

Key Points
The covalent BTK inhibitor evobrutinib blocks B cell and myeloid cell activation.
Evobrutinib is efficacious in mouse models for RA, SLE, and cutaneous anaphylaxis.
Covalent binding allows modeling the relationship between BTK occupancy and efficacy.
Abstract
Because of its role in mediating both B cell and Fc receptor signaling, Bruton’s tyrosine kinase (BTK) is a promising target for the treatment of autoimmune diseases such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). Evobrutinib is a novel, highly selective, irreversible BTK inhibitor that potently inhibits BCR- and Fc receptor–mediated signaling and, thus, subsequent activation and function of human B cells and innate immune cells such as monocytes and basophils. We evaluated evobrutinib in preclinical models of RA and SLE and characterized the relationship between BTK occupancy and inhibition of disease activity. In mouse models of RA and SLE, orally administered evobrutinib displayed robust efficacy, as demonstrated by reduction of disease severity and histological damage. In the SLE model, evobrutinib inhibited B cell activation, reduced autoantibody production and plasma cell numbers, and normalized B and T cell subsets. In the RA model, efficacy was achieved despite failure to reduce autoantibodies. Pharmacokinetic/pharmacodynamic modeling showed that mean BTK occupancy in blood cells of 80% was linked to near-complete disease inhibition in both RA and SLE mouse models. In addition, evobrutinib inhibited mast cell activation in a passive cutaneous anaphylaxis model. Thus, evobrutinib achieves efficacy by acting both on B cells and innate immune cells. Taken together, our data show that evobrutinib is a promising molecule for the chronic treatment of B cell–driven autoimmune disorders.
Visual Abstract
Introduction
Bruton’s tyrosine kinase (BTK) is a Tec family tyrosine kinase expressed in many cells of hematopoietic origin, including B cells, monocytes, neutrophils, mast cells, and osteoclasts. BTK expression is absent from T cells. BTK participates in both the adaptive and innate arms of the immune response (1, 2) and mediates signaling of several immune receptors, including the BCR and Fc receptor. Upon receptor ligation, kinases such as Syk or the Src kinases Lyn/Fyn can phosphorylate and partially activate BTK. BTK in turn autophosphorylates at Y223, resulting in full activation and consequent phosphorylation of its immediate downstream effector, PLCγ2, ultimately leading to both calcium and MAPK signaling (3, 4).
BTK has demonstrated importance for the function of several immune cell types. In B cells, it is required for maturation, proliferation, Ag presentation, and differentiation to Ab-producing plasma cells. In innate immune cell types, such as monocytes and granulocytes, BTK controls cytokine production, phagocytosis, and production of inflammatory mediators (5). BTK is also involved in several other processes, such as platelet activation via the glycoprotein VI receptor (6), osteoclast differentiation in response to the receptor activator of NF-κB (RANK) signaling (7), and cell migration in response to certain chemokines (8). Spontaneous mutations leading to BTK deficiency in humans result in X-linked agammaglobulinemia, which is characterized by a nearly complete loss of serum Igs and circulating mature B cells, as well as an increased susceptibility to infections. In some cases (∼25%), X-linked agammaglobulinemia patients also present with neutropenia (9, 10). Absence of BTK leads to loss of Ag-induced signaling, BCR internalization, Ag processing and presentation by B cells (11), and incomplete acquisition of central B cell tolerance during development (12) but also protects against breach of B cell tolerance and autoantibody production and blocks Ig class-switching (13). In contrast, booster immunizations with a T cell–dependent Ag produce a near-normal humoral immune response in BTK-deficient mice (14).
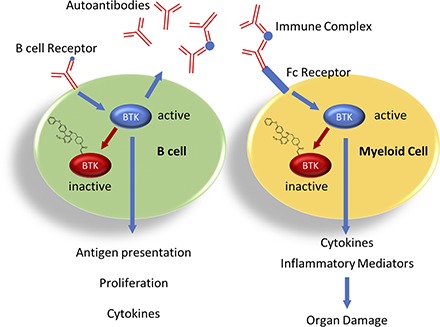
In autoimmune diseases like rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), the strong B cell component is paired with activation of innate immune and stromal cells. In RA patients, B cells may promote disease by several mechanisms, including production of autoantibodies and subsequent complement fixation and Ag presentation to T cells. The efficacy of B cell targeting agents supports a crucial role for this population in RA patients (15). In addition, aberrant osteoclastogenesis and fibroblast activation are believed to contribute to bone destruction in RA (16, 17). In SLE, autoantibodies form immune complexes that, in the case of lupus nephritis, are deposited in the kidney and cause innate immune cell activation and tissue damage (18). The importance of B cells in both diseases is underscored by the efficacy of B cell depletion (15, 19).
However, especially in SLE, B cell–targeting therapies show only limited efficacy (20, 21). In SLE patients treated with rituximab (anti-CD20), those with incomplete B cell depletion are prone to earlier flares, and flares are accompanied by reappearance of memory B cells and plasmablasts (22). In addition, persistent long-lived plasma cells continue to produce autoantibodies, and the resulting immune complexes have been implicated as triggers of flares (23). As BTK is expressed in multiple immune cell types, it may provide therapeutic benefit by blocking not only the B cell component of autoimmune diseases but also the innate component mediating disease pathogenesis and may accomplish what B cell depletion alone cannot. In fact, the efficacy of pharmacological BTK inhibition in mouse models for RA and SLE, including glomerulonephritis, has been demonstrated (24–29). However, a translation of these preclinical findings into the degree of BTK inhibition predicted to provide efficacy in animal models and ultimately in humans is lacking. This would be extremely helpful when developing BTK inhibitors for the treatment of patients with autoimmune diseases.
The BTK inhibitor ibrutinib (Imbruvica), is approved for the treatment of B cell malignancies (chronic lymphocytic leukemia [CLL], Waldenström's macrogammaglobulinemia, marginal zone lymphoma, and Mantle cell lymphoma), but no development for autoimmune disease has been reported. Ibrutinib inhibits a large number of kinases other than BTK, and the substantial off-target activity of the molecule may be prohibitive for its expanded development in autoimmune indications. For example, it has been argued that inhibition of IL-2–inducible T cell kinase (ITK) by ibrutinib may blunt Th2 activation and skew the T cell response toward the Th1 phenotype (30, 31). Although this effect might be helpful in the antitumor response, it would seem undesired when treating autoimmune diseases.
In this study, we characterize a novel, highly specific, and irreversible BTK inhibitor, evobrutinib (Fig. 1A) (32), that may be suitable for the chronic treatment of autoimmune diseases. This molecule binds BTK covalently. Thus, it provides prolonged target inhibition long after the compound has been cleared from the circulation. Evobrutinib potently inhibited BCR- and Fc receptor–mediated signaling. Consequently, evobrutinib prevented B cell and innate immune cell activation in vivo and was highly efficacious in mouse models for RA and SLE at very low doses, in which it prevented joint and kidney damage, respectively. In addition, other parameters that are dysregulated in SLE were normalized in the SLE model, such as lipid and glucose levels, hematocrit, RBC numbers, and hemoglobin. Splenic plasma cell numbers were reduced with evobrutinib treatment, as well, although the effect on anti-dsDNA autoantibodies was mild. Although evobrutinib does not inhibit T cells directly, BTK inhibition by evobrutinib normalized the composition of both B and T cell subsets toward a more naive phenotype. Finally, a pharmacokinetic (PK)/pharmacodynamic (PD)/efficacy model was established with the aim of quantitatively linking the degree of BTK inhibition to its efficacy in preclinical disease models after oral administration. We showed a direct, positive correlation with degree of BTK occupancy, B cell inhibition, and disease inhibition. This work provides a quantitative description of the correlation between BTK inhibition and efficacy in RA and SLE models.
FIGURE 1.

Characterization of evobrutinib as a potent, irreversible, and selective BTK inhibitor in biochemical assays. (A) Structure of evobrutinib, 1-(4-{[6-amino-5-(4-phenoxy-phenyl)-pyrimidin-4-ylamino]-methyl}-piperidin-1-yl)-propenone acid 2-methyl-4-[2-(1-methyl-1H-pyrazol-3-yl)-1H-imidazo[4,5-b]pyridin-7-yl]-benzylamide. (B) Evobrutinib is dependent on covalent binding to C481. The IC50 of evobrutinib on BTK C481S was 1100-fold higher than on WT BTK (left). In contrast, ibrutinib was able to inhibit both WT and C481S BTK with similar potency (right). (C) Selectivity of evobrutinib and ibrutinib was determined in the Millipore KinaseProfiler screen. Results are shown in a kinome tree format. Red circles depict inhibition of a tested kinase, whereas black dots depict no inhibition.
Materials and Methods
All experiments involving animals were conducted in accordance with the local laws and regulations concerning animal welfare, under protocols approved by local authorities.
Kinase assays
The potency of evobrutinib against BTK was determined using purified, full-length recombinant BTK (Carna Biosciences). The BTK protein was diluted in buffer to a final concentration of 0.05 ng/μl with 75 μM ATP and 1 μM KinKDR peptide FITC-AHA-EEPLYWSFPAKKK-NH2 (Tufts Core Facility, Boston, MA). Various concentrations of evobrutinib were also included. Reactions were performed at 25°C for 90 min and halted by addition of stop solution containing 0.5 M EDTA. Plates were then read on the Caliper LabChip 3000 (Caliper Life Sciences, Waltham, MA), and the data were loaded into Genedata Screener for generation of IC50 curves. For the comparison of inhibition of wild-type (WT) BTK versus C481S BTK by evobrutinib or ibrutinib, recombinant proteins covering the kinase domain (BTK WT 328-659 or BTK C481S 328-659) were used. For the jump dilution assay, 20 μl of assay buffer containing 100-fold standard biochemical assay concentration of BTK (0.63 nM) was added to 200 nl of evobrutinib or RN486 at a final concentration of 10-fold IC50 or to negative control (DMSO). After incubation at room temperature for 90 min, 1 μl of the mix solution was diluted into 99 μl of assay buffer containing substrate peptide (sequence FITC-AHA-EEPLYWSFPAKKK-NH2, 1 μM) and ATP (75 μM). The microplate was placed in the Caliper Life Sciences LabChip 3000, and wells were repeatedly sampled for 112 min. Kinase selectivity for evobrutinib and ibrutinib was determined in the KinaseProfiler screening panel (EMD Millipore, Billerica, MA) that tested the inhibitory activity of the compounds at 1 μM against 267 kinases.
BTK phosphorylation in Ramos cells
The effect of evobrutinib on BTK phosphorylation after BCR activation was determined in Ramos B cells. The Ramos Burkitt lymphoma cell line was procured from the American Type Culture Collection (catalog no. CRL1596) and maintained in RPMI 1640 media (Sigma-Aldrich) containing penicillin/streptomycin, 2 mM l-glutamine, and 10% FBS. Ramos cells were seeded into 96-well tissue culture plates at 8 × 106 cells per well. Cells were pretreated with the BTK inhibitor evobrutinib dissolved in DMSO for 30 min at 37°C. After compound treatment, the cells were stimulated with an anti-IgM F(ab′)2 Ab (SouthernBiotech, Birmingham, AL) at a concentration of 5 μg/ml to activate the BCR. The cells were incubated with the anti-IgM for 5 min at 37°C. After treatment, the cells were collected by centrifugation at 500 × g for 5 min. The medium was aspirated, and 150 μl of ice-cold Thermo Scientific Pierce M-PER lysis buffer containing Thermo Scientific Halt protease/phosphatase inhibitor mixture was added to the cells. The cells were resuspended in the lysis buffer, and the lysates were frozen to −80°C for subsequent measurement of BTK phosphorylation. Analysis of BTK phosphorylation was performed by Western blotting using the automated Wes instrument (ProteinSimple, San Jose, CA) according to the manufacturer’s instructions. For the Western blot assay, 6 μl of lysate was used, and a 1:3000 dilution of primary anti-BTK p-Y551 (BD Biosciences, San Diego, CA) or 1:50 anti-BTK p-Y223 (Cell Signaling Technology) was used for detection of phosphorylated BTK.
