

Abstract
Objective
Parkinson's disease (PD) episodic memory impairments are common; however, it is not known whether these impairments are due to hippocampal pathology. Hippocampal Lewy-bodies emerge by Braak stage 4, but are not uniformly distributed. For instance, hippocampal CA1 Lewy-body pathology has been specifically associated with pre-mortem episodic memory performance in demented patients. By contrast, the dentate gyrus (DG) is relatively free of Lewy-body pathology. In this study, we used ultra-high field 7-Tesla to measure hippocampal subfields in vivo and determine if these measures predict episodic memory impairment in PD during life.
Methods
We studied 29 participants with PD (age 65.5 ± 7.8 years; disease duration 4.5 ± 3.0 years) and 8 matched-healthy controls (age 67.9 ± 6.8 years), who completed comprehensive neuropsychological testing and MRI. With 7-Tesla MRI, we used validated segmentation techniques to estimate CA1 stratum pyramidale (CA1-SP) and stratum radiatum lacunosum moleculare (CA1-SRLM) thickness, dentate gyrus/CA3 (DG/CA3) area, and whole hippocampus area. We used linear regression, which included imaging and clinical measures (age, duration, education, gender, and CSF), to determine the best predictors of episodic memory impairment in PD.
Results
In our cohort, 62.1% of participants with PD had normal cognition, 27.6% had mild cognitive impairment, and 10.3% had dementia. Using 7-Tesla MRI, we found that smaller CA1-SP thickness was significantly associated with poorer immediate memory, delayed memory, and delayed cued memory; by contrast, whole hippocampus area, DG/CA3 area, and CA1-SRLM thickness did not significantly predict memory. Age-adjusted linear regression models revealed that CA1-SP predicted immediate memory (beta[standard error]10.895[4.215], p < .05), delayed memory (12.740[5.014], p < .05), and delayed cued memory (12.801[3.991], p < .05). In the fully-adjusted models, which included all five clinical measures as covariates, only CA1-SP remained a significant predictor of delayed cued memory (13.436[4.651], p < .05).
Conclusions
In PD, we found hippocampal CA1-SP subfield thickness estimated on 7-Tesla MRI scans was the best predictor of episodic memory impairment, even when controlling for confounding clinical measures. Our results imply that ultra-high field imaging could be a sensitive measure to identify changes in hippocampal subfields and thus probe the neuroanatomical underpinnings of episodic memory impairments in patients with PD.
Keywords: Parkinson's disease, Episodic memory, Cognitive impairment, MRI, Hippocampus, CA1, 7 Tesla
Highlights
-
•
CA1-SP subfield thickness predicts Parkinson's disease episodic memory impairment.
-
•
The relationship between CA1-SP and memory is beyond the effect of duration and age.
-
•
The relationship between CA1-SP thickness and memory is not due to concomitant AD.
-
•
There was no relationship between whole hippocampus and PD memory impairment.
1. Introduction
Episodic memory impairments are common in Parkinson's disease (PD). Over 20% of newly diagnosed patients express a memory complaint (Breen and Drutyte, 2013) and two large cohorts of newly diagnosed PD patients reported frequent memory impairments on baseline testing (Weintraub et al., 2015; Yarnall et al., 2014). Episodic memory impairments are reported almost as frequently as executive dysfunction (Aarsland et al., 2010), however patients who experience episodic memory impairments are at higher risk of developing dementia than those who experience executive dysfunction alone (Emre et al., 2007; Robbins and Cools, 2014). There are no effective treatments to prevent or delay the memory impairments leading to dementia, in part because the neuroanatomical underpinnings of PD-related episodic memory impairments are not well understood.
Autopsy studies have shown that alpha-synuclein pathology in the hippocampus is common in PD patients, which could be related to memory impairments (Galvin et al., 1999; Adamowicz et al., 2017). According to the Braak staging system in PD, Lewy neurites and Lewy-bodies develop in the hippocampus at stage 4, before Lewy-bodies develop elsewhere in the neocortex (stages 5–6) (Braak et al., 2006; Braak et al., 1998). One clinicopathological study showed that almost 90% of patients identified as Braak stage 4 at autopsy (hippocampal Lewy-bodies without other neocortical Lewy-body pathology) were cognitively impaired prior to death. While these autopsy studies provide invaluable insights into the pathology associated with PD dementia (PDD), they are limited to the end of that pathological process when the brain has been ravaged by disease; and therefore, these studies are insensitive to the earliest brain changes more likely to be associated with mild memory impairments.
Pre-mortem, structural MRI can overcome this challenge. To date, most MRI studies agree that PD patients with dementia have smaller hippocampi than healthy adults but larger hippocampi than Alzheimer's disease (AD) patients (Camicioli et al., 2003; Bruck et al., 2004; Laakso et al., 1996; Novellino et al., 2018); however, studies in early PD patients have been mixed (Camicioli et al., 2003; Bruck et al., 2004; Laakso et al., 1996; Tanner et al., 2017; Pirogovsky-Turk et al., 2015). These conflicting results could have arisen from evaluating the hippocampus as a single structure. This is important to consider because Lewy-body pathology is not found uniformly throughout the hippocampus. For instance, Lewy-body pathology is most prominent in the CA2 subfields (Dickson et al., 1991; Dickson et al., 1994) but is also found throughout CA1(Churchyard and Lees, 1997). By contrast, the dentate gyrus (DG/CA3) is relatively free of Lewy-body pathology. Further, while CA2 and CA1 have a predilection for Lewy-body pathology, a recent autopsy study found that the degree of Lewy-body pathology in CA1, but not CA2, predicted pre-mortem episodic memory impairment in patients with LewyBody Dementia (Adamowicz et al., 2017). The clinical impact of this regional distribution of pathology in the hippocampal subfields, and the CA1 subfield in particular, has not been well studied in earlier PD patients, prior to the onset of dementia.
Here, we sought to determine whether episodic memory impairments in PD can be predicted by specific hippocampal subfields (as an in vivo proxy for regional Lewy-body pathology). To achieve this goal, we leveraged ultra-high field 7-Tesla (7T) MRI to generate images with high enough spatial resolution to visually identify and manually segment and quantify the thickness of sublayers of CA1, along with the area of DG/CA3 and the whole hippocampus (Kerchner et al., 2014; Kerchner et al., 2013; Kerchner et al., 2012). We then determined whether hippocampal subfield microstructural size was related to episodic memory in a cohort of mostly non-demented PD participants.
2. Methods
2.1. Standard protocol approvals, registrations, and patient consents
All study procedures were approved by the Stanford Institutional Review Board. Written informed consent was obtained from all participants in this study.
2.2. Participants
We recruited 38 PD and 10 healthy controls (HC) participants from the Stanford Movement Disorders Clinic and the surrounding community. Inclusion criteria were (1) age between 50 and 79 years, (2) English fluency, (3) right-handedness, (4) no MRI structural abnormalities (including stroke), (5) no past or current history of alcohol abuse, substance abuse, head injury with loss of consciousness ≥15 min, epilepsy, hydrocephalus, or major psychiatric disorder, and 6) no known or potential contraindications to high magnetic field. For PD, additional exclusion criteria were (1) deep brain stimulation and (2) features suggestive of an atypical parkinsonian syndrome (early repeated falls, early symptomatic orthostatic hypotension, or early visual hallucinations).
