

Abstract
The bromodomain‐containing protein 7 (BRD7) is a tumour suppressor protein with critical roles in cell cycle transition and transcriptional regulation. Whether BRD7 is regulated by post‐translational modifications remains poorly understood. Here, we find that chemotherapy‐induced DNA damage leads to the rapid degradation of BRD7 in various cancer cell lines. PARP‐1 binds and poly(ADP)ribosylates BRD7, which enhances its ubiquitination and degradation through the PAR‐binding E3 ubiquitin ligase RNF146. Moreover, the PARP1 inhibitor Olaparib significantly enhances the sensitivity of BRD7‐positive cancer cells to chemotherapeutic drugs, while it has little effect on cells with low BRD7 expression. Taken together, our findings show that PARP1 induces the degradation of BRD7 resulting in cancer cell resistance to DNA‐damaging agents. BRD7 might thus serve as potential biomarker in clinical trial for the prediction of synergistic effects between chemotherapeutic drugs and PARP inhibitors.
Keywords: BRD7, PARP1, PARylation, RNF146, ubiquitination
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Protein post‐translational modifications (PTMs) are known to be essential mechanisms exploited by eukaryotic cells to diversify their protein functions and modulate their cellular signalling networks. Emerging evidence suggests that PTMs play a key role in many cellular processes including protein–protein interactions, protein degradation, gene expression, signal transduction and cell differentiation 1, 2. These modifications including phosphorylation, acetylation, ubiquitination, methylation, glycosylation, oxidation and SUMOylation influence almost all aspects of normal cell biology and pathogenesis. Defect of PTMs leads to human disease including those associated with disorders of cell proliferation, highlighting the importance of PTMs in maintaining normal cellular state 3. Therefore, identifying and understanding PTMs are of biologically and clinically importance.
Poly(ADP‐ribosyl)ation, also termed PARylation, is a unique post‐translational modification of proteins catalysed by ADP‐ribosyltransferases and plays versatile roles in multiple biological processes including chromatin reorganization, DNA damage response, transcriptional regulation, apoptosis and mitosis 4, 5, 6. It causes the formation of poly‐ADP‐ribose (PAR) by transferring the ADP‐ribose moiety from nicotinamide adenine dinucleotide (NAD+) to specific Lys/Glu/Asp/Arg/Cys amino acid residues on substrate proteins 4, 5, 7, 8, 9, 10, 11, 12. Moreover, ADP‐ribosylation is a reversible post‐translational modification, which could be recognized and degraded by ADP‐ribosylhydrolases including Poly(ADP‐ribose) glycohydrolase (PARG), Terminal ADP‐ribose protein glycohydrolase 1(TARG1), ADP‐ribosylhydrolase 1 (ARH1), ARH3, Macro D1 and D2, Nudix‐type‐motif 9 and 16 (NUDT9 and NUDT16) 13, 14, 15, 16, 17. Poly(ADP‐ribose) polymerases (PARPs) composed of 17 members play diverse roles in multiple cellular processes 5, 18. PARP1, the most characterized member of this family, regulates protein–protein interaction, protein stabilization, cellular localization, energy metabolism and cell fate determination through regulation of transcriptional and post‐translational activity of its substrates 19, 20, 21. In response to DNA damage, PARP1 is activated and promotes the formation of poly(ADP‐ribose) polymer (pADPr) on its substrates as well as itself, which provides a signal to recruit DNA double‐strand break proteins to DNA‐damaging site 4, 22. Through modulating histone modification and chromatin structure, PARP1 is also involved in the regulation of transcription process 23, 24, 25, 26, 27. Considering its diverse and important roles, PARP1 inhibitor Olaparib has been used in the treatment of ovarian and breast cancers 28, 29, 30. However, the mechanism that PARP1 inhibitor in combination with DNA‐damaging reagents generates more inhibitory effects is still lacking.
Bromodomain‐containing protein 7 (BRD7) is a member of the family of bromodomain‐containing proteins that contains a single bromodomain involved in multiple cellular processes including cell proliferation, apoptosis and epithelial–mesenchymal transition (EMT) 31, 32, 33. BRD7 has been reported to interact with p53 and is required for p53‐dependent replicative senescence 34, 35. Moreover, as a component of chromatin remodelling of SWI/SNF complex, BRD7 has also been shown to function as either a transcriptional co‐activator or co‐repressor 34, 35, 36, 37. For instance, BRD7 serves as a transcriptional co‐activator through association with tumour suppressors BRCA1 and p53 to regulate downstream gene transcription 34, 35, 37. Conversely, BRD7 can interact with PRMT5 and PRC2 to be involved in transcriptional repression of their target genes 38. BRD7 also plays a critical role in regulation of endoplasmic reticulum (ER) stress and glucose metabolism via association with the p85 regulatory subunit of PI3K, resulting in suppression of PI3K signalling 39, 40. Inactivation or mutation of BRD7 increases sensitivity of tumour cell to interferon‐γ, resulting in elevated sensitivity of cancer cells to PD‐1 blockade, as well as, other forms of immunotherapy treatment 41.
In this study, we identified BRD7 as a novel PARP1‐binding protein and demonstrated that PARP1 directly ribosylated BRD7 and promoted BRD7 degradation, mediated by the E3 ubiquitin ligase RNF146, further enhancing survival of cancer cells. Furthermore, inhibition of PARP1 suppressed cell proliferation and promoted sensitivity of cancer cells to DNA damage chemotherapy through reduction of BRD7 PARylation. Therefore, our study uncovered a novel mechanism that PARP1 modulated resistance to DNA‐damaging agents of cancer cells by promoting PARylation of BRD7, providing new insights into the molecular rationale for combination of chemotherapeutic drugs and PARP inhibitors in clinical treatment, and suggested that BRD7 may be served as a potential biomarker for predicting the synergistic effects of combining chemotherapeutic drugs and PARP inhibitors in clinical trial.
Results
Drug‐ or irradiation‐induced DNA damage promoted BRD7 protein degradation
To study the effect of BRD7 on the sensitivity of genotoxic drug in cancer cell, BRD7 protein levels were examined using cells resistant to either cisplatin (DDP), doxorubicin (ADR) or irradiation (IR). BRD7 protein levels significantly decreased in these cells (Fig 1A). Interestingly, quantitative real‐time PCR data showed that mRNA level of BRD7 was not altered between wild‐type and ADR or IR‐resistant cells (Fig 1B). Notably, data from GEO database also revealed that mRNA level of BRD7 was slightly increased in MCF7/ADR‐resistant cells compared with MCF7 normal cells (Appendix Fig S1). To confirm further these findings, cells were treated with either ADR or CPT for different time intervals. Both ADR and CPT treatments in a time‐dependent manner caused a reduction of BRD7 at the protein levels in MDA‐MB‐468 and MDA‐MB‐231 cell lines (Fig 1C and D). To determine whether downregulation of endogenous BRD7 was not induced by cell death in response to DNA damage, we performed cell death detection assay measured by Fixable Viability Dye eFluor® 455UV reagent (eBioscience) according to manufacturer's instructions. As shown in Appendix Fig S2B–F, there was no dramatic increase in cell death within the treatment of indicated drugs and the ratio of dead cells was very low no more than 2% of total cells in both MDA‐MB‐231 and MDA‐MB‐468 cells. Consistent with the Western blot results, BRD7 fluorescence intensity was diminished dramatically in response to CPT treatment (Fig 1E and F, Appendix Fig S2A). Taken together, these findings suggested that BRD7 was involved in drug‐ and IR‐induced DNA damage and was downregulated at the post‐translational level rather than post‐transcriptionally.
Figure 1. Chemotherapeutic drugs and irradiation deplete BRD7.

-
AWestern blot analysis of BRD7 and γH2AX protein levels in breast cancer cell lines: MCF7 parental or doxorubicin‐resistant MCF7/ADR (ADR, 0.5 μg/ml) and irradiation‐resistant MCF7/IR (IR, 10 Gy), MDA‐MB‐231 parental or doxorubicin‐resistant MDA‐MB‐231/ADR (ADR, 0.5 μg/ml) or cisplatin‐resistant MDA‐MB‐231/DDP (1 μg/ml), non‐small lung cancer cell lines: A549 parental or cisplatin‐resistant A549/DDP (1 μg/ml).
