

SUMMARY
The effector B cell lymphoma-2 (BCL-2) protein BCL-2 ovarian killer (BOK) induces mitochondrial outer membrane permeabilization (MOMP) to initiate apoptosis upon inhibition of the proteasome. How BOK mediates MOMP is mechanistically unknown. The NMR structure of the BCL-2 core of human BOK reveals a conserved architecture with an atypical hydrophobic groove that undergoes conformational exchange. Remarkably, the BCL-2 core of BOK spontaneously associates with purified mitochondria to release cytochrome c in MOMP assays. Alanine substitution of a unique glycine in helix α1 stabilizes BOK, as shown by thermal shift and urea denaturation analyses, and significantly inhibits MOMP, liposome permeabilization, and cell death. Activated BID does not activate WT BOK or the stabilized alanine mutant to promote cell death. We propose that BOK-mediated membrane permeabilization is governed in part by its unique metastability of the hydrophobic groove and helix α1 and not through activation by BH3 ligands.
In Brief
Zheng et al. determined the structure of BOK, revealing intrinsic instability (metastability) in putative regulatory regions and supporting BOK’s independence of activators such as tBID. Structure-based stabilizing mutation in one of the metastable regulatory regions inhibits BOK-mediated liposome permeabilization, MOMP, and apoptosis, establishing a paradigm for apoptosis initiation.
Graphical Abstract
INTRODUCTION
Mitochondrial apoptosis is the most common form of programmed cell death in mammalian health and disease (Hotchkiss et al., 2009). Central to apoptosis is the regulation of mitochondrial outer membrane permeabilization (MOMP) by the B cell lymphoma-2 (BCL-2) family proteins (Chipuk et al., 2010). MOMP is directly affected in diseases such as cancer, wherein pro-apoptotic proteins are downregulated and anti-apoptotic BCL-2 proteins are upregulated (Hanahan and Weinberg, 2011). MOMP is the most robust initiation event in apoptosis that triggers the release of cytochrome c (cyt c), which in turn activates the apoptosome-caspase axis that dismantles cells (Green, 2005). MOMP is executed by the effector BCL-2 family proteins BCL-2 antagonist killer (BAK), BCL-2-associated × protein (BAX), and BCL-2 ovarian killer (BOK) (Czabotar et al., 2014; Llambi et al., 2016; Moldoveanu et al., 2014).
BOK is a non-canonical effector, which appears to act alone to execute MOMP (Brem and Letai, 2016; Llambi et al., 2016). BOK is degraded post-translationally by the endoplasmic reticulum-associated degradation (ERAD)-proteasome system (Llambi et al., 2016). BOK protein turnover is tightly controlled to maintain its levels at low to undetectable in cells expressing BOK mRNA (Echeverry et al., 2013; Llambi et al., 2016). Only when the ERAD-proteasome system is blocked genetically or pharmacologically is the BOK protein stabilized and its role as a MOMP effector is revealed (Llambi et al., 2016). In the absence of BAK and BAX, BOK does not appear to crosstalk with the BCL-2 family protein-protein interaction network to induce MOMP, because it is refractory to antagonism by anti-apoptotic BCL-2 proteins and synergism with BH3-only proteins (Llambi et al., 2016). However, a recent study suggested that BID may facilitate BOK-mediated permeabilization of liposomes (Fernández-Marrero et al., 2017). Deletion of BOK in mice has a minimal effect on organismal homeostasis or the ability of cells derived from bok−/− mice to undergo apoptosis (Carpio et al., 2015; Ke et al., 2012; Llambi et al., 2016), except under some conditions that induce ER stress in cell culture (Carpio et al., 2015). Nonetheless, genetic studies support the idea that BOK is an effector of MOMP in the hematopoietic system (Ke et al., 2013, 2015). BOK may play a role in other cellular processes such as Ca2+ signaling at the ER, where it can interact with inositol 1,4,5-triphosphate receptors (IP3Rs) (Schulman et al., 2013, 2016).
Uncovering the mechanism by which BOK induces MOMP is essential to our understanding of apoptosis. Here, we report the nuclear magnetic resonance (NMR) structure of human BOK and demonstrate that BOK-mediated membrane permeabilization is poorly regulated by weak binding of BH3 ligands to the canonical hydrophobic groove; instead, membrane permeabilization is governed in part by the metastability of BOK.
RESULTS
Enhanced Membrane Recruitment Facilitates Permeabilization by BOK
The MOMP effector function of BOK was studied in liposome permeabilization assays, which monitor dye release from large unilamellar vesicles (LUVs) that mimic the composition of the outer mitochondrial membrane and contain the 1,2-dioleoylsn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid) succinyl] (nickel salt) (DGS-NTA[Ni2+]) lipid for efficient recruitment of His-tagged effectors (Asciolla et al., 2012; Henderson et al., 2007; Llambi et al., 2016; Lovell et al., 2008; Oh et al., 2006, 2010). We previously showed that a transmembrane region-deleted (ΔTM) C-terminal His-tagged (CH) hBOK construct can induce LUV permeabilization (LUVP) in the absence of BH3 peptides (FL [full-length]-BOK-ΔTM-CH, Figure 1A) (Llambi et al., 2016). This is consistent with the transmembrane region (TM) being dispensable for membrane permeabilization by BOK, although it is required for its efficient mitochondrial targeting in cells (Llambi et al., 2016). After several rounds of optimizing BOK protein production in Escherichia coli, based on prior knowledge of folded BCL-2 cores and bioinformatics analysis, a construct amenable to structural characterization was obtained. The optimized BOK construct was truncated at N and C termini (ΔN20-BOK-ΔC35; Figure S1A) to represent the BCL-2 globular core devoid of its C-terminal putative transmembrane-targeting region and the typically unstructured N-terminal region. To improve protein yield and to prevent detrimental oxidation in vitro, several amino acids were substituted, such as C67A, P100R, C137A, and C152A, with and without C177A (PR3CA or PR4CA, respectively; Figures 1A, S1A, and S1B). For biochemical characterization, the CH variant ΔN-BOK-ΔTM-CH was purified. For NMR analysis, the His tag was removed from PR3CA NH (N-terminal His tagged)-ΔN-BOK-ΔTM with tobacco etch virus (TEV) protease to generate the PR3CA BCL-2 core of BOK (Figures 1A, S1A, and S1B).
Figure 1. BOK-Mediated Membrane Permeabilization Correlates with Its Metastability.

(A) Human BOK constructs used in this study. The BCL-2 homology regions (BH1–BH4), TM, residue numbers, and tags are indicated for BOK constructs.
(B) LUVP measured as fluorescent dye release by FL-BOK-DTM-CH was normalized relative to permeabilization by 2.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS). Normalized data were analyzed by Simpson’s rule AUC at 32 min. Data represent the average and SD for one of three experiments performed in triplicate.
(C) Combined average and SE AUC at 32 min for the BOK constructs. Related to Figure S1C.
(D) Combined average and SE of BOK concentration required to achieve half-maximal AUC at 32 min for the BOK constructs (related to Figure S1D).
(E) MOMP assays of purified bak−/− mitochondria were performed for 1 hr at 37°C for monitoring cyt c release and protein loading.
(F) Thermal denaturation measured by TSA at 0.25 mg/mL protein was performed using the manufacturer’s buffer conditions. Data represent average melting temperature (Tm) and SD of replicate performed in quadruplicate.
See also Figure S1.
Similar to FL-BOK-ΔTM-CH (Llambi et al., 2016), the N-terminal truncated ΔN-BOK-ΔTM-CH construct with yield-promoting PR4CA substitution (PR4CA-CH; Figures 1A and S1A) was active (Figures 1B–1D, S1C, and S1D). For functional comparison, LUVP data were analyzed by Simpson’s rule to calculate the area under the curve (AUC) (Burden and Faires, 2000). AUC analysis at 32 min showed that the PR4CA-CH was slightly more active than FL-BOK-ΔTM-CH (Figures 1C and S1C). AUC plotted by BOK concentration (Figure S1D) had a sigmoidal profile that plateaued at 200 nM, with a half-maximal change of ~40 nM (Figure 1D), suggesting that the original and improved constructs have similar activities in LUVP assays.
PR4CA-CH-BOK and an analogous construct BAK-ΔTM-CH were tested in MOMP assays, using mitochondria purified from bak−/− mouse liver. PR4CA-CH released cyt c completely at 5 mM, whereas BAK-ΔTM-CH was inactive (Figures 1E and S1E), which is consistent with our previous report (Moldoveanu et al., 2006). Ponceau S staining of the supernatant (S) and pellet (P) of the MOMP reactions indicated that more than half of the PR4CA-CH associated with the mitochondria, but BAK-ΔTM-CH did not (Figures 1E and S1E). To permeabilize membranes, effectors must undergo conformational changes from soluble to membrane-associated species. The spontaneous membrane association suggests that unlike BAK, the transition of BOK to the active conformation may occur even without help from direct activators. We attribute this transition in conformation to the intrinsic instability of BOK, which was supported by the observation that the melting temperature (Tm) for BOK was 10°–15°C lower than for BAK-ΔTM-CH in thermal shift assays (TSAs; Figure 1F).
Intrigued by the apparent correlation between membrane permeabilization and instability, we investigated single mutants of the PR4CA in ΔN-BOK-ΔTM-CH. Only the P100R, C67A, and C152A mutants but not the C137A and C177A expressed and were purified to homogeneity (Figure S1F). In LUVP assays, the wild-type (WT) and PR4CA ΔN-BOK-ΔTM-CH were similarly active, as indicated by AUC analysis at 32, 16, and 8 min (Figures S1G and S1H). In contrast, the point mutants P100R, C67A, and C152A were significantly slower kinetically, achieving lower AUC levels compared to the WT (Figures S1G and S1H). In TSAs, P100R, C152, and PR4CA mutants exhibited significantly higher Tm values compared to WT, suggesting their stabilization of the fold at two different pH values (Figure S1I). In addition to close to physiological pH 6.8, we performed TSAs at pH 5.0 to mimic the conditions at which the PR4CA construct was most stable for NMR-based structure determination. Accordingly, we observed a decrease in Tm values for the WT and all of our mutants at pH 6.8 compared to pH 5.0, supporting the stabilization of the domain at low pH. We did not obtain enough protein for analysis of the C67A because of its low expression yield. Our point mutant analysis suggests that P100R and C152A substitutions significantly stabilize the fold, thereby slowing down the kinetics of membrane permeabilization. The additive effect of these mutations in stabilizing the PR4CA mutant is possibly counteracted by the point cysteine mutants that we could not produce. We sought to determine the structure of BOK to shed light on the molecular basis of its intrinsic instability, which we refer to as metastability.
