

Abstract
Adoptive cellular therapy involving genetic modification of T cells with chimeric antigen receptor (CAR) transgene offers a promising strategy to broaden the efficacy of this approach for the effective treatment of cancer. Although remarkable antitumor responses have been observed following CAR T‐cell therapy in a subset of B‐cell malignancies, this has yet to be extended in the context of solid cancers. A number of promising strategies involving reprogramming the tumor microenvironment, increasing the specificity and safety of gene‐modified T cells and harnessing the endogenous immune response have been tested in preclinical models that may have a significant impact in patients with solid cancers. This review will discuss these exciting new developments and the challenges that must be overcome to deliver a more sustained and potent therapeutic response.
Keywords: CAR, adoptive cell therapy, solid tumors, T cells
Introduction
Evidence highlighting the importance of harnessing the immune system for cancer control has become increasingly apparent and is in part attributed to the efficacy of checkpoint blockade therapy such as α‐PD‐1 and α‐CTLA‐4 antibodies.1 More recently, the success of CD19‐targeted chimeric antigen receptor (CAR) T‐cell therapy in some haematological malignancies, including B‐ALL and non‐Hodgkin's lymphoma, has further accentuated the potent antitumor potential of T cells.2, 3, 4 However, the impressive clinical success seen with CAR T cells in B‐cell malignancy patients has yet to be translated beyond CD19+ malignancies. Both preclinical studies and multicenter clinical trials have been conducted and are still ongoing to test the efficacy of CAR T‐cell therapy targeting different antigens in solid tumors. Results thus far have indicated that CAR T‐cell therapy for epithelial cancers has not matched the remarkable clinical responses observed in patients with B‐cell malignancies.5 Although not completely understood, the discrepancy between CAR T‐cell effectiveness in CD19+ haematological and solid cancers may be due to several factors that include the immunosuppressive tumor environment in solid tumors, the lack of CAR T‐cell trafficking and penetration into the solid mass, tumor antigen heterogeneity, as well as the lack of full complement of activation signals required for optimal functional responses by CAR T cells. In addition, the use of CAR T cells in solid tumors faces additional challenges in terms of safety and, similarly as with haematological malignancies, the high costs associated with generating a personalised CAR T‐cell product. To overcome these challenges, a substantial amount of work has focused on investigating novel strategies to augment CAR T‐cell therapeutic efficacy and applicability, including combining CAR T cells with immunomodulatory antibodies, manipulation of the tumor microenvironment (TME), induction of immune responses against antigen‐negative tumors, targeting of intracellular tumor antigens, safety and development of ‘universal’ CAR T‐cell strategies. Each of these key aspects will be discussed in this review.
Targeting tumor microenvironment (TME) in CAR T‐cell therapy
The efficacy of CAR T‐cell therapy is influenced by both the environment to which CAR T cells are exposed and also the intrinsic functional parameters of CAR T cells which determine whether they can generate effective antitumor responses. The solid tumor landscape presents multiple barriers that can ultimately neutralise CAR T‐cell activity. In order to successfully eliminate the tumor cells, CAR T cells must successfully traffic to the tumor site. This could be challenging in the case where there is a mismatch between tumor‐derived chemokines and chemokine receptors on T cells. Subsequently, if these CAR T cells do get to the tumor site, the next challenge is to successfully infiltrate the stromal elements for CAR T cells to induce antitumor cytotoxic effects. Therefore, strategies to degrade extracellular matrix in an attempt to improve tumor infiltration by T cells have been explored, for example by engineering CAR T cells to express the heparanase enzyme.6 Finally, even after successful trafficking and infiltration to the tumor, CAR T cells must then overcome multiple obstacles created by the tumor and/or the host cells in the TME, including the presence of immunosuppressive soluble factors, cytokines and immune cells.7 Attempts to overcome these obstacles have led to the development of various strategies involving manipulation of the TME.
Using immunomodulatory antibodies to enhance CAR T‐cell antitumor responses
Checkpoint inhibitors such as α‐CTLA‐4 and α‐PD‐1 antibodies in cancer therapy work by blocking the inhibitory mechanisms in T cells, consequently leading to further T‐cell activation and tumor killing. Remarkable results from clinical trials using α‐CTLA‐4 and α‐PD‐1 antibodies led to their approval by the Food and Drug Administration (FDA) in 2011 and 2014, respectively.8, 9, 10 Given the clinical success in enhancing T‐cell function and antitumor activity, administration of checkpoint inhibitors makes an ideal partner for tumor‐targeted engineered CAR T cells. Preclinical studies have tested the combination of CAR T‐cell therapy and α‐PD‐1 mAb against a number of cancers, either through systemic administration of α‐PD‐1 mAb or genetic modification of CAR T cells to express α‐PD‐1 single‐chain variable fragment (scFv). Combination therapy using α‐PD‐1 demonstrated superior antitumor efficacy in an in vivo model compared to conventional CAR T cells that correlated with enhanced effector function of the CAR T cells such as granzyme B and IFNγ upon PD‐1 blockade.11 More recently, a study involving the combination of CAR T cells, α‐PD‐1 mAb and additionally an A2AR antagonist that blocks the adenosine immunosuppressive pathway reported an even greater antitumor response in a preclinical model.12 The clinical translation of CAR T‐cell and α‐PD‐1 mAb is now underway with multiple clinical trials currently recruiting patients.13 In addition to checkpoint inhibitors, agonistic monoclonal antibodies that activate T‐cell costimulatory receptors have also advanced in their development, including, for example, α‐4‐1BB and α‐OX40 mAbs.14, 15 Inclusion of 4‐1BB and/or OX40 domains directly in the CAR construct as costimulatory signals has been investigated and demonstrated potent ability to support CAR T‐cell activation. Notably, these costimulatory domains significantly impact on T‐cell cytokine secretion and proliferation function.16 Both 4‐1BB‐ and/or OX40‐containing CAR T cells have been tested in various preclinical studies; however, comparisons between the two domains remain inconclusive in terms of overall antitumor effect observed given variability in the models used from different groups.16, 17 In the context of costimulation using exogenous antibodies, a recent preclinical study tested the combination of Her2‐specific CAR T cells with α‐4‐1BB therapy against Her2‐expressing solid tumors. The combination treatment resulted in significantly enhanced tumor regression compared to CAR T‐cell therapy alone or control T cells in combination with α‐4‐1BB mAb.18 This study highlights the potential of using an agonistic antibody to improve CAR T‐cell efficacy in solid tumors, and therefore, testing of other agonistic antibodies in this context is warranted.
Previous studies have combined the use of both immune checkpoint inhibitors and agonistic antibodies in preclinical cancer models for increasing the endogenous antitumor immune response (Figure 1). Some of these studies reported increased antitumor effects following the combination of α‐PD‐1 and α‐4‐1BB antibodies in a number of murine cancer models,19, 20, 21 and α‐PD‐1 and α‐OX40 antibodies in an ID8 murine ovarian cancer model.22 However, more recently other studies have reported opposing effects. Two different studies reported that the concurrent addition of α‐PD‐1 mAb markedly reduced the therapeutic response of α‐OX40 mAb.23, 24 Interestingly, however, a study by Messenheimer et al. found that when α‐OX40 and α‐PD‐1 antibodies were administered sequentially by treating MMTV‐PyMT tumor‐bearing mice with α‐OX40 mAb before α‐PD‐1 mAb, the sequential combination therapy resulted in augmented antitumor efficacy. However, this improved effect was not observed when α‐PD‐1 mAb was administered before α‐OX40 mAb, highlighting the importance of timing and sequence of such treatments.23 Further, another study that tested the combination of α‐4‐1BB and α‐PD‐1 antibodies in a murine spontaneous B‐cell lymphoma model found that simultaneous use of α‐PD‐1 mAb diminished the antitumor activity of α‐4‐1BB mAb alone. The mechanism for this effect was thought to be due to a dramatic reduction in the function of effector CD8+ T cells in the presence of α‐PD‐1 mAb, potentially through induced apoptosis.25 The reason for the discrepancies observed between different studies involving checkpoint inhibitor and immune agonist combination is not completely understood; however, the dose and timing of the immune‐modulating antibody administration may be of high importance. Together, all these observations reveal that whilst supporting T‐cell activation using combination of immune agonists and checkpoint inhibitors could generate synergistic antitumor effects, caution is necessary in choosing the optimal timing and sequence in order to achieve maximal therapeutic efficacy particularly in the context of adoptive transfer of gene‐modified T cells.