-
BMCF7, MCF7/ADR and MCF7/IR, MDA‐MB‐231 and MDA‐MB‐231/ADR cells were harvested and mRNA levels of BRD7 were determined by real‐time PCR (n = 3). Values are mean ± SEM.
-
C, DWestern blot analysis of BRD7 protein levels in MDA‐MB‐468 and MDA‐MB‐231 cell after treatment with ADR (5 μM) or camptothecin (CPT) (1 μM) for different intervals (n = 3).
-
ERepresentative images of endogenous BRD7 (green) and γH2AX foci (red) in paraformaldehyde‐fixed MDA‐MB‐468 cells after treatment with CPT (1 μM) for different intervals. Visualized by immunofluorescence using anti‐BRD7 and Alexa Fluor 555 anti‐γH2AX antibodies. DNA staining with DAPI; Scale bars, 2 μm.
-
FQuantification of average fluorescence intensity of BRD7 of cells in (E). Error bars indicate SEM; n > 100.
Source data are available online for this figure.
BRD7 interacted with PARP1
To identify proteins that interact with BRD7 in response to DNA damage, proteomic analysis was performed. SFB‐tagged (S‐protein, Flag and streptavidin‐binding peptide) BRD7 was stably expressed in HEK293T cell. After tandem affinity purification (TAP), proteins associated with BRD7 were subjected to silver staining and mass spectrometry (Fig 2A). In addition to known BRD7‐binding proteins, such as TP53 34 and PIK3R 40, we identified PARP1 as a novel binding partner of BRD7 (Fig 2B, Dataset EV1). PARP1 is a key enzyme involved in DNA damage repair and cell survival 42. To confirm the interaction between BRD7 and PARP1, we performed transient transfection and co‐IP experiments. As shown in Fig 2C and D, a complex containing BRD7 and PARP1 was clearly detected using either Flag agarose or Myc agarose in HEK293T cells expressing both Flag‐tagged PARP1 and Myc‐tagged BRD7. To examine further the interaction between endogenous BRD7 and PARP1, HeLa and MDA‐MB‐231 whole‐cell extracts were prepared and subjected to immunoprecipitation assays in the presence of either control IgG, anti‐BRD7 or anti‐PARP1 antibody. Both BRD7 and PARP1 were clearly detected in the immunoprecipitated complex (Fig 2E and F). To further determine whether the interaction of BRD7 with PARP1 dependent on its poly‐ADP‐ribose (PAR) chains, we detected the association of BRD7 to PARP1 with or without PARP1 inhibitor Olaparib. As shown in Fig 2G, there was significant inhibition of the interaction of PARP1 with BRD7 upon Olaparib treatment, indicating that the PARylation of PARP1 is required to interact with BRD7. Most importantly, the interaction between BRD7 and PARP1 was greatly enhanced following exposure to CPT (Fig 2H and I). These data together suggest that BRD7 is a PAR‐binding protein.
Figure 2. BRD7 interacts with PARP1.
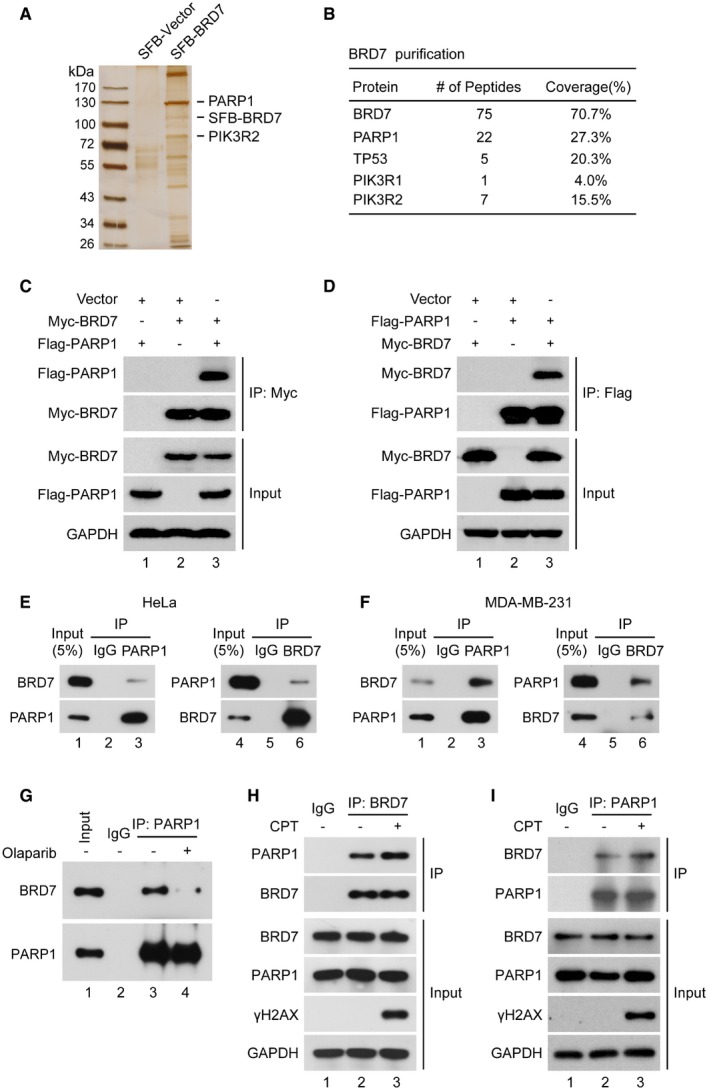
-
ASilver staining of the BRD7 complex separated by SDS–PAGE. HEK293T cells stably expressing SFB‐tagged BRD7 were used for tandem affinity purification (TAP) of protein complexes. BRD7‐interacting proteins, including PARP1 and PIK3R2, are indicated.
-
BTable summarizes proteins identified by mass spectrometry analysis.
-
C, DHEK293T cells transiently transfected with Flag‐PARP1 and Myc‐BRD7 for 24 h were lysed with RIPA buffer. Followed by immunoprecipitation (IP) using antibodies to either Myc (C) or Flag (D) conjugated to agarose followed by Western blot with the indicated antibodies (n = 3).
-
E, FHeLa and MDA‐MB‐231 cells were lysed with RIPA buffer, and lysates were subjected to immunoprecipitation using either anti‐IgG, or BRD7 or PARP1 antibodies, and analysed by Western blot (n = 3).
-
GMDA‐MB‐231 cells were treated first with Olaparib (10 μM) for 6 h and lysed with RIPA buffer, and lysates were subjected to immunoprecipitation using either anti‐IgG or PARP1 antibodies, and analysed by Western blot (n = 3).
-
H, IAssociation of endogenous BRD7 with PARP1 in HeLa cells was performed by co‐immunoprecipitation using anti‐BRD7 or anti‐PARP1 antibody. HeLa cell was treated with CPT (1 μM, 1 h), followed by IP using indicated antibodies, and Western blot was performed. γH2AX was used as a marker of DNA damage induced by CPT (n = 3).
Source data are available online for this figure.
Next, a serial of deletion mutants was generated to map the region of interaction of BRD7 and PARP1. PARP1 contains a DNA‐binding domain, a BRCA1 C terminus, a tryptophan–glycine–arginine domain (WGR) and a catalytic domain (CA) (Appendix Fig S3A). As shown in Appendix Fig S3B, both the N‐terminal DNA‐binding/automodification domain and C‐terminal catalytic domain of PARP1 were capable of binding BRD7. BRD7 has a conserved bromodomain (BD, 128–238 amino acids) which specifically binds to acetylated lysines on histones, and an uncharacterized conserved domain (361–651 amino acids) (Appendix Fig S3C). Deletion of individual regions of BRD7 demonstrated that the N‐terminal bromodomain was not required for BRD7 binding to PARP1, but depletion of 361–651 domain significantly decreased the binding ability to PARP1 (Appendix Fig S3D). These results reinforce the hypothesis that BRD7 physically and specifically interacts with PARP1.