B cell activation in PBMCs and whole blood
The ability of evobrutinib to block BCR signaling was determined using B cells in whole blood or purified PBMCs. Human blood was collected with citrate as an anticoagulant. For the PBMC assay, PBMCs were isolated over a Ficoll gradient, and 2.5 × 105 cells per well were seeded into a 96-well plate. For the whole blood assay, 90 μl of blood per well was directly transferred to 96-well plates. Cells were pretreated for 60 min at 37°C with dilutions of evobrutinib before activation with goat anti-human IgM F(ab′)2 (Dianova), added to a final concentration of 20 μg/ml, and incubated at 37°C overnight. After activation, cells were stained for 45 min with anti-CD69–allophycocyanin (BD Biosciences) and anti-CD19–PerCP-Cy5.5 and then lysed using FACS lysis solution (BD Biosciences) and resuspended in PBS prior to FACS analysis. FACS analysis was performed on the FACSCanto II instrument. Cells were first gated on CD19, and the percentage of CD19+ cells that were also positive for CD69 was determined.
B cell proliferation, cytokine release, and plasmablast differentiation assays
CD19+ B cells were isolated from PBMCs of healthy volunteers by negative selection using the B cell purification kit II (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s instructions. Purified B cells were incubated with evobrutinib for 1 h and stimulated with 10 μg/ml goat F(ab′)2 anti-IgM (SouthernBiotech) and 10 ng/ml recombinant human IL-4 (ImmunoTools, Friesoythe, Germany) for 4 d. One microcurie of [3H]thymidine (Perkin Elmer, Shelton, CT) was added for the last 18 h of culture. Proliferation was measured using a multiplate β counter (Perkin Elmer). For the cytokine release assay, CD19+ B cells isolated from PBMCs of healthy volunteers were incubated with evobrutinib for 1 h and stimulated with 10 μg/ml rabbit anti-human IgA + IgG + IgM (H+L) (The Jackson Laboratory, Bar Harbor, ME), 3 μg/ml CpG oligodeoxynucleotide 2006 (InvivoGen, San Diego, CA), and 8000 IU/ml recombinant human IFN-α for 48 h. Cytokines in the supernatants were measured with Cytometric Bead Array kits (BD Biosciences). For Ig production, isolated B cells were stimulated with 20 U/ml IL-2, 100 ng/ml IL-10, 10 μg/ml inactivated Staphylococcus aureus Cowan, and various concentrations of evobrutinib. After 10 d of culture, IgG and IgM levels in the supernatant were measured by ELISA.
Inhibition of FcγR signaling
U937 NF-κB–Luc reporter cells were maintained as an adherent culture at 37°C in a CO2-regulated tissue culture incubator. The day of the experiment, cells were collected, counted, and plated in a 96-well tissue plate. Evobrutinib was added at concentrations ranging from 5 nM to 10 μM. Cells were incubated with evobrutinib for 30 min in a 37°C tissue culture incubator. The cells were then transferred to fresh plates coated with anti-CD64 and stimulated for 4 h. Luciferase activity in cell lysates was measured using an EnVision plate reader.
Basophil inhibition assay
The ability of evobrutinib to block Fc receptor signaling was determined using basophils in whole blood. Human blood was collected with citrate as an anticoagulant and transferred to 96-well plates. Blood was pretreated for 30 min at 37°C with dilutions of evobrutinib before activation with anti-IgE (Beckman Coulter, Brea, CA), added to a final concentration of 2 μg/ml, and incubated at 37°C for 5 min. After activation, cells were stained for 15 min with anti-CD63–FITC (BD Biosciences), and then PBS–EDTA (20 mM) was added, followed by fixative/lysing buffer. Cells were fixed in formaldehyde prior to FACS analysis. The mean fluorescence intensity (MFI) for CD63 expression was determined after first gating for CD123+HLA-DR− cells using the FACSCanto II instrument (BD Biosciences).
BioMAP profiling
The biological selectivity of evobrutinib and ibrutinib was assessed in vitro using primary human cells with BioMAP profiling by BioSeek. The activity of the compound was assessed using a concentration range of 370 nM–10 μM in 12 different primary cell coculture assay systems according to previously published detailed methods (33, 34).
Ex vivo B cell stimulation in mouse whole blood
Evobrutinib was administered by oral gavage to female C57BL/6 mice (five per group) at indicated doses and time points before heparinized whole blood was obtained and divided into two aliquots. One aliquot was incubated with anti-IgD as stimulation and another with PBS as basal control. CD69 MFI on the B cell surface was measured by flow cytometry. The difference between stimulated and basal levels of MFI was calculated and expressed as Δ MFI. Percent inhibition was calculated according to the following formula: percent inhibition = (1− [Δ MFIevobrutinib/Δ MFIvehicle]) × 100.
PK/PD model and occupancy assay for whole blood
To build a PK/PD relationship for evobrutinib, DBA/1J female mice aged 11–12 wk were dosed with the compound. BTK occupancy in the blood and plasma concentrations were measured over time. Evobrutinib was formulated in a vehicle solution of 20% kleptose and 50 mM citrate, pH 3, and mice were administered the compound via oral gavage. At various time points after dosing, mice were euthanized, and blood was collected into heparinized tubes via the vena cava. For determination of BTK occupancy, a previously described method that uses a biotinylated BTK-binding probe and a streptavidin-capture ELISA was used (35). Plasma samples were analyzed by liquid chromatography–tandem mass spectrometry for determination of evobrutinib concentrations.
Passive cutaneous anaphylaxis in mice
C57BL/6 mice were sensitized intradermally in the back with 250 ng of anti-DNP IgE (catalog no. D-8406; Sigma, St. Louis, MO) or anti-OVA IgE (sham mice, catalog no. MCA2259; ABD Serotec). Twenty-four hours later, all mice were challenged i.v. with 0.5 mg of DNP–human serum albumin (catalog no. A6661; Sigma) in the presence of 0.5% Evans blue (catalog no. E-2129; Sigma). Mice were sacrificed 30 min after challenge, back skin was harvested, and the Evans blue was extracted in formamide for 24 h at 55°C. OD was measured at 620 nm and compared with a standard curve of known Evans blue concentrations. The results are expressed as nanograms of dye per milligram of tissue.
Collagen-induced arthritis in mice
All mouse collagen-induced arthritis (CIA) studies were performed at Bolder BioPATH. DBA/1OlaHsd mice (12–15 animals per group) were anesthetized with isoflurane, shaved at the base of the tail, and injected intradermally with 150 μl of CFA (Sigma) containing bovine type II collagen (2 mg/ml; Elastin Products, Owensville, MO) at the base of the tail on day 0 and again on day 21. On study day 18, mice were randomized by body weight into treatment groups. Treatment was initiated after enrollment and continued daily (once daily at 24-h intervals) through study day 33. Animals were dosed by the oral route with vehicle (20% hydroxy-propyl-β cyclodextrin in H2O) or evobrutinib at various doses or the reference compound methotrexate (MTX; 0.5 mg/kg). On study day 34, the studies were terminated. Daily clinical scores were given for each of the paws (right front, left front, right rear, and left rear) on arthritis days 18–34 using the following criteria: 0 = normal; 1 = one hind or forepaw joint affected or minimal diffuse erythema and swelling; 2 = two hind or forepaw joints affected or mild diffuse erythema and swelling; 3 = three hind or forepaw joints affected or moderate diffuse erythema and swelling; 4 = four hind or forepaw joints affected or marked diffuse erythema and swelling; and 5 = entire paw affected, severe diffuse erythema and severe swelling, and unable to flex digits. Histopathological scoring was performed on forepaws, hind paws, and knees from mice. Inflammation, pannus formation, cartilage damage, and bone resorption were scored separately. Scores for all four parameters were added for each individual ankle or knee. Mean scores for all six tissues were calculated for each animal, and mean scores for each group are shown.
SLE model
Ten-week-old female NZB/W F1 mice (The Jackson Laboratory) were given two i.v. injections on day 0 and day 1 of 1 × 108 IU/100 μl of adenovirus with mmIfna5_v1 insert (CoA.NL.09.160; Biofocus) in saline or left untreated (sham). Drug treatments were initiated at 2 wk after delivery of adenovirus with mmIfna5_v1 insert and continued until the end of the experiment (at 10 wk). Mice (10 per group) were treated once daily with evobrutinib at indicated doses or mycophenolate mofetil (CellCept; Roche) at 300 mg/kg by oral gavage. Serum and urine samples were collected for anti-dsDNA Ab determination (by ELISA) and urinary protein creatinine ratio (UPCR; measured on ADVIA 1800) determination, respectively, on the days indicated. Proteinuria was defined as UPCR > 3. In addition, serum was analyzed for clinical chemistry parameters including urea nitrogen, albumin, total protein, and cholesterol on an ADVIA1800 chemistry analyzer on the final day of the experiment. Hematological analysis was performed on whole blood on the final day using a Sysmex XT-2000iV analyzer. Spleen cells were analyzed for B and T cell subsets on the final day using flow cytometry. The gating strategy is shown in Fig. 9. BTK occupancy in splenocytes was measured as described by Honigberg and colleagues (25).
FIGURE 9.

Gating strategy to identify B and T cell subsets by flow cytometry. The example shows spleen cell stainings. Black arrows show Ag being stained. Red arrows show gating strategy. Bmem, memory B cells; CM, central memory CD8 T cells; EM, effector memory CD8 T cells; GC, germinal center B cells; MZ, marginal zone B cells; PC, plasma cells; T1, transitional type 1 B cells; T2, transitional type 2 B cells.
Histopathology analysis of kidney
Paraffin-embedded, H&E-stained 4-μm tissue sections were blindly scored as the sum of three systems, including inflammation, glomerular damage, and sclerosis. Inflammation: Grade 1, mild focal interstitial inflammation; Grade 2, multifocal areas of mild interstitial inflammation; Grade 3, moderate multifocal interstitial inflammation; and Grade 4, significant multifocal interstitial inflammation. Glomerular damage: Grade 1, initial glomerular lesions characterized by increased cellular components in a single to a few glomeruli with cellular proliferation and thickening of basement membrane (membranoproliferative/mesangiocapillary); Grade 2, multifocal areas of glomerular lesions; Grade 3, multifocal areas of glomerular lesions with significant damage, characterized by proliferation of epithelial cells of capsule of Bowman with compression of glomerular capillaries; and Grade 4, pronounced multifocal to generalized glomerular damage (i.e., Grade 3 plus obliteration of Bowman space, glomerular compression, and hyalinosis). Glomerular sclerosis: Grade 1, focal mild glomerular sclerosis; Grade 2, multifocal mild glomerular sclerosis; Grade 3, severe focal glomerular sclerosis; and Grade 4, severe multifocal glomerular sclerosis.
Detection of Ab-secreting cells
ELISPOT plates (Merck Millipore, Billerica, MA) were coated with 10 μg/ml goat anti-mouse IgG (Calbiochem). Spleen cells were harvested and titrated starting with 300,000 cells per well. Cells were incubated for 5 h after washing and incubation with secondary HRP-conjugated anti-mouse IgG (Sigma). ELISPOTs were developed with the AEC Staining Kit (Sigma). ELISPOTs were counted using an ELISPOT reader (AID, Strassberg, Germany).
Determination of anti-dsDNA level
ELISA plates were coated with 10 μg of thymic DNA (Sigma). Serum of each animal was titrated to determine anti-dsDNA Ab levels. The levels for each individual were calculated as arbitrary units compared with a pooled standard serum from 22-wk-old MRLlpr/lpr mice. Detection was performed using goat anti-mouse IgG HRP coupled Abs and 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich).
PK/PD modeling of in vivo BTK occupancy in mice
The PK/PD analysis was performed to evaluate the PK-driven time course of BTK occupancy in WBCs following oral administration of evobrutinib in mice. PK parameters were estimated from a two-compartment model with first-order absorption and were fixed in the PK/PD model. All dose PD data were simultaneously fit by the irreversible turnover model and described by the following differential equation (Eq. 1):
| (1) |
where Cp is evobrutinib plasma concentration; BTK0 is the baseline unoccupied BTK (set to 1); kdeg is the first-order degradation rate of BTK protein; kirrev is the second-order irreversible binding rate constant; and BTK is the time-dependent fraction of unoccupied BTK.