Figure 1.
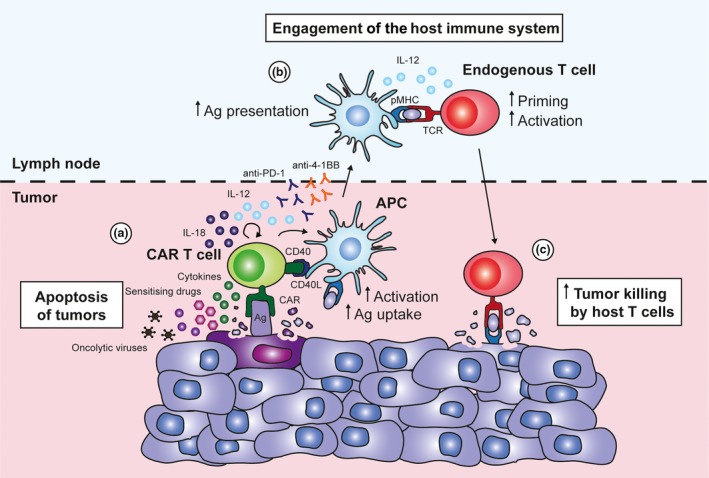
Enhancing tumor killing by engaging host immunity during chimeric antigen receptor (CAR) T‐cell therapy. (a) CAR T cells lyse tumor cells in an antigen‐specific manner and secrete pro‐inflammatory cytokines IFNγ and TNFα at the tumor site. Apoptosis‐sensitising drugs can be used in combination with CAR T‐cell therapy to enhance tumor lysis through TNF‐mediated bystander killing. Oncolytic viruses can directly lyse tumor cells, engage host responses and act as carriers of transgenes that facilitate CAR T‐cell antitumor activity. Immune‐stimulatory antibodies such as anti‐4‐1BB or anti‐PD‐1 can be used to increase tumor killing by CAR T cells whilst improving responses by host immune cells. CAR T cells modified to secrete IL‐12 or IL‐18 not only increase their effector function but also modulate various endogenous immune cell types such as Tregs and myeloid cells at the tumor site. (b) CAR T cells can be engineered to express CD40 ligand, which interacts with CD40 on antigen‐presenting cells such as DCs to enhance their maturation, antigen uptake and presentation to endogenous T cells. (c) This may result in enhanced tumor killing by endogenous T cells, thereby improving the overall efficacy of CAR T‐cell therapy.
Localised expression of immune‐stimulatory molecules by CAR T cells in the TME
The area of synthetic biology is vastly developing and has provided us with the technology to perform customised engineering of cellular pathways, enabling various modifications in cell response behaviours required for more effective T cell‐based therapies.26, 27, 28, 29 Strategies to modulate the local TME have led to the generation of ‘armored’ CAR T cells that provide localised expression of pro‐inflammatory cytokines or costimulatory ligands to improve CAR T‐cell function within tumors. CAR T cells engineered to secrete IL‐12 have resulted in enhanced in vivo efficacy in several preclinical models including CD19+ B‐cell lymphoma and MUC16‐expressing ovarian cancer. In these studies, CAR T cell‐secreted IL‐12 augmented their cytotoxic function and alleviated regulatory T cell (Treg)‐mediated suppression.30, 31, 32 Using a similar approach, CAR T cells secreting IL‐18 demonstrated improved antitumor activity, increased proliferation and persistence in an in vivo model.33, 34 Other systems involving cytokine‐mediated enhancement of CAR T cells include the genetic modification of these cells to express a form of membrane‐bound chimeric IL‐15, which gave rise to a population of CAR T cells that possessed a T memory stem cell phenotype and a better memory potential even in the absence of antigen stimulation.35
Chimeric antigen receptor T cells have also been modified to express immune‐stimulatory molecules to influence their interaction with other cell types within the local TME. Constitutive expression of CD40 ligand by CAR T cells not only resulted in their enhanced killing and pro‐inflammatory cytokine production but also led to increased maturation and IL‐12 secretion by dendritic cells (DCs) (Figure 1). Furthermore, CD40 ligand directly engaged CD40‐expressing tumor cells to alter their immunogenicity through the upregulation of surface receptors including MHC molecules and Fas ligand.36 In other studies, CAR T cells co‐expressing 4‐1BB ligand and CD80 provided auto‐costimulation and induced an additional trans‐costimulatory effect on bystander T cells, overcoming the lack of immune‐stimulatory signals within the TME that resulted in the eradication of large tumors in preclinical models.37 A recent study by Rafiq et al.38 demonstrated that CAR T cells secreting α‐PD‐1 scFv provided localised delivery of the immune checkpoint inhibitor and blocked T‐cell PD‐1 and tumor PD‐L1 binding, enhancing the efficacy of these T cells in both syngeneic and xenograft tumor models. Although not in the context of CAR T‐cell therapy, an alternate attempt to overcome immunosuppressive signals involved adoptively transferred T cells overexpressing a dominant‐negative TGF‐β receptor type II in the treatment of EBV+ Hodgkin's lymphoma. These cells were found to be immune to the immunosuppressive effects of TGF‐β in vitro, expanded significantly in vivo and resulted in a complete response in 3 of 7 patients.39 Overall, these studies suggest that therapeutic responses against solid tumors can potentially be augmented by engineering CAR T cells to express additional mediators that boost their local effector function and alter their interaction with surrounding cells in the TME.
Targeting the chemokine milieu to enhance CAR T‐cell therapy
Given that trafficking and penetration of CAR T cells into tumor mass are major obstacles that have to be overcome in solid cancers, manipulation of chemokine signalling is another approach that is currently under intense investigation. A recent study by Adachi et al. utilised the cytokine modulation approach in combination with chemokine modulation for CAR T cells, combining the overexpression of IL‐7 cytokine and CCL19 chemokine in CD20‐targeted CAR T cells. In two different murine CD20‐overexpressing tumor models, P815 mastocytoma and 3LL Lewis lung carcinoma, CCL19 and IL‐7‐secreting CAR T cells were proven superior in promoting tumor clearance in pre‐established solid tumors compared to conventional CAR T cells. This superior performance by CCL19 and IL‐7‐secreting CAR T cells was associated with a marked increase in CAR T‐cell and DC infiltration into tumor tissues.40
Solid tumors can secrete various chemokines which are able to prevent effective T‐cell trafficking into the tumor such as CXCL5 and CXCL12.41, 42 Moreover, chemokine receptors expressed on T cells frequently do not match the tumor chemokine signature, leading to limited trafficking to the tumor site.43 Therefore, matching chemokine receptor(s) expressed on CAR T cells to the tumor chemokine milieu is also an attractive strategy as it may allow for a higher frequency of CAR T cells to traffic to the tumor site. Indeed, a preclinical study by Kershaw et al. demonstrated that T cells engineered with CXCR2 were able to migrate towards various tumor cells expressing the cognate chemokine CXCL1.44 A similar effect has also been observed in other studies utilising CCR2b‐bearing CAR T cells in neuroblastoma and mesothelioma xenografts, as well as CCR4‐bearing CAR T cells in Hodgkin's lymphoma45, 46, 47 (Figure 2a). One alternative to this strategy may also be to match the tumor chemokine profile to more closely resemble the CAR T‐cell chemokine receptor profile.48, 49, 50 For instance, whilst CXCR3 is highly expressed on most activated CD8+ T cells and is critical for their trafficking to the tumor site,51 intratumoral expression of the CXCR3 ligands CXCL9, CXCL10 and CXCL11 is generally low. However, a recent study utilising a xenograft model of ovarian cancer demonstrated that co‐administration of T cells with the EZH2 inhibitor, GSK126, and DNA methyltransferase inhibitor, 5‐AZA‐dC, led to a significant enhancement in CXCR3‐dependent trafficking and antitumor efficacy.52 This regime was shown to augment T‐cell trafficking by inducing tumor cells to express the epigenetically silenced chemokines CXCL9 and CXCL10 (Figure 2b), suggesting that modulating tumor chemokine profile through epigenetic modification may be a viable mechanism to overcome poor trafficking into solid tumors. Alternatively, PD‐1 blockade has been shown to significantly enhance adoptive T‐cell trafficking to tumors53 as well as CAR T‐cell activation.11 In the context of adoptive cell therapy (ACT), it was found that enhanced T‐cell trafficking was caused by an IFNγ‐dependent increase of intratumoral CXCL10.53 Taken together, based on promising observations from these preclinical studies investigating cytokine and/or chemokine genetic manipulation approach, it is anticipated that more cytokines and chemokines involved in promoting immune cell responses will be explored in the context of CAR T‐cell therapy. Importantly, given the chemokine signature may also be influenced by the tumor location, stroma and surrounding cytokine milieu,54 these factors should be taken into consideration when designing appropriate chemokine receptors for CAR T cells.