BRD7 was ADP‐ribosylated by PARP1 in vitro and in vivo
Function of PARP1 is attaching the pADPr chain to specific glutamate, aspartate, arginine, lysine or cysteine residues of target proteins 4, 5, 7, 8, 9, 10, 11, 12. We tested if BRD7 is ribosylated by PARP1 directly. First, we used anti‐PAR antibody to immunoprecipitate the PARylated proteins in the absence and presence of CPT. As a positive control, we detected increased ribosylated PARP1 in response to CPT (Fig 3A). As expected, PARsylation of BRD7 was also greatly increased after 3 h CPT exposure (Fig 3A). Second, we used anti‐BRD7 antibody to immunoprecipitate BRD7 followed by immunoblotting for PAR and further confirmed that BRD7 is ribosylated in vivo (Fig 3B). Moreover, to rule out the possibility of indirect binding of BRD7 to PARylated proteins, we performed a denaturing immunoprecipitation using either anti‐BRD7 antibody or anti‐PAR antibody. As shown in Appendix Fig S4A and B, a clear band of PARylated BRD7 was detected and suggested that BRD7 is covalently modified by poly‐ADP‐ribose (PAR) in vivo. To determine whether exogenous BRD7 could also be ribosylated, we used anti‐myc agarose to immunoprecipitate myc‐tagged BRD7. Consistently, exogenous BRD7 was also ribosylated, and the ribosylation could be enhanced in the cells treated with either CPT or ADR (Fig 3C and D). Next, we examined if BRD7 could be ribosylated by PARP1 in vivo. As shown in Fig 3E, depletion of PARP1 profoundly decreased BRD7 ribosylation levels in vivo. Moreover, we identified nine ADP‐ribosylation sites between aspartic acid and glutamic acid resides on BRD7 by mass spectrometric analysis (Fig EV1 and Dataset EV2). Hence, a novel post‐translational modification of BRD7 has been discovered.
Figure 3. BRD7 is ADP‐ribosylated by PARP1 in vitro and in vivo.

- HeLa cells were untreated or treated with CPT (1 μM) for 1 h followed by lysing with RIPA buffer, and lysates were then immunoprecipitated using anti‐IgG or anti‐PAR antibodies and immunoblotted with the indicated antibodies (n = 3).
- HeLa cells were untreated or treated with CPT (1 μM) for 1 h, and cellular lysates were immunoprecipitated using anti‐IgG or anti‐BRD7 antibodies and immunoblotted using the indicated antibodies (n = 3).
- HeLa and 293T cells transfected with Myc‐BRD7 plasmid for 24 h were lysed with RIPA buffer. Lysates were then immunoprecipitated using anti‐Myc agarose and immunoblotted using the indicated antibodies. Ribosylation levels of exogenous BRD7 were detected using anti‐PAR antibody (n = 3).
- HeLa cells transfected with Myc‐BRD7 plasmid. After 24 h, cells were treated with either CPT (1 μM) or ADR (5 μM) combined with MG132 (10 μM) for indicated times. Cellular lysates were immunoprecipitated using anti‐Myc agarose and immunoblotted using the indicated antibodies (n = 3).
- HeLa PARP1 wild‐type and PARP1 knockout cells were transfected with Myc‐BRD7 for 24 h, and lysates were subjected to immunoprecipitation using anti‐Myc agarose and analysed by Western blot (n = 3).
- HeLa was transfected with BRD7 wild‐type and various BRD7‐mutant plasmids for 24 h, lysed with RIPA, followed by anti‐Myc IP and Western blot with indicated antibody (n = 3).
- Ribosylation of BRD7 by PARP1 in vitro. Recombinant BRD7 was subjected to in vitro ribosylation either in absence or presence of biotin‐labelled NAD+. Recombinant proteins were detected by indicated antibodies, and ribosylated proteins were determined with anti‐biotin antibody (n = 3).
- PAR‐binding motif of BRD7 is required for its ribosylation by PARP1. Recombinant Myc‐BRD7‐WT and Myc‐BRD7‐mutant were subjected to in vitro ribosylation assay and analysed by Western blot as indicated (n = 3).
Source data are available online for this figure.
Figure EV1. The PARylation of BRD7 was analysed by mass spectrometry.

- HeLa cells stably expressing SFB‐BRD7 were treated with 10 μM of PARG inhibitor PDD0017273 for 6 h, and SFB‐BRD7 was pulled down by Flag beads from cell lysates and subjected to mass spectrometry analysis.
- BRD7 PARylation was analysed by mass spectrometry.
- Distribution of ADP‐ribosylation sites of BRD7 between aspartic acid (Asp) and glutamic acid (Glu) resides. Asp‐ and Glu‐ADP‐ribosylation is susceptible to NH2OH attack, generating a hydroxamic acid (H) derivative, allowing easy localization of the ADP‐ribosylation sites. The site of modification is indicated by an H.m.
The above data demonstrated in vivo PAR‐binding activity of BRD7 prompted us to search for potential PAR‐binding motif in BRD7 (Fig 2G). PAR‐binding proteins commonly contain a conserved PAR‐binding motif, consisting of eight amino acids [HKR]‐X‐X‐[AIQVY]‐[KR]‐[KR]‐[AILV]‐[FILPV] 43. Through sequence alignment, we identified three highly conserved residues 222Lys/223Lys, 545Arg/546Lys and 613Arg/614Lys in the BRD7 protein as potential PAR‐binding motifs (Appendix Fig S4C and D). To investigate whether the interaction of BRD7 against PARP1 and subsequent ribosylation of BRD7 through its potential PAR‐binding motif, co‐IP was performed. Unlike wild‐type BRD7, each mutant of these three candidate sites decreased the binding affinity for PARP1 and subsequent suppression of its ribosylation, and 613Arg/614Lys may be the major PAR‐binding motif responsible for association of BRD7 with PARP1 (Fig 3F).
To verify further that whether BRD7 could be ribosylated by PARP1 in vitro, an in vitro ribosylation assay was performed using recombinant BRD7 and biotin‐labelled NAD+. As shown in Fig 3G, PARP1 ribosylated not only itself but also BRD7. Indeed, we generated 6A point mutations (222/223/545/546/613/614A) on the PAR‐binding motif of BRD7 (BRD7‐6A) and the ribosylation on BRD7‐6A by PARP1 was significantly inhibited both in vivo and in vitro (Fig 3H and Appendix Fig S4C), caused by dramatic reduction of binding affinity for BRD7 against PARP1. Taken together, BRD7 binds PARylated PARP1 non‐covalently via its PAR‐binding motifs and this interaction enables PARP1 to covalently PARylate BRD7 on one (or more) of the sites identified in the mass spec experiment, but PARP1 does not PARylate BRD7's PAR‐binding motif. These data suggest that BRD7 is a bona fide substrate of PARP1.
PARP1 regulated BRD7 degradation and ubiquitination
Poly(ADP‐ribosyl)ation, as a crucial post‐translational modification, has been reported to regulate protein stabilization, cellular localization and transcriptional activity 44. Therefore, we tested whether PARP1 contributed to BRD7 degradation. As shown in Fig 4A, overexpression of PARP1 markedly promoted degradation of BRD7 protein. MG132, a specific proteasome inhibitor, substantially rescued the decline of BRD7 protein caused by overexpression of PARP1 in HeLa cells. Moreover, four different siRNA duplexes specifically targeting different PARP1 coding regions markedly increased BRD7 protein levels (Fig 4B). This negative regulation of BRD7 by PARP1 did not occur at the transcriptional level, as the mRNA level of BRD7 was not altered with silencing PARP1 in U2OS cells (Appendix Fig S5B). Moreover, this negative correlation between BRD7 and PARP1 was noted in multiple cancer cell lines including MDA‐MB‐231 cells (Appendix Fig S5A).
Figure 4. PARP1 regulates BRD7 protein stability through ubiquitination‐dependent pathway.