NMR Structure of Human BOK Reveals a Typical Globular BCL-2 Core
The 15N-1H transverse relaxation optimized spectroscopy (TROSY) spectrum of 13C,15N-PR3CA BOK suggested that it is well folded, showing a 1H chemical shift dispersion of 6–10 ppm (Figure 2A). The backbone was assigned, and Cα chemical shift was plotted to predict the secondary structure of BOK (Figure S2A). The BCL-2 core of BOK contains eight a helices similar to those of folded BCL-2 proteins (Figure S2A) (Chipuk et al., 2010; Moldoveanu et al., 2006). The backbone of the stabilizing mutant G35A PR3CA (described in detail later) also was assigned, which is globally similar to WT PR3CA, as observed by the localized chemical shift perturbation (CSP) observed between them (Figures 2A and S2A).
Figure 2. NMR Structure of Human BOK Reveals a Typical BCL-2 Core.

(A) Overlay of 1H-15N TROSY spectra of the BCL-2 core of BOK, PR3CA ΔN20-BOK-ΔC35, and the G35A mutant, with some assignments illustrating chemical shift perturbation between the two.
(B) Cartoon representation of the NMR structure of the BCL-2 core of BOK. BH1–BH4 regions are colored as in Figure 1A.
(C) Protein dynamics analyses including backbone 1H-15N transverse (R2) and longitudinal (R1) relaxation and hetNOE analysis of WT and G35A PR3CA revealed two flexible regions, the α1–α2 and the α2–α4 connecting regions, both having hetNOE values lower than the domain average. The R2, R1, and τc analyses indicated that the α1–α2 and α2–α4 connecting regions had faster and slower motions than the domain average, respectively, suggesting nanosecond to picosecond dynamics for the former and chemical (conformational) exchange microsecond to millisecond dynamics for the latter. The unique properties of the α2–α4 connecting region, which also harbors the short helix α3, likely contribute to the overall meta-stability of BOK. The stabilizing G35A mutant in helix a1 reduces but does not abolish the dynamics in the α2–α4 connecting region, suggesting a global effect on BOK metastability.
See also Figure S2.
The NMR structure of the BCL-2 core of BOK (Figures S2B and S2C) revealed that it adopts a fold typical of globular BCL-2 family proteins, consisting of seven amphipathic α helices that wrap around the core hydrophobic helix α5 (Figure 2B) (Petros et al., 2004). Helices α1 and α2 are linked by a 15-residue loop that showed no secondary structure and exhibited disorder in the 20 NMR structures (Figures S2A and S2B). This region exhibits divergence in sequences among species (Figure S2D). We refer to the α1–α2 region (residues 21–80) as the N-terminal bundle (Moldoveanu et al., 2006). The N-terminal bundle precedes a stretch of seven residues (RPSVYRN) that orient a short helix α3 toward the core helix α5 (Figure 2B). The C-terminal bundle, made up of helices α3–α6 of the BCL-2 core, shows the characteristic packing seen in other BCL-2 proteins, with well-defined short turns linking the four a helices. The BH2 region, made up of helices α7 and α8, seals the circular arrangement of helices by stabilizing the hydrophobic core with two tryptophan residues that are conserved among globular BCL-2 proteins (Figures 2B and S2D).
Conformational Exchange in the Hydrophobic Groove Governs the Dormant State of BOK
Examination of the hydrophobic groove in BOK revealed a unique conformation imposed by the region between helices α2 and α4 (Figure 2B). To determine the role of the hydrophobic groove conformation in BOK regulation, protein dynamics were analyzed by NMR. Backbone 15N-1H heteronuclear nuclear Overhauser effects (hetNOEs) and longitudinal (spin-lattice) and transverse (spin-spin) relaxation analyses suggested that the region between helices α2 and α4 (P82–E99) exhibits flexibility and slower motion (milliseconds) than the domain average (Figures 2C). This region had lower {1H}−15N hetNOE values (~0.68) than the domain average (~0.75), suggesting intrinsic flexibility (Kleckner and Foster, 2011). The higher-than-average R2 values and the similar-to-domain average R1 values indicate chemical exchange on the microsecond to millisecond timescale and suggest the presence of conformational exchange in this region (Kleckner and Foster, 2011). The rotational correlation time (τc), which also reports on internal motions, was calculated based on the T1:T2 ratio and corroborates the unique dynamic properties of this region (Figure 2C). Residues in the P82–E99 regions exhibit much higher τc values than the domain average, and Y85 exhibited the highest τc. In contrast, the α1–α2 loop region (L48–R62) showed a difference in dynamic properties with {1H}−15N hetNOEs (~0.48) and below-average R2 (~7 Hz) and τc values, supporting its disorder and much faster internal motion than the domain average (nanoseconds to picoseconds). Similar to Y85 on the N-terminal side of helix α3, E99, found between α3 and α4, exhibits the second-highest R2 and τc values, indicating conformational exchange on the microsecond to millisecond timescale for this region (Figures 2B and 2C). The P100R mutant forms a salt bridge with E99 and may stabilize this region, corroborating its higher Tm values in TSAs compared to the WT (Figure S1I). Our results suggest that the dynamics in the hydrophobic groove of BOK may contribute to metastability and gate BH3 ligand binding through conformational exchange.
Atypical Hydrophobic Groove of BOK Gates Access of BH3 Ligands
To gain insights into its structural similarity with family members, pairwise structural alignments of BOK were performed with mammalian globular BCL-2 proteins identified using top primary sequence alignment predictions by the HHpred homology detection and structure prediction server (Biegert et al., 2006). The BCL-2 core of BOK aligned most closely with that of BAX, followed by those of BCL-xL, BCL-w, MCL-1, and BCL-2 (root-mean-square deviation <2.5Å), and aligned more distantly with those of BAK and A1 (root-mean-square deviation >4Å, Figures 3A and S3). We found it interesting that BOK more closely resembled the BID BH3-bound BCL-2 core of BAK than apo-BAK (Figure S3C). All BCL-2 proteins show large deviations in alignment of the region between helices α2 and α4, which adopts a unique conformation in BOK to define an atypical canonical hydrophobic groove.
Figure 3. Structural Comparison Reveals Possible Gating of BH3 Ligand Access to the Atypical Hydrophobic Groove of BOK.

(A) Alignment of the BCL-2 core of BOK (green) and that of BAX, BCL-xL, and BCL-w completed in PyMOL. The root-mean-square deviation is indicated for the total number of atoms used in the alignment. Top: cartoon representations indicate major deviations in alignments in the region connecting helices α2–α4, which defines the atypical groove of BOK. Bottom: cartoon-and-stick representations of BOK protruding through the surface representation of the aligning partner. Clashes with incoming hydrophobic ligands are predicted throughout the hydrophobic groove of BOK.
(B) The atypical hydrophobic groove of BOK is shown in the apo configuration or interacting with the BID BH3 from the BID SAHB-BAK complex (2m5b.pdb). Left: the electrostatic potential plotted on the surface of BOK. Middle and right: the putative clashes of the BID BH3 ligand at the groove of BOK. The chemical staple of the BID SAHB was excluded (right). The putative hydrophobic pockets are designated as P0–P5.
(C) The alignment of BOK and the BAX-BIM SAHB complex showing a view of the activation site of BAX engaged by BIM SAHB. The predicted structural clashes and electrostatic incompatibilities of BIM SAHB and BOK at this site are illustrated in the surface-cartoon representation, suggesting that their interaction is disfavored. See also Figure S3.
The atypical canonical groove of BOK appears collapsed and occluded, distantly resembling the occlusion observed in apo-BAK (Moldoveanu et al., 2006). In contrast, the occlusion of the groove in BAX is achieved by binding of the TM region (Suzuki et al., 2000). BCL-2 hydrophobic grooves are highly adaptable to ligand binding. In the presence of BH3 peptides, these grooves reorganize upon binding to present up to six hydrophobic pockets (designated P0–P5; Figure 3B), which accommodate six to seven hydrophobic residues of peptide ligands. Helix α3 provides extensive lateral support for BH3 ligands within the canonical grooves. In BOK, helix α3 occludes the P0–P1 pockets, possibly gating canonical BH3 ligand access. The collapse of helix α3 toward the core helix α5 generates a deep hydrophobic pocket in BOK that centers on the P2, where the putative conserved leucine residue in BH3 ligands typically binds. The bulky Y85 residue obstructs the P3–P4 sites (Figure 3B). This overall atypical architecture of the hydrophobic groove predicted clashes (P0–P1 and P3–P4) and lack of interactions (P2), thereby posing challenges to the binding of canonical BH3 ligands. These unique features were highlighted for the interaction between BID BH3 and BOK, which was modeled based on the structural alignment of BOK and the BAK-BID BH3 complex (Figures 3B and S3C) (Moldoveanu et al., 2013).
Full-length BAX accommodates BIM BH3 at the non-canonical groove at an interface defined by helices α1 and α6, and the α1–α2 loop (Gavathiotis et al., 2008). Structural and electro-static features of the non-canonical groove are not conserved between BOK and BAX (Figures 3C and S3B). When BOK and the non-canonical complex between BAX and BIM BH3 are aligned, clashes throughout the length of BIM BH3 at the putative non-canonical BH3-binding groove in BOK are more austere than at the canonical groove, with the two C-terminal turns of the BH3 helix overlapping the α1–α2 loop of BOK (Figure 3C). Our structural comparison suggests that the non-canonical site may be less likely than the canonical site to bind BH3 ligands, and therefore we tested BH3 ligand binding to BOK by NMR.
Weak BH3 Binding at the Canonical Groove Promotes Liposome Permeabilization within a Narrow Dose Range
Titration of 15N-labeled WT and the yield-promoting mutant G35A PR3CA with BH3 peptides, including the stabilized α helix of BCL-2 protein (SAHB) peptides with enhanced helicity, followed by CSP analysis (Schumann et al., 2007), indicated similar, weak binding of BID BH3 to either version of BOK (Figures 4A–4C, S4A, S4B, S5A, and S5B). Although BOK BH3 induced visible precipitation that precluded NMR analysis, we detected small CSP (<0.04 ppm) with BOK SAHB throughout the globular domain of WT and G35A PR3CA, which indicates much weaker binding compared to BID BH3 (Figures S4A and S4B). In contrast, BID BH3 and BID SAHB induced CSP mostly in the fast (traceable peaks; <0.3 ppm) and intermediate exchange (disappearing peaks) NMR regimes that mapped extensively to the canonical groove (Figures 4A–4C, S4A and S4B). Our observations support weak binding of BH3 peptides at the canonical groove.
Figure 4. Weak Binding of BID BH3 at the Canonical Hydrophobic Groove of BOK Promotes Membrane Permeabilization at Specific Dosing Combinations.

(A) Unstapled and stapled BH3 peptides. Residues highlighted in black are identical between BID and BOK BH3. X, pentenyl-Ala; B, norleucine.