2.3. Clinical assessments
A board-certified neurologist with specialty training in movement disorders completed a history, general physical examination and neurological examination. Participants with PD performed the Movement Disorders Society Unified Parkinson's Disease Rating Scale, part III motor exam (MDS-UPDRS-III) (Goetz et al., 2008) in the clinically-defined off dopaminergic state, according to published protocols (Poston et al., 2016), and were diagnosed with probable PD using the UK Parkinson's Disease Society Brain Bank clinical diagnostic criteria (Litvan et al., 2003). A trained psychometrician administered comprehensive neuropsychological testing to all participants. The neurologist and psychometrician where blinded to the MRI segmentation data. As recommended by the Movement Disorder Society commissioned task force (Litvan et al., 2012), the neuropsychological battery included at least two tests in each of five cognitive domains (episodic memory, attention/working memory, executive, visuospatial, and language) and an additional test of general cognition (Montreal Cognitive Assessment) (Nasreddine et al., 2005). Based on our central hypothesis that hippocampal subfield microstructural size is related to memory performance in PD and based on our prior publications (Kerchner et al., 2012), we focused the analysis on episodic memory cognitive measures taken from the California Verbal Learning Test-II (CVLT-II) (Delis et al., 2000). Specifically, we studied (1) immediate memory (CVLT-II short delay, free recall), (2) delayed memory (CVLT-II long delay, free recall), (3) delayed cued memory (CVLT-II long delay, cued recall), and (4) learning (CVLT-II trials 1–5 summation). We included delayed cued memory since several studies suggest that in PD patients poor cued recall is more likely due to a memory impairment per se, whereas poor free recall could be due to either memory or executive impairments (Novellino et al., 2018; Edelstyn et al., 2015; Costa et al., 2014).
2.4. Image acquisition
All 48 enrolled participants were scanned with a 7T GE Healthcare Discovery MR950 MRI whole-body scanner (GE Healthcare, Waukesha, WI) using a 32-channel radiofrequency receive head coil contained within a quadrature transmit coil (Nova Medical, Inc., Wilmington, MA). Sixteen oblique coronal images oriented perpendicular to the longitudinal axis of the hippocampus were acquired with a T2-weighted fast spin echo sequence: echo time 47 milliseconds; repetition time 5–8 s (cardiac gated); acquired voxel size was 0.22 × 0.22 × 1.5 mm3 with a slice gap of 0.5 mm, interpolated by zero filling to 0.166 × 0.166 × 1.5 mm3.
To minimize head motion, we stabilized each participant's head by positioning inflatable and/or foam cushions between the surface of the head and the inner surface of the receive coil. For subjects exhibiting tremor, we placed soft, weighted bags atop the limbs to reduce the potential transmission of tremor motion to the head. We placed a plethysmograph on one index finger to monitor peripheral pulse. PD participants were scanned in the typical on-medication state (Ng et al., 2017).
2.5. 7T image analysis
We traced regions-of-interest (ROIs) using FSL-view, a component of the FSL 5.0.10 software package (http://www.fmrib.ox.ac.uk/fsl/), and OsiriX 8.5 software package (http://www.osirix-viewer.com/). We used previously validated semi-automated methods for subfield identification and derivation of subfield metrics (Kerchner et al., 2013; Kerchner et al., 2012).
We focused our analysis on the thickness of the CA1 stratum pyramidale (CA1-SP) and stratum radiatum lacunosum moleculare (CA1-SRLM) layers, and the cross-sectional area of the DG/CA3 and the whole hippocampus (Fig. 1). We choose CA1-SP as the primary variable of interest because Lewy-bodies are found in the cell bodies of the CA1 subfield post-mortem (Braak et al., 2006), which has been correlated with pre-mortem memory performance (Adamowicz et al., 2017). We included DG/CA3 as a ‘control’ region because it is relatively spared of Lewy-bodies post-mortem. We included the whole hippocampus for comparison to prior publications. While Lewy-bodies are also prominent in the CA2 subfield at autopsy, this region is not visually discernable even with high-resolution imaging and prior attempts to manually segment CA2 have shown poor inter-study reliability (Yushkevich et al., 2015), therefore we did not include CA2 measures. For all analyses, we separately estimated then averaged the right and left hemispheres. We used raw metrics, rather than adjust for head size, based on our previous data that strongly suggests that these raw measures are likely to reflect the presence of pathology and do not vary with intracranial volume (Kerchner et al., 2010).
Fig. 1.
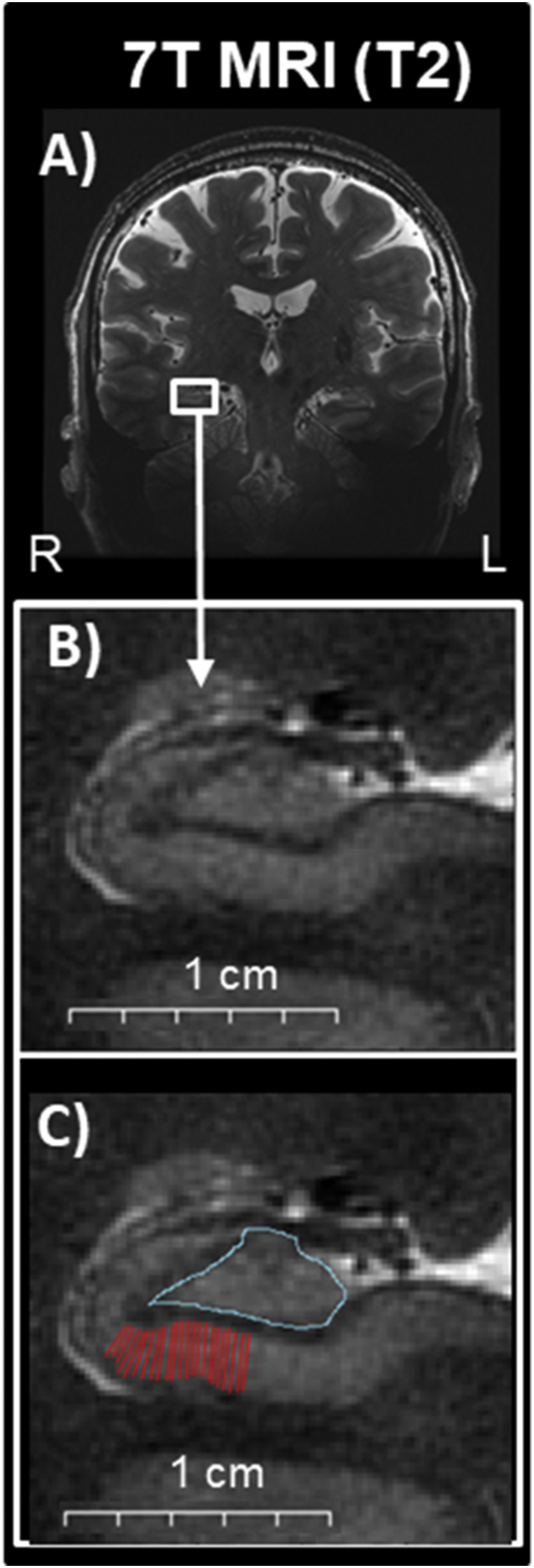
Hippocampal subfields illustrations on images obtained at 7T MRI. A) Oblique coronal view of a 7T T2-weighted MRI scan. B) Magnified area of the hippocampus where subfields are readily visualized and thus able to be manually traced. C) Superimposed manual tracing of the CA1 (red) and DG/CA3 (turquoise). Abbreviations: R: right, L: left.