-
AHeLa cells were transfected with Flag‐PARP1 and Myc‐BRD7 for 24 h, then treated with MG132 (10 μM) for additional 4 h, and proteins were detected by Western blot with indicated antibodies (n = 3).
-
BHeLa cells were transfected with either scrambled or PARP1 siRNAs for 48 h, and protein levels were detected by Western blot with the indicated antibodies (n = 3).
-
CExpression of BRD7, p21 and PARP1 measured in PARP1 wild‐type and knockout HeLa cells using PARP1 sgRNA by Western blot (n = 3).
-
DMDA‐MB‐231 cell was treated with different concentration of PARG inhibitor PDD0017273 for 48 h, and cell lysates were analysed by Western blot (n = 3).
-
EMDA‐MB‐231 cell was treated with 10 μM of PDD0017273 for indicated times, and cell lysates were analysed by Western blot (n = 3).
-
F, GWild‐type and PARP1 knockout HeLa cells were incubated with 10 μg/ml cycloheximide (CHX) for the indicated times. Lysates were harvested and analysed by Western blot. Quantification of BRD7 protein is shown in (G) (n = 3), and results represent mean ± SEM. Relative amounts normalized to the BRD7 protein level at 0 h of Control or sgPARP1 cells, respectively.
-
HPARP1 wild‐type and knockout HeLa cells were transfected with HA‐ubiquitin for 24 h; MG132 (10 μM) was added for an additional 4 h, and cells were lysed with RIPA buffer, followed by anti‐BRD7 IP and analysed by Western blot with the indicated antibodies (n = 3).
-
ISFB‐BRD7 stably overexpressing HeLa cells transfected with scramble or PARP1 siRNA for 24 h were transfected with vector or HA‐ubiquitin (Lys48 or Lys63 only) for 24 h, and MG132 (10 μM) was added for an additional 4 h, lysed with RIPA, subjected to IP using S tag beads followed by Western blot (n = 3).
-
JHeLa cells were co‐transfected either wild‐type or BRD7 mutant with Flag‐PARP1 for 24 h and analysed by Western blot (n = 3).
-
KHeLa cells were co‐transfected with either wild‐type or BRD7 mutant plus HA‐ubiquitin for 24 h, and MG132 (10 μM) was added for an additional 4 h and lysed with RIPA buffer, followed by anti‐Myc agarose IP and analysed by Western blot with the indicated antibodies (n = 3).
Source data are available online for this figure.
To confirm further these results, CRISPR/Cas9 gene‐editing was used to generate a HeLa cell line specifically defective in PARP1 (Fig 4C). Similar with the siRNA‐treated HeLa cells, either depletion or inhibition of PARP1 led to BRD7 stabilization (Fig 4C, Appendix Fig S5D). PARP2 is a conserved homolog of PARP1, silencing PARP2 with siRNA did not change levels of BRD7 protein (Appendix Fig S5C). Consistently, PARG inhibitor PDD00017273, an inhibitor of poly(ADP‐ribose) glycohydrolase (PARG) which catalyses the degradation of the poly(ADP)ribose chain (an inverse step of poly‐ADP‐ribosylation) 45, 46, markedly decreased the protein level of BRD7 in a concentration or time‐dependent manner (Fig 4D and E). Consistent with this notion, the half‐life of endogenous and exogenous BRD7 protein became longer in PARP1 knockout cells in the presence of cycloheximide, an inhibitor of protein synthesis (Fig 4F and G, Appendix Fig S5E and F). Collectively, these results indicate that PARP1 negatively regulates BRD7 stability through the proteasome‐dependent pathway.
Ubiquitination‐mediated proteasomal degradation of proteins is a most common mechanism that regulates protein stability. Ribosylated proteins lead to their degradation via a proteasome‐dependent pathway 47, 48, 49, 50, 51. Here, we examined whether PARP1‐mediated degradation of BRD7 occurs through poly‐ubiquitination. As shown in Fig 4H, less ubiquitination of BRD7 was observed in PARP1‐depleted cells compared to PARP1‐wild‐type cells, indicating that PARP1 may enhance BRD7 polyubiquitylation and promote it ubiquitin‐proteasome‐dependent degradation. In general, polyubiquitylation via either K48 or K11 commits the substrate to degradation by the 26S proteasome, whereas either monoubiquitylation or K63‐linked polyubiquitylation does not accelerate protein degradation 52. To elucidate the styles of Ub chain linkages of BRD7, in vivo ubiquitination assay was performed and showed that K48‐linked polyubiquitylation of BRD7 decreased and K63‐linked polyubiquitylation was little change in PARP1‐depleted cells (Fig 4I). In addition, overexpression of PARP1 led to decreased levels of wild‐type BRD7 but not BRD7‐6A mutant (mutant of three PAR‐binding motif of BRD7) (Fig 4J). Consistent with this notion, half‐life of BRD7‐mutant protein increased in HeLa cells (Appendix Fig S5G and H). An in vivo ubiquitination assay showed that PARP1 can efficiently promote ubiquitination of wild‐type BRD7 but not the BRD7 mutant (Fig 4K). Taken together, these results indicate that PARP1 is involved in regulation of ubiquitylation and degradation of BRD7.
E3 ligase RNF146 promotes ubiquitination and degradation of BRD7
Recent studies reported that the E3 ligase RNF146 was responsible for interacting with PARsylated proteins and promoting their degradation 47, 53, 54, 55. Moreover, RNF146 has been shown to translocate from cytoplasm to nucleus in response to laser‐induced damage, and further bind and ubiquitinate both PARsylated and PAR‐binding proteins, promoting these proteins for ubiquitin proteasomal degradation 56.
We tested whether RNF146 can also act as an E3 ubiquitin ligase for BRD7. First, depletion of RNF146 led to BRD7 stabilization (Fig 5A). Half‐life of endogenous BRD7 protein increased after silencing RNF146 in HeLa cells in the presence of cycloheximide (Fig 5B and C). As expected, BRD7 became more unstable in RNF146 overexpressing cells (Fig 5D and E). Knockdown of RNF146 significantly inhibited endogenous BRD7 ubiquitination levels (Fig 5F). Similarly, the poly‐ubiquitination level of BRD7 was upregulated dramatically by addition of exogenous RNF146 (Fig 5G). Collectively, these results indicated that RNF146 is an E3 ligase that promotes BRD7 poly‐ubiquitination and degradation via a proteasome‐dependent pathway.
Figure 5. E3 ligase RNF146 promotes BRD7 ubiquitination and degradation.

- HeLa and MDA‐MB‐231 cells were transfected with either scrambled or RNF146 siRNAs (48 h). Protein levels were detected by Western blot. PTEN, which is a known RNF146‐interacting protein, was included as a positive control (n = 3).
- HeLa cells were transfected with either scrambled or RNF146 siRNAs for 48 h, followed by incubation with 10 μg/ml cycloheximide (CHX) for the indicated periods of time. Lysates were harvested and analysed by Western blot (n = 3).
- Quantification of BRD7 protein levels from (B), n = 3. Error bars indicate SEM. Relative amounts normalized to the BRD7 protein level at 0 h.
- Control and RNF146 stably overexpressing HeLa cells were transfected with Myc‐BRD7‐WT for 24 h, followed by incubation with 10 μg/ml cycloheximide (CHX) for indicated periods of time. Lysates were subjected to Western blot analysis (n = 3).
- Quantification of BRD7 protein levels from (D), n = 3. Error bars indicate SEM. Relative amounts normalized to the BRD7 protein level at 0 h.
- HeLa cells transfected with either RNF146 or control siRNA for 24 h followed by transfection of HA‐ubiquitin for another 24 h; MG132 (10 μM) was added for 4 h and lysed with RIPA, followed by anti‐BRD7 IP and analysed by Western blot with the indicated antibodies (n = 3).
- HeLa cells stably expressing RNF146 were transfected with HA‐ubiquitin for 24 h; MG132 (10 μM) was added for an additional 4 h, followed by anti‐BRD7 IP and Western blot (n = 3).
Source data are available online for this figure.