Disease progression PD-driven modeling in CIA and lupus disease models
PD/efficacy analysis was performed to evaluate the PD-driven inhibition of disease progression in an accelerated NZB/W F1 SLE mouse lupus model. Mouse BTK occupancy PK/PD drug and system parameters were estimated as described above and were fixed in the PD/efficacy model. All dose/efficacy data were simultaneously fit by a modified disease model (36) described by the following differential equation (Eq. 2). The model was modified to exclude the time-dependent, non–compound-mediated remission of disease, as this was not demonstrated in the preclinical data set and, thus, could not be parameterized.
| (2) |
where kin is the progression of disease (protein/creatinine ratio for lupus model; clinical score for RA model); D is the disease score; Dmax is the maximal disease score; K is the second-order rate constant for BTK occupancy–mediated inhibition of disease progression; BTKRO is the fractional BTK occupancy; and Tonset is the onset of disease.
Once the PD/efficacy model was parameterized, efficacy simulations were performed across a wide dose range that generated an occupancy span of 0–100% BTK occupancy at PD steady state. The area under the effect curves (AUEC) of the simulations were calculated with noncompartmental analysis. Inhibition of disease progression was estimated as from the following equation (Eq. 3):
| (3) |
where AUECtreatment is the AUEC with evobrutinib treatment; AUECbaseline is the AUEC of disease score prior to induction of disease; and AUECvehicle is the AUEC of disease progression without evobrutinib treatment intervention.
Results
Biochemical characterization of evobrutinib
During lead optimization, evobrutinib (32) was identified as a potent, selective, irreversible, and orally available inhibitor of BTK (Fig. 1A). Evobrutinib inhibits BTK enzymatic activity with an IC50 of 9 nM (Supplemental Fig. 1A). Evobrutinib is an irreversible inhibitor, as shown by jump dilution analysis (Supplemental Fig. 1B). The covalent bond of the molecule to C481 is responsible for the irreversible inhibitory activity and potency of evobrutinib. The IC50 for inhibition of a BTK mutant in which Cys481 is mutated to serine is ∼1100-fold higher than for WT BTK (7854 versus 7.14 nM) (Fig. 1B). In contrast, ibrutinib inhibits both WT and C481S BTK with comparable potency, as the IC50 value is <3-fold higher for inhibition of BTK C481S (2.45 versus 0.93 nM). This indicates that even without covalent binding, ibrutinib’s scaffold is already a potent BTK inhibitor. The absolute dependence of evobrutinib on covalent binding to a conserved cysteine, which is found in only 11 kinases in the kinome (37), is largely responsible for the high kinase selectivity of evobrutinib. Indeed, selectivity was confirmed in a kinase screen, and evobrutinib showed much greater selectivity compared with ibrutinib (Fig. 1C). Out of a panel of 267 kinases, evobrutinib inhibited BTK, BMX, and TEC by 90, 93, and 82%, respectively. Additional kinases that were weakly inhibited were TXK, RSK1, PAK3, and Her4 (36, 31, 30, and 29% inhibition, respectively). Inhibition of the binding of sunitinib to these kinases by evobrutinib correlated with inhibition of kinase activity. A determination of IC50 values revealed that evobrutinib inhibited BTK and BMX with IC50 values of 0.058 and 0.02 μM, respectively, whereas TEC was inhibited with a much higher IC50 of 7.3 μM (Supplemental Table I).
Inhibition of BCR- and Fc receptor–dependent B cell and basophil cell activation by evobrutinib
Following BCR or Fc receptor ligation, BTK is partially activated by phosphorylation of Y551 by Src family tyrosine kinases such as Syk or Lyn. This enables autophosphorylation of Y223 and full activation of BTK (4). In the human Ramos B cell line, evobrutinib inhibited autophosphorylation of Y223 but not the phosphorylation of Y551 by an upstream kinase (Fig. 2A). Stimulation of B cells via the BCR leads to upregulation of the activation marker CD69 within hours. The effect of evobrutinib on CD69 upregulation on CD19+ cells after BCR ligation was measured by flow cytometry both in human PBMCs and in whole blood. Evobrutinib inhibited B cell activation in PBMCs with a mean IC50 of 15.8 nM (Fig. 2B, Supplemental Table I) and in whole blood with an IC50 of 84.1 nM (Supplemental Table I). The percentage of B cells within the PBMCs was not reduced, suggesting that evobrutinib was not toxic (Supplemental Fig. 1C). Upregulation of the costimulatory CD86 was inhibited, as well, whereas MHC class II expression was neither increased by stimulation nor diminished by treatment with evobrutinib (Fig. 2C) In a set of assays using purified B cells, the impact of BTK inhibition on B cell function was tested. Purified primary human B cells were stimulated via the BCR and, depending on the assay, different costimuli. As shown in Fig. 2D and Supplemental Table I, proliferation and cytokine production were inhibited with low nanomolar IC50 values. In addition, evobrutinib potently inhibited the differentiation of plasmablasts after stimulation of B cells with S. aureus Cowan I strain, as measured by inhibition of IgG and IgM production (Supplemental Table I).
FIGURE 2.

Evobrutinib inhibits BTK autophosphorylation and B cell activation. (A) Evobrutinib inhibits phosphorylation of BTK Y223 but not Y551 after BCR stimulation. Ramos cells were preincubated with indicated concentrations of evobrutinib and stimulated with anti-IgM for 5 min. Cell lysates were separated by Protein Wes and stained for BTK–p-Y223 or BTK–p-Y551. Duplicates are shown. Total protein was measured and adjusted for each sample to load comparable amounts of BTK. (B) PBMCs from healthy donors were incubated with indicated concentrations of evobrutinib and stimulated with anti-IgM. After 18 h of incubation, levels of CD69 expression were measured using flow cytometry and used to calculate the IC50. Data shown are representative of over 30 experiments with PBMCs from different donors. (C) Cells were stimulated as described in (B), and surface expression of CD69, CD86, and HLA-DR was measured. Data are representative of two donors. Open symbols, no stimulation; gray symbols, stimulation in the presence of DMSO only. (D) Purified B cells from healthy donors were stimulated with anti-IgM for 4 d in the presence of indicated amounts of evobrutinib. Proliferation was quantified by addition of [3H]thymidine during the last 18 h of culture. Data are representative of four different donors. Unstimulated cells (blue triangle) and stimulated cells treated with DMSO only (black diamond) are shown. (E) U937 NF-κB–Luc reporter cells were stimulated with anti-CD64 in the presence of various concentrations of evobrutinib. Luciferase activity was measured after 4 h. Data representative of two experiments are shown. (F) Inhibition of basophil activation. Data are representative of seven experiments with different donors. cpd, compound; cpm, counts per minute; cps, counts per second; Ctrl, control.
Inhibition of FcγR signaling by evobrutinib was studied in the U937 monocytic cell line transfected with an NF-κB/luciferase reporter. FcγR (CD64) signaling was activated using a CD64-specific Ab. Evobrutinib inhibited NF-κB activation and, therefore, FcγR signaling with an IC50 of 78 nM (Fig. 2E). To study the effect of BTK inhibition on FcεR signaling, we stimulated primary human basophils with anti-IgE. Activation of basophils via FcεR is being increasingly recognized as a contributor to SLE pathophysiology. In addition, IgE has been described to activate plasmacytoid dendritic cells and augment the IFN-α response to TLR9 ligands (38–40). Inhibition of basophil activation was demonstrated with ibrutinib in vivo (41). In our studies, human whole blood was treated with anti-IgE in the presence of evobrutinib, and basophil degranulation was assessed by measuring surface CD63 levels on basophils by flow cytometry. It was found that evobrutinib inhibited basophil activation with an average IC50 of 1.66 μM (Fig. 2F, Supplemental Table I). Thus, evobrutinib is a potent inhibitor of BCR and FcγR signaling, whereas FcεR signaling is inhibited much less potently in vitro.
Evobrutinib selectively inhibits B cell responses in a panel of different activated primary human cell types
After having demonstrated its intended effect on BCR- and Fc receptor–dependent cell activation, we tested evobrutinib in a broader panel of primary human cell cultures to determine its full range of biological activity. The BioMAP Diversity Plus panel includes 12 different assay systems containing human primary cell types such endothelial cells, fibroblasts, bronchial epithelial cells, keratinocytes, macrophages, smooth muscle cells, PBMCs, and B cells (42). Cells were either cultured alone or cocultured (see Fig. 3A) and stimulated with various factors to activate cells under different conditions modeling disease-related inflammation and tissue remodeling biology. For example, the BT system contained PBMCs and purified B cells and was stimulated with anti-IgM and submitogenic concentrations of a mixture of superantigens for TCR and MHC class II engagement), thus modeling B cell stimulation in the context of T cell activation. In other cultures, combinations of cytokines, growth factors, and TLR ligands were used to stimulate each system. No stimuli that activate Fc receptor are included in the BioMAP panel. Compound effects on various cell bound and soluble protein-based biomarkers were measured in each system, and changes are expressed as log ratio of the vehicle controls (see Supplemental Table III for a detailed list of measured parameters). As shown in Fig. 3A, addition of evobrutinib at concentrations ranging from 370 nM to 10 μM showed clear concentration-dependent inhibitory effects in the BT system, as evidenced by reduced proliferation and decreased production of the soluble cytokines IL-2, TNF-α, IL-17A and IL-17F, and IL-6. As expected, Ig production was strongly impacted by exposure to evobrutinib (Fig. 3B). No significant effects were observed in any of the other cell systems, again demonstrating the exquisite specificity of evobrutinib (Fig. 3A). In contrast, an overlay of effects of evobrutinib versus ibrutinib showed that, in addition to expected effects in the BT system, a concentration of 1 μM ibrutinib was also active in several other cellular systems, including systems without any immune cells present (e.g., CASM3C and HDF3CGF) (Fig. 3C). Specifically, ibrutinib increased epidermal growth factor receptor (EGFR) expression in dermal fibroblasts in the HDF3CGF system. This activity is also observed with EGFR kinase inhibitors erlotinib and gefitinib (Fig. 3D, right). Moreover, increased EGFR levels in the BioMAP HDF3CGF systems is a sentinel activity of all EGFR kinase inhibitors and correlates with a skin rash in responsive non–small cell lung cancer patients (43). A similar skin rash was reported for ibrutinib in a CLL patient (44) and supports the polypharmacology of ibrutinib. BTK-specific impact of evobrutinib, ibrutinib, and another covalent BTK inhibitor, AVL-292 (also known as CC-292 or spebrutinib), were overlapping in the BT system, whereas erlotinib and gefitinib were inactive in this system (Fig. 3D, left). Overall, these data show more restricted biological effects of evobrutinib and are consistent with the more selective kinase profile of evobrutinib compared with ibrutinib.
FIGURE 3.

Evobrutinib specifically inhibits B cell activation in a panel of primary human coculture systems. (A) Evobrutinib was tested in increasing concentrations in the BioMAP Diversity Plus panel. For each culture system, included cell types are shown in green boxes and simulated biology in blue boxes. Levels of proteins were measured by ELISA and presented as log expression ratios (log10[parameter value with test compound / parameter value of 0.1% DMSO]). The gray area represents the 95% prediction interval of the 0.1%-DMSO data. Significantly altered readouts were observed only in the BT system, indicating B cell inhibition. (B) Blowout of BT system. Evobrutinib reduced proliferation, IgG production, and production of IL-17A, IL-17F, IL-2, IL-6, and TNF-α. (C) Overlay of evobrutinib and ibrutinib at 1.1 μM in the BioMAP panel. Ibrutinib (blue line) showed activity in every cellular system. (D) Overlay of different BTK and EGFR inhibitors in the BT and HDF3CGF systems. AVL-292 is a synonym for CC-292.
PD activity of evobrutinib in mice
To test the inhibition of B cells in vivo, different doses of evobrutinib were administered to mice orally. One hour later, animals were sacrificed, and whole blood was stimulated with anti-IgD or left unstimulated for 4 h. The expression of CD69 on B cells was then measured by flow cytometry. As shown in Fig. 4A, evobrutinib inhibited B cell activation in a dose-dependent manner, reaching almost 90% inhibition at a plasma concentration of 21.9 ng/ml after administration of a 1 mg/kg dose. Next, we tested the duration of B cell inhibition. Mice were administered a single dose of 12 mg/kg evobrutinib, and B cell inhibition was measured as before at different time points. As shown in Fig. 4B, 16 h after dosing, B cell activation was still inhibited by roughly 50%. Because evobrutinib is a covalent irreversible inhibitor, the recovery of B cell activation is reflective of new BTK protein synthesis.
FIGURE 4.