To estimate the thickness of CA1-SP, we manually traced curvilinear ROIs on the hyperintense region of the CA1-SP in each slice within the delimiting boundaries of the hippocampal body (Fig. 2). We then estimated the thickness of the CA1-SP using an in-house semi-automated method measuring the widths of derived orthogonal vectors from a computed spline. Specifically, the in-house semi-automated algorithm derives orthogonal vectors along a spline generated from the user-specified line of hyperintensity. We used a distance-from-spline function with each signal intensity-distance averaged with its corresponding reverse function to obtain a true midpoint of the structure of interest. We estimated the thickness of the CA1-SP as the distance between the peak and the trough of the derivative of the signal intensity-distance function (Kerchner et al., 2012).
Fig. 2.

Estimation of CA1-SP thickness using in-house semi-automated algorithm. Top: Magnification of the CA1-SP region and the re-centered orthogonal vectors to the spline of the traced ROI used in the CA1-SP thickness estimation. Yellow squares show traced ROIs, red lines show orthogonal planes to the spline, and white arrows show the re-centered orthogonal vectors where thickness is estimated. Bottom: CA1-thickness is estimated as the average distance between the peak and the trough of the 1st derivative of the intensity plot. Methods described in detail by Kerchner et al. Neuroimage Kerchner et al., 2012.
Similarly, we estimated the thickness of the CA1-SRLM using the curvilinear ROI tracings, but on the hypointense band between the more intense DG and CA1-SP cell layers (Kerchner et al., 2012). This was followed by an akin implementation of the semi-automated algorithm and estimation of thickness as the distance between the peak and trough of the derivative of the signal intensity-distance function.
As part of our quality checks, we did not draw an ROI when there was (1) insufficient image contrast/clarity for identification of the superior and inferior boundaries of the CA1, (2) excessive blurring of these boundaries due to motion, (3) inadequate coverage of the hippocampus, either from non-optimal slice prescription or signal drop-out, or (4) distorted CA1 uniformity due to the presence of a hippocampal sulcal remnant cyst.
To estimate cross-sectional area of the DG/CA3 and whole hippocampus, we manually traced ROIs on each slice within the delimiting boundaries of the hippocampal body.(Kerchner et al., 2012) We combined DG and CA3 areas because no visible boundary can be confidently identified, even on our high-resolution images. CA4 was considered part of CA3.
All manual ROI tracings required a minimum of 4 measurable slices to be included for analysis. We excluded 11 participants (9 PD and 2 HC) that did not meet these criteria, resulting in a final cohort of 29 PD and 8 HC included in the 7T analysis (Table 1).
Table 1.
Demographics and clinical characteristics of participants included in 7T MRI analysis.
| HC | PD | p | |
|---|---|---|---|
| Total | 8 | 29 | |
| Age, y | 67.9 ± 6.8 (57–76) | 65.5 ± 7.8 (52–79) | .41 |
| Female, n (%)† | 5 (62.5%) | 15 (51.7%) | .23 |
| Education, y | 17.3 ± 2.1 (14–20) | 16.7 ± 2.7 (12−20) | .58 |
| Disease duration, y | n/a | 4.5 ± 3.0 (1–15) | |
| n (%) PD 0–5 y | 19 (65.5%) | ||
| n (%) PD 6–10 y | 9 (31.0%) | ||
| n (%) PD 11–16 y | 1 (3.5%) | ||
| MDS-UPDRS-III (Off) | n/a | 33.7 ± 13.6 (6–62) | |
| Cognitive diagnosis | |||
| n (%) NC | 8 (100%) | 18 (62.1%) | |
| n (%) PD-MCI | 8 (27.6%) | ||
| n (%) PDD | 3 (10.3%) | ||
| MoCA†† | 27.6 ± 2.2 (24–29) | 25.5 ± 3.9 (14–29) | .14 |
| CVLT†† | |||
| Immediate memory | 10.9 ± 2.4 (7–15) | 9.1 ± 3.8 (0–15) | .23 |
| Delayed memory | 11.8 ± 2.3 (9–16) | 9.4 ± 4.6 (0–16) | .22 |
| Delayed cued memory | 12.4 ± 1.5 (10–16) | 11.0 ± 3.7 (2–16) | .36 |
| Learning | 51.6 ± 4.7 (45–59) | 44.8 ± 12.6 (14–67) | .15 |
Mean ± SD (range).
Abbreviations: y = years, MDS-UPDRS-III = Movement Disorders Society Unified Parkinson's Disease Rating Scale, part III (motor exam), n (%) PD = number (percent) of Parkinson's disease participants with a disease duration in that range, n (%) NC = number (percent) of participants with cognition in the normal range, n (%) PD-MCI = number (percent) of Parkinson's disease participants with mild cognitive impairments, n (%) PDC = number (percent) of Parkinson's disease participants dementia, n/a = not applicable.
All between group p-values represent t-tests, except where indicated by † for Pearson χ2 test between dichotomous variable, †† for Mann–Whitney U for continuous variables with unequal variance.
2.6. Validation of 7T semi-automated CA1-SP and CA1-SRLM thickness measurement
Our semi-automated hippocampal segmentation technique has been validated in healthy older adults (Carr et al., 2017), and in amnestic mild cognitive impairment and AD (Kerchner et al., 2014; Kerchner et al., 2013; Kerchner et al., 2012). Because this is the first application of this technique in PD, we performed a validation step with two independent and blinded raters, who performed the manual tracing and semi-automated CA1-SP and CA1-SRLM thickness estimations. Data from rater 1 were used for statistical analyses and data from raters 1 and 2 were used to calculate interrater reliability. We applied a two-way-mixed level ANOVA and computed the absolute intraclass correlation coefficient (ICC) (Shrout and Fleiss, 1979). We found an absolute ICC value of 0.86 for CA1-SP mean thickness values (95% CI: 0.70–0.93) and 0.77 for CA1-SRLM mean thickness values (95% CI: 0.54–0.89).