The E3 ligase RNF146 interacted with BRD7 depending on PARP1‐mediated ribosylation of BRD7
A complex containing RNF146 and BRD7 was clearly detected in HEK293T cells containing exogenous RNF146 and BRD7 (Fig 6A and B) using co‐IP assay. To confirm this interaction, an endogenous complex containing RNF146 and BRD7 was also detected (Fig 6C). Interestingly, interaction between RNF146 and BRD7 was significantly enhanced with CPT treatment (Fig 6D). Knockout of PARP1 significantly inhibited the interaction of BRD7‐RNF146 (Fig 6E). Mutant BRD7 without of ribosylation failed to interact with RNF146 (Fig 6F). These results suggested that PARP1‐mediated ribosylation was required for the interaction between BRD7 and RNF146.
Figure 6. E3 ligase RNF146 interacted with BRD7 in a PARP1‐dependent manner.

-
A, BHeLa cells transfected with the indicated plasmids; after 24 h, cells were lysed with RIPA buffer followed by immunoprecipitation (IP) using either anti‐Myc or anti‐Flag agarose and Western blot with indicated antibody (n = 3).
-
CHeLa and MDA‐MB‐231 cells were lysed with RIPA buffer, and lysates were subjected to IP using either anti‐IgG, anti‐BRD7 or anti‐RNF146 antibodies followed by analysis with Western blot (n = 3).
-
DAssociation of endogenous BRD7 with RNF146 in HeLa cells was performed by co‐IP using anti‐RNF146 antibody. Cell lysates were incubated with protein G agarose beads conjugated with indicated antibodies, and Western blot was performed (n = 3).
-
EHeLa cells with wild type or PARP1 knockout were transfected with Myc‐BRD7‐WT for 24 h, and their lysates were subjected to IP using anti‐Myc agarose and analysed by Western blot with indicated antibodies (n = 3).
-
FHeLa cells stably expressing Flag‐RNF146 were transfected with either Myc‐BRD7‐WT or Myc‐BRD7‐mutant for 24 h, and lysates were subjected to IP using anti‐Flag agarose and analysed by Western blot with indicated antibodies (n = 3).
Source data are available online for this figure.
According to the protein structural analysis, RNF146 contains a PAR recognition domain (WWE) and an E3 ligase activity domain (RING). Deletions of these two domain were generated (RNF146‐ΔWWE and RNF146‐ΔRING) (Appendix Fig S6A). We observed that wild‐type RNF146 and the RNF146‐ΔRING mutant associated with BRD7, but the RNF146‐ΔWWE deletion mutant failed to do so (Appendix Fig S6B), indicating that the PAR recognition domain (WWE) of RNF146 was required for the interaction of RNF146 with BRD7. To illustrate further the interaction between BRD7 and RNF146, the domain of BRD7 critical for the interaction with RNF146 was determined (Appendix Fig S6C). As shown in Appendix Fig S6D, RNF146 interacted with the N‐terminal part of BRD7 bromodomain (residues 128–238) and the C‐terminal part of BRD7 (residues 361–651), but not the N‐terminal region of BRD7 (residues 1–128).
To determine the effect of the PAR‐binding motif of BRD7 on RNF146‐mediated BRD7 ubiquitination and degradation, we transfected wild‐type BRD7 or the indicated BRD7‐mutant plasmids into HeLa cells stably expressing SFB‐RNF146. As indicated in Appendix Fig S6E, wild‐type BRD7 associated with RNF146 obviously, mutants of 613Arg/614Lys greatly prevent BRD7‐RNF146 interaction, in consistent with previous data that R613/K614 may be the major PAR‐binding motifs. Similarly, in vivo ubiquitination assays revealed that RNF146 triggered the ubiquitination of wild‐type BRD7 significantly but not R613/K614 mutant (Appendix Fig S6F), again supporting the notion that RNF146‐mediated BRD7 ubiquitination and degradation depend on the ribosylation of BRD7.
PARylation of BRD7 mediated by PARP1 conferred resistance of tumour cells to chemotherapy
Accumulating evidence demonstrates that BRD7 plays vital roles in cell proliferation and tumorigenesis through modulating crucial signalling pathway, such as Ras/Raf/MEK/ERK and PI3K/AKT signalling 40, 57, 58, 59, 60. Thus, we investigated if PARP1 can regulate cell survival through BRD7. Indeed, decrease levels of BRD7 increased cell proliferation of MDA‐MB‐231 cells, but silencing PARP1 in BRD7‐depleted cells did not significantly reduce proliferation (Fig 7A and B), suggesting that PARP1 controlled cell proliferation at least in part in a BRD7‐dependent manner.
Figure 7. Inhibiting PARylation of BRD7 enhanced sensitivity of cancer cells to DNA‐damaging agents.

- MDA‐MB‐231 cells were transfected twice with indicated siRNAs, and the absorbance values of indicated cell lines were measured at different time points by CCK8 assay (n = 3).
- Cell lysates treated in (A) were analysed by Western blot with indicated antibodies (n = 3).
- MDA‐MB‐231 cells were treated with Olaparib (10 μM) combined with ADR (0.5 μM), CPT (0.5 μM) or VP16 (10 μM) for 72 h, and CCK8 activity of the cells was detected (n = 3).
- MDA‐MB‐231 cells were treated with Olaparib (10 μM) combined with ADR (0.5 μM) for 72 h, and their lysates were subjected to IP using anti‐BRD7 antibodies and analysed by Western blot with indicated antibodies (n = 3).
- MDA‐MB‐231 cells were transfected twice with BRD7 siRNAs, and these cells were subjected to CCK8 assay after treatment with Olaparib (10 μM) combined with either ADR (0.5 μM) or VP16 (10 μM) for 72 h (n = 3).
- Resistance of BRD7‐depleted cells to Olaparib was reversed by the expression of wild‐type BRD7 but not mutant BRD7. shRNA‐resistant BRD7 wild‐type and BRD7‐mutant plasmids were transduced into BRD7‐depleted MDA‐MB‐231 cells, and cell viability was determined as indicated above (n = 3).
Olaparib is a widely used PARP1 inhibitor which effectively blocks PARP1 catalytic activity 45. The sensitivity of cancer cells to chemotherapy, including VP16, CPT and ADR, was significantly increased by PARP1 inhibitor Olaparib (Fig 7C and Appendix Fig S7A). Western blot analysis further confirmed that DNA damage drug combined with Olaparib could reverse the BRD7 stabilization caused by downregulation of BRD7 ribosylation (Fig 7D). However, its effect was greatly compromised after depletion of BRD7 (Fig 7E and Appendix Fig S7B). Moreover, rescue experiments using shRNA‐resistant wild‐type and mutant BRD7 into BRD7‐depleted cells reversed cell viability to a normal level after treatment with ADR alone; however, combining Olaparib with ADR treatment suppressed cell viability only in cells expressing wild‐type BRD7 but not those expressing the mutant BRD7 (Fig 7F and Appendix Fig S7C). Similar results were obtained by measuring the protein levels of BRD7 (Appendix Fig S7D). Collectively, these results indicate that PARP1 inhibition may be effective only in treating tumour cells expressing wild‐type BRD7.
Discussion
Accumulating evidence suggests BRD7 functions in regulating cell proliferation, apoptosis, oncogene‐induced senescence and epithelial–mesenchymal transition 33, 34, 58, 61, 62, 63. BRD7 behaves as a potent tumour suppressor in a number of human cancers 34, 35, 40, 61, 63, 64. BRD7 has been documented to inhibit cell growth and induce premature senescence through various mechanisms 33, 35. Indeed, BRD7 regulates cell cycle inhibitor p21 in response to both p53 and Smad2/3/4, suggesting that BRD7 may interact with multiple transcription factor 33, 34, 35. For instance, BRD7 is recruited to the promoter regions of p53 target genes to mediate p53‐dependent oncogene‐induced senescence and cell cycle arrest. Moreover, BRD7 forms a complex with the p85α, a regulatory subunit of p85 family, and induces nuclear translocation of p85α, thereby attenuating PI3K signalling, suggesting that the BRD7‐p85α complex is distinct from the PBAF complex 40. Notably, BRD7 forms a complex with Smad3/4 to potentiate TGF‐β induced transcriptional responses independent of the cellular p53 status, indicating that the BRD7/p53 complex and BRD7/Smad complex are distinct. Taken together, these data indicate that BRD7 has alternative mechanisms contributing to the inhibition of cell proliferation.