(B) Overlay of 1H-15N TROSY spectra of 15N-labeled G35A PR3CA titration with BID BH3 at 303 K and pH 5.0.
(C) CSP magnitudes in G35A PR3CA induced by 200 μM BID BH3 at 303 K and pH 5.0 (Figure S4B) were plotted over the cartoon putty representation of WT PR3CA. Putty thickness correlates with CSP magnitude.
(D) MOMP assay of purified bak−/− mitochondria by WT PR4CA-CH in the presence of unstapled and stapled BH3 peptides and the DMSO vehicle control for monitoring cyt c release and protein loading.
(E) Combined average and SE of AUC at 32 min for WT and mutant PR4CA-CH in the presence of unstapled and stapled BH3 peptides and DMSO vehicle control for the normalized LUVP in Figure S4G. Data were corrected for LUVP induced by peptides and vehicle alone (Figure S4F). Peptide concentrations were empirically selected to induce low or no LUVP and MOMP in the absence of BOK.
See also Figure S4.
When the activation of BOK by BID and BOK BH3 peptides was tested in MOMP assays with bak−/− mitochondria and the PR4CA-CH construct, the level of cyt c release and mitochondrial association by BOK was similar to that for the DMSO vehicle control (Figures 4D and S4C). To test the activation of BOK by the same panel of peptides in liposome permeabilization, LUVP analysis was performed with WT, G35A, Y85A, and Y85F PR4CA-CH (Figures 4E and S4D–S4G). At 200 nM, the WT and mutant PR4CA-CH promoted similar LUVP according to the AUC at 32 min. At earlier times, however, G35A and Y85A mutants induced significantly less and more LUVP compared to the WT, respectively, whereas Y85F exhibited no significant difference (Figures S4D and S4E). LUVP analysis at specific peptide and protein doses that did not individually induce LUVP (Figure S4F) demonstrated activation of WT and mutant PR4CA-CH (50 nM) upon the addition of excess BID BH3 (10 mM). BID BH3-induced BOK activation also was observed at 100 nM and 200 nM WT and mutant PR4CA-CH, although the effect was less pronounced than at 50 nM (Figures 4E and S4G). It is interesting that mutagenesis of the occluding Y85 residue did not significantly affect activation by BH3 peptides in LUVP assays, perhaps supporting the conformational exchange exhibited by this residue. Y85 may adopt a different conformation that does not participate directly in the binding of BH3 ligands to the canonical groove.
BID BH3 Mutagenesis Supports Weak Binding and Sequence-Specific Activation of BOK-Dependent Liposome Permeabilization
We produced five unstapled BID BH3 peptides with single substitutions at I86 (P1), L90 (P2), and A91, and with double substitutions (Figures 5A and 5B). The LUVP analysis showed that at 5 μM, but not at lower or higher concentrations, three of the mutant peptides promoted significantly less BOK-dependent liposome permeabilization compared to WT BID BH3 (Figures 5C, 5D, and S5D). Common among these three peptides is the P1 I86A substitution, suggesting its significant contribution to binding-induced activation of BOK in liposome permeabilization. In contrast, P2 L90A or A91W substitutions did not significantly affect LUVP, suggesting that these residues do not contribute significantly to the activation of BOK in liposome permeabilization.
Figure 5. BID BH3 Engages the Canonical Groove of BOK Similar to that of BAK to Promote Liposome Membrane Permeabilization at Specific Dosing Combinations.

(A) Unstapled WT and mutant BID BH3 peptides. Residues highlighted in black are conserved in most BH3-only proteins. Mutations are color coded. B, norleucine.
(B) Cartoon-surface representation of BID BH3 bound to BOK, modeled based on the alignment of BOK NMR structure over the BID BH3-BAK complex (2m5b.pdb). Residues in BID were mutated according to the substitutions in (A). BOK residues that participate in binding have been labeled.
(C) Combined average and SE of AUC at 32 min for WT PR4CA-CH in the presence of unstapled WT and mutant BID BH3 peptides for the normalized LUVP in Figure S5D.
(D) Combined average and SE of AUC at 32 min for WT and mutant BID BH3 peptides alone (Figure S5D).
(E) Overlay of 1H-15N TROSY spectra of 15N-labeled G35A PR3CA titration with WT and mutant BID BH3 at 303 K and pH 6.8.
(F) CSPs plotted against concentration for indicated residues were fitted to a hyperbola to determine the KD for the interaction of G35A PR3CA and the WT BID BH3 peptide. Mutant BID BH3 peptides exhibited weaker binding to BOK compared to the WT.
See also Figure S5.
NMR titrations with the mutant peptides indicated loss of binding compared to WT BID BH3. We monitored eight residues found in the hydrophobic groove of BAK that exhibit CSPs upon BID BH3 binding (Figures 5B and 5E). Three of these residues exhibited intermediate exchange, indicated by the disappearance of peaks upon BID BH3 binding (H93, G121, and K121; Figure 5E). Five of these residues exhibited fast exchange on binding, and their CSPs were used to estimate the binding affinity of BID BH3 for G35A PR3CA BOK construct (KD of ~226 ± 41 μM, at 303 K and pH 6.8; Figures 5E and 5F). The P1 I86A single mutant peptide showed the most significant loss in binding affinity to BOK by NMR, followed by A91W and the P2 L90A (Figures 5E and 5F). The double mutant peptides containing the P1 I86A substitution exhibited an additive effect in binding affinity to BOK, which supports a major role for the I86 interaction at P1 in stabilizing the BID BH3-BOK complex. We performed titrations with BH3 ligands at neutral pH (Figure 5E) and pH 5.0 for both WT and G35A PR3CA constructs (Figures 4B, S4A, S5A, and S5E). WT PR3CA was unstable and precipitated at pH 6.8 precluding the analysis, whereas the G35A was well behaved. At pH 5.0 and 298 K (25°C), both constructs bound BID BH3 with similar weak binding affinity (KD ~2.6 mM; Figure S5C). The binding affinity of BID BH3 for G35A PR3CA increased by an order of magnitude at 303 K (30°C) compared to 298 K, and to a lesser extent at the same temperature but pH 6.8 (Figures 5F, S5C, and S5F). Overall, our data suggest that binding of BID BH3 at the canonical groove of BOK is weak but specific and possibly contributes to BOK-mediated membrane permeabilization at low BOK concentration.
Truncated BID Does Not Activate BOK in Cells
To test activation of BOK by activated BID, we transiently transfected truncated BID (tBID) in bak−/− bax−/− mouse embryonic fibroblasts (MEFs) reconstituted with the inducible Tet-On 3G system (Clontech) to stably express Venus-2A-HA-(mouse) mBOK (Figures 6 and S6) (Llambi et al., 2016). Upon stimulation with doxycycline (Dox), the mRNAs of Venus and BOK express at equimolar amounts, but only Venus protein is produced, because BOK protein is turned over by the ERAD gp78-protea-some system; MG132 inhibition of the proteasome results in BOK protein stabilization (Figure 6A). By transiently transfecting tBID, it is theoretically possible to recapitulate a similar experimental setting to that of in vitro liposome permeabilization by low levels of BOK activated by BID BH3 peptides. We used previously tested FLAG-tBID and tBID-mCherry constructs (Llambi et al., 2011) in transient transfection assays (Figure S6A) and observed robust expression by the latter (25%–50% transfection efficiency) but undetectable expression by the former, as judged by western blotting (Figure S6D) and IncuCyte imaging (Videos S1 and S2). We monitored cell death hourly by IncuCyte imaging of uptake of the cell-impermeable fluorescent dye SYTOX Green, which binds to the nuclear DNA of dying cells. We report an insignificant difference in the amount of cell death induced by Dox addition to cells transfected with vector alone, mCherry, or tBID-mCherry (Figures 6B, 6C, S6B, and S6C). Our results indicate that under these conditions tBID does not influence BOK-mediated cell death.
Figure 6. tBID Does Not Promote BOK-Mediated Cell Death.

(A) Schematic of the Tet-On 3G stable expression system of Venus-2A-HA-mouse BOK in bak−/− bax−/− MEFs. We typically preincubate cells with Dox for up to 24 hr, which initiates the production of BOK mRNA, and induce cell death with MG132, which inhibits the proteasome, thereby stabilizing BOK protein.
(B) Transient transfection of vectors expressing mCherry (mC) and tBID-mCherry (tBID-mC) was performed according to the scheme in Figure S6A. After recovery in the respective doses of Dox, cell death was induced with MG132 for 18 hr. Western blotting of samples treated with the caspase inhibitor qVD was conducted sequentially with BID, GFP, BOK, and actin antibodies. The blue arrow in the BID blot indicates cross-reactivity by the BID antibody with a band that migrates slightly faster than tBID-mCherry. Weak production of tBID alone also is detected, perhaps being generated through proteolysis of tBID-mCherry. The banding pattern indicates potential BOK ubiquitination.
(C) Cell death induced by WT mBOK in (B) was monitored hourly by IncuCyte imaging of SYTOX Green staining. The 18 hr time point indicates insignificant differences between the mCherry and tBID-mCherry samples at each Dox concentration. Data represent average and SD of triplicate. See also Figure S6.
Unique Glycine in Helix α1 Contributes to Metastability and MOMP
One of the most striking primary sequence differences between BOK and folded BCL-2 family proteins is the occurrence of a glycine residue (G35) in the middle of helix α1. We reasoned that G35 may contribute to the metastability of BOK. G35A substitution in PR4CA-CH or ΔN-BOK-ΔTM-CH exhibited higher protein expression yields in E. coli compared to the respective WT constructs. Compared to the WT constructs, G35A mutants consistently promoted less LUVP (Figures 7A, S4D, S4E, S7A, and S7B) and melted at a significantly higher temperature in TSAs (Figure 7B). In addition, G35A PR3CA unfolded at a higher urea concentration compared to the WT (4–6 M versus 3–4 M) as monitored by TROSY NMR titrations (Figures 7C and S7C). We examined WT and G35A PR3CA CSP induced by urea and identified urea-susceptible hotspots mostly within the N-terminal bundle and the atypical groove of BOK (Figure S7D). We did not detect major differences between WT and G35A CSP, suggesting that their BCL-2 cores fall apart similarly in urea independent of helix α1 stability. Moreover, G35A PR4CA-CH significantly induced less MOMP (Figures 7D and S7E). Together, these results support a destabilizing contribution of G35 to the BOK fold, which is crucial in promoting membrane permeabilization.
Figure 7. Intrinsic Instability in Helix a1 Facilitates Membrane Permeabilization to Promote Cell Death.

(A) Average and SE of LUVP AUC at 32 min induced by WT and G35A mutant of PR4CA-CH and ΔN-BOK-ΔTM-CH for three independent experiments performed in triplicate.