Evobrutinib inhibits B cells and mast cells in vivo and shows a prolonged PD effect despite its short t1/2. (A) Dose-dependent inhibition of B cell activation. Mice were administrated evobrutinib PO at the indicated doses (filled circles). Whole blood was drawn 1 h later and stimulated with anti-IgD or left unstimulated (empty circles). The differences in MFI of the activation marker CD69 (Δ MFI CD69) between stimulated and unstimulated samples is shown, as well as percent inhibition compared with vehicle and plasma exposure. One-way ANOVA followed by Dunnett posttest indicated statistically significant inhibition at 3 mg/kg. (B) Time-dependent inhibition of B cell activation. Mice were dosed PO with 12 mg/kg evobrutinib, and B cells were stimulated ex vivo at the indicated time points. Plasma exposure is indicated. At 16 and 24 h, evobrutinib was nd. (C and D) Time course of BTK occupancy (C) and plasma exposure (D) in DBA/1 mice at different doses. The detection limit for evobrutinib was 0.1 ng/ml. Values below LLOQ were visualized as 0.1 ng/ml in the graph. (E) Dose-dependent inhibition of mast cell degranulation in mice by evobrutinib. Animals were dosed 1 h before the DNP challenge. Six to seven animals per group were used. Percent inhibition relative to the vehicle group is indicated. (F) Time-dependent inhibition of mast cell degranulation. Animals were dosed with 39.5 mg/kg evobrutinib at indicated time points. Percent inhibition is indicated. Statistical analysis was performed as in (E). *p < 0.05, **p < 0.01, ****p < 0.0001, one-way ANOVA with Dunnett posttest for treated groups compared with vehicle. ####p < 0.0001 compared with sham. LLOQ, lower limit of quantification; nd, no longer detectable; PO, per os (orally).
The covalent binding of evobrutinib provided an opportunity to directly measure target engagement over time and to correlate these data to exposure and B cell inhibition. We therefore developed a method to quantify BTK occupancy using a biotin-labeled probe, which is itself another irreversible BTK inhibitor tagged with biotin. The probe only binds to free BTK (i.e., BTK not already occupied by evobrutinib). The probe/BTK complex was pulled down from cell lysates using streptavidin-coated plates, and BTK was detected using a specific Ab. Mice were orally administered different doses of evobrutinib, and animals were sacrificed at different time points. BTK occupancy was measured using isolated WBCs, and evobrutinib concentrations were measured in the plasma. The percent BTK occupancy was calculated in relation to vehicle-treated animals (0% occupancy). At all three doses tested, maximal exposure was achieved at the first time point (0.5 h) and then quickly dropped. Even at the highest dose of 5 mg/kg, evobrutinib was no longer detectable 18 h after administration (Fig. 4D). In contrast, maximal BTK occupancy was reached around 4–5 h after dosing and was sustained for 18 h for the 1 and 5 mg/kg doses. In the 5 mg/kg dose group, 40% BTK occupancy was measured 48 h after dosing. These data clearly demonstrate a prolonged target engagement (Fig. 4C) even after the compound has been cleared from the circulation. Like for B cell inhibition, BTK occupancy eventually declines because new BTK protein is being synthesized.
Next, we studied the dose- and time-dependent effects of the irreversible BTK inhibitor evobrutinib on Fc receptor signaling in vivo using a passive cutaneous anaphylaxis model in mice. Mice were sensitized with anti-DNP IgE Abs and challenged 24 h later with DNP–human serum albumin in the presence of Evans blue dye. Evans blue extravasation into the tissue was measured 30 min after the challenge. In the first experiment, mice were dosed orally with three different doses (3.95, 19.8, and 39.5 mg/kg) of evobrutinib 1 h before challenge. This treatment resulted in a dose-dependent reduction in mast cell degranulation. The dose necessary to achieve full inhibition of mast cell degranulation was significantly higher than that required to inhibit B cell activation (Fig. 4A, 4E). This is reflective of the different IC50 values determined for basophil versus B cell inhibition (Supplemental Table I). Next, we tested whether evobrutinib would exert a prolonged PD effect on mast cell inhibition, as well. Animals were treated with 39.5 mg/kg evobrutinib at 1, 4, 16, or 24 h before the challenge. As shown in Fig. 4F, treatment with 39.5 mg/kg evobrutinib led to a 94, 98, 67, and 45% reduction in dye extravasation at 1, 4, 16, and 24 h postdose, respectively, demonstrating a long-lasting effect of treatment with the irreversible BTK inhibitor, even though the compound was only detectable in the plasma up to 4 h postdose.
Evobrutinib inhibits disease progression in a mouse RA model
Having demonstrated inhibition of BCR and Fc receptor signaling in vivo, we evaluated the ability of evobrutinib to treat disease in the CIA model. Male DBA/1OlaHsd mice were immunized with collagen in CFA on days 0 and 21. The animals were treated by once-daily oral administration of evobrutinib on days 18–33. As shown in Fig. 5A, evobrutinib demonstrated dose-dependent efficacy in the CIA model, as determined by reduction of clinical arthritis scores, according to which a dose of 3 mg/kg almost completely prevented the development of arthritis symptoms. Area under the curve (AUC) analysis showed a 69 and 92% disease inhibition at 1 and 3 mg/kg, respectively, whereas treatment with MTX inhibited disease only by 13% (Fig. 5B). In addition, the incidence of disease was reduced from 100% in control animals to 33% at study termination in animals treated with 3 mg/kg evobrutinib (Fig. 5C). Histopathology of the affected joints confirmed the benefit of evobrutinib on inflammation, cartilage destruction, and bone resorption associated with the disease. High-dose groups had significant (63 and 89%) reductions in six-joint (four paws and two knees) mean animal composite scores as compared with disease controls (Fig. 5D). Individual histopathological scores for inflammation, cartilage destruction, and bone resorption were reduced in a comparable manner (data not shown). Treatment with evobrutinib did not affect the levels of anti-collagen Abs measured at study termination (Fig. 5E).
FIGURE 5.

Evobrutinib reduced paw scores and joint damage in a mouse CIA model. (A) Arthritis was induced in DBA/1 mice by collagen injection, and animals were treated with the indicated doses of evobrutinib or vehicle alone starting on day 18. Twelve animals were used per group. Mean paw scores are shown. MTX at 0.5 mg/kg was used as reference treatment. Evobrutinib reduced paw scores in a dose-dependent fashion. Two-way ANOVA with Bonferroni multiple comparison was performed. Days on which the treatment groups were significantly different from vehicle control are indicated. (B) AUC of paw scores. The percent reduction of paw score AUC compared with vehicle is indicated. One-way ANOVA, followed by Dunnett posttest, was performed for treated groups compared with vehicle. (C) Evobrutinib reduced the percent incidence of disease in a dose-dependent fashion. Any animal with a score of at least one for at least one paw was counted as being positive. (D) Histopathology scores (mean ± SEM) of six joints (four paws plus two knees), combining inflammation, cartilage destruction, pannus formation, and bone resorption. The percent reduction compared with vehicle group is indicated. Statistical analysis was performed as in (B). (E) Serum concentration of anti-collagen IgG2a and IgG2b was measured by ELISA on day 34. No statistically significant differences between treatments were found. *p < 0.05, ****p < 0.0001. n.d., not detected.
Evobrutinib inhibits kidney damage in the accelerated–IFN-α NZB/W F1 lupus model
SLE pathophysiology is driven by inappropriate activation of B cells, leading to production of autoantibodies that form immune complexes that activate innate immune cells. Innate immune cell activation results in damage to various tissues, such as the kidney. BTK inhibitors block both BCR-mediated activation of B cells and immune complex–mediated activation of Fc receptors on innate immune cells and should, therefore, be efficacious in this disease. We tested evobrutinib for efficacy in the IFN-α–accelerated NZB/W F1 lupus model. Lupus-prone NZB/W F1 mice were injected with a replication-deficient adenovirus encoding IFN-α to promote disease development and accelerate its onset. The adenovirus triggers a rapid but transient increase in IFN-α, and 14 d postinfection, levels are normalized again (Supplemental Fig. 1D). Evobrutinib was administered orally once daily at 0.1, 0.3, 1, and 3 mg/kg, starting on day 14 after adenovirus injection and continuing for the duration of the study until termination. As a positive control, mycophenolate mofetil (CellCept) was used once daily at 300 mg/kg.
Urinary protein and creatinine levels were measured weekly. Incidence of proteinuria, as defined by a UPCR > 3, reached 80% in the vehicle-treated group. The incidence of proteinuria was reduced in a dose-dependent manner by evobrutinib, and at the highest dose of 3 mg/kg, the incidence was 0%. CellCept reduced the incidence of proteinuria to 20% at the end of the study (Fig. 6A). The efficacy of evobrutinib was also reflected in the mean UPCR measured over time. Whereas the mean UPCR in the vehicle-treated group ultimately was >40, this was reduced in a dose-dependent manner, with the UPCR in the group treated with 3 mg/kg evobrutinib always remaining under 3. The mean UPCR in the group treated with CellCept reached 10 toward the end of the experiment (Fig. 6B). AUC calculations of individual proteinuria over time showed a significant decrease by evobrutinib starting at the 0.3 mg/kg dose. The mean AUC was lower in the 1 and 3 mg/kg groups compared with CellCept-treated animals (Fig. 6C). Histopathological analysis of the kidneys showed mild to moderate multifocal interstitial inflammation, glomerular lesions with significant damage, characterized by proliferation of epithelial cells of capsule of Bowman, and mild focal or multifocal sclerosis in the vehicle-treated mice (Fig. 6E). Treatment with evobrutinib reduced the histopathology score in a dose-dependent manner. These changes were significantly different from the vehicle group starting at a dose of 0.3 mg/kg. In terms of reducing pathology scores, the 3 mg/kg dose was more efficacious than CellCept (Fig. 6D). Overall, by all measures, evobrutinib significantly reduced kidney damage and compared favorably to the clinically used drug CellCept.
FIGURE 6.

Evobrutinib reduced proteinuria and kidney damage in the mouse accelerated NZB/W lupus model. (A) Incidence of proteinuria (defined as protein/creatinine ratio > 3). Log-rank test comparing each curve to vehicle was performed. (B) Mean UPCR over time. Two-way ANOVA, followed by Bonferroni posttest, comparing treatment groups to vehicle was performed. (C) Mean and individual AUC of UPCR. The mean percent reduction compared with vehicle is shown. One-way ANOVA followed by Dunnett posttest was performed. (D) Mean and individual histopathology scores for kidney damage. Statistical analysis was performed as in (C). (E) Representative H&E stained kidney sections from a vehicle-treated animal (upper row) and an animal treated with 1 mg/kg evobrutinib (lower row). *p < 0.05, **p < 0.01, ***p < 0.001.
Treatment with evobrutinib resulted in preservation of kidney function and normalization of hematologic manifestations of SLE
In the NZB/W F1 model, as in lupus patients, kidney damage leads to increased blood urea nitrogen and decreased serum albumin and total protein levels compared with normal control mice. This indicates a defect in glomerular filtration. Treatment with evobrutinib prevented the changes in blood urea nitrogen and albumin normally associated with disease in this model. Evobrutinib was more efficacious than CellCept in restoring albumin levels (Fig. 7A). In addition, cholesterol levels are elevated in diseased NZB/W mice compared with younger, healthy control NZB/W mice. An elevation in total cholesterol can also be observed in lupus patients with kidney involvement and may be indicative of adverse renal outcomes (45). Treatment with evobrutinib reduced the cholesterol levels to those found in healthy mice (Fig. 7A). Anemia is often found in SLE patients, and it manifests in low hemoglobin levels, low RBC numbers, and low hematocrit (46). We found all of the above signs of anemia in diseased NZB/W mice. Treatment with evobrutinib resulted in dose-dependent increases and normalization of RBCs, hemoglobin, and hematocrit. Interestingly, treatment with CellCept did not normalize these parameters (Fig. 7B).
FIGURE 7.

Treatment with evobrutinib normalized clinical chemistry and hematology parameters. (A) Urea nitrogen, albumin, and cholesterol levels in NZB/W mice treated with evobrutinib, CellCept, or vehicle on day 56. Normal control (NC), young NZB/W mice not yet showing signs of proteinuria. (B) Numbers of RBCs, hemoglobin, and hematocrit in the same animals. One-way ANOVA with Dunnett posttest was performed for treated groups compared with vehicle. *p < 0.05, **p < 0.01, ***p < 0.001.