In this study, we demonstrated that BRD7 was a novel PARP1 substrate containing three conserved PAR‐binding motifs, which could promote PARsylation of BRD7 by PARP1 in vitro and in vivo (Figs 2, 3, 4), consistent with recent finding by Gibson et al 18. We noted that ribosylated BRD7 was recognized by the PAR‐binding E3 ligase RNF146 which poly‐ubiquitinated BRD7 leading to its degradation (Figs 5 and 6). Importantly, targeting PARP1 with Olaparib significantly enhanced sensitivity of cancer cells to chemotherapy only in cells expressing wild‐type BRD7 but not those BRD7‐depleted cells or expressing the mutant BRD7 (Fig 7).
PARP1 is an abundant nuclear protein with weak enzyme activity that can be activated by DNA damage, reactive oxygen species and inflammatory signals. The activation of PARP1 promotes the poly(ADP‐ribosyl)ation of its target proteins, which leads to the modulation of stability of target proteins, as well as localization and formation of new protein interaction scaffolds 7, 8. Our data show that PARP1 regulates the suppressive function of cancer cells through BRD7 poly(ADP‐ribosyl)ation (Figs 2, 3, 4 and 7). However, as shown in Fig 3E, poly(ADP‐ribosyl)ation of BRD7 still occurred in PARP1 knockout cells, suggesting that other PARP(s) may also be involved in poly(ADP‐ribosyl)ation of BRD7. Tankyrase 1 (TNKS1/PARP5a) and Tankyrase 2 (TNKS2/PARP5b) are members of the poly(ADP‐ribose) polymerase (PARP) family with diverse functions 49, 65, 66. Indeed, previous studies of tankyrases showed it regulated cell proliferation by targeting axin for degradation in an ubiquitin‐proteasome‐dependent manner, thus suppressing the WNT signalling pathway 48, 49. Interestingly, we found that BRD7 contained a highly conserved motif similar to the RXXXDG binding motif (RRKPDG) of tankyrase, and our preliminary co‐IP results showed that TNKS1 and TNKS2 were associated with BRD7 (data not showed), indicating that tankyrases may be important for function and regulation of BRD7. Further studies will be needed to elucidate the relationship between these proteins.
TGF‐β signalling pathway takes part in numerous cellular processes, including cell proliferation, apoptosis and epithelial–mesenchymal transition (EMT) 67. Liu et al showed that BRD7 forms a TGF‐β‐inducible complex with Smad3/4 to enhance Smad transcriptional activity, and further potentiated TGF‐β‐mediated EMT responses 33. Notably, PARP1 can dissociate Smad complexes from DNA through poly(ADP‐ribosyl)ating Smad3 and Smad4, therefore attenuating Smad‐mediated transcription and inhibiting TGF‐β‐induced EMT transition 68. We have demonstrated PARP1 directly ribosylated BRD7 and promoted its degradation (Figs 2, 3, 4), and PARP1 enhanced this process through promoting degradation of BRD7.
Many tumour cells have defective DNA repair pathways. For instance, BRCA1/2 mutations occur frequently in breast and ovarian cancers 69, 70, ARID1A (AT‐rich interactive domain 1A) mutations are present in multiple cancer types 71. Targeting DNA repair pathways is a promising strategy for achieving synthetic lethality of cancers. PARP1 plays critical roles in DNA damage response. A number of PARP1 inhibitors are either FDA‐approved (Olaparib, Rucaparib) or under clinical development (Veliparib) for treatment of cancer including BRCA1/2‐deficient ovarian or breast cancers 29, 30. Nevertheless, there is still a substantial fraction of BRCA deficient breast cancers do not respond to PARP inhibition 72, raising the possibility that another uncovered mechanism involved in this process. Our results showed that the combination of Olaparib with chemotherapy efficiently inhibited proliferation of cancer cells; however, these effects were greatly compromised after silencing BRD7 (Fig 7E and F). Phosphoinositide 3‐kinase (PI3K) activity is important for regulating cell growth, survival and motility via activation of the protein‐Ser/Thr kinase, AKT 73. It has been documented that BRD7 forms a complex with PI3K regulatory subunit p85α, and induces the nuclear translocation of p85α through its NLS, resulting in instability of p110 monomers and attenuation of PI3K signalling 39, 40. In accordance with previous finding that BRD7 can attenuate the survival signals resulting from PI3K/AKT activation 40, our study revealed that PARP1 promotes activation of AKT phosphorylation at least in part in a BRD7‐dependent manner through which PARP1 directly ribosylated BRD7 and enhanced it ubiquitination by RNF146 (data not shown), indicating that BRD7 might play a crucial role in coordination of PARP1‐induced AKT activation.
In summary, our study uncovered for the first time that BRD7 is directly ribosylated by PARP1 which is then ubiquitinated by a PAR‐binding E3 ubiquitin ligase RNF146, leading to degradation of BRD7 and survival of cancer cells (Fig 8). Inhibition of PARP1 suppressed cell proliferation and promoted sensitivity of cancer cells to DNA damage chemotherapy through reduction of BRD7 PARylation. Our finding uncovered a novel mechanism that PARP1 modulated resistance to DNA‐damaging agents of cancer cells, and suggested that BRD7 may be served as a potential biomarker for predicting the synergistic effects of combining chemotherapeutic drugs and PARP inhibitors in clinical trial.
Figure 8. Schematic model of the PARP1‐RNF146‐BRD7 pathway in response to DNA damage.

BRD7 was ribosylated by PARP1 after DNA damage, promoting recognization by E3 ligase RNF146 which targeted BRD7 for ubiquitination–proteasome‐dependent degradation enhancing cell survival.
Materials and Methods
Cells culture and transfection
HeLa, U2OS, HEK293T, MDA‐MB‐468 and MDA‐MB‐231 cells were obtained from ATCC and cultured in DMEM (Life Technologies) supplemented with 10% foetal bovine serum (FBS, Life Technologies) with 5% CO2 at 37°C. Non‐small lung cancer cell line A549 and cisplatin‐resistant A549/DDP (1 μg/ml) cells were cultured in RPMI1640 (Life Technologies) supplemented with 10% FBS 74. Breast cancer cell line MCF7, MCF7/doxorubicin (ADR, 0.5 μg/ml)‐resistant cell and MCF7/irradiation (IR, 10 Gy)‐resistant cell line were gifts from Prof. Herui Yao (Sun Yat‐Sen University Memorial Hospital, Guangzhou, China), and breast cancer cell line MDA‐MB‐231, MDA‐MB‐231/doxorubicin (ADR, 0.5 μg/ml)‐resistant cell and MDA‐MB‐231/cisplatin (DDP, 1 μg/ml)‐resistant cell line were gifts from Prof.Qiang Liu (Sun Yat‐Sen University Memorial Hospital, Guangzhou, China). All the above breast cancer cell lines were maintained in RPMI 1640 supplemented with 10% FBS.
Plasmid transfections were performed using Lipofectamine 2000 (Life Technologies). siRNA transfection was performed using Lipofectamine RNAiMAX reagent (Life Technologies) according to its protocol. 48 h post‐transfection, cells were harvested and subjected to Western blot. The PARP1 siRNA sequences were as follows 75, 76: #1: 5′‐AAGCCTCCGCTCCTGAACAAT‐3′, #2: 5′‐AAGATAGAGCGTGAAGGCGAA‐3′, #3: 5′‐GGCCAGGATGGAATTGGTA‐3′, #4: 5′‐CCAATAGGCTTAATCCTGT‐3′; the BRD7 siRNA sequences were as follows 33, 40: 5′‐GTCCCTCATACAGAGAAAT‐3′, 5′‐GACTCGTGAGGAAGGAATG‐3′, targeting the 3′ untranslated region (UTR); the RNF146 siRNA sequences were as follows: #1: 5′‐GCACGTTTTCTGCTATCTA‐3′, #2: 5′‐GGCTAGACTGTGATGCTAA‐3′, #3: 5′‐GCTTTGCTGTCTAGTCTTATA‐3′, #4: 5′‐GGACGTCGCAGGAAGATTA‐3′, #5: 5′‐GGATGTATCTGCAGTTGTT‐3′. PARP2 siRNA sequence was as follows 77: 5′‐AAGATGATGCCCAGAGGAACT‐3′.