2.7. CSF analysis
Comorbid Alzheimer's disease (AD) pathology in our PD cohort could confound the interpretation of our hippocampal measurements. In prior work, we demonstrated CA1-SP and CA1-SRLM thinning in amnestic mild cognitive impairment and in patients with mild AD relative to healthy controls, and this thinning was highly correlated to memory performance. Therefore, in a subset of participants, we obtained CSF by lumbar puncture between the L3/L4 or L4/L5 intervertebral space and collected in 20-mL polypropylene tubes. We centrifuged CSF samples for 10 min at 1800 × g at 4 °C within 2 h of collection and divided into aliquots in polypropylene tubes of 0.5 or 1 mL at −80 °C until analysis. We measured Aβ1–42 and p-tau using commercially available ELISA kit according to the manufacturer's protocol. Because reference values for these proteins can vary, we determined the cutoff for low Aβ1–42 and elevated p-tau using an independent cohort of 21 healthy controls and 14 clinical AD participants, who had CSF processed at the same time. We used an ROC analysis to determine that a cutoff of Aβ1–42 ≤ 660 and p-tau ≥60 gave 100% sensitivity with 92% specificity (AUC = 0.98) differentiating AD from the healthy controls. We then dichotomize PD participants who have CSF in this range (Aβ1–42 ≤ 660 and p-tau ≥60) as ‘possible dual PD/AD’.
2.8. Statistical analysis
We used the Statistical Package for the Social Sciences (IBM SPSS Statistics, version 24; IBM Corp, https://www.ibm.com/) for all statistical analyses and used two-tailed p values. For between-group analysis, we calculated two-sample t-tests for continuous variables with equal variance, Mann–Whitney U for continuous variables with unequal variance, and chi-square for dichotomous variables. For univariate correlational analyses between continuous variables, we calculated Pearson coefficients. To assess whether episodic memory in PD is best predicted by hippocampal subfield metrics or clinical measures, we performed three linear regression models in the whole PD cohort, with episodic memory cognitive measures as the dependent variable and hippocampal subfield metrics, age, disease duration, education, gender, and CSF (dichotomous for possible dual PD/AD pathology) as independent variables. In model 1, we performed a linear regression with only the primary variable of interest (CA1-SP) as the independent variable (unadjusted model). In model 2, we included the minimal adjustment by adding age into the model (age-adjusted model). We chose age because it is the strongest predictor of memory in healthy aging and in PD (Cholerton et al., 2013; Reid et al., 2011). In model 3, we performed a fully-adjusted model with all clinical measures associated with memory impairment in PD (age, disease duration, education, gender, and CSF) as independent variables (fully-adjusted model). We repeated these regression analyses for CA1-SRLM, DG/CA3, and whole hippocampus.
3. Results
3.1. Behavioral results
See Table 1 for complete demographic characteristics. The PD and HC participants did not differ in age, gender, or education. On formal neuropsychological testing, all HC participants were within 1.5 standard deviations of age- and education-matched normative values. For the PD participants enrolled, 18 had cognition in the normal range (PD-NC), 8 had mild cognitive impairment (PD-MCI), and 3 had dementia (PDD), according to published criteria (Emre et al., 2007; Litvan et al., 2012). For this analysis we used memory as a continuous variable rather than subdividing PD by cognitive group because (1) less than half of the cohort were cognitively impaired, with very few diagnosed as PDD and (2) the designation of PD-MCI is not based on any one domain and requires two abnormal tests, therefore PD-MCI can represent highly heterogeneous patients. As a result, some patients can be designated PD-MCI without any deficits in episodic memory and others can be designated PD-NC with an episodic memory deficit that is found on only one test.
CSF protein analysis was available for 26 participants (18 PD and 8 HC) (Table 2). Using our criteria above, 5 PD participants were categorized as possible dual PD/AD. Compared to the remaining 24 PD participants (13 with negative AD CSF biomarkers and 11 with unknown AD CSF biomarkers), the 5 participants with possible dual PD/AD pathology were older (63.6 ± 6.7 and 74.2 ± 5.7, p = .003), had more severe MDS-UPDRS-III off (30.2 ± 11.1 and 48.8 ± 12.3, p = .002), and lower MoCA (26.7 ± 2.3 and 21.0 ± 5.4, p = .001).
Table 2.
Participants with CSF.
| HC | PD-NC | PD-MCI | PDD | Total PD | |
|---|---|---|---|---|---|
| Total participants | 8 | 18 | 8 | 3 | 29 |
| Participants with CSF | 8 | 10 | 5 | 3 | 18 |
| Positive AD CSF biomarkers | 0 | 1 | 2 | 2 | 5 |
| Negative AD CSF biomarkers | 8 | 9 | 3 | 1 | 13 |
| Unknown AD CSF biomarkers | 0 | 8 | 3 | 0 | 11 |
Based on an independent cohort of 21 healthy controls and 14 clinical AD participants we used a cutoff of Aβ1–42 ≤ 660 and p-tau ≥60 to determine possible dual PD/AD pathology due to positive AD CSF biomarkers.
Abbreviations: AD = Alzheimer's disease, CSF = cerebral spinal fluid, HC = Healthy Controls, PD-NC = PD with cognition in the normal range, PD-MCI = PD with mild cognitive impairment, PDD = PD with dementia.
3.2. 7T hippocampal correlations and linear regression
As an initial exploratory analysis that did not control for other factors, we used univariate analysis by Pearson's correlation to test the relationship between memory performance in PD and our independent variables, including hippocampal metrics and clinical variables that have previously been associated with poor memory performance in PD patients (Cholerton et al., 2013) (Table 3). We found that poorer immediate memory (r = 0.557, p = .005), delayed memory (r = 0.554, p = .005), delayed cued memory (r = 0.626, p = .001), and learning (r = 0.618, p = .001) were associated with thinner CA1-SP but not CA1-SRML, DG/CA3 area, or total hippocampus (Fig. 3; Table 3). We then tested to see if correlations differed across regions. For each of the three measures of memory performance, the correlation with CA1-SP was significantly greater than the correlation with DG/CA3 and with CA1-SRLM, whereas the correlation with CA1-SP versus the correlation with total hippocampus did not statistically differ (Table 3). Regarding clinical measures associated with memory, we found poorer immediate memory (r = −0.401, p = .031), delayed memory (r = −0.410, p = .027), delayed cued memory (r = −0.385, p = .039), and learning (r = −0.430, p = .020) were also associated with older age, but not with disease duration or education. Importantly, given the modest sample size, we note that some caution is warranted in interpreting the effect sizes of these significant relationships.
Table 3.
Brain regions showing significant functional connectivity changes (post-treatment minus pre-treatment) with right hippocampus and bilateral ACC as seeds.
| Immediate Memory |
Delayed Memory |
Delayed Cued Memory |
|
|---|---|---|---|
| r (p) | r (p) | r (p) | |
| Clinical Characteristics | |||
| Age | -0.40 (0.031)* | -0.41 (0.027)* | -0.39 (0.039)* |
| Duration | -0.29 (0.123) | -0.27 (0.152) | -0.27 (0.165) |
| Education | -0.08 (0.698) | -0.06 (0.767) | -0.11 (0.556) |
| 7T MRI Metrics | |||
| CA1-SP | 0.56 (0.005)* | 0.55 (0.005)* | 0.63 (0.001)* |
| CA1-SRLM | 0.06 (0.779) | 0.01 (0.966) | -0.084 (0.683) |
| DG | 0.02 (0.913) | 0.02 (0.905) | 0.03 (0.866) |
| Total Hippocampus | 0.27 (0.155) | 0.35 (0.066) | 0.29 (0.125) |
| z (p) | z (p) | z (p) | |
| Between correlation differences | |||
| CA1-SP vs CA1-SRLM | 1.9 (0.06) | 2.02 (0.04)* | 2.71 (0.007)* |
| CA1-SP vs DG | 2.07 (0.04)* | 2.05 (0.04)* | 2.39 (0.02)* |
| CA1-SP vs Total Hippocampus | 1.19 (0.23) | 0.90 (0.37) | 1.48 (0.14) |
Abbreviations: r: Pearson's correlation coefficient, z: Fisher r-to-z transformation, CA1-SP = CA1 stratum pyramidale layer, CA1-SRLM = CA1 stratum radiatum lacunosum moleculare, DG/CA3 = dentate gyrus and CA3 layer.
