

Abstract
Objectives
Multiple invasive group A Streptococcus (GAS) infections were reported to public health by a skilled nursing facility (facility A) in Illinois between May 2014 and August 2016. Cases continued despite interventions including antibiotic prophylaxis for all residents and staff. Two other geographically close facilities reported contemporaneous outbreaks of GAS. We investigated potential reasons for ongoing transmission.
Methods
We obtained epidemiologic data from chart review of cases and review of facility and public health records from previous investigations into the outbreak. Infection control practices at facility A were observed and evaluated. Whole genome sequencing followed by phylogenetic analysis was performed on available isolates from the three facilities.
Results
From 2014 to 2016, 19 invasive and 60 noninvasive GAS infections were identified at facility A occurring in three clusters. Infection control evaluations during clusters 2 and 3 identified hand hygiene compliance rates of 14% to 25%, appropriate personal protective equipment use in only 33% of observed instances, and deficient wound-care practices. GAS isolates from residents and staff of all three facilities were subtype emm89.0; on phylogenetic analysis, facility A isolates were monophyletic and distinct.
Conclusions
Inadequate infection control and improper wound-care practices likely led to this 28-month-long outbreak of severe infections in a skilled nursing facility. Whole genome sequencing and phylogenetic analysis suggested that intrafacility transmission of a single highly transmissible GAS strain was responsible for the outbreak in facility A. Integration of genomic epidemiology tools with traditional epidemiology and infection control assessments was helpful in investigation of a facility-wide outbreak.
Keywords: Group A, Intrafacility transmission, Invasive, Outbreak, Single strain, Streptococcal disease
Introduction
Invasive group A Streptococcus (GAS) infections such as pneumonia, sepsis, necrotizing fasciitis and streptococcal toxic shock syndrome are an important cause of morbidity and mortality in the United States, with the highest rates among older adults [1]. Risk factors for invasive GAS infections (including advanced age, crowded living conditions, wounds and the presence of comorbidities such as diabetes) are common among residents of long-term care facilities (LTCFs). Incidence of invasive GAS infections among older adults residing in LTCFs is three- to eightfold higher than among community residents of the same age [2]. Outbreaks of GAS infections among residents of LTCFs have been described, with the bacteria introduced into these facilities by residents, staff or visitors and often spread by poor infection control practices and crowding [2,3].
Genotyping of GAS isolates through pulsed-field gel electrophoresis, emm typing and multilocus sequence typing [4] (MLST) complement epidemiologic investigations of outbreaks at LTCFs by ascertaining whether infections are caused by similar strains. However, the discriminatory power of these techniques is limited; two GAS isolates with the same pulsed-field gel electrophoresis profile, emm or MLST type might indicate reintroduction from the surrounding community rather than a common source of infection within a facility. This is especially true for outbreaks caused by strains within common GAS clonal complexes, which often share identical genotyping profiles despite being separated from a common ancestor by decades [5]. In prolonged GAS outbreaks in LTCFs, it is important to determine whether cases are part of a single propagated outbreak related to continuing intrafacility transmission of a single strain or represent repeated introductions of closely related GAS strains from the community. The first suggests failure to detect and control infection sources within the facility, while the second suggests a need for screening and control measures to target new residents and visitors. A higher-resolution genetic analysis is needed in such outbreaks. Whole genome sequencing (WGS) allows detection of single nucleotide polymorphisms (SNPs) for increased resolution of GAS outbreak isolates [3,5,6].
We describe the investigation of a large, prolonged GAS outbreak at an LTCF and the use of WGS to elucidate potential disease transmission patterns in support of the epidemiologic investigation and disease control efforts.
Methods
Illinois Department of Public Health and Centers for Disease Control and Prevention (CDC) institutional review board approval was not required because the activity was carried out as a response under public health authority.
Setting
Facility A is a large skilled nursing facility in city A in Illinois, accepting residents needing long-term care, short-term rehabilitation and skilled nursing. From May to July 2014, a cluster (cluster 1) of GAS infections due to subtype emm89.0 among residents at facility A was reported to local public health authorities. From February to April 2015, a second cluster of infections due to emm89.0 (cluster 2) was identified. Cases ceased briefly following facility-wide chemoprophylaxis of all residents and staff between 28 April and 2 May 2015. Multiple emm89.0 GAS infections recurred among residents beginning 30 June 2015 (cluster 3).