Plasmids construction
Full‐length cDNAs coding for BRD7, PARP1 and RNF146 were obtained from cDNA of human HEK293 cell by PCR and were then cloned into either pcDNA3.1 or pcDNA6B‐myc/his vector with indicated tag sequence. SFB‐BRD7 and SFB‐RNF146 were first subcloned into pDONR201 entry vector and then transferred into destination vectors with the indicated SFB tag using Gateway Technology (Invitrogen, Camarillo, CA, USA). HA‐tagged ubiquitin was used previously 78; HA‐Ubk48 and HA‐Ubk63 were gifts from Prof. Ronggui Hu (Institute of Biochemistry and Cell Biology, SIBS, CAS). Mutations were introduced using the Quick‐Change Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA), and all mutations were verified by DNA sequencing.
Antibodies and reagents
Human anti‐BRD7 (A302‐304A) antibody was from Bethyl Laboratories. Human anti‐PARP1 (SC‐8007), anti‐RNF146 (SC‐132440, Western blot), anti‐Rad51 (SC‐8349) and anti‐PTEN (SC‐7974) antibodies were from Santa Cruz Biotechnology. Human anti‐RNF146 (ARP43340‐T100, immunoprecipitation) was from Aviva Systems Biology. Human anti‐PAR (4336‐BPC‐100) antibody was from Trevigen. Human anti‐Flag (#8146), anti‐Myc (#2278), anti‐biotin (#5571S) and anti‐HA (#3724) antibodies were from Cell Signaling Technology. Human anti‐p21 (554262), anti‐γH2AX antibodies (560443, Western blot) and Alexa Fluor 555 anti‐γH2AX (560446, Immunofluorescence) were from BD biosciences. Human anti‐GAPDH (HC301‐02) and anti‐tubulin (HC101‐02) antibodies were from TransGen Biotechnology. Rucaparib, Olaparib, BMN673, MG132, doxorubicin (ADR) and camptothecin (CPT) were purchased from Selleck Company. Cycloheximide (CHX) was purchased from Sigma Chemical Co. PDD00017273 was purchased from TOCRIS.
CRISPR‐Cas9 to generate PARP1 knockout HeLa cells
For CRISPR‐Cas9 knockout of human PARP1 in HeLa cells, following small guide RNAs (sgRNAs) were used: GAGTCGAGTACGCCAAGAGC. gRNA sequences were cloned into the vector pX330‐CRISPRv2 cloning vector according to the standard protocol 79. Twenty‐four hours after transfection with gRNA/Cas9 expression construct, HeLa cells were cultured in the presence of puromycin (2 μg/ml). Approximately 2 weeks later, puromycin‐resistant clones were picked and expanded. Lack of PARP1 protein expression in HeLa cells was confirmed by Western blot with PARP1 antibody.
Tandem affinity purification of SFB‐tagged BRD7
For affinity purification, ten plates of HEK293T cells stably expressing tagged proteins were lysed with NETN buffer (20 mM Tris–HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, and 0.5% Nonidet P‐40 and a protease and phosphatase inhibitor cocktail (Bimake, China)) for 20 min. Supernatants were cleared at 15,000 g to remove debris followed by incubation with streptavidin‐conjugated beads (Amersham Biosciences) for 12 h at 4°C. Beads were washed three times with NETN buffer, and then bead‐bound proteins were eluted with NETN buffer containing 1 mg/ml biotin (Sigma). Elute was incubated with S‐protein beads (Novagen) for 2 h at 4°C. Beads were washed three times with NETN buffer and subjected to SDS–PAGE. Protein bands were excised and digested, and peptides were analysed by mass spectrometry (MS).
For identifying ADP‐ribosylation sites of BRD7, we used PARG inhibitor to enhance the accumulation of PARylated BRD7 and sample preparation for mass spectrometric analysis was performed as previously described 10.
Western blot and co‐immunoprecipitation
Cells were lysed in RIPA buffer (50 mM Tris–HCl [pH 8.0], 5 mM EDTA, 150 mM NaCl, 0.5% Nonidet P‐40 and a protease and phosphatase inhibitor cocktail (Bimake, China)). The clarified lysates were resolved by SDS–PAGE and transferred to PVDF membranes for Western blot using ECL detection reagents (Beyotime, China). Immunoblots were processed according to standard procedures using indicated antibodies. For immunoprecipitation, supernatants were first incubated with either anti‐Flag or anti‐Myc agarose (Sigma Chemical Co.) overnight at 4°C, followed by precipitation being washed three times with NETN buffer. For denaturing immunoprecipitation, cells were lysed with RIPA denaturing buffer (50 mM Tris–HCl [pH 8.0], 5 mM EDTA, 150 mM NaCl, 0.5% Nonidet P‐40, 0.5% deoxycholate, 0.5% SDS) containing PARG inhibitor PDD00017273 (10 μM) and protease inhibitor (Bimake, China). Lysates were then immunoprecipitated using control or anti‐BRD7 antibodies followed by Western blotting as indicated. To detect endogenous interaction, the cell lysates were divided into two parts for incubation with anti‐IgG or anti‐PARP1, anti‐BRD7 or anti‐RNF146 antibodies for 2 h and then protein G agaroses (Life Technologies) overnight. After washing three times with NETN buffer, samples were boiled in 2× SDS loading buffer and resolved on SDS–PAGE. Membranes were blocked in 5% milk in TBST buffer and then probed with antibodies as indicated.
Establishment of shRNAs cell lines
pLKO.1 puro was a gift from Bob Weinberg (Addgene plasmid #8453). The following oligonucleotides for human BRD7 were as follows: 5′‐GACTCGTGAGGAAGGAATG‐3′, targeting the 3′ untranslated region (UTR). The non‐silencing control sequence is 5′‐CCCATAAGAGTAATAATA‐3′. The shRNAs were packaged into lentiviruses by cotransfecting with packaging plasmids pMD2G and pSPAX2 (kindly provided by Songyang Zhou, Baylor College of Medicine) into 293T cells. Forty‐eight hours after transfection, the supernatant was collected for infection of MDA‐MB‐468 cells. Infected cells were selected with media containing puromycin (2 μg/ml) for 72 h.
In vivo ubiquitination assay
This procedure was performed as previously described 80. Briefly, HeLa cells were transfected with either indicated plasmids or siRNAs for 24–48 h and then treated with 10 μM MG132 for additional 6 h prior to harvesting. Cells were lysed in RIPA buffer with a cocktail of protease and phosphatase inhibitors (Bimake, China). Endogenous BRD7 was immunoprecipitated using anti‐BRD7 antibody for 12 h at 4°C. Poly‐ubiquitinated BRD7 was detected using an anti‐HA antibody.
In vitro PARylation assay
This procedure was performed as previously described with minor modification 47. Briefly, Flag‐PARP1 was obtained by immunoprecipitation using anti‐Flag agarose from lysates of HEK293T cells overexpressing Flag‐PARP1. Myc‐BRD7‐WT and Myc‐BRD7‐mutant were synthesized in vitro using S30 T7 High‐Yield Protein Expression System (Promega). Flag‐PARP1 and recombinant Myc‐BRD7‐WT, and Myc‐BRD7‐mutant were incubated in 100 μl of PARP reaction buffer (50 mM Tris–HCl [pH 8.0], 4 mM MgCl2, 0.2 mM dithiothreitol) either with or without 25 μM biotinylated NAD+ (Trevigen) for 30 min at 25°C. Reactions were terminated by addition of 2 × sample buffer and analysed by immunoblotting using an anti‐biotin HRP antibody.