(B) TSA of WT and G35A mutant of PR4CA-CH and ΔN-BOK-ΔTM-CH. Red dotted lines are at 5°C above the WT Tm. Data represent average Tm and SD of replicate performed in quadruplicate.
(C) Overlay of 1H-15N TROSY spectra of 15N-labeled WT and G35A PR3CA titration with urea indicates a transition at higher urea concentration for the stabilizing mutant G35A compared to WT.
(D) MOMP assay of purified bak−/− mitochondria by WT and G35A PR4CA-CH for monitoring cyt c release and protein loading.
(E) Western blot of MEFs expressing mBOK probed for BOK, GFP, and actin. Banding pattern indicates potential BOK ubiquitination.
(F) Cell death of WT and G35A mBOK, induced at 24 hr according to IncuCyte imaging of SYTOX Green staining and FACS of PI staining. Data represent average and SD of triplicate.
(G) Transient transfection with empty vector (EV) and tBID-mCherry (tBID-mC) of mBOK-expressing cells was performed according to the scheme in Figure S6A. After recovery in the respective doses of Dox, cell death was induced with MG132 for 18 hr. Cell death induced by WT mBOK was monitored hourly by IncuCyte imaging of SYTOX Green staining. The 18 hr time point indicates insignificant differences between the EV and tBID-mCherry samples at the higher Dox concentration. Transient transfection of tBID-mC was more toxic than that by the EV control, as indicated by significant cell death in the absence of Dox (Dox 0). Black and red arrows show comparable levels of BOK as determined in (H). Data represent average and SD of triplicate.
(H) Western blotting of samples treated with the caspase inhibitor qVD in (G) was conducted sequentially with BID, GFP, BOK, and actin antibodies. Weak production of tBID alone also is detected, perhaps being generated through the proteolysis of tBID-mCherry. Banding pattern indicates potential BOK ubiquitination. Black and red arrows show comparable levels of BOK, which have been integrated and normalized to G35A. Their values are indicated below the arrows.
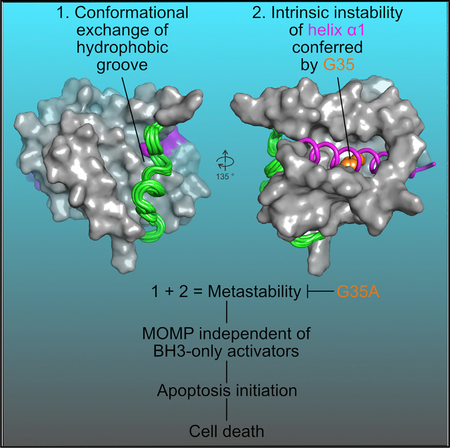
(I) BOK-mediated MOMP is achieved by inhibition of the ERAD gp78-proteasome system, which stabilizes BOK protein and results in its accumulation. Built-in metastability of BOK is governed in part by (1) conformational exchange in the hydrophobic groove and (2) intrinsic instability conferred by G35 in helix a1 (purple rod), which is critical in driving (3) spontaneous membrane association, MOMP, and cell death initiation. G35A mutation blocks BOK-mediated cell death. Helix a1 dissociation from the rest of the BCL-2 core is shown for emphasis, but it was not demonstrated. The color change of BOK as it interacts with the OMM indicates a different unknown conformation than soluble BOK.
See also Figure S7.
G35A mBOK is inactive in the Tet-On 3G-based cell death system using bak−/− bax−/− MEFs (Figures 6A, 7F, and 7G). We titrated Dox in the presence of a constant concentration of MG132 in WT and G35A mBOK expressing MEFs and observed a dose-dependent increase in the levels of BOK and Venus but not of actin (Figures 7E and 7H). Accordingly, we observed a dose-dependent increase in apoptosis induced by WT mBOK, monitored by IncuCyte imaging of SYTOX Green uptake and by fluorescence-activated cell sorting (FACS) of propidium iodide (PI) uptake (Figures 7F and 7G). Compared to the WT, even maximum doses of Dox could not induce similar levels of cell death in G35A mBOK-expressing MEFs, although the levels of this mutant were similar to those of WT BOK that induced significant cell death (Figures 7E and 7H). We transiently transfected tBID-mCherry in cells reconstituted with inducible WT and G35A mBOK, but we did not observe any stimulation of cell death beyond levels achieved by Dox+MG132 with the empty vector or in the absence of transfection (Figures 7G, 7H, S7F, and S7G and Videos S3, S4, S5, and S6). The apoptosis results are consistent with those from in vitro experiments supporting a role for G35 in the metastability of BOK. G35A substitution thus inhibits cell death by its remarkable stabilization of BOK, which diminishes the extent of MOMP.
DISCUSSION
Our structure-function data support the role of BOK as an effector of MOMP, whose underlying intrinsic metastability drives the permeabilization of membranes. BOK has a typical BCL-2 core and is structurally most similar to BAX, BCL-xL, and BCL-w (>21% homology). BH3 peptides bind weakly to the canonical hydrophobic groove of BOK, and they promote BOK-mediated liposome permeabilization but not MOMP with C-terminal TM-truncated BOK. Another study concluded that BID is able to synergize with BOK-ΔTM in giant unilamellar vesicle permeabilization but not in MOMP (Fernández-Marrero et al., 2017). In that study, the concentration of BOK-ΔTM (200 nM) was below that needed to observe release. We previously showed that ≥2 μM BAK-DTM activated by activated BID protein (N/C BID) could induce MOMP (Moldoveanu et al., 2006). Here, we show that ~5 mM BOK-ΔTM is required to induce MOMP from purified mitochondria. We show that the most potent BH3-only effector activator, tBID, was unable to promote cell death by full-length BOK, suggesting that the weak activation by BH3 ligands observed in vitro may be irrelevant in our cellular system.
The NMR structure of BOK represents the dormant BOK conformation, which is not active in MOMP. Several features hold BOK in the dormant conformation: (1) the α2–α4 connecting loop in the groove is undergoing slow motion conformational exchange to regulate metastability; (2) the P0 and P1 canonical hydrophobic pockets are partially occluded by a novel conformation of a short helix α3; (3) the P2 pocket is wide and deep and does not significantly affect access of the conserved leucine of BH3 ligands; and (4) the central P3 and P4 pockets are occupied by Tyr85, a residue exhibiting the most pronounced conformational exchange in the domain. The built-in conformational exchange may have evolved to overcome the need for ligand binding at the hydrophobic groove for BOK activation.
Although binding of BH3 ligands to the canonical groove of BOK appears to be similar to that of the effector BAK, the binding affinity of the best ligand BID BH3 is 30- to 300-fold lower for BOK compared to BAK (Moldoveanu et al., 2013), being pH and temperature dependent. Weaker binding affinity for BOK may be a result of the combined contributions by the P2 leucine and the salt bridge contact by the conserved aspartate of BID BH3 to K122 in the BH1 region, which is unique to BOK. The more electropositive arginine, present in all other BCL-2 proteins (except Boo/Diva) at this position provides a major stabilizing salt bridge interaction to the conserved aspartate of all BH3 ligands. Mutagenesis of the BID BH3 indicated a contribution by the P1 I86 to BOK binding and a moderate contribution by the P2 L90. BID BH3 peptides bind weakly to the canonical hydrophobic groove of BOK in a sequence-specific fashion to promote BOK-mediated liposome permeabilization only at specific dosing combinations, but they have no direct activation effect in MOMP assays using purified mitochondria and added C-terminal TM-truncated BOK.
Expression of BOK protein in cells induced dose-dependent cell death only upon proteasome inhibition, as previously reported (Llambi et al., 2016). We did not detect activation of BOK by tBID in our inducible cell-based system, suggesting that the weak activation by BH3 ligands observed in vitro may not be physiologically relevant in our cellular system. BOK protein level upregulation is dominant in the Tet-On 3G system+MG132, making it potentially difficult to detect weak activation events, as observed in vitro. Analysis of full-length BCL-2 proteins in vitro has been difficult in general because of poor protein expression and stability, and we do not exclude the possibility of introducing functional artifacts through the use of truncated effectors lacking the TM region. We therefore always strive to test concepts in cells with full-length proteins. Based on our in-depth analysis, we conclude that BH3-ligand binding does not appear to contribute to BOK-mediated cell death induced upon proteasome inhibition.
BOK autoactivation likely does not involve BOK BH3 binding to the canonical groove, which is supported by our binding and functional analysis and a divergent BOK BH3 that contains arginine residues instead of the P0 and P5 hydrophobic residues that are found in most BH3 regions. Remarkably, BOK alone could release cyt c in MOMP assays, and this activity correlated with enhanced outer mitochondrial membrane (OMM) association compared to BAK. We attribute these properties to the metastability of BOK, whose BCL-2 core melted at >15°C lower Tm than that of BAK. By stabilizing helix α1, which is critical for autoinhibition of effectors (Alsop et al., 2015; Brouwer et al., 2017; Llambi et al., 2011; Zheng et al., 2016), the point mutation G35A was sufficient to significantly increase the Tm, reduce MOMP, and block BOK-dependent cell death in cells compared to WT.
Effectors exist in dormant and active conformations. Compared to BOK, BAK and BAX are more stable and therefore require BH3 binding for activation. BAX activation is induced by sequential BH3-binding events involving (1) an accessible surface site at the interface between helices α1 and α6 and the α1–α2 loop (trigger site), followed by (2) the canonical groove, which is masked by the C-terminal TM region. BH3 binding of the trigger site allosterically unmasks the canonical groove. Engagement of the canonical groove is common to BAK and BAX and promotes the conformational changes that are required for membrane association and permeabilization (Moldoveanu et al., 2014; Zheng et al., 2016). It is possible that BH3 binding to the trigger site in BAX induces helix α1 metastability, as seen in BOK.
Our structure-function study confirms that BOK is an effector of MOMP able to associate with and permeabilize membranes even in the absence of interactions with BH3 ligands, in part by virtue of its metastability, which is governed by a dynamic hydrophobic groove prone to conformational exchange and an unstable helix α1 containing a helix-breaking glycine (Figure 7I). Understanding autoactivation, membrane association (Fernández-Marrero et al., 2017), and BOK-mediated MOMP mechanistically remain critical to deciphering how mitochondrial apoptosis is initiated in health and disease.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tudor Moldoveanu (tudor.moldoveanu@stjude.org)
MODEL SYSTEMS AND PERMISSIONS
XL10-Gold E. coli
Site-directed mutagenesis and ligation-independent cloning procedures were carried out using XL10-Gold competent cells (Agilent Technologies) transformed according to the manufacturer or published protocols, respectively, and grown on LB medium and LB-agar plates at 37°C overnight (Savitsky et al., 2010).