Figure 2.
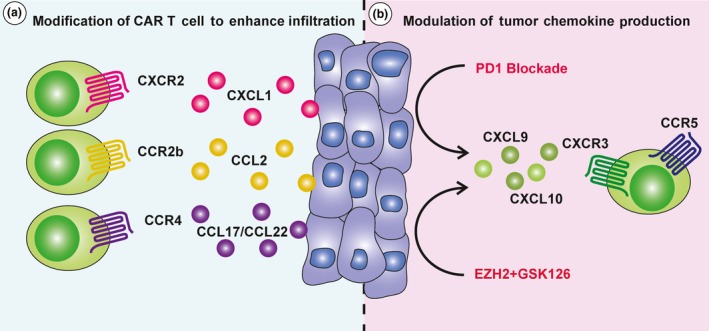
Approaches for enhancing intratumoral chimeric antigen receptor (CAR) T‐cell infiltration. Commonly, the chemokine receptors endogenously expressed by CAR T cells do not match the chemokines present in the tumor microenvironment. (a) One approach to improve CAR T‐cell infiltration therefore is to transduce them with chemokine receptors such as CXCR2, CCR2b or CCR4, better matching the chemokine profile of the tumor microenvironment. (b) Alternatively, both PD‐1‐blockade and epigenetic modifiers, EZH2 and GSK126, have been shown to increase intratumoral expression of chemokines CXCL9 and CXCL10, leading to enhanced CXCR3‐dependent trafficking of adoptively transferred T cells.
Inducing tumor eradication beyond CAR T‐cell antigen recognition
Treatment with engineered CAR T cells has predominantly focused on targeting one single tumor antigen.45, 55, 56, 57 This has been an effective strategy in the case where the target antigen is ubiquitously expressed by tumor cells, which is the case for CD19 in B‐cell malignancies. However, even in the most successful case of CD19‐CAR T cells targeting B‐cell malignancies, a significant fraction of patients relapse not long after T‐cell infusion. A proportion of the relapsed patients experience antigen‐negative disease recurrence, manifested by the outgrowth of CD19− tumor cells.58, 59 This phenomenon therefore highlights the important subject of tumor antigen heterogeneity, even when the target antigen is uniformly expressed on all tumor cells. Investigation into the mechanisms leading to this antigen loss phenomenon revealed an alternative splicing mechanism of the CD19 mRNA, resulting in the loss of the cognate epitope on the CD19 protein required for recognition by CD19‐CAR T cells.60 Similar evidence of this splice variant process was reported in the context of melanoma cells resistant to vemurafenib, together indicating splice‐based adaptations by tumor cells as an escape mechanism that consequently leads to outgrowth of tumor variants.61 These findings suggest that CAR T‐cell therapy, or any other targeted therapy more generally, may promote the outgrowth of tumor escape variants. Antigen loss renders CAR T cells ineffectual and may have significant ramifications for the wider success of CAR T‐cell therapy. Thus, the ability to induce additional antitumor responses recognising alternative tumor antigens, especially in the context of solid tumors where antigen expression is more heterogeneous, will be pivotal for CAR T cells to have a universally efficacious antitumor effect.
Several strategies have been developed in an attempt to overcome the problem of antigen‐negative relapse. For example, the generation of dual CAR T cells targeting two different antigens, CD19 and CD22, has been shown to increase antitumor effects compared to CAR T cells targeting either single antigen, where mice were co‐inoculated with a mixture of CD19− CD22+ and CD19+ CD22− NALM6 leukaemia cells. Importantly, it was demonstrated that having two CARs in the T‐cell population was able to offset antigen escape.62, 63 Other dual and even trivalent CAR T cells have also been investigated in preclinical studies, including CAR T cells targeting CD19 and CD123 in a leukaemia model,64 and CAR T cells targeting Her2, IL13Rα2 and EphA2 in a glioblastoma model,65 respectively. Despite the promising potential of this approach, however, it may be difficult for most malignancies to find multiple different tumor antigens on one tumor cell that can be targeted by CAR T cells in a safe and effective manner.
Alternatively, an antitumor response against a wider range of targets may be achieved through the recruitment and stimulation of the endogenous immune response, as this involves various effector cells with a broad spectrum of recognition capabilities and antitumor functions. Indeed, engagement of endogenous cellular immunity has been reported to induce antigen‐negative tumor cell killing following antigen‐specific T‐cell therapy.59 One strategy to potentially promote tumor killing beyond a CAR T‐cell antigen‐specific response has been through further modification of these cells to secrete pro‐inflammatory cytokines (Figure 1). A study by Chmielewski et al. elegantly demonstrated the capacity of CAR T cells engineered to secrete the IL‐12 cytokine that enabled responses against both antigen‐positive and antigen‐negative tumor cells. This effect was accompanied by accumulation of macrophages, which were shown to be a critical facilitator for the observed antitumor response.66 One other potential mechanism to increase the targeting of antigen‐negative tumors may also be to combine CAR T‐cell treatment with apoptosis‐sensitising drugs such as birinapant, leading to enhanced TNF‐mediated bystander killing of tumor cells67 (Figure 1). Further, given previous preclinical studies suggesting that the combination of CAR T cells with CD40 ligand or α‐4‐1BB mAb works in part through the activation of host DCs, approaches that engage the endogenous immune system may be an effective approach to induce antitumor responses beyond CAR T‐cell target antigen18, 36 (Figure 1). Interestingly, in the context of PD‐1 blockade, PD‐1‐blocking scFv secreted by CAR T cells in a preclinical model was shown to bind to bystander tumor‐specific T cells, resulting in an overall enhanced antitumor effect.38
Another approach to enhance the efficacy of CAR T‐cell therapy is in combination with oncolytic viruses (OVs). OVs can help to overcome the immunosuppressive tumor microenvironment by providing pathogen‐associated molecular patterns, upregulating MHC class I machinery as well as directly lysing tumor cells, releasing danger‐associated molecular patterns and tumor‐associated antigens (TAAs) that help prime host antigen‐presenting cells and engage endogenous T‐cell responses.68, 69, 70 Moreover, OVs can be modified to express transgenes that improve the antitumor responses of CAR T cells. By administering gp100‐expressing recombinant vaccinia virus vaccination with dual‐specific T cells comprising gp100‐specific TCR and anti‐Her2 CAR, one group reported significant antitumor responses against both antigen‐positive and antigen‐negative tumors, suggesting the potential of epitope spreading following combination therapy.71 OVs engineered to express cytokines IL‐2 and TNFα, chemokines such as CXCL11, or in some cases a combination of both IL‐15 and RANTES/CCL5 have also been shown to increase recruitment of CAR T cells to the tumor and enhancement of antitumor activity in preclinical models.72, 73, 74 Likewise, CAR T cells displayed superior therapeutic efficacy when combined with OVs expressing localised anti‐PD‐L1 minibody in comparison with systemically delivered anti‐PD‐L1 antibody.75 Another study combined antifolate receptor alpha (FRα) CAR T cells with OVs expressing bispecific T‐cell engager targeting a second tumor antigen epidermal growth factor receptor (EGFR) and showed that both endogenous and CAR T cells were successfully redirected against EGFR+ FRα− tumors, overcoming tumor heterogeneity and prolonging survival of mice.76
Taken together, it is well established that tumors can enhance their capacity for immune escape by loss of a targeted antigen or epitope. This makes antigen heterogeneity a major challenge that urgently needs to be overcome in the context of antigen‐targeted therapy, more specifically CAR T‐cell therapy. Furthermore, in CAR T‐cell clinical trials for solid malignancies, eligibility criteria for patients to be enrolled often requires only partial expression of the CAR target antigen on a patient's tumor. For example, enrolment criteria in one of the Lewis Y‐specific CAR T‐cell clinical trials specified Lewis Y to be expressed on a minimum of 20% of tumor blast cells, meaning that the majority of the tumor cells would not be recognised by the CAR T cells in some patients.77 Therefore, engagement of endogenous antitumor immunity appears to be a promising approach to increase the likelihood of epitope spreading, which may potentially lead to eradication of antigen‐negative tumor cells, and subsequently decreased risk of antigen escape variants emerging.