To assess the effects of evobrutinib on autoantibody production, serum was analyzed for anti-dsDNA IgG on days 14 and 41 and on day 58, the final day of the experiment. During the course of the experiment, the anti-dsDNA levels increased up to 100-fold in the vehicle group. In animals treated with CellCept or 3 mg/kg evobrutinib, the anti-dsDNA titers still increased but to a smaller degree (16- and 21-fold for CellCept and evobrutinib, respectively). However, because of considerable variability, this reduction was not statistically significant (Fig. 8A). In accordance with dampening the increase in anti-dsDNA titers, evobrutinib had direct effects on the composition of key B cell subsets that play a role in SLE. The percentage of splenic CD138+ plasma cells was diminished by treatment with evobrutinib. In addition, the percentage of follicular B cells in the spleen was reduced more efficiently than by CellCept (Fig. 8B). The changes of immune cell subsets after evobrutinib treatment extended to T cells, as well, which do not express BTK. Increasing doses of evobrutinib led to normalization of T cell activation, as evidenced by increased percentages of naive CD4 and CD8 T cells (Fig. 8D) and reduced percentages of memory CD4 and CD8 T cells (Fig. 8C). These effects were comparable to treatment with CellCept.
FIGURE 8.

Effects of evobrutinib on autoantibodies and immune cell subsets. (A) Levels of anti-dsDNA on day 56. Data are expressed as fold change in autoantibody levels compared with day 14. (B–D) Percentage of plasma cells, follicular B cells, and memory and naive CD4 and CD8 T cells compared with total splenocytes. Statistical analysis was performed as described in Fig. 7. *p < 0.05, **p < 0.01, ***p < 0.001.
To assess whether changes in immune cell subsets represented a normalization or a loss of certain cell types, we performed another experiment with the same treatment as described above and added a sham group of NZB/W mice that did not receive the IFN-α adenovirus. At the end of the experiment, various immune cell subsets in the spleen were quantified. The gating strategy is shown in Fig. 9. Sham-treated animals did not develop proteinuria, and evobrutinib reduced proteinuria in a dose-dependent manner (Supplemental Fig. 1E). As shown in Fig. 10, the absolute numbers of total and follicular B cells were increased 4–5-fold in vehicle-treated versus sham-treated mice. Treatment with 0.3, 1, and 3 mg/kg evobrutinib significantly reduced the numbers of these cells, with the highest dose bringing the absolute numbers down to levels observed in proteinuria-free sham-treated mice. Plasma cell numbers were reduced by evobrutinib, and the numbers of memory and germinal center B cells showed a trend toward reduction in a dose-dependent manner. Interestingly, evobrutinib had no effect on marginal zone B cell numbers. Effects on transitional B cells were less clear. Both transitional type 1 and transitional type 2 B cell numbers were increased in diseased NZB/W mice over sham-treated animals. Evobrutinib reduced numbers of transitional type 2 B cells, but this effect was not dose-dependent. Evobrutinib showed a trend toward normalizing transitional type 1 B cell numbers at the highest dose.
FIGURE 10.

Evobrutinib normalizes the numbers of various immune cell subsets. NZB/W mice were either left completely untreated (sham) or injected with IFN-α adenovirus to induce disease. Animals were treated with vehicle, CellCept, or various doses of evobrutinib as indicated. At the end of the experiment, spleen cells were stained, as detailed in Fig. 9. Statistical analysis was performed as described in Fig. 7. Bmem, memory B cells; GC, germinal center B cells; MZ B cells, marginal zone B cells; T1, transitional type 1 B cells; T2, transitional type 2 B cells. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Memory CD4+ T cell numbers were normalized as well, and a trend toward reduction of memory CD8+ T cell numbers was observed. Naive CD4+ or CD8+ T cell numbers were not affected by evobrutinib.
Next, we explored the correlation of B cell inhibition and BTK occupancy with disease amelioration in the NZB/W model. On day 29, we isolated whole blood 1 h after dosing with evobrutinib and activated B cells by treatment with anti-IgD. After 4 h of incubation, B cell activation was assessed by measuring CD69 upregulation compared with unstimulated controls using flow cytometry. As shown in Fig. 11A and 11B, cell activation was inhibited in a dose-dependent manner by evobrutinib, reaching a maximum inhibition of 77% at 3 mg/kg. CellCept did not significantly inhibit B cell activation. Additionally, we measured BTK occupancy in splenocytes isolated upon study termination, 4 h after the last dose. Occupancy increased dose-dependently, and 100% occupancy was achieved at 3 mg/kg (Fig. 11B). Next, the mean percent BTK occupancy at this single time point was calculated for each dose group and plotted against mean B cell inhibition (shown in Fig. 11A) as well as mean inhibition of proteinuria (shown in Fig. 6C). As shown in Fig. 11C, BTK occupancy of 100% in splenocytes, measured 4 h after dosing, correlated with complete inhibition of proteinuria, whereas B cell activation was inhibited only up to 80%.
FIGURE 11.

Correlation of BTK occupancy, B cell inhibition, and disease inhibition in the NZB/W lupus model. (A) Evobrutinib inhibits ex vivo B cell activation in the NZB/W lupus model. Whole blood was isolated on day 28, 1 h after dosing, and B cells were stimulated. B cell activation is expressed as difference of MFI of the activation marker CD69 between stimulated and nonstimulated cells (ΔMFI CD69). Mean percent inhibition of B cell activation compared with vehicle-treated animals is indicated. One-way ANOVA and Dunnett posttest were performed. (B) Individual and mean BTK occupancy in the spleen of NZB/W mice treated with evobrutinib on day 56, 4 h after dosing. (C) Correlation of BTK occupancy with inhibition of B cell activation and inhibition of proteinuria. Mean BTK occupancy for each dose group was calculated and correlated to the mean percent B cell inhibition (Fig. 9A) as well as the mean inhibition of proteinuria (Fig. 6C). *p < 0.05, ***p < 0.001.
PK/PD modeling of BTK occupancy in mice
We next established a relationship between exposure, target occupancy, and efficacy in preclinical disease models. PK parameters were estimated from a two-compartment model with first-order absorption for male DBA/1OlaHsd and for female NZB/W mice, which were used in the RA and SLE models, respectively. Strain-specific PK parameters may be found in Supplemental Table II. The corresponding exposure data are shown in Supplemental Fig. 1F and 1G.
Once evobrutinib has engaged BTK, the PD effect depends largely on the resynthesis and degradation rates of BTK in vivo rather than the systemic exposure of evobrutinib. This is due to the covalent binding of evobrutinib to BTK and illustrated in Fig. 12A. BTK occupancy PK/PD modeling was performed for male DBA/1OlaHsd mice using the BTK occupancy and exposure data shown in Fig. 4C and 4D. PK parameters from this strain were used to parameterize the second-order rate constant kirrev, which describes binding of evobrutinib to BTK. The plasma concentration and BTK occupancy time course following oral administration of evobrutinib were fit to a PK/PD model adapted from Abelö et al. (47) and shown schematically in Fig. 12A. The selected PD model consisted of an indirect response model that describes both the second-order rate constant describing the irreversible binding of evobrutinib to BTK (kirrev) and the mouse degradation rate of the BTK protein (kdeg). The estimated PK/PD parameters in mouse WBCs are reported in Supplemental Table II, and the occupancy dose–response time course is shown in Fig. 12B. These drug (kirrev) and system (kdeg) parameters were integrated into a model describing fluctuation of BTK occupancy at steady state after daily dosing. As shown in Fig. 12C, this model predicts that daily dosing with 1 mg/kg of evobrutinib in mice results in a maximum BTK occupancy of 70% and a minimum occupancy of 50% at steady state. A daily dose of 5 mg/kg results in maximum and minimum BTK occupancy of 97 and 70%, respectively. Higher doses of evobrutinib are predicted to have a marginal effect on the BTK occupancy time course in mice. This is in line with the PD effect being entirely driven by the turnover of BTK protein in vivo rather than exposure with evobrutinib.
FIGURE 12.

PK/PD/efficacy modeling of evobrutinib. (A) Schematic showing the PD model describing BTK turnover and irreversible binding of evobrutinib. (B) BTK occupancy dose response time course in DBA/1 mice. (C) Modeling of fluctuation in BTK occupancy after multiple once-daily dosing with evobrutinib in mice. Raw data are from Fig. 4C. (D) Curve fitting of time versus arthritis scores in the mouse CIA model. Data from two independent experiments were integrated. (E) Simulation of efficacy in the CIA model versus average BTKOSS (%BTKOSS). (F) Curve fitting of time versus proteinuria in the mouse NZB/W lupus model. Data from the experiment shown in Fig. 6B were integrated. (G) Simulation of efficacy in the NZB/W lupus model versus average BTKOSS (%BTKOSS). BTKO, BTK occupancy; BTKOSS, BTKO at steady state.
Preclinical PD efficacy modeling for RA and SLE
A quantitative relationship between BTK occupancy and inhibition of preclinical disease progression was used to provide a translational framework to propose the extent and duration of BTK occupancy required to achieve clinical efficacy in RA and SLE patients. The BTK occupancy PD-driven efficacy model (Eq. 2 in Materials and Methods) was used to fit the dose-dependent efficacy time course data across all dose levels tested in both RA and SLE preclinical disease models. For the RA model, the estimated PD/efficacy parameters for the PD-driven inhibition of disease progression are reported in Supplemental Table II, and the observed and predicted arthritis scores versus time profiles (curve fitting) are shown in Fig. 12D. For the SLE model, modeling results and predicted disease progression are shown in Supplemental Table II and Fig. 12F, respectively.
These parameters were then used to simulate the efficacy response over a wide range of BTK occupancy. Using this simulated dose–response data and Eq. 3, we created a correlation between average BTK occupancy and percent inhibition of disease for both the RA and lupus preclinical disease models. For both disease models (Fig. 12E, RA, and Fig. 12G, lupus), near-maximal inhibition of disease activity is predicted when an average occupancy of >80% at PD steady state is reached.
Discussion
Targeting BTK is a promising approach to treat autoimmune disorders with aberrant B cell responses, such as SLE or RA. For this purpose, we have developed a novel, covalent, and highly specific BTK inhibitor, evobrutinib. This compound blocked BTK enzymatic activity and autophosphorylation at Y223. Evobrutinib did not prevent phosphorylation of Y551, which is the target of upstream kinases such Syk and Lyn/Fyn Src kinases. Potent inhibition of BTK was dependent on covalent binding to Cys481, as evobrutinib was unable to inhibit a BTK C481S mutant. This was in contrast to another covalent BTK inhibitor, ibrutinib, which, in our hands, was not absolutely dependent on Cys481 for the inhibition of BTK kinase activity and, in fact, inhibited the mutant with comparable potency. These results differ to some extent from other reports, in which the C481S mutation was identified in a small group of patients that had developed resistance to ibrutinib treatment. These reports showed a loss of potency of ibrutinib when Cys481 was mutated to serine (48).
The discrepancy may be explained by the use of slightly different assay conditions. We used a truncated recombinant BTK protein and measured phosphorylation of a peptide optimized for certain Tec kinases. In contrast, Woyach et al. (48) used full-length BTK and a nonspecific substrate. In addition, both Furman et al. (49) and Woyach et al. (48) showed reduced capacity of ibrutinib to inhibit Y223 autophosphorylation in the BTK C481S mutant in a cellular system using transfected HEK293T cells. Furthermore, even though Woyach et al. (48) observed increased IC50 values of ibrutinib on the C481S mutant, the compound was still able to eventually inhibit kinase activity and autophosphorylation (49). In contrast, evobrutinib is more dependent on covalent binding, and this may explain in part its increased selectivity over ibrutinib. We also compared the activity of a reversible BTK inhibitor, RN486 (50), in this assay. RN486 was able to inhibit both WT and C481S BTK equally well, with IC50 values of 1.7 and 1.2 nM, respectively.