BL21 Star (DE3), T7 Express, and T7 Express lysY/Iq E. coli
For protein expression BL21 Star (DE3) (Thermo Fisher Scientific), T7 Express, and T7 Express lysY/Iq (New England Biolabs) E. coli containing BOK constructs in pRL574 or pNIC28-Bsa4 vectors were grown in LB medium (Thermo Fisher Scientific) supplemented with 50 mg/L kanamycin (Gold Biotechnology) to an OD600 of 0.8–1.2 before induction with 1 mM isopropyl β-D-1-thiogalactopyrano-side (IPTG) (Gold Biotechnology) at 18–22°C overnight. For isotope labeling WT or G35A NH–PR3CA expressing E. coli were grown in MOPS-based minimal media supplemented with 15NH4Cl and unlabeled glucose or 15NH4Cl and 13C-glucose (Neidhardt et al., 1974) under similar growth and induction conditions.
Bak−/− bax−/− MEFs
Immortalized SV40 bak−/− bax−/− primary MEFs were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin and streptomycin, 1 mM sodium pyruvate, and nonessential amino acids (Llambi et al., 2016).
Bak−/− mice
The use of mice for preparation of liver mitochondria for mitochondrial outer membrane permeabilization (MOMP) assays has been reviewed and approved by the St. Jude IACUC. We purify mitochondria from bak−/− C57BL/6 (B6) mice (Jackson Laboratory) (Lindsten et al., 2000). All aspects of the program for procurement, conditioning/quarantine, housing, management, veterinary care and disposal of carcasses follow the guidelines set down in the 8th Edition of the Guide for Care and Use of Laboratory Animals (NRC). Breeding is done by skilled technicians. Other care is provided by the veterinary staff in the Animal Resource Center at St. Jude. We use both sexes and different ages (4 months – 1-year. old) of bak−/− mice.
METHOD DETAILS
Molecular cloning
For protein production, the hBOK gene was codon optimized for E. coli expression and cloned in the pET-based vector pRL574 as previously described (FL-BOK-ΔTM-CH, Figures 1a and S1a) (Llambi et al., 2016). N- and C-termini–truncated variants starting at S21 and ending at C177 or T181 were cloned into the ligation-independent pNIC28-Bsa4 vector by amplification from the FL–BOK–ΔTM–CH construct with or without the C-His8 tag to generate NH-ΔN-BOK-ΔTM and NH-ΔN-BOK-ΔTM-CH, respectively (Figures 1a and S1a) (Savitsky et al., 2010). The latter was processed with TEV protease to remove the N-His tag (Figure 1a). Point mutations were introduced into different BOK constructs by using the QuikChange II XL kit (Agilent Technologies).
Protein expression and purification and peptide production
Escherichia coli T7 Express pLac I/Y cells (New England Biolabs) were transformed with the human BOK plasmids as described above. Cultures were grown in Luria–Bertani medium at 37°C. After induction with 100 mg/L IPTG, cells were allowed to express for 16–18 h at 18–22°C. Cells were harvested by centrifugation, resuspended in lysis buffer, and lysed by passage through a microfluidizer. Proteins were first purified by batch affinity using Ni2+ beads, followed by fast protein liquid chromatography on S200GL or S200HR size-exclusion columns and Mono S cation-exchange columns using AKTA Pure (GE Healthcare). Whenever necessary, the N-His tag was removed after the Ni2+ affinity step. Fractions containing BOK were pooled, and the buffer was exchanged with 20 mM sodium acetate pH 5.0, 100 mM NaCl before flash-freezing in liquid nitrogen for storage. The BAK-ΔTM-CH construct was expressed and purified as previously described (Moldoveanu et al., 2006). All purified BOK proteins were judged to be more than 95% pure by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and their molecular masses were confirmed by intact mass spectrometry (Proteomics Center at the St. Jude Hartwell Center for Bioinformatics). BH3 peptides were produced in the Hartwell Center for Biotechnology at St. Jude Children’s Research Hospital.
NMR spectroscopy resonance assignment and NOE-based structure determination
NMR spectroscopy experiments were performed in 20 mM sodium acetate pH 5.0, 150 mM NaCl, 10% D2O at 303 K on a Bruker Avance 600-MHz or 800-MHz spectrometer equipped with a 5-mm triple resonance cryoprobe and a single-axis pulse field gradient. Sequence-specific assignments of backbone resonances were made by analyzing HNCACB, CBCA(CO)NH, HNCO spectra for purified 13C,15N-labeled WT and G35A PR3CA ΔN20-BOK-ΔC35 proteins (Sattler et al., 1999). Side-chain resonances were assigned from the combined information content of 15N-edited TOCSY-HSQC, 15N-edited NOESY, HCCH-13C-edited TOCSY, HCC(CO)HN, (HB)CB(CGCD)HD, and (HB)CB(CGCDCE)HE spectra (Sattler et al., 1999). NMR data were processed by the Topspin 3.2 software and analyzed by CARA (Keller, 2004). 93% of the 1H resonances and 100% of the backbone 15N and 13C resonances were assigned. Data have been deposited in the Bioorganic Magnetic Resonance Bank.
NOE distance restraints were obtained from 15N-edited NOESY-HSQC and 13C-edited aliphatic and aromatic NOESY-HSQC spectra at a mixing time of 120 ms. Backbone dihedral angle restraints were predicted by the TALOS+ software (Cornilescu et al., 1999). α-helical hydrogen bonds were introduced for the amides that were missing water-exchange cross-peaks in 15N-edited NOESY spectra. Structures were initially calculated by the program UNIO (Guerry and Herrmann, 2012), using CYANA for energy minimization, and final calculations were made manually using CYANA (Güntert, 2004). The quality of the final 20 lowest-energy conformers from 100 calculated structures was verified using MOLMOL (Koradi et al., 1996) for NOE violations and PROCHECK (Laskowski et al., 1996) for Ramachandran statistics. The structure ensemble was deposited in Protein Data Bank. The accession numbers for the 20 lowest-energy conformers of the human PR3CA ΔN20-BOK-ΔC35 reported in this paper are PDB: 6CKV.pdb and Biomolecular Magnetic Resonance Bank: 304023.
NMR titrations
15N- and 13C,15N-labeled PR3CA ΔN20-BOK-DC35 and the Gly35Ala mutant were generated using the MOPS-based protocol (Neidhardt et al., 1974) and purified in the same manner as unlabeled BOK, including TEV cleavage of the N-His6 tag. Titrations of 25–50 μM PR3CA, or 50 μM G35A with unstapled or stapled BOK and BID BH3 peptides, or urea were performed in 20 mM NaOAc pH 5.0 or 20 mM HEPES pH 6.8, and 100 mM NaCl at 303K and 298K. Data were analyzed and plotted using CARA (Keller, 2004). CSP analysis was done according to published protocols (Schumann et al., 2007), and CSPs were plotted onto the structure by ProtSkin (Ritter et al., 2004) and Pymol (https://pymol.org/2/).
NMR relaxation experiments
The 15N-R1, 15N-R2 and {1H}−15N heteronuclear NOE (hnNOE) were measured on 0.2 mM WT and G35A PR3CA ΔN20-BOK-ΔC35 using standard Bruker pulse programs at 298K in 20 mM NaOAc pH 5.0, 150 mM NaCl. Each 15N-R1 were measured with delay times of 50, 100, 200, 400, 800 and 1200 ms and recycle time of 3 s. Each 15N-R2 were measured with delay times of 16.7, 33.4, 50.1, 66.8 and 83.5 ms and recycle time of 3 s. The R1 and R2 relaxation rate constants and rotational correlation time (τc) were determined using the protein dynamics center (Bruker, Germany), assuming a mono-exponential decay of the peak intensities for T1 and T2. The steady-state hnNOE experiment was carried out in duplicate in an interleaved manner at 298K, with an interscan delay of 3 s and a saturation time of 3 s.
Membrane permeabilization assays
LUVP assays were performed as previously described (Asciolla et al., 2012; Henderson et al., 2007; Llambi et al., 2016; Lovell et al., 2008; Oh et al., 2006; Oh et al., 2010). Briefly, lipid films with molar compositions similar to those of the mitochondrial outer membrane were prepared, including 7.1% cardiolipin, 40.9% phosphatidylcholine, 26.6% phosphatidylethanolamine, 9.1% phosphatidylinositol, 8.3% phosphatidylserine, and 8.0% Ni2+-affinity lipid 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) [DGS NTA(Ni)] (Avanti Polar Lipids). Films were suspended in 10 mM HEPES pH 6.8, 200 mM KCl, and 5 mM MgCl2 buffer containing 8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt (ANTS) and the p-xylene-bis-pyridinium bromide (DPX) (Avanti Polar Lipids) fluorophore–quencher pair by sonication and then extruded through a 0.2-μm membrane (Avanti Polar Lipids) at least 10 times to generate a homogeneous population of LUVs. LUVs were then purified by fast protein liquid chromatography on a S500 gel filtration column and stored under nitrogen at 4°C in the dark. For LUVP assays, reactions were prepared on ice before being monitored kinetically at 37°C every 2 min for 32 min in a CLARIOstar monochromator microplate reader (BMG LABTECH).
LUVP data analysis involved normalization of ANTS fluorescence relative to the maximum and minimum fluorescent readings induced by 2.5% CHAPS and the buffer control, respectively. Area under the curve (AUC) for normalized LUVP data was approximated by numerical integration using Simpson’s rule over even numbers of 2-minute intervals at 8, 16, and 32 min. The LUVP AUC at 32 min was plotted against BOK concentrations, and data were fitted to a Hill equation to derive the concentrations at half-maximal values. Data manipulation, analysis, and presentation were done in Excel (Microsoft Office) and GraphPad Prism v7 (GraphPad Software). LUVP experiments for each BOK construct and their mutants were performed at least 3 times in triplicate.
MOMP assays were performed according to our previously published protocol, using purified mitochondria from bak−/− mouse liver (Llambi et al., 2011; Moldoveanu et al., 2013). Briefly, mitochondria were isolated in mitochondrial isolation buffer containing 200 mM mannitol, 68 mM sucrose, 10 mM HEPES (pH 7.4), 10 mM KCl, and 1 mM EDTA, and supplemented with Complete Protease Inhibitor (Roche). The 50 μL MOMP reactions proceeded for 1 h at 37°C and contained combinations of WT and mutant BOK and WT BAK proteins, with or without BH3 peptides or DMSO vehicle, and appropriate single reagent controls. Pellets and supernatants were separated by centrifugation, and both fractions were quenched by adding SDS sample buffer. To monitor the extent of permeabilization, both fractions were immunoblotted with anti–cyt c monoclonal antibody (BD Biosciences). The blot was stained with Ponceau S (G-Biosciences) immediately after transfer to assess protein loading.