Bold indicates significance of p < 0.05
Fig. 3.

Hippocampal and clinical correlations with episodic memory in Parkinson's disease. Scatter plots of delayed cued memory, delayed memory, immediate memory, and learning versus CA1 stratum pyramidale layer (CA1-SP) width in mm (A, C, E and G respectively) and age in years (B, D, F and H respectively). Regression lines only include the three PD groups (circles: PD with normal cognition in blue, PD with mild cognitive impairment in yellow, and PD with dementia in green). Healthy Control data (×) are included for reference only.
We then performed linear regression analysis to determine the effect of the independent variables on memory performance in the PD participants. In all age-adjusted models, CA1-SP remained a significant predictor of immediate (p = .012), delayed (p = .011), delayed cued (p = .004) memory, and learning (p = .003) (Supplemental Table 1, Supplemental Table 2, Table 4, and Supplemental Table 3, respectively). In the fully-adjusted model, CA1-SP remained a significant predictor of delayed cued memory (p = .010) and learning (p = .026), but also trended toward significance for immediate (p = .075) and delayed (p = .058) memory. When comparing all models, the fully-adjusted model most strongly predicted delayed cued memory in PD; this model explained 50.6% of the variance (R2) and only CA1-SP remained a significant predictor (p < .05, Table 4).
Table 4.
Predictors of delayed cued memory in Parkinson's disease.
| Covariates | Model 1 unadjusted | Model 2 age-adjusted | Model 3 fully-adjusted |
|---|---|---|---|
| CA1-SP | 14.00 (3.72)⁎⁎ | 12.80 (3.99)⁎ | 13.44 (4.65)⁎ |
| Age | −0.08 (0.09) | −0.10 (0.12) | |
| Duration | −0.36 (0.30) | ||
| Education | −0.48 (0.30) | ||
| Gender | 1.71 (1.58) | ||
| Possible dual PD/AD | 1.54 (2.44) | ||
| R2 = 0.391 | R2 = 0.412 | R2 = 0.506 | |
| p = .001 | p = .004 | p = .039 |
Values represent beta regression coefficients (standard error). In model 1, only the covariate of interest (CA1-SP) was entered into the model. In model 2, CA1-SP was adjusted for age only (age-adjusted). In model 3, all clinical covariates were entered (fully-adjusted).
Covariates contributing to the model, ⁎⁎p = .001, ⁎p < .05, all others p > .1.
Abbreviations: CA1-SP = CA1 stratum pyramidale layer.
Bold indicates significance of p < 0.05
When comparing the 29 PD and 8 HC participants, there were no between-group differences in CA1-SP (p = .26), CA1-SRLM (p = .56), DG/CA3 (p = .77) or total hippocampal (p = .61) metrics.
4. Discussion
In this study we found that the best predictor of episodic memory performance in mostly non-demented PD patients was thinning of the CA1-SP layer of the hippocampus, as measured using 7T MRI. In our regression analyses, CA1-SP remained the only significant predictor of memory and learning after adjusting for other clinical measures associated with poor memory in PD. Together, these data are the first in living patients to clearly demonstrate that the CA1-SP hippocampal subfield is linked to memory performance in patients with PD.
4.1. Episodic memory in PD
The frequency and severity of episodic memory impairments in patients with PD has a significant impact on quality of life. Even at the stage of initial motor diagnosis, episodic memory is as commonly impaired as executive function (Weintraub et al., 2015; Yarnall et al., 2014). Additionally, the proportion of patients with episodic memory impairment increases as the disease progresses and is a major component of dementia in PD patients (Emre et al., 2007), markedly increasing patient disability, loss of employment, institutionalization, and mortality (Hely et al., 2005; Hely et al., 2008).
Despite this, the neural underpinning of episodic memory impairments in PD remains unclear (Das et al., 2018); thus, there are almost no symptom-specific therapies available for patients, and no therapies to halt or slow the progression of impairment leading to dementia. Episodic memory impairments in PD were traditionally considered ‘subcortical’ and secondary to dopamine deficiency and subsequent fronto-striatal dysfunction, with inefficient use of strategies during encoding or recall (Massman et al., 1990). However, more recent studies have challenged the notion that memory impairment in PD is purely subcortical and only secondary to underlying executive dysfunction, and instead hypothesized additional hippocampal mechanisms (Robbins and Cools, 2014). For instance, PD patients show loss of dopaminergic intervention to the hippocampus from the ventral tegmental area (German et al., 1989). This ventral tegmental area-hippocampal loop disruption has particular implications for motivationally significant memories (Lisman and Grace, 2005; Shohamy and Adcock, 2010) and dopaminergic input has been shown to influence hippocampal long-term potentiation (Castro-Hernandez et al., 2017). Another potential mechanism underlying PD episodic memory impairment is direct hippocampal pathology (Das et al., 2018), which is supported by our data. Further, our data argue that, within the hippocampus, abnormalities in the CA1 region could specifically contribute to mild memory impairments in PD patients prior to the onset of dementia.
4.2. The hippocampus and CA1 in PD
Prior studies assessing the relationship between total hippocampal volume and PD episodic memory performance have been conflicting, likely due to different methods used to acquire images and measures of the hippocampus. In general, early studies agreed that PD patients with dementia have smaller hippocampal volumes than healthy age-matched control subjects but larger hippocampal volumes than patients with clinical AD (Camicioli et al., 2003; Bruck et al., 2004; Laakso et al., 1996). Visual medial temporal lobe grading on a standardized 5-point atrophy severity scale showed atrophy was less severe in non-demented PD compared to AD (Tam et al., 2005) and that medial temporal lobe volumes could readily discriminate pathologically confirmed AD compared to Lewy Body Dementia (Burton et al., 2009). However, this 5-point atrophy severity scale was not sensitive enough to identify any correlation between changes in medial temporal lobe volume and severity of cognitive impairment in PD patients, either with or without dementia. Other studies that used more sophisticated hippocampal tracing techniques did find a correlation between decreases in memory and hippocampal volume (Bouchard et al., 2008; Junque et al., 2005), but only when including PD patients with dementia. In our study, we did not find a relationship between whole hippocampal volume/area and memory impairment in PD using 7T MRI, likely because our subjects were younger, mostly non-demented, and had a far shorter average disease duration than prior studies (duration of motor symptoms 4.5 ± 3.0 years versus 10–13 years). Thus, our results suggest that in early PD the CA1 region shows structural differences associated with memory impairments before other hippocampal structures. This is important, since biomarkers sensitive to early, mild changes are needed to identify patients at risk for developing future dementia. Further studies assessing whether hippocampal subfield size predicts worsening episodic memory with resultant PD dementia are therefore needed.