Evobrutinib showed very good selectivity in a kinase panel comprising 267 kinases. Besides BTK, only two additional kinases were inhibited >80% at 1 μM. IC50 measurements revealed that only BMX nonreceptor tyrosine kinase was inhibited with an IC50 within 10-fold of BTK. Ibrutinib inhibited 25 off-targets >80% at 1 μM. These included the EGFR and ITK, among others. Inhibition of ITK has been claimed to be potentially beneficial in the treatment of cancer because ITK is involved in promoting Th2 differentiation, and its inhibition might strengthen Th1 differentiation and, therefore, the antitumor response (30, 31). However, if Th1 skewing is part of the mechanism of action of ibrutinib, this effect would be undesirable in autoimmune diseases. Skin rashes are a frequent side effect of treatment with ibrutinib (51) and might be attributed to its inhibition of EGFR, because similar side effects are known for other EGFR inhibitors (52). Again, although tolerable in oncology indications, such effects would be prohibitive for development of chronic treatments in autoimmune indications, particularly SLE, which often involves skin.
The biochemical selectivity of evobrutinib translated into mechanism-specific effects in a panel of primary human cell cocultures designed to model diverse states of immune activation, tissue remodeling, and disease biology. Evobrutinib only showed effects in a B cell activation system but not in 11 other coculture systems that recapitulate inflammation and matrix remodeling responses in T cells, monocytes, and macrophages, as well as endothelial, epithelial, smooth muscle, and fibroblast cell–based systems, Conversely, in line with its broader kinase profile, ibrutinib displayed activity in almost every cell system tested. Interestingly, in a dermal fibroblast culture system, ibrutinib’s profile mimicked that of EGFR inhibitors. It needs to be pointed out that none of the coculture systems reflect Fc receptor activation. Thus, inhibition of this pathway by evobrutinib is not captured in the BioMAP panel.
In addition to B cell activation as measured by CD69 and CD86 upregulation, evobrutinib inhibited a range of B cell functions induced by BCR stimulation, including proliferation, cytokine production, and Ig production. These effects should lead to a reduced differentiation of B cells into plasma cells, which are a source of autoantibodies in diseases like RA and SLE. Indeed, plasma cell numbers were reduced in the NZB/W SLE model.
Evobrutinib inhibited autophosphorylation at Y223 but not phosphorylation of Y551 in Ramos cells. This is consistent with reports for ibrutinib and CC-292, another covalent BTK inhibitor (8). In this context, it is noteworthy that the IC50 of evobrutinib on FcεR-mediated basophil activation was much higher compared with that for B cells (Supplemental Table I). Furthermore, complete inhibition of passive cutaneous anaphylaxis, which is mast cell driven, required more than 10-fold higher doses and plasma exposure compared with B cell inhibition in vivo (compare Fig. 4B, 4F). A similar split between inhibition of BCR- and FcεR-mediated cell activation was observed for ibrutinib and CC-292 but not for two reversible BTK inhibitors, RN486 and CGI-1746 (Supplemental Table I). Interestingly, the two reversible inhibitors have been reported to inhibit phosphorylation of BTK Y551 (8). This may indicate a distinct role for this phosphorylation site in FcεR-mediated signaling. Thus, it seems that the differential inhibitory activity against the BCR and FcεR pathways is not an assay artifact but is instead a real difference in how BTK acts in the different cell types.
In the mouse CIA model, evobrutinib almost completely inhibited disease development at 3 mg/kg. Both incidence and severity of clinical disease signs were reduced in a dose-dependent fashion. Histologically, all hallmark signs of RA, including bone resorption, inflammation, and pannus formation, were reduced, as well. Interestingly, the levels of anti-collagen Abs at the end of the experiment were not affected by treatment. This contrasts with BTK-deficient K/BxN mice, which show both reduced arthritis severity and reduced autoantibody levels (53). Furthermore, BTK deficiency has been shown to reduce autoantibodies in other models for autoimmune diseases, such as glomerulonephritis and diabetes (54–56). However, in the CIA experiment, treatment started only 18 d after the first immunization with collagen, and B cell differentiation to plasma cells may already have been committed. Other BTK inhibitors have been reported to reduce anti-collagen Abs in CIA models on different background strains to various degrees (57, 58). It is unclear why this is not the case with evobrutinib. Perhaps the high selectivity of evobrutinib or differences in mouse strains used may play a role. However, even in published studies with other BTK inhibitors, autoantibody levels were only partially reduced, yet disease was fully inhibited. This suggests that BTK inhibitors may affect arthritis development by inhibiting additional B cell functions, such as inflammatory cytokine production or Ag presentation to T cells (11, 57). Indeed, we found the ratio between naive and memory Th cells shifted toward a more naive phenotype in the accelerated NZB/W model.
In addition, inhibitory effects on the innate immune system likely play a role in the efficacy of evobrutinib in the RA model. Evobrutinib inhibits Fc receptor signaling, and FcγR-deficient mice are protected from disease in the CIA model, despite still producing autoantibodies (59, 60). Furthermore, BTK-deficient animals are defective in osteoclast differentiation (61, 62). This effect might protect against bone resorption in the CIA model. Several BTK inhibitors have been reported to inhibit disease in the collagen Ab–induced arthritis model (50, 58, 63, 64). The collagen Ab–induced arthritis model does not depend on lymphocytes for disease development and instead depends on Fc receptors (65, 66). Di Paolo et al. (58) published data showing inhibition of arthritis using a selective reversible BTK inhibitor, CGI-1706, in a K/BxN serum transfer model into lymphocyte-deficient SCID mice. In contrast, BTK-deficient mice on a NOD background are not protected in the same model (53). One might also need to consider a possible role of off-targets that might be inhibited by evobrutinib. BMX was the only kinase that was inhibited by evobrutinib with an IC50 comparable to BTK. BMX-deficient mice are protected from arthritis in the K/BxN serum transfer model. However, protection is lost when only the kinase domain is mutated (67). A recent publication shows efficacy of GDC-0853, a reversible BTK inhibitor with high selectivity that does not hit BMX, in the rat CIA model (68).
Taking these findings together, we conclude that BTK kinase inhibition was sufficient for disease inhibition in the CIA model. This was independent of autoantibody reduction. Thus, inhibition of Fc receptor–mediated innate immune cell activation likely plays a major role. Inhibition of additional B cell functions such as Ag presentation cannot be excluded either. A better understanding of the processes involved may be possible using tissue-specific and induced BTK-deficient models in the future (69).
Evobrutinib showed excellent potency in the accelerated NZB/W SLE model, in which it reduced proteinuria in a dose-dependent fashion, completely preventing it at a dose of 3 mg/kg. It is noteworthy that evobrutinib was far more efficacious than mycophenolate mofetil, which is a standard of care for more severe forms of SLE and was administered at a very high dose of 300 mg/kg. Consistent with the reduction in proteinuria, it was found that tissue damage in the kidney was also reduced with treatment. Several other covalent or noncovalent BTK inhibitors have been tested in the NZB/W lupus model. These include covalent inhibitors HM71224, PF-06250112, ABBV-105, and BTK-BI-1 (28, 70–72) and the reversible inhibitors G-744 and RN486 (27, 73). G-744 and ABBV-105 were tested in an IFN-α–accelerated NZB/W model, whereas the other molecules were tested in spontaneously developing lupus. All molecules reduced proteinuria severity and incidence as well as histological kidney damage. Likewise, deletion of BTK protected NZB/W mice from developing anti-dsDNA Abs and proteinuria (74).
Several hematological and clinical chemistry parameters, such as cholesterol levels, RBC numbers, hemoglobin, and hematocrit, are often reported to be aberrant in SLE patients. Importantly, these were all normalized by treatment with evobrutinib, but only cholesterol levels were normalized by mycophenolate mofetil. These data show for the first time, to our knowledge, that BTK inhibitors can normalize clinically relevant parameters beyond proteinuria.
Evobrutinib treatment reduced the total number of B cells, which is elevated in diseased NZB/W mice. Within the B cell compartment, follicular B cells, memory B cells, germinal center B cells, and plasma cells were clearly affected. An increase in marginal zone cell numbers has been described for NOD mice, in which these cells are moderately reduced again by BTK knockout (75). In diseased NZB/W mice, marginal zone B cells were increased, as well, but their numbers were not affected significantly by evobrutinib treatment. The lack of effect of evobrutinib on marginal zone B cells is in line with the observation that an inducible deletion of BTK in adult animals does not affect these cells (69). In contrast, published data for the covalent reversible BTK inhibitor PF-06260112 showed an 11-fold and 20-fold reduction of marginal zone and B1 B cells, respectively, in a nonaccelerated NZB/W lupus model. However, only limited information on the selectivity of this molecule was presented, suggesting that perhaps an off-target was responsible for this effect (28). Likewise, treatment with BI-BTK-1 was reported to reduce marginal zone B cell numbers, but for this molecule, neither selectivity data nor the precise structure was published (72).
It is important to note that the numbers of affected B cell subsets never dropped below those observed in sham-treated animals. This indicates that evobrutinib, rather than depleting B cells, normalizes their numbers, which are increased in diseased animals. BTK-BI-1 also showed normalization rather than depletion of various B cell subsets (72). BTK-deficient mice have severe defects in the B cell compartment. However, in these mice, BTK is absent throughout development. Experiments in which BTK was knocked down after B cell subsets were established showed that follicular B cell numbers dropped within 5 wk of BTK depletion to the low levels observed in global BTK knockouts (69). The fact that evobrutinib normalized follicular B cell numbers without signs of depletion may be explained by the remaining scaffolding function of BTK, even when its kinase activity is blocked by evobrutinib. Certain B cell subsets, such as B1 and An1 cells, have been described as autoimmune-prone. Whereas B1 cells did not depend on BTK, An1 cells, sometimes termed T3, are reduced in global and conditional BTK-deficient mice (69, 76). Based on these published data, it will be interesting to determine potential effects of evobrutinib or other BTK inhibitors on An1/T3 B cell subsets in future studies.
Interestingly, both CD4+ and CD8+ T cell compartments were skewed toward a more naive phenotype, demonstrating an indirect effect on the T cell response, as well. Autoantibodies were reduced in a dose-dependent fashion, but toward the end of the experiment, anti-dsDNA was still ∼10-fold higher than at the beginning of the experiment in animals treated with the highest dose of evobrutinib. A similar effect on autoantibodies was published for ABBV-105 and PF-06250112 (28, 70). We did not quantify plasma cells in the bone marrow, and, therefore, we do not know whether they were reduced in a fashion comparable to the spleen. However, plasma cells were still detectable in the spleen, although their numbers were significantly reduced at the highest dose. This may imply that a portion of B cells escape BTK inhibition and differentiate into plasma cells. Alternatively, the anti-dsDNA Abs detectable in the 3-mg/kg dose group may have been derived from long-lived plasma cells that no longer depend on BTK.
We did not measure total levels of IgG or IgM. Evobrutinib normalized B cell numbers rather than depleting them. Furthermore, long-lived plasma cells do not express BTK. Thus, an effect on total Ig levels after 2 mo of treatment in this model is unlikely. Treatment with PF-06250112 for 12 wk in the nonaccelerated NZB/W lupus model reduced total levels of IgM, IgG2a, and IgG3 but not IgG1, IgG2b, or IgA. In all cases, except for IgG3, absolute levels remained above baseline at the end of treatment. This supports the conclusion that a strong drop in total Ig levels is not expected with pharmacological BTK inhibition (28). Interestingly, treatment of CLL patients with ibrutinib, while depleting leukemic B cells, led to a slight increase of polyreactive IgG and IgM, possibly representing recovery of the nonleukemic B cells (77).
Although many of our analyses in the NZB/W model focused on the consequences of BTK inhibition in B cells, the inhibition of innate immune cell function is likely a major driver for efficacy in this model, as well. Anti-dsDNA levels were reduced but not abrogated. Thus, immune complexes may still have been available to trigger a myeloid response. The fact that proteinuria was still completely inhibited suggests that myeloid cell inhibition by evobrutinib is an important mechanism in addition to B cell inhibition in this model. Although the normalization of B cell subsets is likely a direct effect on B cells, the normalization of T cell subsets may be a result of both B cell and myeloid cell inhibition. In line with this interpretation, the BTK inhibitor G-744 reduced myeloid gene expression signature in the kidney of NZB/W mice (73). In addition, evobrutinib inhibits the differentiation of proinflammatory M1 macrophages in vitro (78). We do not know at present to what extent this mechanism is relevant in the NZB/W model, but it may contribute to the observed efficacy.