Thermal shift analysis
TSAs were performed by using the Protein Thermal Shift Starter Kit (ThermoFisher), according to the manufacturer’s protocol, using 0.25 mg/mL purified BOK and BAK. Briefly, for each protein sample, a 90 μL MasterMix was prepared by using the buffer and the fluorescent dye ROX, vortexed at low speed, and 4 aliquots of 20 μL each were dispensed in 4 individual wells of Microamp Fast Optical 96-well reaction plates (ThermoFisher) for analysis in an Applied Biosystems 7500 thermal cycler (ThermoFisher). The temperature gradient was 25°C–99°C at a rate of 1°C/min. Data were analyzed with Protein Thermal Shift Software v1.3 (ThermoFisher) to determine the melting temperature, using the first derivative and Boltzmann equation non-linear regression. Dye-only controls were included in each plate. Experiments were performed at least 2 times in quadruplicates for each protein sample at the 2 protein concentrations. More recently, TSA assays were performed in 20 mM NaOAc pH 5.0 or 20 mM HEPES pH 6.8 and 150 mM NaCl, and 384-well plates in an Applied Biosystems 7900HT Fast Real-Time PCR System (ThermoFisher). Final data analysis and presentation were done in Microsoft Excel and GraphPad Prism v7 (GraphPad Software).
Mammalian cell expression of BOK and tBID
Mouse BOK was stably expressed in bak−/−bax−/− DKO MEFs, using a modified Tet–On 3G retroviral system (Llambi et al., 2016). Briefly, the Tet3G cassette of the pRetroX–Tet3G vector was transferred into the pMX–IRES–Blast (blastidicin selection) and the Venus–2A–HA-(mouse) mBOK cassette was cloned into the multiple cloning site of the pRetroX–TRE3G vector, in which the PuroR cassette was replaced with a ZeoR cassette. Using standard retrovirus-based generation of stable cell lines, the pMX–IRES–Tet3G–Blast was first introduced in DKO MEFs and cells expressing the Tet–On 3G transactivator protein were selected using 20 μg/mL blasticidin. Next, the pRetroX-TREG-Venus-2A-HA-mBOK-Zeo was introduced into the Tet3G-expressing DKO MEFs and cells expressing the complete Tet–On 3G system were selected by using 20 μg/mL blasticidin and 200 μg/mL zeocin. To enrich for a more homogeneous population of cells expressing BOK, 2 rounds of FACS for Venus-expressing cells were performed by treating the blasticidin- and zeocin-resistant populations with doxycycline (Dox), which stabilizes the Tet–On 3G transactivator protein to drive Venus and mBOK expression. To stabilize the BOK protein, cells were treated with the proteasome inhibitor MG132 (Sigma), which blocks BOK turnover. Protein expression levels were measured by western blotting.
Transient transfection of tBID–mCherry pEGFP N1 and FLAG–tBID pcDNA3.1 constructs (Llambi et al., 2011) in bak−/−bax−/− DKO MEFs expressing Tet-On 3G inducible WT or G35A mBOK, were done using lipofectamine 3000. Briefly, lipofectamine 3000/P3000/DNA complexes were prepared according to the manufacturer’s protocol, and were mixed with trypsinized cells for 5–10 min prior to dilution to 125,000 cells per 24-well in OPTI-MEM. 8h after plating the medium was changed to phenol-free DMEM +10% FBS ± Dox, and the cells were allowed to express tBID for 18–24h. Cell death was induced by addition of Dox + MG132 ± qVD in fresh phenol-free DMEM for 18h and was monitored by IncuCyte imaging. Western blot was performed on the samples containing qVD. Mock controls were done in the absence of lipofectamine 3000/P3000/DNA complexes.
Cell death analysis
BOK-dependent apoptosis of DKO MEFs was induced with 2.5 μM MG132 after overnight transactivation of the WT and G35A Venus–2A–HA-mBOK with up to 2000 ng/mL Dox. Cell death was monitored by imaging the uptake of the nucleic acid stain SYTOX Green (Invitrogen) over 24 h using IncuCyte FLR or Zoom instruments (Essen BioScience). SYTOX Green–positive cells were quantified using the IncuCyte image analysis software (Essen BioScience). Data were normalized as the ratio of positive events (SYTOX Green counts) to confluence. At least 3 independent IncuCyte experiments were performed in triplicate or quadruplicate for each condition. At 18 or 24 h, IncuCyte samples were trypsinized, washed once with PBS, and stained with PI (P-3566, Life Technologies). FACS data were immediately collected on a Scan II flow cytometer (BD Biosciences) and later analyzed using FlowJo (https://www.FlowJo.com).
QUANTIFICATION AND STATISTICAL ANALYSIS
LUVP AUC, IncuCyte, FACS, and TSA data were analyzed by one-way ANOVA using Dunnett and Tukey tests in GraphPad Prism. These experiments were repeated at least three times and were done in triplicate or quadruplicate. The p values are 0.1234 (ns), 0.0332 (*), 0.0021 (**), 0.0002 (***), < 0.0001 (****). We identify the condition used to compare against by making the bar black and pointing a line to it.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the 20 lowest-energy conformers of the human PR3CA DN20-BOK-DC35 reported in this paper are PDB: 6CKV.pdb and Biomolecular Magnetic Resonance Bank: 304023. Structural statistics are summarized in Figure S2c. All software packages used in this study are available commercially or free of charge, and are summarized in the KEY RESOURCES TABLE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental Models: Strains/Cell Lines | ||
| Escherichia coli strain: XL110-Gold | Agilent Technologies | Cat# C404010 |
| Escherichia coli strain: BL21 Star (DE3) | Thermo Fisher Scientific | Cat# C601003 |
| Escherichia coli strain: T7 Express | New England Biolabs | Cat# C2566I |
| Escherichia coli strain: T7 Express lysY/Iq | New England Biolabs | Cat# C3013I |
| Phoenix Ampho Packaging Cells | ATCC | Cat# CRL-3213 |
| Immortalized SV40 bak−/−bax−/− Mouse Embryonic Fibroblasts | (Llambi et al., 2016) | PMID 26949185 |
| Bak−/− mice | Jackson Laboratories | B6.129-Bak1tm1Thsn/J |
| Peptides | ||
| hBID_BH3: EDIIRNIARHLAQVGDSBDRSI (B = Nle) | This paper | N/A |
| hBID_BH3a: EDIIRNAARHLAQVGDSMDRSI | This paper | N/A |
| hBID_BH3b: EDIIRNIARHAAQVGDSMDRSI | This paper | N/A |
| hBID_BH3c: EDIIRNAARHAAQVGDSMDRSI | This paper | N/A |
| hBID_BH3d: EDIIRNIARHLWQVGDSMDRSI | This paper | N/A |
| hBID_BH3e: EDIIRNAARHLWQVGDSMDRSI | This paper | N/A |
| hBID SAHB: EDIIRNIARHLAXVGDXBDRSI (X = pentenyl alanine) | This paper | N/A |
| hBOK BH3: RLAEVCAVLLRLGDELEBIR | This paper | N/A |
| hBOK SAHB: RLAEVSAVLLXLGDXLEBIR | This paper | N/A |
| Recombinant DNA | ||
| pRetroX-TRE3G | Clontech | Cat# 631188 |
| pcDNA3.1 | Invitrogen | Cat# V79020 |
| pEGFP N1 | Clontech | Cat# 6085–1 |
| pNIC28-Bsa4 | (Savitsky et al., 2010) | Addgene #26103 |
| Human BOK and mutants in pNIC28-Bsa4 | This paper | N/A |
| Mouse BOK and mutants in pRetroX-TRE3G | (Llambi et al., 2016); this paper | PMID 26949185; N/A |
| FLAG-human tBID and mutants in pcDNA3.1 | (Llambi et al., 2011); this paper | PMID 22036586; N/A |
| Human tBID-mCherry in pEGFP N1 | (Llambi et al., 2011) | PMID 22036586 |
| Chemicals | ||
| Fetal Bovine Serum - Premium Select | Atlanta Biologicals | Cat# S11550 |
| DMEM, high glucose, no phosphates | Thermo Fisher Scientific | Cat# 11971025 |
| GIBCO L-glutamine | Thermo Fisher Scientific | Cat# 25030081 CAS# 56-85-9 |
| GIBCO Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140122 CAS# 69-57-8, 57-92-1 |
| GIBCO Sodium Pyruvate | Thermo Fisher Scientific | Cat# 11360070 CAS# 113-24-6 |
| GIBCO MEM Non-Essential Amino Acids Solution | Thermo Fisher Scientific | 11140050 |
| GIBCO 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985023 CAS# 60-24-2 |
| 0.25% Trypsin, 0.1% EDTA in HBSS w/o Calcium, Magnesium and Sodium Bicarbonate | Corning | 25053CI |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000015 |
| Zeocine | Thermo Fisher Scientific | Cat# R25001 |
| Blastocidine S HCl powder | Thermo Fisher Scientific | Cat# R21001 |
| Doxycycline | Clontech | Cat# 631311 CAS# 24390-14-5 |
| Z-Leu-Leu-Leu-al (MG132) | Sigma Aldrich | Cat# C2211 |
| CAS# 133407-82-6 | ||
| SYTOX Green | Thermo Fisher Scientific | Cat# S7020 CAS# 163795-75-3 |
| Q-VD-Oph hydrate | APExBIO | Cat# A1901 |
| Propidium Iodide | Invitrogen | Cat# P3566 CAS# 25535-16-4 |
| L-α-phosphatidylcholine (Egg, Chicken) | Avanti Polar Lipids | Cat# 840051C CAS# 97281-44-2 |
| L-α-phosphatidylinositol (Liver, Bovine) (sodium salt) | Avanti Polar Lipids | Cat# 840042C CAS# 383907-33-3 |
| L-α-phosphatidylserine (Brain, Porcine) (sodium salt) | Avanti Polar Lipids | Cat# 840032C CAS# 383907-32-2 |
| Cardiolipin (Heart, Bovine) (sodium salt) | Avanti Polar Lipids | Cat# 840012C CAS# 383907-10-6 |
| 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) | Avanti Polar Lipids | Cat# 790404C CAS# 231615-77-3 |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | Avanti Polar Lipids | Cat# 850725C CAS# 4004-05-1 |
| ANTS (8-aminonpaphthalene-1,3,6-trisulfonic acid, disodium salt) | Molecular Probes | Cat# 1278701 CAS# 5398-34-5 |
| DPX (p-xylene-bis-pyridiunium bromide) | Molecular Probes | Cat# X1525 CAS# 14208-10-7 |
| 15N Ammonium Chloride | Cambridge Isotope Laboratories | Cat# NLM-467–10 CAS# 12125-02-9 |
| 13C Glucose | Cambridge Isotope Laboratories | Cat# CLM-1396–1 CAS# 50-99-7 |
| Deuterated DTT | Cambridge Isotope Laboratories | Cat# DLM-2622–1 |
| Deuterium Oxide | Sigma Aldrich | Cat# 617385–1 CAS# 7789-20-0 |
| Deuterated DMSO | Sigma Aldrich | Cat# 151874 |
| Ponceau S | G-Biosciences | Cat# 786–576 |
| cOmplete, Mini Protease Inhibitor Cocktail | Roche | Cat# 11836153001 |
| Antibodies | ||
| Anti-cyt c monoclonal antibody | BD Biosciences | Cat# 556433; RRID:AB_396417 |