Case definitions
GAS infections occurring from May 2014 were considered cases. Among both residents and staff, we defined an invasive case as GAS cultured from a normally sterile site (e.g. blood) and a noninvasive case as signs and symptoms consistent with GAS infection in someone from whom GAS is cultured from a nonsterile site (e.g. throat, wound) or detected from the throat by a rapid antigen detection test. Asymptomatic residents or staff members from whom GAS was isolated from a nonsterile site were considered colonized. A recurrent case was defined as more than one invasive or noninvasive infection in the same individual identified >1 month apart.
Epidemiologic investigations
We reviewed hospital records from large hospitals in city A to augment case finding. Facility A staff and residents were screened for GAS colonization to identify and treat potentially unrecognized but persistent sources of GAS transmission. We swabbed the oropharynx, any wounds and insertion sites of invasive devices (e.g. central lines, urinary catheters) of residents and the oropharynx and any self-identified skin lesions of staff. Swabs were cultured for GAS using standard methods. During investigations into clusters 2 and 3, teams from the Illinois Department of Public Health and the CDC completed infection control evaluations involving direct observations of activities, walk-throughs of areas at the facility and review of policy documents and facility records.
Investigation of contemporaneous cluster
Two LTCFs in city B, 40 miles from city A, reported outbreaks of invasive GAS infections caused by subtype emm89.0 beginning in May 2015 and involving residents and a staff member associated with both city B facilities. Because of the geographic and temporal proximity of these outbreaks to the facility A outbreak, we searched for staff, consultants or residents with links to both facility A and one of the city B facilities.
Characterization of isolates
Available GAS isolates from ill and colonized residents and staff from the facility A and a convenience sample of isolates from city B LTCFs, patients with invasive and noninvasive GAS infections hospitalized in city A between December 2014 and November 2015 (but with no known links to facility A) and GAS-infected patients from other Illinois cities were emm sequence typed at the CDC Streptococcus lab [7]. For genotyping, WGS and library construction was performed using the MiSeq Personal Sequencer Instrument (Illumina, San Diego, CA, USA) as previously described [8].
Bioinformatic analysis
We analysed the sequence data using an automated GAS typing pipeline (https://github.com/BenJamesMetcalf) that determines multiple strain features including emm subtype, MLST type, resistance features, presence or absence of several virulence-related genes, and chromosomal regulatory alterations. Sequence data from facility A isolates were compared with data from isolates described above and isolates from patients with invasive GAS infections in 2015 from 10 states as reported through the Active Bacterial Core surveillance (ABCs), as follows: California (3-county San Francisco Bay area), Colorado (5-county Denver area), Connecticut, Georgia (20-county Atlanta area), Maryland (6-county Baltimore area), Minnesota, New Mexico, New York (15-county Rochester and Albany areas), Oregon (3-county Portland area), and Tennessee (20 urban counties) [9]. A core SNP phylogenetic tree was generated for the above genomes using methods described in the Supplementary Methods. To track the evolution of the isolates over the course of the prolonged outbreak in facility A, we performed a time-scaled phylogenetic analysis (Supplementary Methods).
Results
Epidemiologic investigation
From May 2014 through August 2016, 19 invasive and 36 noninvasive (30 wound infections, five pharyngitis and one urinary tract infection) GAS infections were identified among 50 residents at facility A (Fig. 1) (Supplementary Table S1), leading to four deaths. Five residents had recurrent infections (one resident had two invasive infections, two had an invasive and noninvasive infection, and one had two noninvasive infections). From February 2015 through February 2016 (clusters 2 and 3), 24 staff were identified as having GAS pharyngitis. No data were available on staff illnesses during cluster 1. Across the three clusters, three colonization surveys conducted among residents and four among staff identified 11 residents and 18 staff colonized with GAS (Supplementary Table S1).
Fig. 1.