We were able to correlate BTK occupancy in splenocytes to inhibition of B cell stimulation ex vivo and to inhibition of proteinuria. Interestingly, although complete BTK occupancy measured in the spleen correlated to almost complete inhibition of proteinuria, this was not the case for B cell activation, which was only inhibited up to an average of 77%, although complete inhibition was achieved in a few animals (Fig. 11A). These data underscore the idea that BTK inhibitors reduce disease activity via other mechanisms in addition to mere B cell inhibition, such as myeloid cell inhibition. In the mouse spleen, the measured BTK levels will mostly be derived from B cells, with additional smaller contributions from macrophages and NK cells. In human PBMCs, however, B cells make up only a small percentage of leukocytes, with a more significant contribution of monocytes. Measuring total BTK occupancy in PBMCs is, therefore, preferable over cellular assays focusing on single cell types.
As evobrutinib is a covalent inhibitor of BTK, the duration of inhibition of both B cells and mast cells by evobrutinib, as well as BTK occupancy in vivo, was disconnected from the exposure profile. This is to be expected from a covalent inhibitor and was likewise demonstrated previously with CC-292 (79). BTK occupancy and functional inhibition of B cells and mast cells eventually decline. Because evobrutinib is a covalent irreversible BTK inhibitor, the reappearance of free BTK, as well as the recovery of cellular function after a single administration of evobrutinib, is a result of BTK resynthesis.
Taking these findings together, we have shown that evobrutinib is a potent and specific BTK inhibitor. Evobrutinib inhibited BCR and Fc receptor signaling. This inhibition translated into efficacy in animal models for the B cell–driven autoimmune diseases RA and SLE. In the RA model, evobrutinib prevented structural damage in the joints. In the SLE model, evobrutinib prevented proteinuria and reduced histological kidney damage. In addition, features of SLE such as dysregulated cholesterol and hemoglobin levels were normalized. To our knowledge, this is the first report describing these effects of a BTK inhibitor in an animal model for SLE. B cell activation and plasma cell numbers were reduced, and the balance of memory versus naive T cells was shifted toward a more naive phenotype. Our data show that evobrutinib’s efficacy is due to direct effects on both B cells and myeloid cells, which sets BTK inhibitors apart from treatment modalities targeting only B cells. Finally, we have established a PK/PD model describing the time course of BTK occupancy with daily dosing and its link to efficacy in mice. Treatment with evobrutinib in the SLE and RA models was performed prophylactically. To what extent the efficacy observed in this study translates into clinical efficacy after therapeutic treatment is a subject of ongoing investigation.
Supplementary Material
Acknowledgments
We thank the following individuals (all employees of EMD Serono Inc.): Albertina Pereira, Carole Jond-Necand, Corine Gillieron, Ursula Koehler, Ute Felz-Gleitz, Caroline Waltzinger, Nicole Wienandt, Annika Brunner, Alexandra Hohenstein, Felix Jaeger, and Pierre Juillard for expert technical assistance; Maria Stella Lombardi and Hong Ji for the immunoglobulin release assay and in vivo B cell inhibition assay, respectively; Julia Bruttger for analysis of B cell activation markers; and Brian Hodous for contributing to the design of evobrutinib.
This work was supported by EMD Serono Inc. (a business of Merck KGaA, Darmstadt, Germany).
The online version of this article contains supplemental material.
- AUC
- area under the curve
- AUEC
- area under the effect curve
- BTK
- Bruton’s tyrosine kinase
- CIA
- collagen-induced arthritis
- CLL
- chronic lymphocytic leukemia
- EGFR
- epidermal growth factor receptor
- ITK
- IL-2–inducible T cell kinase
- MFI
- mean fluorescence intensity
- MTX
- methotrexate
- PD
- pharmacodynamic
- PK
- pharmacokinetic
- RA
- rheumatoid arthritis
- SLE
- systemic lupus erythematosus
- UPCR
- urinary protein creatinine ratio
- WT
- wild-type.
Disclosures
P.H. and L.B. are employees of Merck KGaA, Darmstadt, Germany. M.C. was an employee of Merck Serono (a business of Merck KGaA), Geneva, Switzerland, at the time of the study. N.N., F.M., and P.S. were employees at the time of the study, and L.L.-B., J.H., S.C.Z., A.T.B., and R.G. are employees of EMD Serono (a business of Merck KGaA), Billerica, MA. A.O. is an employee of Eurofins DiscoverX, South San Francisco, CA, and has provided consulting services to Merck KGaA.
References
- 1.Bradshaw J. M. 2010. The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell. Signal. 22: 1175–1184. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt U., Boucheron N., Unger B., Ellmeier W. 2004. The role of Tec family kinases in myeloid cells. Int. Arch. Allergy Immunol. 134: 65–78. [DOI] [PubMed] [Google Scholar]
- 3.Qiu Y., Kung H. J. 2000. Signaling network of the Btk family kinases. Oncogene 19: 5651–5661. [DOI] [PubMed] [Google Scholar]
- 4.Wahl M. I., Fluckiger A. C., Kato R. M., Park H., Witte O. N., Rawlings D. J. 1997. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton’s tyrosine kinase via alternative receptors. Proc. Natl. Acad. Sci. USA 94: 11526–11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.López-Herrera G., Vargas-Hernández A., González-Serrano M. E., Berrón-Ruiz L., Rodríguez-Alba J. C., Espinosa-Rosales F., Santos-Argumedo L. 2014. Bruton’s tyrosine kinase--an integral protein of B cell development that also has an essential role in the innate immune system. J. Leukoc. Biol. 95: 243–250. [DOI] [PubMed] [Google Scholar]
- 6.Quek L. S., Bolen J., Watson S. P. 1998. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr. Biol. 8: 1137–1140. [DOI] [PubMed] [Google Scholar]
- 7.Shinohara M., Koga T., Okamoto K., Sakaguchi S., Arai K., Yasuda H., Takai T., Kodama T., Morio T., Geha R. S., et al. 2008. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 132: 794–806. [DOI] [PubMed] [Google Scholar]
- 8.Hendriks R. W., Yuvaraj S., Kil L. P. 2014. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 14: 219–232. [DOI] [PubMed] [Google Scholar]
- 9.Tsukada S., Saffran D. C., Rawlings D. J., Parolini O., Allen R. C., Klisak I., Sparkes R. S., Kubagawa H., Mohandas T., Quan S., et al. 1993. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 72: 279–290. [DOI] [PubMed] [Google Scholar]
- 10.Vetrie D., Vorechovský I., Sideras P., Holland J., Davies A., Flinter F., Hammarström L., Kinnon C., Levinsky R., Bobrow M., et al. 1993. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. [Published erratum appears in 1993 Nature 364: 362.] Nature 361: 226–233. [DOI] [PubMed] [Google Scholar]
- 11.Sharma S., Orlowski G., Song W. 2009. Btk regulates B cell receptor-mediated antigen processing and presentation by controlling actin cytoskeleton dynamics in B cells. J. Immunol. 182: 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samuels J., Ng Y. S., Coupillaud C., Paget D., Meffre E. 2005. Human B cell tolerance and its failure in rheumatoid arthritis. Ann. N. Y. Acad. Sci. 1062: 116–126. [DOI] [PubMed] [Google Scholar]
- 13.Halcomb K. E., Musuka S., Gutierrez T., Wright H. L., Satterthwaite A. B. 2008. Btk regulates localization, in vivo activation, and class switching of anti-DNA B cells. Mol. Immunol. 46: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan W. N., Alt F. W., Gerstein R. M., Malynn B. A., Larsson I., Rathbun G., Davidson L., Müller S., Kantor A. B., Herzenberg L. A., et al. 1995. Defective B cell development and function in Btk-deficient mice. Immunity 3: 283–299. [DOI] [PubMed] [Google Scholar]
- 15.Lee Y. H., Bae S. C., Song G. G. 2011. The efficacy and safety of rituximab for the treatment of active rheumatoid arthritis: a systematic review and meta-analysis of randomized controlled trials. Rheumatol. Int. 31: 1493–1499. [DOI] [PubMed] [Google Scholar]
- 16.Huber L. C., Distler O., Tarner I., Gay R. E., Gay S., Pap T. 2006. Synovial fibroblasts: key players in rheumatoid arthritis. Rheumatology (Oxford) 45: 669–675. [DOI] [PubMed] [Google Scholar]
- 17.Schett G., Gravallese E. 2012. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 8: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pathak S., Mohan C. 2011. Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models. Arthritis Res. Ther. 13: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan V. S., Tsang H. H., Tam R. C., Lu L., Lau C. S. 2013. B-cell-targeted therapies in systemic lupus erythematosus. Cell. Mol. Immunol. 10: 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey P. R., Gordon C. 2013. B-cell targeted therapies in systemic lupus erythematosus: successes and challenges. BioDrugs 27: 85–95. [DOI] [PubMed] [Google Scholar]
- 21.Mok C. C. 2014. Update on B-cell targeted therapies for systemic lupus erythematosus. Expert Opin. Biol. Ther. 14: 773–788. [DOI] [PubMed] [Google Scholar]
- 22.Vital E. M., Dass S., Buch M. H., Henshaw K., Pease C. T., Martin M. F., Ponchel F., Rawstron A. C., Emery P. 2011. B cell biomarkers of rituximab responses in systemic lupus erythematosus. Arthritis Rheum. 63: 3038–3047. [DOI] [PubMed] [Google Scholar]
- 23.Hiepe F., Dörner T., Hauser A. E., Hoyer B. F., Mei H., Radbruch A. 2011. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat. Rev. Rheumatol. 7: 170–178. [DOI] [PubMed] [Google Scholar]
- 24.Chen J. S., Chang L. C., Huang S. J., Cheng C. W. 2014. Targeting spleen tyrosine kinase-Bruton’s tyrosine kinase axis for immunologically mediated glomerulonephritis. Biomed Res. Int. 2014: 814869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honigberg L. A., Smith A. M., Sirisawad M., Verner E., Loury D., Chang B., Li S., Pan Z., Thamm D. H., Miller R. A., Buggy J. J. 2010. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 107: 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutcheson J., Vanarsa K., Bashmakov A., Grewal S., Sajitharan D., Chang B. Y., Buggy J. J., Zhou X. J., Du Y., Satterthwaite A. B., Mohan C. 2012. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res. Ther. 14: R243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mina-Osorio P., LaStant J., Keirstead N., Whittard T., Ayala J., Stefanova S., Garrido R., Dimaano N., Hilton H., Giron M., et al. 2013. Suppression of glomerulonephritis in lupus-prone NZB × NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 65: 2380–2391. [DOI] [PubMed] [Google Scholar]
- 28.Rankin A. L., Seth N., Keegan S., Andreyeva T., Cook T. A., Edmonds J., Mathialagan N., Benson M. J., Syed J., Zhan Y., et al. 2013. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J. Immunol. 191: 4540–4550. [DOI] [PubMed] [Google Scholar]
- 29.Whang J. A., Chang B. Y. 2014. Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov. Today 19: 1200–1204. [DOI] [PubMed] [Google Scholar]
- 30.Dubovsky J. A., Beckwith K. A., Natarajan G., Woyach J. A., Jaglowski S., Zhong Y., Hessler J. D., Liu T. M., Chang B. Y., Larkin K. M., et al. 2013. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 122: 2539–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sagiv-Barfi I., Kohrt H. E., Czerwinski D. K., Ng P. P., Chang B. Y., Levy R. 2015. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA 112: E966–E972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.World Health Organization 2016. International nonproprietary names for pharmaceutical substances (INN). WHO Drug Inf. 30: 277. [Google Scholar]
- 33.Haselmayer P., Camps M., Muzerelle M., El Bawab S., Waltzinger C., Bruns L., Abla N., Polokoff M. A., Jond-Necand C., Gaudet M., et al. 2014. Characterization of novel PI3Kδ inhibitors as potential therapeutics for SLE and lupus nephritis in pre-clinical studies. Front. Immunol. 5: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berg E. L., Yang J., Polokoff M. A. 2013. Building predictive models for mechanism-of-action classification from phenotypic assay data sets. J. Biomol. Screen. 18: 1260–1269. [DOI] [PubMed] [Google Scholar]