| Anti-human BID monoclonal antibody | Santa Cruz | Cat# SC-56025; RRID:AB_781628 |
| Anti-human BOK monoclonal antibody | Abcam | Cat# ab186745 |
| Anti-GFP monoclonal antibody | Santa Cruz | Cat# SC-9996; RRID:AB_627695 |
| Anti-actin monoclonal antibody | Millipore Sigma | Cat# MAB1501; RRID:AB_2223041 |
| Critical Commercial Assays | ||
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200522 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| Plasmid Plus Midi Kit | QIAGEN | Cat# 12945 |
| Protein Thermal Shift Starter Kit | Thermo Fisher Scientific | Cat# 4462263 |
| Supersignal West Dura | Thermo Fisher Scientific | Cat# 34075 |
| Deposited Data | ||
| Human BOK structure | This paper | PDB: 6CKV.pdb; BioMagResBank: 304023 |
| Software and Algorithms | ||
| TopSpin v3.2 | Bruker BioSpin | http://www.bruker.com |
| CARA v1.9.1.7 | (Keller, 2004) | http://cara.nmr.ch/doku.php |
| TALOS+ | (Cornilescu et al., 1999) | https://spin.niddk.nih.gov/bax/software/TALOS/ |
| UNIO’10 v2.0.1 | (Guerry and Herrmann, 2012) | http://perso.ens-lyon.fr/torsten.herrmann/Herrmann/Software.html |
| CYANA v2.1 | (Guntert, 2004) | http://www.cyana.org |
| MOLMOL v2.1–2.6 | (Koradi et al., 1996) | https://sourceforge.net/p/molmol/wiki/Home/ |
| PROCHECK v3.4.3 | (Laskowski et al., 1996) | https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/ |
| ProtSkin | (Ritter et al., 2004) | http://www.mcgnmr.mcgill.ca/ProtSkin/ |
| MacPyMOL v1.7.6.3 | Schrodinger, LLC | https://pymol.org/2/ |
| ImageJ | NIH, USA | https://www.imagej.nih.gov/ij |
| IncuCyte v2016A | Essen BioScience | https://www.essenbioscience.com |
| IncuCyte 2011A Rev2 v20111.3.4288 | Essen BioScience | https://www.essenbioscience.com |
| FlowJo Xv10.0.7r2 | FlowJo, LLC | https://www.flowjo.com |
| Microsoft Excel | Microsoft | https://products.office.com/excel?legRedir=true&CorrelationId=129ae579-ccf4–43ae-89ae-a241f392c557 |
| Prism v7.0a | GraphPad Software | https://www.graphpad.com |
| Protein Thermal Shift Software v1.3 | Thermo Fisher Scientific | Cat# 4466037 |
| ABI 7900HT Sequence Detection Systems v 2.4.1 | Thermo Fisher Scientific | Cat# 4350490 |
| ClarioSTAR Microplate Reader Software V5.40 Edition 2; Reader Control v5.40; Firmware v1.20; MARS Data Analysis v3.30 | BMG Lab Tech | https://www.bmglabtech.com/en/products/software/ |
| Other | ||
| Nickel agarose beads (high density) | Gold Biotechnology | Cat# H-320–500 |
| Amicon Ultra 15-mL 3K MWCO centrifugal filter | Millipore Sigma | Cat# UFC900324 |
| Amicon Ultra 15-mL 10K MWCO centrifugal filter | Millipore Sigma | Cat# UFC800324 |
| MonoS 5/50 GL column | GE Healthcare | Cat# 17-5168-01 |
| Superdex200 Increase 10/300 GL column | GE Healthcare | Cat# 28990944 |
| HiPrep 16/60 Sephacryl S-500 HR column | GE Healthcare | Cat# 28935606 |
Supplementary Material
Highlights.
BOK exhibits conformational exchange in the hydrophobic regulatory groove
BID weakly binds BOK, promoting liposome permeabilization but not MOMP
BOK does not require truncated activated BID (tBID) to initiate apoptosis
Destabilization in regulatory helix a1 is essential for MOMP and apoptosis
ACKNOWLEDGMENTS
This study was funded by the American Lebanese Syrian Associated Charities (ALSAC) and St. Jude Children’s Research Hospital Comprehensive Cancer Center NIH grant 5 P30CA021765–36 (to T.M.). We thank Dr. Vani Shanker for scientific editing of our manuscript. We thank Dr. Douglas Green, the Immunology Department chair at St. Jude, for critical evaluation of our manuscript and insightful discussions. We thank Drs. Mao Yang and Douglas Green of St. Jude for providing bak−/− mice for MOMP assays.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the NMR structure of human BOK reported in this paper are PDB: 6CKV and Biological Magnetic Resonance Bank: 304023.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and six videos and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.04.060.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alsop AE, Fennell SC, Bartolo RC, Tan IK, Dewson G, and Kluck RM (2015). Dissociation of Bak α1 helix from the core and latch domains is required for apoptosis. Nat. Commun 6, 6841. [DOI] [PubMed] [Google Scholar]
- Asciolla JJ, Renault TT, and Chipuk JE (2012). Examining BCL-2 family function with large unilamellar vesicles. J. Vis. Exp (68), 4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegert A, Mayer C, Remmert M, Söding J, and Lupas AN (2006). The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 34, W335–W339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem EA, and Letai A (2016). BOK: oddball of the BCL-2 family. Trends Cell Biol. 26, 389–390. [DOI] [PubMed] [Google Scholar]
- Brouwer JM, Lan P, Cowan AD, Bernardini JP, Birkinshaw RW, van Delft MF, Sleebs BE, Robin AY, Wardak A, Tan IK, et al. (2017). Conversion of Bim-BH3 from activator to inhibitor of Bak through structure-based design. Mol. Cell 68, 659–672.e9. [DOI] [PubMed] [Google Scholar]
- Burden RL, and Faires JD (2000). Numerical Analysis 7th ed (Brooks/Cole; ). [Google Scholar]
- Carpio MA, Michaud M, Zhou W, Fisher JK, Walensky LD, and Katz SG (2015). BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc. Natl. Acad. Sci. USA 112, 7201–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, and Green DR (2010). The BCL-2 family reunion. Mol. Cell 37, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, and Bax A (1999). Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, and Adams JM (2014). Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol 15, 49–63. [DOI] [PubMed] [Google Scholar]
- Echeverry N, Bachmann D, Ke F, Strasser A, Simon HU, and Kaufmann T (2013). Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ. 20, 785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Marrero Y, Bleicken S, Das KK, Bachmann D, Kaufmann T, and Garcia-Saez AJ (2017). The membrane activity of BOK involves formation of large, stable toroidal pores and is promoted by cBID. FEBS J. 284, 711–724. [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, and Walensky LD (2008). BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR (2005). Apoptotic pathways: ten minutes to dead. Cell 121, 671–674. [DOI] [PubMed] [Google Scholar]
- Guerry P, and Herrmann T (2012). Comprehensive automation for NMR structure determination of proteins. Methods Mol. Biol 831, 429–451. [DOI] [PubMed] [Google Scholar]
- Güntert P (2004). Automated NMR structure calculation with CYANA. Methods Mol. Biol 278, 353–378. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Henderson MP, Billen LP, Kim PK, and Andrews DW (2007). Cell-free analysis of tail-anchor protein targeting to membranes. Methods 41, 427–438. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Strasser A, McDunn JE, and Swanson PE (2009). Cell death. N. Engl. J. Med 361, 1570–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke F, Voss A, Kerr JB, O’Reilly LA, Tai L, Echeverry N, Bouillet P, Strasser A, and Kaufmann T (2012). BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ. 19, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke F, Bouillet P, Kaufmann T, Strasser A, Kerr J, and Voss AK (2013). Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis. 4, e650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke F, Grabow S, Kelly GL, Lin A, O’Reilly LA, and Strasser A (2015). Impact of the combined loss of BOK, BAX and BAK on the hematopoietic system is slightly more severe than compound loss of BAX and BAK. Cell Death Dis. 6, e1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R (2004). The Computer Aided Resonance Assignment Tutorial (CANTINA Verlag). [Google Scholar]
- Kleckner IR, and Foster MP (2011). An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 1814, 942–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi R, Billeter M, and Wüthrich K (1996). MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph 14, 51–55, 29–32. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, and Thornton JM (1996). AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. (2000). The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, and Green DR (2011). A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 44, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, Parsons MJ, Zheng JH, Brown SA, Pelletier S, et al. (2016). BOK Is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 165, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, and Andrews DW (2008). Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084. [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, and Gehring K (2006). The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol. Cell 24, 677–688. [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, and Green DR (2013). BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol 20, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Follis AV, Kriwacki RW, and Green DR (2014). Many players in BCL-2 family affairs. Trends Biochem. Sci 39, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, and Smith DF (1974). Culture medium for enter-obacteria. J. Bacteriol 119, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh KJ, Barbuto S, Pitter K, Morash J, Walensky LD, and Korsmeyer SJ (2006). A membrane-targeted BID BCL-2 homology 3 peptide is sufficient for high potency activation of BAX in vitro. J. Biol. Chem 281, 36999–37008. [DOI] [PubMed] [Google Scholar]