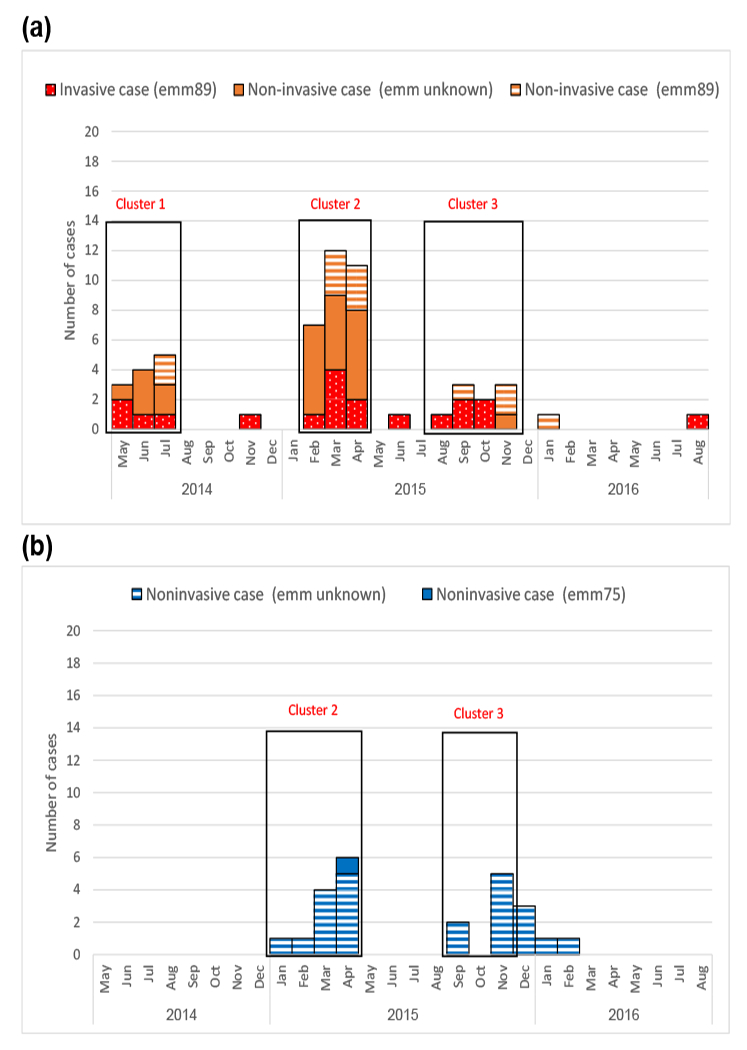
(a) Epidemic curve of cases of GAS infections among residents at facility A, May 2014 to August 2016. (b) Epidemic curve of cases of GAS infections among staff at facility A, May 2014 to August 2016. GAS, group A Streptococcus.
Assessment of infection control at facility A
Lapses identified on infection control evaluation during cluster 2 included a hand hygiene compliance rate of 14%, no policy for promotion of alcohol-based hand-rub dispensers and lack of knowledge among staff of appropriate use of personal protective equipment. During cluster 3, hand hygiene compliance was 25% (three noted of 12 observed opportunities for hand hygiene during resident care activities), personal protective equipment use was appropriate in two (33%) of six use instances while caring for patients with contact precautions, and no clear separation of dirty or contaminated supplies from clean supplies was noted during observed wound-care activities. Our case-finding activities identified seven staff with GAS pharyngitis from May to October 2015 that were not captured by the facility’s employee infection tracking log.
Investigation of contemporaneous GAS cluster
No facility A staff reported having worked at either of the facilities from the two-facility cluster in city B. However, we could not entirely rule out residents or staff at facility A having any epidemiologic links to the facilities at city B.
Characterization of isolates
The CDC Streptococcus lab received isolates from all 19 invasive cases and 12 of 36 noninvasive cases among residents from facility A; all were identified to be emm89.0. The single isolate received from one of the 24 staff pharyngitis cases was emm75.0. All isolates received from colonization surveys (including isolates from all 11 colonized residents and nine of 18 colonized staff) were emm89.0. We sequenced 59 isolates from the 51 facility-linked cases, with good assembly data available on 58 of 59 isolates. A good assembly was defined as having less than 150 contigs, a N50 greater than 30,000 and an assembly size greater than 1,650,000 bases. All 58 emm89.0 isolates shared identical genomic typing pipeline features, including pilus type T89 and MLST sequence type (ST) 101. They were uniformly positive for surface protein genes mrp, enn, fbaA, prtF2 and sof, and exotoxin genes speG and smeZ; negative for the capsule biosynthesis gene hasA; and contained an nga operon promoter sequence (Pnga-3).