- 35.Bender A. T., Pereira A., Fu K., Samy E., Wu Y., Liu-Bujalski L., Caldwell R., Chen Y. Y., Tian H., Morandi F., et al. 2016. Btk inhibition treats TLR7/IFN driven murine lupus. Clin. Immunol. 164: 65–77. [DOI] [PubMed] [Google Scholar]
- 36.Liu L., Di Paolo J., Barbosa J., Rong H., Reif K., Wong H. 2011. Antiarthritis effect of a novel Bruton’s tyrosine kinase (BTK) inhibitor in rat collagen-induced arthritis and mechanism-based pharmacokinetic/pharmacodynamic modeling: relationships between inhibition of BTK phosphorylation and efficacy. J. Pharmacol. Exp. Ther. 338: 154–163. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q., Sabnis Y., Zhao Z., Zhang T., Buhrlage S. J., Jones L. H., Gray N. S. 2013. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 20: 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henault J., Riggs J. M., Karnell J. L., Liarski V. M., Li J., Shirinian L., Xu L., Casey K. A., Smith M. A., Khatry D. B., et al. 2016. Self-reactive IgE exacerbates interferon responses associated with autoimmunity. Nat. Immunol. 17: 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pellefigues C., Charles N. 2013. The deleterious role of basophils in systemic lupus erythematosus. Curr. Opin. Immunol. 25: 704–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charles N., Rivera J. 2011. Basophils and autoreactive IgE in the pathogenesis of systemic lupus erythematosus. Curr. Allergy Asthma Rep. 11: 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Advani R. H., Buggy J. J., Sharman J. P., Smith S. M., Boyd T. E., Grant B., Kolibaba K. S., Furman R. R., Rodriguez S., Chang B. Y., et al. 2013. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 31: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berg E. L., Yang J., Melrose J., Nguyen D., Privat S., Rosler E., Kunkel E. J., Ekins S. 2010. Chemical target and pathway toxicity mechanisms defined in primary human cell systems. J. Pharmacol. Toxicol. Methods 61: 3–15. [DOI] [PubMed] [Google Scholar]
- 43.Liu H. B., Wu Y., Lv T. F., Yao Y. W., Xiao Y. Y., Yuan D. M., Song Y. 2013. Skin rash could predict the response to EGFR tyrosine kinase inhibitor and the prognosis for patients with non-small cell lung cancer: a systematic review and meta-analysis. PLoS One 8: e55128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mannis G., Wu D., Dea T., Mauro T., Hsu G. 2015. Ibrutinib rash in a patient with 17p del chronic lymphocytic leukemia. Am. J. Hematol. 90: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tisseverasinghe A., Lim S., Greenwood C., Urowitz M., Gladman D., Fortin P. R. 2006. Association between serum total cholesterol level and renal outcome in systemic lupus erythematosus. Arthritis Rheum. 54: 2211–2219. [DOI] [PubMed] [Google Scholar]
- 46.Monrad S., Kaplan M. 2011. Cellular hematology. In Systemic Lupus Erythematosus, 5th Ed., Lahita R., Tsokos G., Buyon J., Koike T., eds. Elsevier and Academic Press, Amsterdam, p. 905–920. [Google Scholar]
- 47.Abelö A., Eriksson U. G., Karlsson M. O., Larsson H., Gabrielsson J. 2000. A turnover model of irreversible inhibition of gastric acid secretion by omeprazole in the dog. J. Pharmacol. Exp. Ther. 295: 662–669. [PubMed] [Google Scholar]
- 48.Woyach J. A., Furman R. R., Liu T. M., Ozer H. G., Zapatka M., Ruppert A. S., Xue L., Li D. H., Steggerda S. M., Versele M., et al. 2014. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 370: 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furman R. R., Cheng S., Lu P., Setty M., Perez A. R., Guo A., Racchumi J., Xu G., Wu H., Ma J., et al. 2014. Ibrutinib resistance in chronic lymphocytic leukemia. [Published erratum appears in 2014 N. Engl. J. Med. 370: 2547.] N. Engl. J. Med. 370: 2352–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu D., Kim Y., Postelnek J., Vu M. D., Hu D. Q., Liao C., Bradshaw M., Hsu J., Zhang J., Pashine A., et al. 2012. RN486, a selective Bruton’s tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J. Pharmacol. Exp. Ther. 341: 90–103. [DOI] [PubMed] [Google Scholar]
- 51.Chavez J. C., Sahakian E., Pinilla-Ibarz J. 2013. Ibrutinib: an evidence-based review of its potential in the treatment of advanced chronic lymphocytic leukemia. Core Evid. 8: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Melosky B., Burkes R., Rayson D., Alcindor T., Shear N., Lacouture M. 2009. Management of skin rash during EGFR-targeted monoclonal antibody treatment for gastrointestinal malignancies: Canadian recommendations. Curr. Oncol. 16: 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nyhoff L. E., Barron B. L., Johnson E. M., Bonami R. H., Maseda D., Fensterheim B. A., Han W., Blackwell T. S., Crofford L. J., Kendall P. L. 2016. Bruton’s tyrosine kinase deficiency inhibits autoimmune arthritis in mice but fails to block immune complex-mediated inflammatory arthritis. Arthritis Rheumatol. 68: 1856–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonami R. H., Sullivan A. M., Case J. B., Steinberg H. E., Hoek K. L., Khan W. N., Kendall P. L. 2014. Bruton’s tyrosine kinase promotes persistence of mature anti-insulin B cells. J. Immunol. 192: 1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crofford L. J., Nyhoff L. E., Sheehan J. H., Kendall P. L. 2016. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 12: 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whyburn L. R., Halcomb K. E., Contreras C. M., Lowell C. A., Witte O. N., Satterthwaite A. B. 2003. Reduced dosage of Bruton’s tyrosine kinase uncouples B cell hyperresponsiveness from autoimmunity in lyn-/- mice. J. Immunol. 171: 1850–1858. [DOI] [PubMed] [Google Scholar]
- 57.Park J. K., Byun J. Y., Park J. A., Kim Y. Y., Lee Y. J., Oh J. I., Jang S. Y., Kim Y. H., Song Y. W., Son J., et al. 2016. HM71224, a novel Bruton’s tyrosine kinase inhibitor, suppresses B cell and monocyte activation and ameliorates arthritis in a mouse model: a potential drug for rheumatoid arthritis. Arthritis Res. Ther. 18: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di Paolo J. A., Huang T., Balazs M., Barbosa J., Barck K. H., Bravo B. J., Carano R. A., Darrow J., Davies D. R., DeForge L. E., et al. 2011. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 7: 41–50. [DOI] [PubMed] [Google Scholar]
- 59.Díaz de Ståhl T., Andrén M., Martinsson P., Verbeek J. S., Kleinau S. 2002. Expression of FcgammaRIII is required for development of collagen-induced arthritis. Eur. J. Immunol. 32: 2915–2922. [DOI] [PubMed] [Google Scholar]
- 60.Monach P. A., Benoist C., Mathis D. 2004. The role of antibodies in mouse models of rheumatoid arthritis, and relevance to human disease. Adv. Immunol. 82: 217–248. [DOI] [PubMed] [Google Scholar]
- 61.Lee S. H., Kim T., Jeong D., Kim N., Choi Y. 2008. The tec family tyrosine kinase Btk regulates RANKL-induced osteoclast maturation. J. Biol. Chem. 283: 11526–11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shinohara M., Chang B. Y., Buggy J. J., Nagai Y., Kodama T., Asahara H., Takayanagi H. 2014. The orally available Btk inhibitor ibrutinib (PCI-32765) protects against osteoclast-mediated bone loss. Bone 60: 8–15. [DOI] [PubMed] [Google Scholar]
- 63.Chang B. Y., Huang M. M., Francesco M., Chen J., Sokolove J., Magadala P., Robinson W. H., Buggy J. J. 2011. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res. Ther. 13: R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gillooly K. M., Pulicicchio C., Pattoli M. A., Cheng L., Skala S., Heimrich E. M., McIntyre K. W., Taylor T. L., Kukral D. W., Dudhgaonkar S., et al. 2017. Bruton’s tyrosine kinase inhibitor BMS-986142 in experimental models of rheumatoid arthritis enhances efficacy of agents representing clinical standard-of-care. PLoS One 12: e0181782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Christensen A. D., Haase C., Cook A. D., Hamilton J. A. 2016. K/BxN serum-transfer arthritis as a model for human inflammatory arthritis. Front. Immunol. 7: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kagari T., Tanaka D., Doi H., Shimozato T. 2003. Essential role of Fc gamma receptors in anti-type II collagen antibody-induced arthritis. J. Immunol. 170: 4318–4324. [DOI] [PubMed] [Google Scholar]
- 67.Gottar-Guillier M., Dodeller F., Huesken D., Iourgenko V., Mickanin C., Labow M., Gaveriaux S., Kinzel B., Mueller M., Alitalo K., et al. 2011. The tyrosine kinase BMX is an essential mediator of inflammatory arthritis in a kinase-independent manner. J. Immunol. 186: 6014–6023. [DOI] [PubMed] [Google Scholar]
- 68.Crawford J. J., Johnson A. R., Misner D. L., Belmont L. D., Castanedo G., Choy R., Coraggio M., Dong L., Eigenbrot C., Erickson R., et al. 2018. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early clinical development. J. Med. Chem. 61: 2227–2245. [DOI] [PubMed] [Google Scholar]
- 69.Nyhoff L. E., Clark E. S., Barron B. L., Bonami R. H., Khan W. N., Kendall P. L. 2018. Bruton’s tyrosine kinase is not essential for B cell survival beyond early developmental stages. J. Immunol. 200: 2352–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goess C., Harris C. M., Murdock S., McCarthy R. W., Sampson E., Twomey R., Mathieu S., Mario R., Perham M., Goedken E. R., Long A. J. 2018. ABBV-105, a selective and irreversible inhibitor of Bruton’s tyrosine kinase, is efficacious in multiple preclinical models of inflammation. Mod. Rheumatol. DOI: 10.1080/14397595.2018.1484269. [DOI] [PubMed] [Google Scholar]
- 71.Kim Y. Y., Park K. T., Jang S. Y., Lee K. H., Byun J. Y., Suh K. H., Lee Y. M., Kim Y. H., Hwang K. W. 2017. HM71224, a selective Bruton’s tyrosine kinase inhibitor, attenuates the development of murine lupus. Arthritis Res. Ther. 19: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chalmers S. A., Wen J., Doerner J., Stock A., Cuda C. M., Makinde H. M., Perlman H., Bosanac T., Webb D., Nabozny G., et al. 2018. Highly selective inhibition of Bruton’s tyrosine kinase attenuates skin and brain disease in murine lupus. Arthritis Res. Ther. 20: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Katewa A., Wang Y., Hackney J. A., Huang T., Suto E., Ramamoorthi N., Austin C. D., Bremer M., Chen J. Z., Crawford J. J., et al. 2017. Btk-specific inhibition blocks pathogenic plasma cell signatures and myeloid cell-associated damage in IFNα-driven lupus nephritis. JCI Insight 2: e90111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Steinberg B. J., Smathers P. A., Frederiksen K., Steinberg A. D. 1982. Ability of the xid gene to prevent autoimmunity in (NZB X NZW)F1 mice during the course of their natural history, after polyclonal stimulation, or following immunization with DNA. J. Clin. Invest. 70: 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kendall P. L., Moore D. J., Hulbert C., Hoek K. L., Khan W. N., Thomas J. W. 2009. Reduced diabetes in btk-deficient nonobese diabetic mice and restoration of diabetes with provision of an anti-insulin IgH chain transgene. J. Immunol. 183: 6403–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tanwar S., Dhar A., Varanasi V., Mukherjee T., Boppana R., Basak S., Bal V., George A., Rath S. 2017. Mediation of transitional B cell maturation in the absence of functional Bruton’s tyrosine kinase. Sci. Rep. 7: 46029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun C., Tian X., Lee Y. S., Gunti S., Lipsky A., Herman S. E., Salem D., Stetler-Stevenson M., Yuan C., Kardava L., et al. 2015. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood 126: 2213–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alankus B. Y., Grenningloh R., Haselmayer P., Bender A., Bruttger J. 2018. Inhibition of Bruton’s tyrosine kinase (BTK) prevents inflammatory macrophage differentiation: a potential role in RA and SLE. Arthritis Rheumatol. 70(Suppl. 10) (Abstr.). [Google Scholar]
- 79.Evans E. K., Tester R., Aslanian S., Karp R., Sheets M., Labenski M. T., Witowski S. R., Lounsbury H., Chaturvedi P., Mazdiyasni H., et al. 2013. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 346: 219–228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.