CAR T cells targeting intracellular tumor antigens
Designing a treatment that is effective in facilitating tumor destruction whilst sparing healthy cells is considered the ‘holy grail’ in cancer immunotherapy. Discovery of tumor antigens that are only expressed by tumors but not by healthy cells remains a key aspect towards improving the specificity and safety of immunotherapies. Tumor antigens can be classified into several categories, namely TAAs such as overexpressed self‐antigens (Her2, CD19), tissue‐specific antigens (CEA), as well as tumor‐specific antigens including mutated antigens (neoantigens) and viral antigens. Most of the tumor antigens targeted by immunotherapies to date are those overexpressed on tumor cells but are also present on healthy cells to a lesser extent. Examples of these include the Her2, CEA, GD2 and Lewis Y antigens that have been used as targets for CAR T‐cell therapy.57, 78, 79, 80, 81 Given that these antigens are found on healthy cells, their use as immunotherapeutic targets is attributable solely to their preferential expression on tumors. However, caution has to be taken as on‐target off‐tumor side effects can in certain cases pose a significant limitation on this approach.82, 83
Careful selection and design of CAR T‐cell constructs may potentially alleviate some of the issues associated with differences in antigen expression between healthy and tumor tissues. A recent study by Majzner et al.84 demonstrated that CAR T cells targeting the pan‐cancer antigen B7‐H3 mediated cytolysis of high antigen‐expressing tumor cells whilst displaying minimal reactivity towards low antigen‐expressing cells. This suggests that antigens that are found on normal tissues may still serve as safe targets, provided that their expression on tumor cells is sufficiently distinguishable by CAR T cells. Nonetheless, extensive pre‐evaluation will be required to determine therapeutic efficacy versus safety as antigen density can vary widely across individual patients’ normal and tumor tissues.
Another factor is the suitability of tumor antigens as targets for CAR T‐cell technology. CAR‐engineered T cells are only able to recognise antigens that are expressed on the cell surface. However, a number of tumor antigens are found intracellularly and are therefore considered nontargetable by conventional CAR T cells. One innovative approach to circumvent this problem is the development of CAR T cells using antibody fragments that recognise intracellular tumor antigens based on their surface presentation as peptide epitopes on MHC molecules. These TCR‐mimic CAR T cells are designed to specifically engage MHC–peptide complexes found on the surface of target cells. Willemsen et al.85, 86 first reported the engineering of CAR T cells using a phage display‐derived MAGE‐A1/HLA‐A1‐specific Fab, which induced in vitro target lysis and cytokine production against MAGE‐A1 expressing HLA‐A1+ melanoma cells. These studies provided a conceptual framework for the development of similar CAR T cells targeting tumor antigens NY‐ESO‐1 and proteinase 3 peptide PR1.87, 88 More recently, two groups have individually described the generation of TCR‐mimic CAR T cells targeting intracellular Wilm's tumor 1 antigen in the context of HLA‐A2 in preclinical models, with one of these studies demonstrating in vivo therapeutic efficacy of these CAR T cells against antigen‐expressing leukaemia and ovarian tumors.89, 90 Based on these promising developments, there is the potential to expand the repertoire of tumor antigens targetable by CAR T‐cell therapy.
Generating universal ‘off‐the‐shelf’ CAR T cells
The two recently approved CD19‐CAR T‐cell products, Kymriah™ and Yescarta™, although highly effective, are very expensive treatments, priced at US $475 000 and US $373 000 for a one‐time treatment, respectively.91 These high costs are partly attributed to the fact that the process from T‐cell collection, genetic modification, to CAR T‐cell reinfusion is patient specific. To make CAR T‐cell therapy more broadly applicable to diverse patient populations, strategies to generate universal off‐the‐shelf CAR T‐cell products that can be safely and effectively delivered to multiple recipients will be a key issue to address.92 At present, there have been a number of clinical trials evaluating the potential use of allogeneic CAR T cells.93
It is anticipated that the use of allogeneic rather than the more personalised autologous CAR T cells will significantly reduce manufacturing costs, as bulk manufacturing of CAR T cells can be achieved in a time‐efficient and less labour‐intensive manner. Indeed, the feasibility of using ‘off‐the‐shelf’ T cells in humans has now been well demonstrated to be both an effective and safe treatment for a number of viral diseases.94, 95 However, to permit such an approach, two inherent challenges associated with allogeneic cell transfer must be overcome. These include graft‐versus‐host disease (GVHD) and rejection of the infused CAR T cells by the host. Studies have been conducted to address these issues, as exemplified by the development of an approach using Transcription Activator‐Like Effector Nucleases (TALEN™) to simultaneously inactivate both the endogenous TCR and CD52 of the adoptively transferred T cells. This approach decreased GVHD risk due to elimination of the endogenous TCR, whilst allowing for persistence of the infused CAR T cells due to depletion of the host T cells upon α‐CD52 mAb administration.96, 97 Another report utilised CAR T cells that were additionally transduced with an ER retention signal‐containing scFv specific for the CD3ε component of the TCR. This resulted in the surface downregulation of endogenous TCRs on the CAR T cells and a reduced occurrence in GvHD.98 A recently developed alternative to remove TCR expression on the transferred CAR T cells is by targeting the CAR transgene insertion into the native TCR alpha chain (TRAC) locus, either using TRC1‐2 nuclease or CRISPR/Cas9 technology. It was demonstrated that the transduced CD19‐specific CAR T cells lacked the endogenous TCR whilst exhibiting robust antitumor responses due to reduced tonic CAR signalling and T‐cell exhaustion.99 In a mouse model of ALL, enhanced tumor rejection was observed following treatment with CD19‐CAR T cells directed to the TRAC locus.100 Others have reported similar genomic modification strategies combining the basis of TALEN DNA binding with meganucleases (megaTAL) to insert a CAR transgene into the CCR5 locus of primary human T cells.101 In addition, Cooper and colleagues demonstrated the utility of zinc finger nucleases to specifically disrupt endogenous TCR and HLA genes in T cells, increasing the prospects of generating allogeneic CAR T cells for individuals of disparate HLA.102, 103
Given rapid advances in gene‐editing technology such as CRISPR and other site‐specific endonucleases, such genetic modification strategies may be achieved in an efficient and precise manner, allowing for widespread clinical use.99 Other potential avenues to generate off‐the‐shelf antigen‐specific effector cells have also been investigated, and a promising strategy involved the incorporation of a CAR transgene into NK cells instead of T cells. The use of CAR‐expressing NK cells potentially obviates the GVHD issue from allogeneic donors as they do not induce GVHD. Several groups have reported preclinical evaluation of these CAR‐expressing NK cells with promising results demonstrated,104, 105 and clinical trials investigating this approach are currently underway.106
In addition to improving the widespread applicability of allogeneic CAR T cells, current efforts have also been focused on generating universal CARs to provide greater flexibility for antigen recognition by CAR T cells. These methods generally involve the engineering of a generic receptor on the extracellular portion of the CAR which can then be coupled with a soluble ligand‐conjugated, antigen binder of choice. Using the biotin–avidin system, Urbanska et al.107 generated CAR T cells containing extracellular avidin and showed that these cells could elicit antigen‐specific effector functions against EpCAM+ ovarian tumors in an in vivo model when conjugated with a biotinylated anti‐EpCAM antibody. Similarly, another group administered CAR T cells containing an extracellular high‐affinity Fc receptor‐binding CD16 variant and demonstrated significantly enhanced antitumor efficacy of rituximab or trastuzumab, respectively, against CD20+ or HER2+ expressing tumors in mice.108 Exploring further on this concept, Cho et al. developed split, universal and programmable (SUPRA) CARs in which CAR T cells express an extracellular leucine zipper (zipCAR), which can bind to scFvs containing leucine zippers (zipFvs). Using this approach, zipCARs can simultaneously be endowed with multiple specificities based on the variety of scFvs present, and signal strengths can be adjusted depending on each individual zipper binding affinities. This potentially enables the production of CAR T cells targeting a broad range of antigens without having to further engineer the CARs and may also help to address the issue of tumor escape and toxicity.109
Safety of CAR T‐cell therapy
In cases where CAR T cells are directed against nontumor‐specific TAAs, potential toxicity due to CAR T‐cell recognition of low levels of the target antigen on healthy cells remains an important issue to be addressed. Thus, multiple strategies to mitigate the on‐target off‐tumor effects are currently being investigated. One approach has been to ensure target selectivity by dual CAR T‐cell recognition of two different TAAs on the same tumor cell. In this setting, the two CARs are designed to either induce a ζ‐chain signal or a CD28 costimulatory signal, allowing for superior T‐cell activation upon simultaneous antigen engagement of the two CARs. As a result, this may be a safer approach restricting CAR T‐cell full activity to only tumor cells expressing the two antigens at the same time, whereas the potency of signals delivered into the CAR T cells via only one CAR engagement remains below the activation threshold and hence rendered ineffective.110 A similar approach of dual T‐cell recognition involves engineering a T‐cell circuit whereby a synthetic Notch receptor for one antigen leads to subsequent expression of a CAR specific for a second antigen. These T cells are only activated when both antigens are present on the tumor cells. Both of these approaches may be suitable for controlling potential on‐target off‐tumor effects, as dual antigen recognition may allow for more selective tumor elimination, whilst sparing healthy cells that express only a single antigen.