GAS isolates recovered from 35 sporadic cases (14 invasive and 21 noninvasive) from hospitals in city A comprised 19 different emm types; the only two isolates identified as emm89.0 were sequenced. We also sequenced emm89.0 isolates cultured from ten cases from the two-facility cluster in city B, five cases from other cities in Illinois and 68 cases reported from various ABCs sites in 2015. The isolates from city B and non—facility A—linked isolates from city A shared identical pipeline features as facility A isolates, with the exception of being additionally positive for the speC exotoxin gene. The 68 emm09.0 ABCs isolates included in the phylogenetic analysis also shared the same pipeline features except for some variation within exotoxin profiles, and two ABCs isolates were negative for the mrp gene query.
Core SNP phylogeny of outbreak and contemporary emm89.0 isolates
We assessed the relatedness between the emm09.0 isolates from the facility A outbreak and the two-facility city B cluster using a core SNP phylogenetic tree comprising the isolates described above. Non-outbreak isolates were included to provide the background level of genetic diversity found within a broad sampling of closely related contemporary emm89.0 strains, allowing the genetic relatedness of outbreak isolates to be placed within proper phylogenetic context. All but one of the facility A isolates clustered on a single branch of the tree, were highly related with an average pairwise difference of 3.42 SNPs and were clearly separate from the city B isolates that formed a distinct clade (Fig. 2).
Fig. 2.
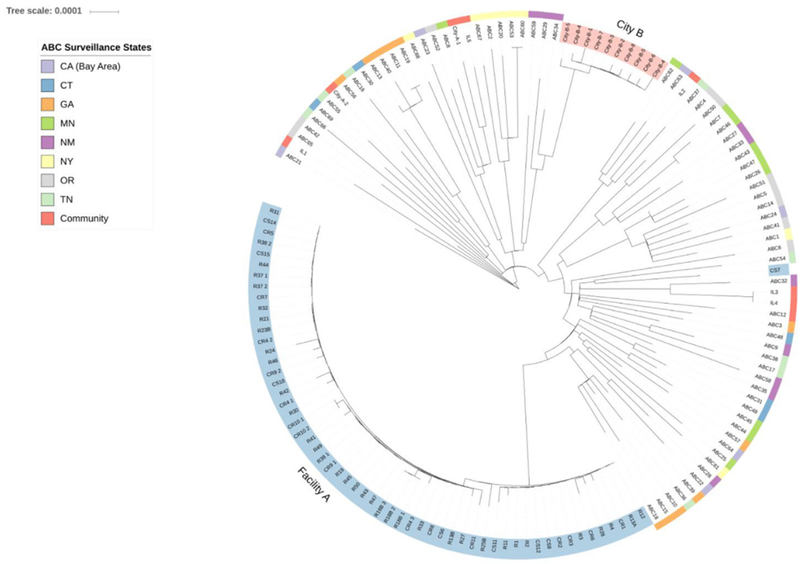
Phylogenetic tree comprising emm89.0 isolates from facility A and two-facility cluster at city B, along with other emm89.0 isolates from Illinois (community) and other ABCs sites: CA, California; CT, Connecticut; GA, Georgia; MN, Minnesota; NM, New Mexico; NY, New York; OR, Oregon; TN, Tennessee. Sample information is encoded within tip labels. Tip labels shaded blue correspond to facility A and those shaded pink correspond to city B. For samples from facility A, labels that begin with ‘C indicate GAS-colonized sample, while ‘S’ and ‘R’ designation represent staff and resident, respectively. Number after ‘S’ or ‘R’ is staff or resident index number. Cases where multiple samples were taken from individual at different time points are coded by letter after index number. When multiple samples were taken from same individual at same time point, numeric identifier is assigned following underscore. Isolates from city B facilities are labeled as city-B-X. Isolates from city A but not linked to facility A are labeled as city-A-X. Isolates from rest of Illinois are labeled as ILx and ABCs isolates as ABCx. GAS, group A Streptococcus.