- Oh KJ, Singh P, Lee K, Foss K, Lee S, Park M, Lee S, Aluvila S, Park M, Singh P, et al. (2010). Conformational changes in BAK, a pore-forming proapoptotic Bcl-2 family member, upon membrane insertion and direct evidence for the existence of BH3-BH3 contact interface in BAK homo-oligomers. J. Biol. Chem 285, 28924–28937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros AM, Olejniczak ET, and Fesik SW (2004). Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 1644, 83–94. [DOI] [PubMed] [Google Scholar]
- Ritter B, Denisov AY, Philie J, Deprez C, Tung EC, Gehring K, and McPherson PS (2004). Two WXXF-based motifs in NECAPs define the specificity of accessory protein binding to AP-1 and AP-2. EMBO J. 23, 3701–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, and Griesinger C (1999). Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spec-trosc 34, 93–158. [Google Scholar]
- Savitsky P, Bray J, Cooper CD, Marsden BD, Mahajan P, Burgess-Brown NA, and Gileadi O (2010). High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol 172, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman JJ, Wright FA, Kaufmann T, and Wojcikiewicz RJ (2013). The Bcl-2 protein family member Bok binds to the coupling domain of inositol 1,4,5-trisphosphate receptors and protects them from proteolytic cleavage. J. Biol. Chem 288, 25340–25349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman JJ, Wright FA, Han X, Zluhan EJ, Szczesniak LM, and Wojcikiewicz RJ (2016). The stability and expression level of Bok are governed by binding to inositol 1,4,5-trisphosphate receptors. J. Biol. Chem 291, 11820–11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann FH, Riepl H, Maurer T, Gronwald W, Neidig KP, and Kalbitzer HR (2007). Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR 39, 275–289. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, and Tjandra N (2000). Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103, 645–654. [DOI] [PubMed] [Google Scholar]
- Zheng JH, Viacava Follis A, Kriwacki RW, and Moldoveanu T (2016). Discoveries and controversies in BCL-2 protein-mediated apoptosis. FEBS J. 283, 2690–2700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the 20 lowest-energy conformers of the human PR3CA DN20-BOK-DC35 reported in this paper are PDB: 6CKV.pdb and Biomolecular Magnetic Resonance Bank: 304023. Structural statistics are summarized in Figure S2c. All software packages used in this study are available commercially or free of charge, and are summarized in the KEY RESOURCES TABLE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental Models: Strains/Cell Lines | ||
| Escherichia coli strain: XL110-Gold | Agilent Technologies | Cat# C404010 |
| Escherichia coli strain: BL21 Star (DE3) | Thermo Fisher Scientific | Cat# C601003 |
| Escherichia coli strain: T7 Express | New England Biolabs | Cat# C2566I |
| Escherichia coli strain: T7 Express lysY/Iq | New England Biolabs | Cat# C3013I |
| Phoenix Ampho Packaging Cells | ATCC | Cat# CRL-3213 |
| Immortalized SV40 bak−/−bax−/− Mouse Embryonic Fibroblasts | (Llambi et al., 2016) | PMID 26949185 |
| Bak−/− mice | Jackson Laboratories | B6.129-Bak1tm1Thsn/J |
| Peptides | ||
| hBID_BH3: EDIIRNIARHLAQVGDSBDRSI (B = Nle) | This paper | N/A |
| hBID_BH3a: EDIIRNAARHLAQVGDSMDRSI | This paper | N/A |
| hBID_BH3b: EDIIRNIARHAAQVGDSMDRSI | This paper | N/A |
| hBID_BH3c: EDIIRNAARHAAQVGDSMDRSI | This paper | N/A |
| hBID_BH3d: EDIIRNIARHLWQVGDSMDRSI | This paper | N/A |
| hBID_BH3e: EDIIRNAARHLWQVGDSMDRSI | This paper | N/A |
| hBID SAHB: EDIIRNIARHLAXVGDXBDRSI (X = pentenyl alanine) | This paper | N/A |
| hBOK BH3: RLAEVCAVLLRLGDELEBIR | This paper | N/A |
| hBOK SAHB: RLAEVSAVLLXLGDXLEBIR | This paper | N/A |
| Recombinant DNA | ||
| pRetroX-TRE3G | Clontech | Cat# 631188 |
| pcDNA3.1 | Invitrogen | Cat# V79020 |
| pEGFP N1 | Clontech | Cat# 6085–1 |
| pNIC28-Bsa4 | (Savitsky et al., 2010) | Addgene #26103 |
| Human BOK and mutants in pNIC28-Bsa4 | This paper | N/A |
| Mouse BOK and mutants in pRetroX-TRE3G | (Llambi et al., 2016); this paper | PMID 26949185; N/A |
| FLAG-human tBID and mutants in pcDNA3.1 | (Llambi et al., 2011); this paper | PMID 22036586; N/A |
| Human tBID-mCherry in pEGFP N1 | (Llambi et al., 2011) | PMID 22036586 |
| Chemicals | ||
| Fetal Bovine Serum - Premium Select | Atlanta Biologicals | Cat# S11550 |
| DMEM, high glucose, no phosphates | Thermo Fisher Scientific | Cat# 11971025 |
| GIBCO L-glutamine | Thermo Fisher Scientific | Cat# 25030081 CAS# 56-85-9 |
| GIBCO Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140122 CAS# 69-57-8, 57-92-1 |
| GIBCO Sodium Pyruvate | Thermo Fisher Scientific | Cat# 11360070 CAS# 113-24-6 |
| GIBCO MEM Non-Essential Amino Acids Solution | Thermo Fisher Scientific | 11140050 |
| GIBCO 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985023 CAS# 60-24-2 |
| 0.25% Trypsin, 0.1% EDTA in HBSS w/o Calcium, Magnesium and Sodium Bicarbonate | Corning | 25053CI |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000015 |
| Zeocine | Thermo Fisher Scientific | Cat# R25001 |
| Blastocidine S HCl powder | Thermo Fisher Scientific | Cat# R21001 |
| Doxycycline | Clontech | Cat# 631311 CAS# 24390-14-5 |
| Z-Leu-Leu-Leu-al (MG132) | Sigma Aldrich | Cat# C2211 |
| CAS# 133407-82-6 | ||
| SYTOX Green | Thermo Fisher Scientific | Cat# S7020 CAS# 163795-75-3 |
| Q-VD-Oph hydrate | APExBIO | Cat# A1901 |
| Propidium Iodide | Invitrogen | Cat# P3566 CAS# 25535-16-4 |
| L-α-phosphatidylcholine (Egg, Chicken) | Avanti Polar Lipids | Cat# 840051C CAS# 97281-44-2 |
| L-α-phosphatidylinositol (Liver, Bovine) (sodium salt) | Avanti Polar Lipids | Cat# 840042C CAS# 383907-33-3 |
| L-α-phosphatidylserine (Brain, Porcine) (sodium salt) | Avanti Polar Lipids | Cat# 840032C CAS# 383907-32-2 |
| Cardiolipin (Heart, Bovine) (sodium salt) | Avanti Polar Lipids | Cat# 840012C CAS# 383907-10-6 |
| 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) | Avanti Polar Lipids | Cat# 790404C CAS# 231615-77-3 |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | Avanti Polar Lipids | Cat# 850725C CAS# 4004-05-1 |
| ANTS (8-aminonpaphthalene-1,3,6-trisulfonic acid, disodium salt) | Molecular Probes | Cat# 1278701 CAS# 5398-34-5 |
| DPX (p-xylene-bis-pyridiunium bromide) | Molecular Probes | Cat# X1525 CAS# 14208-10-7 |
| 15N Ammonium Chloride | Cambridge Isotope Laboratories | Cat# NLM-467–10 CAS# 12125-02-9 |
| 13C Glucose | Cambridge Isotope Laboratories | Cat# CLM-1396–1 CAS# 50-99-7 |
| Deuterated DTT | Cambridge Isotope Laboratories | Cat# DLM-2622–1 |
| Deuterium Oxide | Sigma Aldrich | Cat# 617385–1 CAS# 7789-20-0 |
| Deuterated DMSO | Sigma Aldrich | Cat# 151874 |
| Ponceau S | G-Biosciences | Cat# 786–576 |
| cOmplete, Mini Protease Inhibitor Cocktail | Roche | Cat# 11836153001 |
| Antibodies | ||
| Anti-cyt c monoclonal antibody | BD Biosciences | Cat# 556433; RRID:AB_396417 |
| Anti-human BID monoclonal antibody | Santa Cruz | Cat# SC-56025; RRID:AB_781628 |
| Anti-human BOK monoclonal antibody | Abcam | Cat# ab186745 |
| Anti-GFP monoclonal antibody | Santa Cruz | Cat# SC-9996; RRID:AB_627695 |
| Anti-actin monoclonal antibody | Millipore Sigma | Cat# MAB1501; RRID:AB_2223041 |
| Critical Commercial Assays | ||
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200522 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| Plasmid Plus Midi Kit | QIAGEN | Cat# 12945 |
| Protein Thermal Shift Starter Kit | Thermo Fisher Scientific | Cat# 4462263 |
| Supersignal West Dura | Thermo Fisher Scientific | Cat# 34075 |
| Deposited Data | ||
| Human BOK structure | This paper | PDB: 6CKV.pdb; BioMagResBank: 304023 |
| Software and Algorithms | ||
| TopSpin v3.2 | Bruker BioSpin | http://www.bruker.com |
| CARA v1.9.1.7 | (Keller, 2004) | http://cara.nmr.ch/doku.php |
| TALOS+ | (Cornilescu et al., 1999) | https://spin.niddk.nih.gov/bax/software/TALOS/ |
| UNIO’10 v2.0.1 | (Guerry and Herrmann, 2012) | http://perso.ens-lyon.fr/torsten.herrmann/Herrmann/Software.html |
| CYANA v2.1 | (Guntert, 2004) | http://www.cyana.org |
| MOLMOL v2.1–2.6 | (Koradi et al., 1996) | https://sourceforge.net/p/molmol/wiki/Home/ |
| PROCHECK v3.4.3 | (Laskowski et al., 1996) | https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/ |
| ProtSkin | (Ritter et al., 2004) | http://www.mcgnmr.mcgill.ca/ProtSkin/ |
| MacPyMOL v1.7.6.3 | Schrodinger, LLC | https://pymol.org/2/ |
| ImageJ | NIH, USA | https://www.imagej.nih.gov/ij |
| IncuCyte v2016A | Essen BioScience | https://www.essenbioscience.com |
| IncuCyte 2011A Rev2 v20111.3.4288 | Essen BioScience | https://www.essenbioscience.com |
| FlowJo Xv10.0.7r2 | FlowJo, LLC | https://www.flowjo.com |
| Microsoft Excel | Microsoft | https://products.office.com/excel?legRedir=true&CorrelationId=129ae579-ccf4–43ae-89ae-a241f392c557 |
| Prism v7.0a | GraphPad Software | https://www.graphpad.com |
| Protein Thermal Shift Software v1.3 | Thermo Fisher Scientific | Cat# 4466037 |
| ABI 7900HT Sequence Detection Systems v 2.4.1 | Thermo Fisher Scientific | Cat# 4350490 |
| ClarioSTAR Microplate Reader Software V5.40 Edition 2; Reader Control v5.40; Firmware v1.20; MARS Data Analysis v3.30 | BMG Lab Tech | https://www.bmglabtech.com/en/products/software/ |
| Other | ||
| Nickel agarose beads (high density) | Gold Biotechnology | Cat# H-320–500 |
| Amicon Ultra 15-mL 3K MWCO centrifugal filter | Millipore Sigma | Cat# UFC900324 |
| Amicon Ultra 15-mL 10K MWCO centrifugal filter | Millipore Sigma | Cat# UFC800324 |
| MonoS 5/50 GL column | GE Healthcare | Cat# 17-5168-01 |
| Superdex200 Increase 10/300 GL column | GE Healthcare | Cat# 28990944 |
| HiPrep 16/60 Sephacryl S-500 HR column | GE Healthcare | Cat# 28935606 |