Another strategy employs the principle of an inhibitory CAR (iCAR), a receptor designed to counteract the CAR T‐cell activation signal induced by the conventional CAR. This approach involves a signalling combination of two different CARs in the engineered T cell, whereby the iCAR, upon engagement by a specific antigen expressed only on healthy cells, induces a dominant inhibitory signal to limit the T‐cell activating signal generated by the conventional CAR. In human T cells, PD‐1 and CTLA‐4 intracellular signalling domains were used in iCARs owing to their ability to reduce TCR signalling, resulting in decreased T‐cell cytokine production and lysis upon antigen stimulation on healthy cells.111 This iCAR T‐cell strategy thus provides a self‐regulating safety switch that allows for distinction between the tumor and healthy cells, resulting in a more selective elimination of tumor cells. One additional area of investigation has been the generation of ‘titratable’ CAR T cells that allow exogenous regulation of T‐cell function. This has been achieved through the generation of ‘On‐switch’ CAR receptors consisting of separate extracellular and intracellular domains containing an FKBP and FRB domain, respectively, that heterodimerise and signal only in the presence of rapamycin analogues.112, 113 Whilst promising, these preclinical models are limited by their reliance on rapamycin analogues that have unfavorable pharmacokinetic characteristics.112
An alternative approach to overcome potential CAR T‐cell therapy toxicity is through incorporation of the so‐called ‘suicide genes’ into transferred cells that allow for their targeted depletion. One key approach involves the incorporation of extracellular markers that can then be targeted by antibodies with pre‐established clinical use. These markers include codon optimised CD20 (CD20op)114 and RQR8,115 both targetable by rituximab; truncated epidermal growth factor (EGRFt),116 targetable by cetuximab; and HSC‐tk, targetable by ganciclovir.117 Arguably the most specific method, however, involves the integration of an inducible caspase 9 (iCasp9) domain into CAR T cells. The iCasp9 gene consists of an intracellular compartment of the human Casp9 protein, which is a pro‐apoptotic molecule, fused to a chemical induction of dimerisation (CID) drug‐binding domain. Upon administration of a CID drug, the drug‐binding domains of the fused iCasp9 protein are cross‐linked, leading to dimerisation of the Casp9 proteins that eventually results in cellular apoptosis induced by the downstream caspase 3 molecule.118, 119 In 2010, Hoyos et al.120 reported the first preclinical study of CAR T cells incorporating the iCasp9 gene. In this study, second‐generation CD19‐CAR T cells expressing iCasp9 were used in vivo and were successfully eliminated within 3 days following administration of a CID drug. In the event of a serious adverse event, this suicide gene strategy may facilitate immediate removal of CAR T cells, alleviating toxicity induced by the CAR T cells. However, it is important to note that there may be other preferred strategies to utilise given that complete removal of CAR T cells may increase the risk of tumor relapse.
Future perspectives
Adoptive T‐cell therapy holds promising potential for being a standard of care treatment option. This is supported by the recent FDA approval of two CD19‐CAR T‐cell products for the treatment of patients with B‐cell ALL and non‐Hodgkin lymphoma who have not responded to, or who have relapsed following at least two other conventional treatments.121, 122 In order for adoptive T‐cell therapy to become a first in‐line treatment option, however, a number of crucial challenges still need to be addressed.
Overall, clinical responses of patients with B‐cell ALL and non‐Hodgkin lymphoma to the FDA‐approved CD19 CAR T cells have been excellent.123 However, a significant proportion of patients treated with Kymriah™ have relapsed after several months, and many of the patients treated with Yescarta™ have exhibited only partial responses that eventually waned by 6 months post‐treatment.124 Thus, despite the remarkable success, there are concerns about long‐term efficacy of CAR T cells, and it remains unknown how long the responses might last. More clinical data with long‐term follow‐up will be required to assess the long‐term benefit of CAR T cells. Furthermore, in the more complex setting of solid cancers, significant clinical responses are yet to be achieved.5 Several factors could potentially have an impact on CAR T‐cell efficacy, including the variable potencies of the CARs themselves. It has been reported that tonic CAR signalling triggered by the clustering of CAR scFvs independent of antigen is capable of inducing CAR T‐cell exhaustion hence limiting antitumor activity. Such activation was observed to varying degrees in multiple CARs studied targeting different antigens, except for the highly efficacious CD19 CAR.125 Another factor that could influence CAR T‐cell effectiveness includes the makeup of the gut microbiome. A recent study demonstrated that the efficacy of adoptive therapy in a mouse model was significantly affected by differences in the composition of gut bacteria.126 Further, the TME is highly immunosuppressive in solid tumors,127 and hence, combination strategies that can alleviate the immunosuppressive environment will be important to test in future clinical trials.
In addition to the TME, other factors related to the CAR T cells themselves can also have significant impacts on the therapeutic outcome. For example, component variability of the CAR T‐cell product such as variable differentiation stages of T cells used for infusion can potentially affect overall CAR T‐cell activity, and thus, a more uniform production method may be important in the future. One approach to address this issue may include determining the optimum formulation of T‐cell subsets to use for infusion. It is well established that each subset of T cells has a unique function and cytokine profile, which influence their respective antitumor response.128, 129 In the clinic, predefined CD4:CD8 T‐cell compositions have been used in clinical trials funded by therapeutic companies including Juno and Celgene.130, 131 Moreover, it is increasingly apparent that the quality and efficacy of T‐cell immunity are a result of the diversification of naïve T cells into a number of phenotypically different subsets, including the highly differentiated effector, tissue resident memory, effector memory, central memory and memory stem T cells.132 Naïve T cells can give rise to long‐lived memory stem and central memory T cells that are capable of self‐renewal and can provide proliferating populations of more differentiated effector T cells, which are relatively short‐lived.129 This fate framework indicates that CAR genetic modification of less differentiated T‐cell subsets may result in achieving a greater and more sustained therapeutic response.133 Indeed, preclinical studies have reported that receptor engineering of T cells selected from naïve and central memory subsets, or expanding naïve T cells in vitro with the addition of factors preventing the differentiation of T cells, can result in cell products possessing superior antitumor effects, proliferation and engraftment following adoptive transfer.134, 135 Further, given that different cytokines commonly used in in vitro culture to maintain T‐cell survival such as IL‐2, IL‐7, IL‐15 and IL‐21 may have different impacts on T‐cell differentiation,136, 137, 138, 139, 140 the cytokines used to culture T cells prior to adoptive transfer require further testing and characterisation. These observations together indicate that determining the optimum formulation of T‐cell subsets with the most superior antitumor potency for uniform use in adoptive transfer may help improve therapeutic outcome. Furthermore, this strategy may additionally reduce product variability between patients, resulting in a more consistent therapeutic response. Taken together, resolving these concerns and challenges will hopefully allow for more widespread application, as well as acceleration of CAR T‐cell therapy to become a standard of care treatment option for various cancer types.
Concluding summary
The approaches described herein to potentially improve CAR T‐cell therapy emphasise the important challenges within both haematological and solid malignancies that need to be overcome. Increasing the specificity and safety of CAR T cells, harnessing the endogenous immune response to extend tumor destruction beyond CAR T‐cell recognition and reducing the manufacturing costs will together accelerate the broad application of CAR T‐cell therapy in various cancer types. Notably, emerging technologies using nonviral gene transfer such as mRNA electroporation and the Sleeping Beauty and PiggyBac transposon/transposase systems are currently being explored as inexpensive alternatives for large‐scale manufacturing of CAR T cells.141, 142 Insights gained from ongoing research will be important to the growing body of knowledge that provides novel strategies to significantly address some of the existing limitations for the treatment of solid malignancies.