Time-scaled phylogeny of isolates from facility A
Time-scaled phylogeny of the 57 facility A isolates which formed a monophyletic clade on the phylogenetic tree (time to most recent common ancestor (tMRCA) analysis; Fig. 3) indicated that the common ancestor emerged around 15 January 2012 (30 April 2008 to 15 October 2013; 95% highest probability density). The substitution rate of 6.86 × 10−7 per site per year (2.72 × 10−7 to 1.14 × 10−6; 95% highest probability density) was consistent with previous estimates of substitution rates in GAS [10]. Isolates obtained after the April—May 2015 chemoprophylaxis intervention (Fig. 3, green line) clustered within one clade (Fig. 3, highlighted in yellow). A number of postprophylaxis strains, including one from an asymptomatic wound-care nurse (CS18), clustered into a sub-clade (Fig. 3, highlighted in blue). To quantify the strength of this ancestral signal, tips of the time-scaled phylogenetic tree were coded as either preprophylaxis or postprophylaxis. The significance of this phylogeny—trait association was tested by BaTS 0.2.3 (Supplementary Table S2). Both the parsimony score and the association index found strong evidence (p <0.005) to support clustering of postprophylaxis samples on the tree.
Fig. 3.
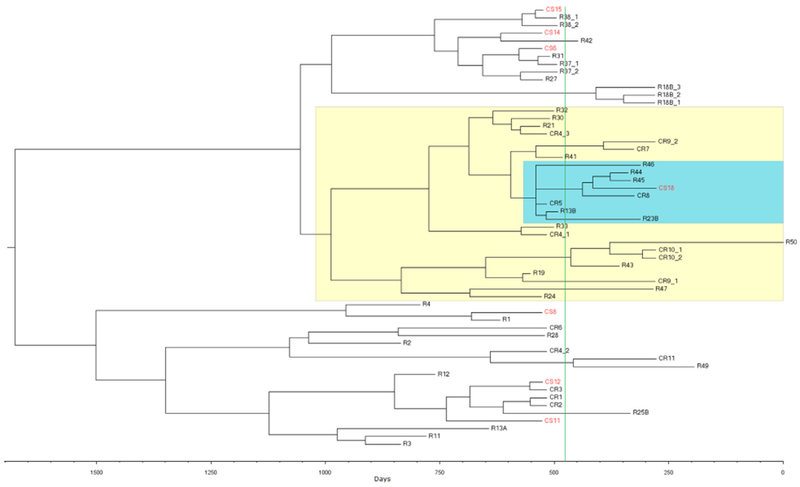
Time-scaled phylogeny of 57 facility A isolates which formed monophyletic clade on phylogenetic tree, generated using methods detailed in Supplementary Materials. Scale axis represents number of days preceding 20 August 2016 sampling date of most recent outbreak isolate. Green vertical line indicates chemoprophylaxis treatment at end of April 2015. Clade highlighted in yellow suggests possible cluster of postchemoprophylaxis isolates. Clade branch highlighted in blue contains samples highly related to wound-care nurse CS18. Sample information is encoded within tip labels. Labels that begin with ‘C indicate GAS-colonized sample, while ‘S’ and ‘R’ designation represent staff and resident, respectively. Staff samples are also highlighted in red. Number after ‘S’ or ‘R’ is staff or resident index number. Cases where multiple samples were taken from same individual at different time points are coded by letter after index number. If multiple samples were taken from same individual at same time point, numeric identifier is assigned after underscore. GAS, group A Streptococcus.