Our focus on hippocampal subfields, and CA1 in particular, is founded on previous autopsy studies that show alpha-synuclein and Lewy-body pathology preferentially affect some regions of the hippocampus more than others. While Lewy-body pathology at autopsy is most prominent in the CA2 subfields (Dickson et al., 1991; Dickson et al., 1994) these regions have been difficult to individually distinguish even at high resolution and have poor inter-study reliability (Yushkevich et al., 2015). Therefore, we concentrated our analysis on the hippocampal subfields where segmentation techniques have been well-validated with consistent inter-rater reliability, namely CA1 and DG/CA3(Kerchner et al., 2014; Kerchner et al., 2013; Kerchner et al., 2012; Kerchner et al., 2010). Of these, only CA1-SP thickness correlated with episodic memory impairment in PD, whereas CA1-SRLM thickness and DG/CA3 area did not. Further, the strength of the memory correlation with CA1-SP was greater than that with DG/CA3. This is consistent with autopsy studies that show Lewy-body pathology is found throughout the cell bodies of CA1(Churchyard and Lees, 1997) and not in DG/CA3. This association suggests Lewy-body pathology in CA1 could be a cause of episodic memory impairments in PD. A recent neuropathological study in Lewy Body Dementia patients also supports this hypothesis, where the severity of Lewy-body pathology in CA1 (but not CA2) predicted pre-mortem episodic memory impairment (Adamowicz et al., 2017). This finding may reflect a sensitive role of CA1 in episodic memory function. Additional studies in PD patients with longitudinal imaging and post-mortem analysis are needed to verify this relationship.
There are several mechanisms by which Lewy-bodies in the hippocampus might disrupt normal memory function. The current study describes structural alteration (thinning) of the CA1-SP presumably due to neuronal loss; however, the presence of alpha-synuclein may also interfere with neurochemical or synaptic signaling within this region. Churchyard and Lees suggested that Lewy neurite formation in CA2 disrupts hippocampal function by interfering with inputs to CA1. In that study, Lewy neurite density in the CA2 subfield of the hippocampus was related to the severity of dementia (Churchyard and Lees, 1997). A more recent study using optical imaging of in vitro mouse hippocampal neurons found that alpha-synuclein accumulation at synaptic membranes of CA1 neurons interfered with synaptic transmission at the level of vesicular recycling (Nemani et al., 2010). Further animal studies showed accumulating alpha-synuclein severely affects hippocampal neurogenesis in a transgenic rat model of PD (Kohl et al., 2016). As with all transgenic PD animal models, it is unclear if these findings hold true in sporadic onset disease in humans. While structural imaging cannot directly probe how these proposed mechanisms cause changes in memory performance, our findings support these transgenic animal models by showing a relationship between human CA1 structure and memory impairment in PD patients. Further studies are required to uncover the downstream sequelae of alpha-synuclein aggregation in the hippocampus.
4.3. Clinical and methodologic considerations
Approximately 80% of PD patients with >10 years disease duration develop dementia (Cholerton et al., 2013; Hely et al., 2008), and the neuropathology underlying dementia in PD is heterogeneous. Specifically, AD-related amyloid plaques and tau tangles frequently co-exists with Lewy-body pathology in PDD patients at autopsy (Emre et al., 2007; Hely et al., 2008). With this in mind, we took several steps to limit the potential confound of dual PD/AD pathology in our study. First, we recruited a cohort where all but one of our PD participants had <10 years disease duration, since younger patients at the earlier stages of disease are less likely to have dual pathology. Second, most of our PD participants (96.5%) were non-demented. Studies have shown that non-demented PD patients less commonly express concomitant AD pathology at autopsy and, when amyloid is present, it typically does not reach a severity to justify a pathological diagnosis of AD (Emre et al., 2007; McKeith et al., 2017). A recent study using amyloid and tau PET imaging in non-demented PD patients, with and without cognitive impairments, found that none of the PD patients had tau PET binding in brain regions reflecting likely AD pathology. The authors concluded that cognitive deficits in non-demented PD do not appear to reflect measurable AD pathology (Winer et al., 2017). Third, we included CSF biomarkers in a subgroup of participants, including all 3 of the participants with dementia, the group the most likely to have dual PD/AD pathology. Therefore, we feel confident that the relationship between CA1-SP thickness and episodic memory in our PD cohort is not due to concomitant AD, but rather a primary feature of PD itself.
Another strength of our study was that we were able to show the correlation between 7T measures of CA1-SP and episodic memory was robust after including other critical clinical predictors of PD cognitive impairment. Indeed, older age and longer duration of motor symptoms, fewer years of education, and male gender, have all been shown to correlate with severity of memory impairments in large PD cohorts (Cholerton et al., 2013). In our correlation analysis, age had a significant relationship with episodic memory and one recent PD study at 3 T showed total hippocampus volume correlated with disease duration when controlling for age (Tanner et al., 2017). In our case, age and duration were not significant in the fully-adjusted regression model and CA1-SP remained a significant predictor of delayed cued memory. Therefore, in mild to moderate PD patients the relationship between CA1-SP thickness and memory appears to be beyond the effects of disease duration and age.
On our 7T high-resolution images we based our thickness estimation procedures on manual tracing of curvilinear CA1-SP ROIs, which could be prone to inter-rater segmentation differences. However, we achieved good inter-rater reliability (ICC = 0.86), comparable to the typical intra-rater ICC range for hippocampal subfields (0.80–0.95).
4.4. Conclusions and future directions
Autopsy studies have recently established a relationship between CA1 Lewy-body pathology and episodic memory impairments in patients with Lewy Body Dementia (Adamowicz et al., 2017); our study demonstrated a similar relationship in patients with PD, many of whom had only mild memory impairments. Further studies investigating whether hippocampal subfield changes are domain-specific or whether they develop before the onset of PD cognitive impairments are warranted. Indeed, the results from our analysis can empower future studies to validate and expand the relationships between hippocampal subfields and cognitive impairments in PD, and ultimately provide clarity to understanding the neuroanatomical underpinnings of this devastating symptom.
Acknowledgments
Acknowledgement
The authors would like to thank Dr. Tony Wyss-Coray and Dr. Eva Czirr for their assistance with the CSF analysis and Sophie YorkWilliams, David Everling, and Anisa Marshall for assistance with participant recruitment and data collection.
Author contributions
C.L. and P.L. contributed equally toward this manuscript, designing the study, analyzing and interpreting the data, and drafting the manuscript. J.D.B., M.A.I.U., B.K.R, and A.D.W. contributed in the design and conceptualization of the study and the analysis and interpretation of the data. M.F., G.K.D. and L.T. contributed to analysis and interpretation of the data. K.L.P., G.A.K., and M.Z. contributed in designing and conceptualizing the study, analyzing and interpreting the data, and drafting and revising the manuscript.