Acknowledgements
This work was funded by a project and programme grant from the National Health and Medical Research Council (NHMRC; Grant number APP1062580 and APP1132373) and a project grant from the Cancer Council of Victoria (APP1143517). PA Beavis was supported by a National Breast Cancer Foundation Fellowship (ECF‐17‐005). PK Darcy was supported by a NHMRC Senior Research Fellowship (APP1136680).
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Paul Andrew Beavis, Email: paul.beavis@petermac.org.
Phillip Kevin Darcy, Email: phil.darcy@petermac.org.
References
- 1. Baumeister SH, Freeman GJ, Dranoff G et al Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol 2016; 34: 539–573. [DOI] [PubMed] [Google Scholar]
- 2. Maude SL, Teachey DT, Rheingold SR et al Sustained remissions with CD19‐specific chimeric antigen receptor (CAR)‐modified T cells in children with relapsed/refractory ALL. ASCO Meeting Abstracts 2016; 34: 3011. [Google Scholar]
- 3. Srivastava S, Riddell SR. Engineering CAR‐T cells: design concepts. Trends Immunol 2015; 36: 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maude SL, Barrett DM, Rheingold SR et al Efficacy of humanized CD19‐targeted chimeric antigen receptor (CAR)‐modified T cells in children and young adults with relapsed/refractory acute lymphoblastic leukemia. Blood 2016; 128: 217.27207794 [Google Scholar]
- 5. Kershaw M, Westwood J, Darcy P. Gene‐engineered T cells for cancer therapy. Nat Rev Cancer 2013; 13: 525–541. [DOI] [PubMed] [Google Scholar]
- 6. Caruana I, Savoldo B, Hoyos V et al Heparanase promotes tumor infiltration and antitumor activity of CAR‐redirected T lymphocytes. Nat Med 2015; 21: 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T‐cell therapy for solid tumors. Molecular Therapy ‐ Oncolytics 2016; 3: 16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raedler LA. Opdivo (Nivolumab): second PD‐1 inhibitor receives FDA approval for unresectable or metastatic melanoma. Am Health Drug Benefits 2015; 8: 180–183. [PMC free article] [PubMed] [Google Scholar]
- 9. Gong J, Chehrazi‐Raffle A, Reddi S et al Development of PD‐1 and PD‐L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer 2018; 6: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lipson EJ, Drake CG. Ipilimumab: an anti‐CTLA‐4 antibody for metastatic melanoma. Clin Cancer Res 2011; 17: 6958–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. John LB, Devaud C, Duong CPM et al Anti‐PD‐1 antibody therapy potently enhances the eradication of established tumors by gene‐modified T cells. Clin Cancer Res 2013; 19: 5636–5646. [DOI] [PubMed] [Google Scholar]
- 12. Beavis PA, Henderson MA, Giuffrida L et al Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest 2017; 127: 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chong EA, Melenhorst JJ, Lacey SF et al PD‐1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: refueling the CAR. Blood 2017; 129: 1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bartkowiak T, Curran MA. 4‐1BB agonists: multi‐potent potentiators of tumor immunity. Frontiers in Oncology 2015; 5: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jensen S, Maston L, Gough M et al Signaling through OX40 enhances antitumor immunity. Semin Oncol 2010; 37: 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28‐OX40 signalling. Int J Cancer 2011; 129: 2935–2944. [DOI] [PubMed] [Google Scholar]
- 17. Quintarelli C, Orlando D, Boffa I et al Choice of costimulatory domains and of cytokines determines CAR T‐cell activity in neuroblastoma. OncoImmunology 2018; 7: e1433518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mardiana S, John LB, Henderson MA et al A Multifunctional role for adjuvant anti‐4‐1BB therapy in augmenting antitumor response by chimeric antigen receptor T cells. Can Res 2017; 77: 1296. [DOI] [PubMed] [Google Scholar]
- 19. Chen S, Lee LF, Fisher TS et al Combination of 4‐1BB agonist and PD‐1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res 2015; 3: 149–160. [DOI] [PubMed] [Google Scholar]
- 20. Shindo Y, Yoshimura K, Kuramasu A et al Combination immunotherapy with 4‐1BB activation and PD‐1 blockade enhances antitumor efficacy in a mouse model of subcutaneous tumor. Anticancer Res 2015; 35: 129–136. [PubMed] [Google Scholar]
- 21. Sánchez‐Paulete AR, Cueto FJ, Martínez‐López M et al Cancer immunotherapy with immunomodulatory anti‐CD137 and anti‐PD‐1 monoclonal antibodies requires Batf3‐dependent dendritic cells. Cancer Discov 2016; 6: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo Z, Wang X, Cheng D et al PD‐1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS ONE 2014; 9: e89350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Messenheimer DJ, Jensen SM, Afentoulis ME et al Timing of PD‐1 blockade is critical to effective combination immunotherapy with Anti‐OX40. Clin Cancer Res 2017; 23: 6165–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shrimali RK, Ahmad S, Verma V et al Concurrent PD‐1 blockade negates the effects of OX40 agonist antibody in combination immunotherapy through inducing T‐cell apoptosis. Cancer Immunol Res 2017; 5: 755–766. [DOI] [PubMed] [Google Scholar]
- 25. McKee SJ, Doff BL, Soon MS et al Therapeutic efficacy of 4‐1BB costimulation is abrogated by PD‐1 blockade in a model of spontaneous B‐cell lymphoma. Cancer Immunol Res 2017; 5: 191–197. [DOI] [PubMed] [Google Scholar]
- 26. Fischbach MA, Bluestone JA, Lim WA. Cell‐based therapeutics: the next pillar of medicine. Sci Transl Med 2013; 5: 179 ps177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bashor CJ, Helman NC, Yan S et al Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science 2008; 319: 1539. [DOI] [PubMed] [Google Scholar]
- 28. Watanabe N, Bajgain P, Sukumaran S et al Fine‐tuning the CAR spacer improves T‐cell potency. OncoImmunology 2016; 5: e1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Juillerat A, Marechal A, Filhol JM et al An oxygen sensitive self‐decision making engineered CAR T‐cell. Sci Rep 2017; 7: 39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chinnasamy D, Yu Z, Kerkar SP et al Local delivery of interleukin‐12 using T cells targeting VEGF receptor‐2 eradicates multiple vascularized tumors in mice. Clin Cancer Res 2012; 18: 1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pegram HJ, Lee JC, Hayman EG et al Tumor‐targeted T cells modified to secrete IL‐12 eradicate systemic tumors without need for prior conditioning. Blood 2012; 119: 4133–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koneru M, Purdon TJ, Spriggs D et al IL‐12 secreting tumor‐targeted chimeric antigen receptor T cells eradicate ovarian tumors. Oncoimmunology 2015; 4: e994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Avanzi MP, van Leeuwen DG, Li X et al IL‐18 secreting CAR T cells enhance cell persistence, induce prolonged B cell aplasia and eradicate CD19+ tumor cells without need for prior conditioning. Blood 2016; 128: 816.27301861 [Google Scholar]
- 34. Hu B, Ren J, Luo Y et al Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL‐18. Cell Rep 2017; 20: 3025–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hurton LV, Singh H, Najjar AM et al Tethered IL‐15 augments antitumor activity and promotes a stem‐cell memory subset in tumor‐specific T cells. Proc Natl Acad Sci 2016; 113: e7788–e7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Curran KJ, Seinstra BA, Nikhamin Y et al Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther 2015; 23: 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stephan MT, Ponomarev V, Brentjens RJ et al T cell–encoded CD80 and 4‐1BBL induce auto‐ and transcostimulation, resulting in potent tumor rejection. Nat Med 2007; 13: 1440. [DOI] [PubMed] [Google Scholar]
