achieved in the P450(BioI) ACP complex. Proc. Natl. Acad. Sci. U. S. A. 105, 15696–15701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nguyen C, Haushalter RW, Lee DJ, Markwick PR, Bruegger J, Caldara-Festin G, Finzel K, Jackson DR, Ishikawa F, O’Dowd B, McCammon JA, Opella SJ, Tsai SC, and Burkart MD (2014) Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature 505, 427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Guy JE, Whittle E, Moche M, Lengqvist J, Lindqvist Y, and Shanklin J (2011) Remote control of regioselectivity in acyl-acyl carrier protein-desaturases. Proc. Natl. Acad. Sci. U. S. A. 108, 16594–16599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Masoudi A, Raetz CR, Zhou P, and Pemble C. W. t. (2014) Chasing acyl carrier protein through a catalytic cycle of lipid A production. Nature 505, 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sarria S, Bartholow TG, Verga A, Burkart MD, and Peralta-Yahya P (2018) Matching protein interfaces for improved medium-chain fatty acid production. ACS Synth. Biol. 7, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Mitchell CA, Shi C, Aldrich CC, and Gulick AM (2012) Structure of PA1221, a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains. Biochemistry 51, 3252–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sundlov JA, Shi C, Wilson DJ, Aldrich CC, and Gulick AM (2012) Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem. Biol. 19, 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Miller BR, Drake EJ, Shi C, Aldrich CC, and Gulick AM (2016) Structures of a nonribosomal peptide synthetase module bound to MbtH-like proteins support a highly dynamic domain architecture. J. Biol. Chem. 291, 22559–22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Drake EJ, Miller BR, Shi C, Tarrasch JT, Sundlov JA, Allen CL, Skiniotis G, Aldrich CC, and Gulick AM (2016) Structures of two distinct conformations of holo-non-ribosomal peptide synthetases. Nature 529, 235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Chen WH, Li K, Guntaka NS, and Bruner SD (2016) Interdomain and intermodule organization in epimerization domain containing nonribosomal peptide synthetases. ACS Chem. Biol. 11, 2293–2303. [DOI] [PubMed] [Google Scholar]
- (54).Liu Y, Zheng T, and Bruner SD (2011) Structural basis for phosphopantetheinyl carrier domain interactions in the terminal module of nonribosomal peptide synthetases. Chem. Biol. 18, 1482–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Tanovic A, Samel SA, Essen LO, and Marahiel MA (2008) Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 321, 659–663. [DOI] [PubMed] [Google Scholar]
- (56).Reimer JM, Aloise MN, Harrison PM, and Schmeing TM (2016) Synthetic cycle of the initiation module of a formylating nonribosomal peptide synthetase. Nature 529, 239–242. [DOI] [PubMed] [Google Scholar]
- (57).Haslinger K, Brieke C, Uhlmann S, Sieverling L, Sussmuth RD, and Cryle MJ (2014) The structure of a transient complex of a nonribosomal peptide synthetase and a cytochrome P450 monooxygenase. Angew. Chem., Int. Ed. 53, 8518–8522. [DOI] [PubMed] [Google Scholar]
- (58).Uhlmann S, Sussmuth RD and Cryle MJ (2013) Cytochrome P450sky interacts directly with the nonribosomal peptide synthetase to generate three amino acid precursors in skyllamycin biosynthesis. ACS Chem. Biol. 8, 2586–2596. [DOI] [PubMed] [Google Scholar]
- (59).Gadda G (2012) Oxygen activation in flavoprotein oxidases: the importance of being positive. Biochemistry 51, 2662–2669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.