Study funding
This research was supported by grants from the Seiger Family Foundation (K.L.P.), the National Institutes of Health (NS075097-K.L.P.; AG047366-K.L.P.; P41EB015891-B.K.R. and M.M.Z.; S10RR026351-B.K.R. and M.M.Z.), National Institute of Mental Health Predoctoral Fellowship (MH15750 M.F.), the Michael J. Fox Foundation for Parkinson's Research (K.L.P. and G.A.K.), the Center for Biomedical Imaging at Stanford (G.A.K.), and GE Healthcare (B.K.R. and M.M.Z.).
Disclosure
G Kerchner is an employee of Genentech. K Poston is a consultant for Allergan. C La, P Linortner, J Bernstein, M A. I. Ua Cruadhlaoich, M Fenesy, G Deutsch, B Rutt, L Tian, and A Wagner report no disclosures.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nicl.2019.101824.
Appendix A. Supplementary data
Supplementary material
References
- Aarsland D., Bronnick K., Williams-Gray C. Mild cognitive impairment in Parkinson disease: a multicenter pooled analysis. Neurology. 2010;75:1062–1069. doi: 10.1212/WNL.0b013e3181f39d0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamowicz D.H., Roy S., Salmon D.P. Hippocampal alpha-synuclein in dementia with Lewy bodies contributes to memory impairment and is consistent with spread of pathology. J. Neurosci. 2017;37:1675–1684. doi: 10.1523/JNEUROSCI.3047-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard T.P., Malykhin N., Martin W.R. Age and dementia-associated atrophy predominates in the hippocampal head and amygdala in Parkinson's disease. Neurobiol. Aging. 2008;29:1027–1039. doi: 10.1016/j.neurobiolaging.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Braak H., de Vos R.A., Jansen E.N., Bratzke H., Braak E. Neuropathological hallmarks of Alzheimer's and Parkinson's diseases. Prog. Brain Res. 1998;117:267–285. doi: 10.1016/s0079-6123(08)64021-2. [DOI] [PubMed] [Google Scholar]
- Braak H., Bohl J.R., Muller C.M., Rub U., de Vos R.A., Del Tredici K. Stanley Fahn lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov. Disord. 2006;21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- Breen K.C., Drutyte G. Non-motor symptoms of Parkinson's disease: the patient's perspective. J. Neural Transm. (Vienna) 2013;120:531–535. doi: 10.1007/s00702-012-0928-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck A., Kurki T., Kaasinen V., Vahlberg T., Rinne J.O. Hippocampal and prefrontal atrophy in patients with early non-demented Parkinson's disease is related to cognitive impairment. J. Neurol. Neurosurg. Psychiatry. 2004;75:1467–1469. doi: 10.1136/jnnp.2003.031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton E.J., Barber R., Mukaetova-Ladinska E.B. Medial temporal lobe atrophy on MRI differentiates Alzheimer's disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis. Brain. 2009;132:195–203. doi: 10.1093/brain/awn298. [DOI] [PubMed] [Google Scholar]
- Camicioli R., Moore M.M., Kinney A., Corbridge E., Glassberg K., Kaye J.A. Parkinson's disease is associated with hippocampal atrophy. Mov. Disord. 2003;18:784–790. doi: 10.1002/mds.10444. [DOI] [PubMed] [Google Scholar]
- Carr V.A., Bernstein J.D., Favila S.E., Rutt B.K., Kerchner G.A., Wagner A.D. Individual differences in associative memory among older adults explained by hippocampal subfield structure and function. Proc. Natl. Acad. Sci. U. S. A. 2017;114:12075–12080. doi: 10.1073/pnas.1713308114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Hernandez J., Adlard P.A., Finkelstein D.I. Pramipexole restores depressed transmission in the ventral hippocampus following MPTP-lesion. Sci. Rep. 2017;7:44426. doi: 10.1038/srep44426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholerton B.A., Zabetian C.P., Quinn J.F. Pacific Northwest Udall Center of excellence clinical consortium: study design and baseline cohort characteristics. J. Park. Dis. 2013;3:205–214. doi: 10.3233/JPD-130189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchyard A., Lees A.J. The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson's disease. Neurology. 1997;49:1570–1576. doi: 10.1212/wnl.49.6.1570. [DOI] [PubMed] [Google Scholar]
- Costa A., Monaco M., Zabberoni S. Free and cued recall memory in Parkinson's disease associated with amnestic mild cognitive impairment. PLoS One. 2014;9 doi: 10.1371/journal.pone.0086233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T., Hwang J.J., Poston K.L. Episodic recognition memory and the hippocampus in Parkinson's disease: a review. Cortex. 2018;113:191–209. doi: 10.1016/j.cortex.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delis D., Kramer J., Kaplan E., Ober B. The Psychological Corporation; San Antonio, TX: 2000. California Verbal Learning Test – Second Edition Manual. [Google Scholar]
- Dickson D.W., Ruan D., Crystal H. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer's disease: light and electron microscopic immunocytochemistry of CA2-3 neurites specific to DLBD. Neurology. 1991;41:1402–1409. doi: 10.1212/wnl.41.9.1402. [DOI] [PubMed] [Google Scholar]
- Dickson D.W., Schmidt M.L., Lee V.M.-Y., Zhao M.-L., Yen S.-H., Trojanowski J.Q. Immunoreactivity profile of hippocampal CA2/3 neurites in diffuse Lewy body disease. Acta Neuropathol. 1994;87:269–276. doi: 10.1007/BF00296742. [DOI] [PubMed] [Google Scholar]
- Edelstyn N.M., John C.M., Shepherd T.A. Evidence of an amnesia-like cued-recall memory impairment in nondementing idiopathic Parkinson's disease. Cortex. 2015;71:85–101. doi: 10.1016/j.cortex.2015.06.021. [DOI] [PubMed] [Google Scholar]
- Emre M., Aarsland D., Brown R. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov. Disord. 2007;22:1689–1707. doi: 10.1002/mds.21507. [DOI] [PubMed] [Google Scholar]
- Galvin J.E., Uryu K., Lee V.M., Trojanowski J.Q. Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. U. S. A. 1999;96:13450–13455. doi: 10.1073/pnas.96.23.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German D.C., Manaye K., Smith W.K., Woodward D.J., Saper C.B. Midbrain dopaminergic cell loss in Parkinson's disease: computer visualization. Ann. Neurol. 1989;26:507–514. doi: 10.1002/ana.410260403. [DOI] [PubMed] [Google Scholar]
- Goetz C.G., Tilley B.C., Shaftman S.R. Movement Disorder Society-sponsored revision of the unified Parkinson's disease rating scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov. Disord. 2008;23:2129–2170. doi: 10.1002/mds.22340. [DOI] [PubMed] [Google Scholar]
- Hely M.A., Morris J.G., Reid W.G., Trafficante R. Sydney Multicenter study of Parkinson's disease: non-L-dopa-responsive problems dominate at 15 years. Mov. Disord. 2005;20:190–199. doi: 10.1002/mds.20324. [DOI] [PubMed] [Google Scholar]