- 38. Rafiq S, Yeku OO, Jackson HJ et al Targeted delivery of a PD‐1‐blocking scFv by CAR‐T cells enhances anti‐tumor efficacy in vivo. Nat Biotechnol 2018; 36: 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bollard CM, Tripic T, Cruz CR et al Tumor‐specific T‐cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J Clin Oncol 2018; 36: 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adachi K, Kano Y, Nagai T et al IL‐7 and CCL19 expression in CAR‐T cells improves immune cell infiltration and CAR‐T cell survival in the tumor. Nat Biotechnol 2018; 36: 346–351. [DOI] [PubMed] [Google Scholar]
- 41. Feig C, Jones JO, Kraman M et al Targeting CXCL12 from FAP‐expressing carcinoma‐associated fibroblasts synergizes with anti‐PD‐L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA 2013; 110: 20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang G, Lu X, Dey P et al Targeting YAP‐dependent MDSC infiltration impairs tumor progression. Cancer Discov 2016; 6: 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harlin H, Meng Y, Peterson AC et al Chemokine expression in melanoma metastases associated with CD8+ T‐cell recruitment. Cancer Res 2009; 69: 3077–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kershaw MH, Wang G, Westwood JA et al Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther 2002; 13: 1971–1980. [DOI] [PubMed] [Google Scholar]
- 45. Craddock JA, Lu A, Bear A et al Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother 2010; 33: 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moon EK, Carpenito C, Sun J et al Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin‐specific chimeric antibody receptor. Clin Cancer Res 2011; 17: 4719–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Di Stasi A, De Angelis B, Rooney CM et al T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009; 113: 6392–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Spranger S, Dai D, Horton B et al Tumor‐residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T‐cell therapy. Cancer Cell 2017; 31: 711–723. e714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Can Res 2014; 74: 7168. [DOI] [PubMed] [Google Scholar]
- 50. Bedognetti D, Spivey TL, Zhao Y et al CXCR3/CCR5 pathways in metastatic melanoma patients treated with adoptive therapy and interleukin‐2. Br J Cancer 2013; 109: 2412–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mikucki ME, Fisher DT, Matsuzaki J et al Non‐redundant requirement for CXCR3 signalling during tumoricidal T‐cell trafficking across tumour vascular checkpoints. Nat Commun 2015; 6: 7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peng D, Kryczek I, Nagarsheth N et al Epigenetic silencing of TH1‐type chemokines shapes tumour immunity and immunotherapy. Nature 2015; 527: 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peng W, Liu C, Xu C et al PD‐1 blockade enhances T‐cell migration to tumors by elevating IFN‐gamma inducible chemokines. Cancer Res 2012; 72: 5209–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bonecchi R, Locati M, Mantovani A. Chemokines and cancer: a fatal attraction. Cancer Cell 2011; 19: 434–435. [DOI] [PubMed] [Google Scholar]
- 55. Brentjens R, Davila M, Riviere I et al CD19‐targeted T cells rapidly induce molecular remissions in adults with chemotherapy‐refractory acute lymphoblastic leukemia. Sci Transl Med 2013; 5: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Till BG, Jensen MC, Wang J et al CD20‐specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4‐1BB domains: pilot clinical trial results. Blood 2012; 119: 3940–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ahmed N, Brawley VS, Hegde M et al Human epidermal growth factor receptor 2 (HER2)—specific chimeric antigen receptor‐modified T cells for the immunotherapy of HER2‐positive sarcoma. J Clin Oncol 2015; 33: 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grupp SA, Maude SL, Shaw P et al T cells engineered with a chimeric antigen receptor (CAR) targeting CD19 (CTL019) have long term persistence and induce durable remissions in children with relapsed, refractory ALL. Blood 2014; 124: 380. [Google Scholar]
- 59. Jackson HJ, Brentjens RJ. Overcoming antigen escape with CAR T‐cell therapy. Cancer Discov 2015; 5: 1238–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sotillo E, Barrett DM, Black KL et al Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART‐19 immunotherapy. Cancer Discov 2015; 5: 1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Poulikakos PI, Persaud Y, Janakiraman M et al RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011; 480: 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hegde M, Corder A, Chow KKH et al Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther 2013; 21: 2087–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Qin H, Nguyen SM, Ramakrishna S et al Novel CD19/CD22 bicistronic chimeric antigen receptors outperform single or bivalent cars in eradicating CD19+CD22+, CD19‐, and CD22‐ Pre‐B leukemia. Blood 2017; 130: 810. [Google Scholar]
- 64. Ruella M, Barrett DM, Kenderian SS et al Dual CD19 and CD123 targeting prevents antigen‐loss relapses after CD19‐directed immunotherapies. J Clin Invest 2016; 126: 3814–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bielamowicz K, Fousek K, Byrd TT et al Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro‐Oncology 2018; 20: 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chmielewski M, Kopecky C, Hombach AA et al IL‐12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen‐independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res 2011; 71: 5697–5706. [DOI] [PubMed] [Google Scholar]
- 67. Michie J, Beavis PA, Freeman AJ et al Antagonism of IAPs enhances CAR T‐cell efficacy. Cancer Immunol Res 2019; 7: 183. [DOI] [PubMed] [Google Scholar]
- 68. Gujar S, Dielschneider R, Clements D et al Multifaceted therapeutic targeting of ovarian peritoneal carcinomatosis through virus‐induced immunomodulation. Mol Ther 2013; 21: 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gujar SA, Pan D, Marcato P et al Oncolytic virus‐initiated protective immunity against prostate cancer. Mol Ther 2011; 19: 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gauvrit A, Brandler S, Sapede‐Peroz C et al Measles virus induces oncolysis of mesothelioma cells and allows dendritic cells to cross‐prime tumor‐specific CD8 response. Cancer Res 2008; 68: 4882–4892. [DOI] [PubMed] [Google Scholar]
- 71. Slaney CY, von Scheidt B, Davenport AJ et al Dual‐specific chimeric antigen receptor T cells and an indirect vaccine eradicate a variety of large solid tumors in an immunocompetent, self‐antigen setting. Clin Cancer Res 2017; 23: 2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nishio N, Diaconu I, Liu H et al Armed oncolytic virus enhances immune functions of chimeric antigen receptor–modified T cells in solid tumors. Can Res 2014; 74: 5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Moon EK, Wang L‐CS, Bekdache K et al Intra‐tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T‐cell therapy and vaccines. OncoImmunology 2018; 7: e1395997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Watanabe K, Luo Y, Da T et al Pancreatic cancer therapy with combined mesothelin‐redirected chimeric antigen receptor T cells and cytokine‐armed oncolytic adenoviruses. JCI Insight 2018; 3: 99573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tanoue K, Rosewell Shaw A, Watanabe N et al Armed oncolytic adenovirus–expressing PD‐L1 mini‐body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Can Res 2017; 77: 2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wing A, Fajardo CA, Posey AD et al Improving CART‐cell therapy of solid tumors with oncolytic virus–driven production of a bispecific T‐cell engager. Cancer Immunol Res 2018; 6: 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. US National Library of Science . ClinicalTrials.gov[online], https://clinicaltrials.gov/ct2/show/NCT01716364. 2010.