Discussion
We report a prolonged outbreak of GAS infections that lasted 28 months and included 19 invasive and 60 noninvasive cases among residents and staff at an LTCF. All facility A isolates except one belonged to subtype emm89.0. Because of its frequency in the United States and the near-identical WGS pipeline features shared between most current emm89.0 isolates, comparisons at the SNP level were essential to discriminate among the many isolates collected during this investigation (Supplementary Fig. S1). Phylogenetic analysis indicated that the isolates from the facility formed a different monophyletic clade compared to a contemporaneous outbreak at two geographically proximal facilities and to a broad sampling of closely related contemporary US isolates. Through Bayesian evolutionary analysis, tMRCA for isolates from the facility A outbreak was calculated to predate the first detected case by approximately 15 months. This strongly suggests that the outbreak resulted from intrafacility transmission of a single strain. If instead there had been multiple separate introductions of GAS emm89.0 into facility A by visitors, staff or new residents, tMRCA would be expected to occur significantly earlier than observed.
Although the duration of the outbreak attests to the strain’s transmissibility and virulence, we believe its persistence at the facility was likely related to poor infection control practices. Despite specific recommendations, lapses in hand hygiene and disinfection were repeatedly observed. We also observed infection control lapses during wound care (e.g. no clear separation of dirty/contaminated supplies from clean supplies), which would have increased the risk for GAS transmission.
In addition to improving infection control, facility A screened for and treated staff and residents colonized with GAS on several occasions through this prolonged outbreak. Failure of these actions to prevent new cases during cluster 2 prompted the facility to initiate mass antibiotic treatment for all staff and residents with a regimen of either benzathine penicillin G + rifampin or cephalexin [11,12]. Unlike other instances where mass prophylaxis led to outbreak control [3,13,14], cases recurred after a brief hiatus at facility A. Temporal phylogenetic analysis and pre- vs. postchemoprophylaxis trait association of facility A isolates supported clustering of postprophylaxis samples into a subclade. The type of clustering can provide insight into potential factors driving postprophylaxis infections, suggesting that chemoprophylaxis was ineffective (Fig. 4a), that it was partially effective with transmission continuing but limited to certain individuals (Fig. 4b) or that a separate strain was introduced (Fig. 4c). The clustering seen here suggests partial impact with continuation of the outbreak confined to a limited number of staff or patients as in Fig. 4b, and not a complete failure of prophylaxis. There could be several reasons for this. Not all staff consented to the prophylaxis. While GAS can also colonize the skin, rectum and vagina, staff who refused prophylaxis were permitted to return to work after only oropharyngeal screening. Further, antibiotic prophylaxis regimens are not 100% effective in eliminating asymptomatic GAS colonization, and some of the treated individuals could have continued to be colonized and remain sources of infection [15].
Fig. 4.

Schematic illustrating how BaTS AI score was used to discern whether chemoprophylaxis treatment had significant impact on stopping spread of infection. (a) Chemoprophylaxis was ineffective. (b) Chemoprophylaxis was partially effective, with transmission continuing but limited to certain individuals. (c) Chemoprophylaxis effective but a separate strain was introduced. Blue and orange nodes indicate pre- and postchemoprophylaxis strains, respectively; chemoprophylaxis event is represented by green vertical line. Each time-measured phylogeny represents different postprophylaxis outcome. In all three cases, there are 14 postprophylaxis strains, but each tree differs by amount of postprophylaxis clustering. AI, association index.
Our use of SNP phylogeny to differentiate between contemporaneous clusters at different LTCFs was similar to a previous report [3]. Additionally, we used genomic data to investigate whether multiple clusters of a single emm type were part of a single outbreak or separate outbreaks caused by multiple, subtly distinct strains and to reflect on effect of mass chemoprophylaxis as a control measure during this particular outbreak.
We tracked the transmission of a single strain of emm89.0 GAS among both vulnerable residents and healthy workers within a single facility. The unusual duration and magnitude of this outbreak highlights the importance of maintaining good infection control practices at skilled nursing care facilities. Besides helping understand transmission at the facility, use of WGS enabled us to differentiate the outbreak from another contemporaneous outbreak with the same emm type, helping guide the public health response to these outbreaks. When possible, facility outbreak investigations could benefit from integrating traditional epidemiology and infection control evaluations with available genomic epidemiologic tools.
Supplementary Material
Acknowledgments
Transparency declaration
Supported in part by the CDC and the Illinois Department of Public Health. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the CDC. All authors report no conflicts of interest relevant to this article.
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.cmi.2018.04.034.