- Hely M.A., Reid W.G., Adena M.A., Halliday G.M., Morris J.G. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov. Disord. 2008;23:837–844. doi: 10.1002/mds.21956. [DOI] [PubMed] [Google Scholar]
- Junque C., Ramirez-Ruiz B., Tolosa E. Amygdalar and hippocampal MRI volumetric reductions in Parkinson's disease with dementia. Mov. Disord. 2005;20:540–544. doi: 10.1002/mds.20371. [DOI] [PubMed] [Google Scholar]
- Kerchner G.A., Hess C.P., Hammond-Rosenbluth K.E. Hippocampal CA1 apical neuropil atrophy in mild Alzheimer disease visualized with 7-T MRI. Neurology. 2010;75:1381–1387. doi: 10.1212/WNL.0b013e3181f736a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner G.A., Deutsch G.K., Zeineh M., Dougherty R.F., Saranathan M., Rutt B.K. Hippocampal CA1 apical neuropil atrophy and memory performance in Alzheimer's disease. Neuroimage. 2012;63:194–202. doi: 10.1016/j.neuroimage.2012.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner G.A., Bernstein J.D., Fenesy M.C. Shared vulnerability of two synaptically-connected medial temporal lobe areas to age and cognitive decline: a seven tesla magnetic resonance imaging study. J. Neurosci. 2013;33:16666–16672. doi: 10.1523/JNEUROSCI.1915-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner G.A., Berdnik D., Shen J.C. APOE epsilon4 worsens hippocampal CA1 apical neuropil atrophy and episodic memory. Neurology. 2014;82:691–697. doi: 10.1212/WNL.0000000000000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl Z., Ben Abdallah N., Vogelgsang J. Severely impaired hippocampal neurogenesis associates with an early serotonergic deficit in a BAC alpha-synuclein transgenic rat model of Parkinson's disease. Neurobiol. Dis. 2016;85:206–217. doi: 10.1016/j.nbd.2015.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakso M.P., Partanen K., Riekkinen P. Hippocampal volumes in Alzheimer's disease, Parkinson's disease with and without dementia, and in vascular dementia: an MRI study. Neurology. 1996;46:678–681. doi: 10.1212/wnl.46.3.678. [DOI] [PubMed] [Google Scholar]
- Lisman J.E., Grace A.A. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Litvan I., Bhatia K.P., Burn D.J. SIC task force appraisal of clinical diagnostic criteria for parkinsonian disorders. Mov. Disord. 2003;18:467–486. doi: 10.1002/mds.10459. [DOI] [PubMed] [Google Scholar]
- Litvan I., Goldman J.G., Troster A.I. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society task force guidelines. Mov. Disord. 2012;27:349–356. doi: 10.1002/mds.24893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massman P.J., Delis D.C., Butters N., Levin B.E., Salmon D.P. Are all subcortical dementias alike? Verbal learning and memory in Parkinson's and Huntington's disease patients. J. Clin. Exp. Neuropsychol. 1990;12:729–744. doi: 10.1080/01688639008401015. [DOI] [PubMed] [Google Scholar]
- McKeith I.G., Boeve B.F., Dickson D.W. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology. 2017;89:88–100. doi: 10.1212/WNL.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasreddine Z.S., Phillips N.A., Bedirian V. The Montreal cognitive assessment, MoCA: a brief screening tool for mild cognitive impairment. J. Am. Geriatr. Soc. 2005;53:695–699. doi: 10.1111/j.1532-5415.2005.53221.x. [DOI] [PubMed] [Google Scholar]
- Nemani V.M., Lu W., Berge V. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng B., Varoquaux G., Poline J.B., Thirion B., Greicius M.D., Poston K.L. Distinct alterations in Parkinson's medication-state and disease-state connectivity. Neuroimage Clin. 2017;16:575–585. doi: 10.1016/j.nicl.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novellino F., Vasta R., Sarica A. Relationship between hippocampal subfields and category cued recall in AD and PDD: a multimodal MRI study. Neuroscience. 2018;371:506–517. doi: 10.1016/j.neuroscience.2017.12.028. [DOI] [PubMed] [Google Scholar]
- Pirogovsky-Turk E., Filoteo J.V., Litvan I., Harrington D.L. Structural MRI correlates of episodic memory processes in Parkinson's disease without mild cognitive impairment. J. Park. Dis. 2015;5:971–981. doi: 10.3233/JPD-150652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston K.L., YorkWilliams S., Zhang K. Compensatory neural mechanisms in cognitively unimpaired Parkinson disease. Ann. Neurol. 2016;79:448–463. doi: 10.1002/ana.24585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid W.G., Hely M.A., Morris J.G., Loy C., Halliday G.M. Dementia in Parkinson's disease: a 20-year neuropsychological study (Sydney multicentre study) J. Neurol. Neurosurg. Psychiatry. 2011;82:1033–1037. doi: 10.1136/jnnp.2010.232678. [DOI] [PubMed] [Google Scholar]
- Robbins T.W., Cools R. Cognitive deficits in Parkinson's disease: a cognitive neuroscience perspective. Mov. Disord. 2014;29:597–607. doi: 10.1002/mds.25853. [DOI] [PubMed] [Google Scholar]
- Shohamy D., Adcock R.A. Dopamine and adaptive memory. Trends Cogn. Sci. 2010;14:464–472. doi: 10.1016/j.tics.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Shrout P.E., Fleiss J.L. Intraclass correlations: uses in assessing rater reliability. Psychol. Bull. 1979;86:420–428. doi: 10.1037//0033-2909.86.2.420. [DOI] [PubMed] [Google Scholar]
- Tam C.W., Burton E.J., McKeith I.G., Burn D.J., O'Brien J.T. Temporal lobe atrophy on MRI in Parkinson disease with dementia: a comparison with Alzheimer disease and dementia with Lewy bodies. Neurology. 2005;64:861–865. doi: 10.1212/01.WNL.0000153070.82309.D4. [DOI] [PubMed] [Google Scholar]
- Tanner J.J., McFarland N.R., Price C.C. Striatal and hippocampal atrophy in idiopathic Parkinson's disease patients without dementia: a morphometric analysis. Front. Neurol. 2017;8:139. doi: 10.3389/fneur.2017.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub D., Simuni T., Caspell-Garcia C. Cognitive performance and neuropsychiatric symptoms in early, untreated Parkinson's disease. Mov. Disord. 2015;30:919–927. doi: 10.1002/mds.26170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer J.R., Maass A., Pressman P. Associations between tau, beta-amyloid, and cognition in Parkinson disease. JAMA Neurol. 2017;75(2):227–235. doi: 10.1001/jamaneurol.2017.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarnall A.J., Breen D.P., Duncan G.W. Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE-PD study. Neurology. 2014;82:308–316. doi: 10.1212/WNL.0000000000000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushkevich P.A., Amaral R.S., Augustinack J.C. Quantitative comparison of 21 protocols for labeling hippocampal subfields and parahippocampal subregions in in vivo MRI: towards a harmonized segmentation protocol. Neuroimage. 2015;111:526–541. doi: 10.1016/j.neuroimage.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material