- 78. Zhang C, Wang Z, Yang Z et al Phase I escalating‐dose trial of CAR‐T therapy targeting CEA+ metastatic colorectal cancers. Mol Ther 2017; 25: 1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Park JR, Digiusto DL, Slovak M et al Adoptive transfer of chimeric antigen receptor re‐directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther 2007; 15: 825–833. [DOI] [PubMed] [Google Scholar]
- 80. Louis CU, Savoldo B, Dotti G et al Antitumor activity and long‐term fate of chimeric antigen receptor‐positive T cells in patients with neuroblastoma. Blood 2011; 118: 6050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ritchie DS, Neeson PJ, Khot A et al Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther 2013; 21: 2122–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Parkhurst MR, Yang JC, Langan RC et al T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011; 19: 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Morgan RA, Yang JC, Kitano M et al Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Majzner RG, Theruvath JL, Nellan A et al CAR T cells targeting B7‐H3, a pan‐cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res 2019. e‐pub ahead of print Jan 17; 10.1158/1078-0432.ccr-18-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Willemsen RA, Ronteltap C, Chames P et al T cell retargeting with MHC class I‐restricted antibodies: the CD28 costimulatory domain enhances antigen‐specific cytotoxicity and cytokine production. J Immunol 2005; 174: 7853–7858. [DOI] [PubMed] [Google Scholar]
- 86. Willemsen RA, Debets R, Hart E et al A phage display selected fab fragment with MHC class I‐restricted specificity for MAGE‐A1 allows for retargeting of primary human T lymphocytes. Gene Ther 2001; 8: 1601–1608. [DOI] [PubMed] [Google Scholar]
- 87. Stewart‐Jones G, Wadle A, Hombach A et al Rational development of high‐affinity T‐cell receptor‐like antibodies. Proc Natl Acad Sci 2009; 106: 10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ma Q, Garber HR, Lu S et al A novel TCR‐like CAR with specificity for PR1/HLA‐A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy 2016; 18: 985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhao Q, Ahmed M, Tassev DV et al Affinity maturation of T‐cell receptor‐like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia 2015; 29: 2238–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rafiq S, Purdon TJ, Daniyan AF et al Optimized T‐cell receptor‐mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia 2016; 31: 1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. de Lima Lopes G, Nahas GR. Chimeric antigen receptor T cells, a savior with a high price. Chin Clin Oncol 2018; 7: 21. [DOI] [PubMed] [Google Scholar]
- 92. The quest for off‐the‐shelf CAR T cells. Cancer Discov 2018; 8: 787–788. [DOI] [PubMed] [Google Scholar]
- 93. Yang Y, Jacoby E, Fry TJ. Challenges and opportunities of allogeneic donor‐derived CAR T cells. Curr Opin Hematol 2015; 22: 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Papadopoulou A, Gerdemann U, Katari UL et al Activity of broad‐spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci Transl Med 2014; 6: 242ra283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tzannou I, Papadopoulou A, Naik S et al Off‐the‐shelf virus‐specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, epstein‐barr virus, and adenovirus infections after allogeneic hematopoietic stem‐cell transplantation. J Clin Oncol 2017; 35: 3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Poirot L, Philip B, Schiffer‐Mannioui C et al Multiplex genome‐edited T‐cell manufacturing platform for “off‐the‐shelf” adoptive T‐cell immunotherapies. Cancer Res 2015; 75: 3853–3864. [DOI] [PubMed] [Google Scholar]
- 97. Qasim W, Zhan H, Samarasinghe S et al Molecular remission of infant B‐ALL after infusion of universal TALEN gene‐edited CAR T cells. Sci Transl Med 2017; 9: 374. [DOI] [PubMed] [Google Scholar]
- 98. Kamiya T, Wong D, Png YT et al A novel method to generate T‐cell receptor–deficient chimeric antigen receptor T cells. Blood Adv 2018; 2: 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. MacLeod DT, Antony J, Martin AJ et al Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene‐edited CAR T cells. Mol Ther 2017; 25: 949–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Eyquem J, Mansilla‐Soto J, Giavridis T et al Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017; 543: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sather BD, Romano Ibarra GS, Sommer K et al Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med 2015; 7: 307ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Torikai H, Reik A, Liu PQ et al A foundation for universal T‐cell based immunotherapy: T cells engineered to express a CD19‐specific chimeric‐antigen‐receptor and eliminate expression of endogenous TCR. Blood 2012; 119: 5697–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Torikai H, Reik A, Soldner F et al Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013; 122: 1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Chu J, Deng Y, Benson DM et al CS1‐specific chimeric antigen receptor (CAR)‐engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014; 28: 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schonfeld K, Sahm C, Zhang C et al Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2‐specific chimeric antigen receptor. Mol Ther 2015; 23: 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kenderian SS, Porter DL, Gill S. Chimeric antigen receptor T cells and hematopoietic cell transplantation: how not to put the CART before the horse. Biol Blood Marrow Transplant 2017; 23: 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Urbanska K, Lanitis E, Poussin M et al A universal strategy for adoptive immunotherapy of cancer through use of a novel T‐cell antigen receptor. Cancer Res 2012; 72: 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kudo K, Imai C, Lorenzini P et al T lymphocytes expressing a CD16 signaling receptor exert antibody‐dependent cancer cell killing. Can Res 2014; 74: 93. [DOI] [PubMed] [Google Scholar]
- 109. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T‐cell responses. Cell 2018; 173: 1426–1438. e1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Grada Z, Hegde M, Byrd T et al TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids 2013; 2: e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Fedorov VD, Themeli M, Sadelain M. PD‐1‐ and CTLA‐4‐based inhibitory chimeric antigen receptors (iCARs) divert off‐target immunotherapy responses. Sci Transl Med 2013; 5: 215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wu C‐Y, Roybal KT, Puchner EM et al Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science 2015; 350: aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Juillerat A, Marechal A, Filhol J‐M et al Design of chimeric antigen receptors with integrated controllable transient functions. Sci Rep 2016; 6: 18950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Vogler I, Newrzela S, Hartmann S et al An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene‐modified T cells for adoptive immunotherapy. Mol Ther 2010; 18: 1330–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Philip B, Kokalaki E, Mekkaoui L et al A highly compact epitope‐based marker/suicide gene for easier and safer T‐cell therapy. Blood 2014; 124: 1277. [DOI] [PubMed] [Google Scholar]
- 116. Paszkiewicz PJ, Fräßle SP, Srivastava S et al Targeted antibody‐mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Investig 2016; 126: 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Barese CN, Krouse AE, Metzger ME et al Thymidine kinase suicide gene‐mediated ganciclovir ablation of autologous gene‐modified rhesus hematopoiesis. Mol Ther 2012; 20: 1932–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Straathof KC, Pule MA, Yotnda P et al An inducible caspase 9 safety switch for T‐cell therapy. Blood 2005; 105: 4247–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Di Stasi A, Tey SK, Dotti G et al Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365: 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hoyos V, Savoldo B, Quintarelli C et al Engineering CD19‐specific T lymphocytes with interleukin‐15 and a suicide gene to enhance their anti‐lymphoma/leukemia effects and safety. Leukemia 2010; 24: 1160–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. (FDA) FaDA . FDA approval brings first gene therapy to the United States. Maryland: Food and Drug Administration (FDA), 2017. [Google Scholar]
- 122. (FDA) FaDA . FDA approves CAR‐T‐cell therapy to treat adults with certain types of large B‐cell lymphoma. Maryland: Food and Drug Administration (FDA), 2017. [Google Scholar]
- 123. Zheng P‐P, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long‐term impacts. Drug Discovery Today 2018; 23: 1175–1182. [DOI] [PubMed] [Google Scholar]
- 124. National Cancer Institute . With FDA approval for advanced lymphoma, second CAR T‐cell therapy moves to the clinic 2017. [e‐pub ahead of print]. Available from: https://www.cancer.gov/news-events/cancer-currents-blog/2017/yescarta-fda-lymphoma.
- 125. Long A, Haso W, Shern J et al 4‐1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Uribe‐Herranz M, Bittinger K, Rafail S et al Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL‐12. JCI Insight 2018; 3: 94952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Shiao SL, Ganesan AP, Rugo HS et al Immune microenvironments in solid tumors: new targets for therapy. Genes Dev 2011; 25: 2559–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Raphael I, Nalawade S, Eagar TN et al T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015; 74: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature 2017; 545: 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rouce RH, Heslop HE. Equal opportunity CAR T cells. Blood 2017; 129: 3275–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Turtle CJ, Hanafi LA, Berger C et al CD19 CAR‐T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016; 126: 2123–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol 2014; 14: 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Busch DH, Frassle SP, Sommermeyer D et al Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol 2016; 28: 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Gattinoni L, Klebanoff CA, Palmer DC et al Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Investig 2005; 115: 1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Berger C, Jensen MC, Lansdorp PM et al Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Investig 2008; 118: 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Lu J, Giuntoli RL 2nd, Omiya R et al Interleukin 15 promotes antigen‐independent in vitro expansion and long‐term survival of antitumor cytotoxic T lymphocytes. Clin Cancer Res 2002; 8: 3877–3884. [PubMed] [Google Scholar]
- 137. Kaartinen T, Luostarinen A, Maliniemi P et al Low interleukin‐2 concentration favors generation of early memory T cells over effector phenotypes during chimeric antigen receptor T‐cell expansion. Cytotherapy 2017; 19: 689–702. [DOI] [PubMed] [Google Scholar]
- 138. Read KA, Powell MD, McDonald PW et al IL‐2, IL‐7, and IL‐15: multistage regulators of CD4+ T helper cell differentiation. Exp Hematol 2016; 44: 799–808. [DOI] [PubMed] [Google Scholar]
- 139. Hinrichs CS, Spolski R, Paulos CM et al IL‐2 and IL‐21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008; 111: 5326–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hinrichs CS, Borman ZA, Cassard L et al Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci USA 2009; 106: 17469–17474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kebriaei P, Singh H, Huls MH et al Phase I trials using sleeping beauty to generate CD19‐specific CAR T cells. J Clin Investig 2016; 126: 3363–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Ramanayake S, Bilmon I, Bishop D et al Low‐cost generation of good manufacturing practice‐grade CD19‐specific chimeric antigen receptor‐expressing T cells using piggyBac gene transfer and patient‐derived materials. Cytotherapy 2015; 17: 1251–1267. [DOI] [PubMed] [Google Scholar]