References
- [1].Centers for Disease Control and Prevention. Active bacterial core surveillance (ABCs), Emerging Infections Program Network, group A Streptococcus surveillance report—2014. Available at: http://www.cdc.gov/abcs/reports-findings/survreports/gas14.pdf.
- [2].Ht Jordan, Richards CL Jr Burton DC, Thigpen MC Van Beneden CA. Group A streptococcal disease in long-term care facilities: descriptive epidemiology and potential control measures. Clin Infect Dis 2007;45:742–52. [DOI] [PubMed] [Google Scholar]
- [3].Chalker VJ, Smith A, Al-Shahib A, Botchway S, Macdonald E, Daniel R, et al. Integration of genomic and other epidemiologic data to investigate and control a cross-institutional outbreak of Streptococcus pyogenes . Emerg Infect Dis 2016;22:973–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Spellerberg B, Brandt C. Laboratory diagnosis of Streptococcus pyogenes (group A streptococci) In: Ferretti JJ, Stevens DL, Fischetti VA, editors. Streptococcus pyogenes: basic biology to clinical manifestations. Oklahoma City, OK: University of Oklahoma Health Sciences Center; 2016. [PubMed] [Google Scholar]
- [5].Ben Zakour NL, Venturini C, Beatson SA, Walker MJ. Analysis of a Streptococcus pyogenes puerperal sepsis cluster by use of whole-genome sequencing. J Clin Microbiol 2012;50:2224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Turner CE, Dryden M, Holden MT, Davies FJ, Lawrenson RA, Farzaneh L, et al. Molecular analysis of an outbreak of lethal postpartum sepsis caused by Streptococcus pyogenes . J Clin Microbiol 2013;51:2089–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li Z, Sakota V, Jackson D, Franklin AR, Beall B. Active Bacterial Core Surveillance/Emerging Infections Program Network. Array of M protein gene subtypes in 1064 recent invasive group A Streptococcus isolates recovered from the active bacterial core surveillance. J Infect Dis 2003;188:1587–92. [DOI] [PubMed] [Google Scholar]
- [8].Metcalf BJ, Chochua S, Gertz RE Jr, Li Z, Walker H, Tran T, et al. Using whole genome sequencing to identify resistance determinants and predict antimicrobial resistance phenotypes for year 2015 invasive pneumococcal disease isolates recovered in the United States. Clin Microbiol Infect 2016;22 1002.e1–e8. [DOI] [PubMed] [Google Scholar]
- [9].Langley G, Schaffner W, Farley MM, Lynfield R, Bennett NM, Reingold A, et al. Twenty years of active bacterial core surveillance. Emerg Infect Dis 2015;21: 1520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Turner CE, Abbott J, Lamagni T, Holden MT, David S, Jones MD, et al. Emergence of a new highly successful acapsular group a Streptococcus clade of genotype emm89 in the United Kingdom. MBio 2015;6:e00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shulman ST, Bisno AL, Clegg HW, Gerber MA, Kaplan EL, Lee G, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis 2012;55:1279–82. [DOI] [PubMed] [Google Scholar]
- [12].Public Health Agency of Canada. Supplement: guidelines for the prevention and control of invasive group A streptococcal disease. Canada Communicable Disease Report 32S2. 2006. Available at: https://www.canada.ca/en/public-health/services/reports-publications/canada-communicable-disease-report-ccdr/monthly-issue/2006-32/canada-communicable-disease-report.html.
- [13].Dooling KL, Crist MB, Nguyen DB, Bass J, Lorentzson L, Toews KA, et al. Investigation of a prolonged group A streptococcal outbreak among residents of a skilled nursing facility, Georgia, 2009–2012. Clin Infect Dis 2013;57: 1562–7. [DOI] [PubMed] [Google Scholar]
- [14].Smith A, Li A, Tolomeo O, Tyrrell GJ, Jamieson F, Fisman D. Mass antibiotic treatment for group A Streptococcus outbreaks in two long-term care facilities. Emerg Infect Dis 2003;9:1260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].DeMuri GP, Wald ER. The group A streptococcal carrier state reviewed: still an enigma. J Pediatric Infect Dis Soc 2014;3:336–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.