Cell death detection assay
The proportion of death cells was measured by Fixable Viability Dye eFluor® 455UV reagent (eBioscience) according to manufacturer's instructions. In brief, breast cancer cells were treated with drugs at indicated time and then washed once with PBS buffer following by trypsin digestion. Then, cells were suspended in Flow Cytometry Staining Buffer (eBioscience). For each sample, 1 μl Fixable Viability Dye was added and incubated for 30 min at 4°C, followed by subjection to FACS analysis. Experiments were repeated for three times.
CCK8 assays
Cells were transfected twice with either control siRNA or siRNAs specifically targeting PARP1 or BRD7. Twenty‐four hours after second transfection, cells were seeded into 96‐well plates for 24 h, treated with Olaparib (5 μM) and either etoposide (VP16, 10 μM), ADR (0.5 μM) or CPT (0.5 μM) for indicated times, then incubated with CCK8 for another 2 h. CCK8 activity of cells was detected by 450 nm absorbance. All data are represented as mean ± SEM of three independent experiments.
Author contributions
DY, ES, HPK and KH designed the experiments and KH wrote the draft manuscript. KH, WW, YL and LL carried out most experiments and analysed data. DC, ZC, YZ, HC, XX, HY and SX assisted in the experiments. YG provided insightful discussion for this manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
This study was supported by National Key Research and Development Program of China (2016YFC1302300); Natural Science Foundation of China (81621004 to ES, 81420108026, 81872140, 81572484, to DY, 81301732, 81772821 to KH, 81402199 to WW); Guangdong Science and Technology Department (2017B030314026); Guangzhou Bureau of Science and Information Technology (201704030036 to DY); and Guangdong Natural Science Foundation (Grant No. 2017A030313471 to KH).
EMBO Reports (2019) 20: e46166
Contributor Information
Erwei Song, Email: songew@mail.sysu.edu.cn.
Dong Yin, Email: yin_dong@yahoo.com.
References
- 1. Grotenbreg G, Ploegh H (2007) Chemical biology: dressed‐up proteins. Nature 446: 993–995 [DOI] [PubMed] [Google Scholar]
- 2. Morrison RS, Kinoshita Y, Johnson MD, Uo T, Ho JT, McBee JK, Conrads TP, Veenstra TD (2002) Proteomic analysis in the neurosciences. Mol Cell Proteomics 1: 553–560 [DOI] [PubMed] [Google Scholar]
- 3. Wang YC, Peterson SE, Loring JF (2014) Protein post‐translational modifications and regulation of pluripotency in human stem cells. Cell Res 24: 143–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wei H, Yu X (2016) Functions of PARylation in DNA damage repair pathways. Genomics Proteomics Bioinformatics 14: 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu C, Yu X (2015) ADP‐ribosyltransferases and poly ADP‐ribosylation. Curr Protein Pept Sci 16: 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ceccaldi R, Rondinelli B, D'Andrea AD (2016) Repair pathway choices and consequences at the double‐strand break. Trends Cell Biol 26: 52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schreiber V, Dantzer F, Ame JC, de Murcia G (2006) Poly(ADP‐ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7: 517–528 [DOI] [PubMed] [Google Scholar]
- 8. Li N, Chen J (2014) ADP‐ribosylation: activation, recognition, and removal. Mol Cells 37: 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, Ahel I, Chang P (2014) Family‐wide analysis of poly(ADP‐ribose) polymerase activity. Nat Commun 5: 4426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y, Wang J, Ding M, Yu Y (2013) Site‐specific characterization of the Asp‐ and Glu‐ADP‐ribosylated proteome. Nat Methods 10: 981–984 [DOI] [PubMed] [Google Scholar]
- 11. Messner S, Altmeyer M, Zhao H, Pozivil A, Roschitzki B, Gehrig P, Rutishauser D, Huang D, Caflisch A, Hottiger MO (2010) PARP1 ADP‐ribosylates lysine residues of the core histone tails. Nucleic Acids Res 38: 6350–6362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Just I, Wollenberg P, Moss J, Aktories K (1994) Cysteine‐specific ADP‐ribosylation of actin. Eur J Biochem 221: 1047–1054 [DOI] [PubMed] [Google Scholar]
- 13. Niere M, Mashimo M, Agledal L, Dolle C, Kasamatsu A, Kato J, Moss J, Ziegler M (2012) ADP‐ribosylhydrolase 3 (ARH3), not poly(ADP‐ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix‐associated poly(ADP‐ribose). J Biol Chem 287: 16088–16102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B et al (2013) Deficiency of terminal ADP‐ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J 32: 1225–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jankevicius G, Hassler M, Golia B, Rybin V, Zacharias M, Timinszky G, Ladurner AG (2013) A family of macrodomain proteins reverses cellular mono‐ADP‐ribosylation. Nat Struct Mol Biol 20: 508–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palazzo L, Thomas B, Jemth AS, Colby T, Leidecker O, Feijs KL, Zaja R, Loseva O, Puigvert JC, Matic I et al (2015) Processing of protein ADP‐ribosylation by Nudix hydrolases. Biochem J 468: 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ueda K, Oka J, Naruniya S, Miyakawa N, Hayaishi O (1972) Poly ADP‐ribose glycohydrolase from rat liver nuclei, a novel enzyme degrading the polymer. Biochem Biophys Res Commun 46: 516–523 [DOI] [PubMed] [Google Scholar]
- 18. Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, Schwede F, Yu Y, Kraus WL (2016) Chemical genetic discovery of PARP targets reveals a role for PARP‐1 in transcription elongation. Science 353: 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lupo B, Trusolino L (2014) Inhibition of poly(ADP‐ribosyl)ation in cancer: old and new paradigms revisited. Biochim Biophys Acta 1846: 201–215 [DOI] [PubMed] [Google Scholar]
- 20. Messner S, Hottiger MO (2011) Histone ADP‐ribosylation in DNA repair, replication and transcription. Trends Cell Biol 21: 534–542 [DOI] [PubMed] [Google Scholar]
- 21. Fouquerel E, Sobol RW (2014) ARTD1 (PARP1) activation and NAD(+) in DNA repair and cell death. DNA Repair 23: 27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krishnakumar R, Kraus WL (2010) The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 39: 8–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ji Y, Tulin AV (2010) The roles of PARP1 in gene control and cell differentiation. Curr Opin Genet Dev 20: 512–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kraus WL (2008) Transcriptional control by PARP‐1: chromatin modulation, enhancer‐binding, coregulation, and insulation. Curr Opin Cell Biol 20: 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kraus WL, Hottiger MO (2013) PARP‐1 and gene regulation: progress and puzzles. Mol Aspects Med 34: 1109–1123 [DOI] [PubMed] [Google Scholar]
- 26. Olabisi OA, Soto‐Nieves N, Nieves E, Yang TT, Yang X, Yu RY, Suk HY, Macian F, Chow CW (2008) Regulation of transcription factor NFAT by ADP‐ribosylation. Mol Cell Biol 28: 2860–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL (2004) NAD+‐dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP‐1. Cell 119: 803–814 [DOI] [PubMed] [Google Scholar]
- 28. Curtin NJ (2005) PARP inhibitors for cancer therapy. Expert Rev Mol Med 7: 1–20 [DOI] [PubMed] [Google Scholar]
- 29. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A et al (2017) Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 377: 523–533 [DOI] [PubMed] [Google Scholar]
- 30. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira‐Frommer R, Safra T et al (2012) Olaparib maintenance therapy in platinum‐sensitive relapsed ovarian cancer. N Engl J Med 366: 1382–1392 [DOI] [PubMed] [Google Scholar]
- 31. Filippakopoulos P, Knapp S (2012) The bromodomain interaction module. FEBS Lett 586: 2692–2704 [DOI] [PubMed] [Google Scholar]
- 32. Mantovani F, Drost J, Voorhoeve PM, Del Sal G, Agami R (2010) Gene regulation and tumor suppression by the bromodomain‐containing protein BRD7. Cell Cycle 9: 2777–2781 [PubMed] [Google Scholar]
- 33. Liu T, Zhao M, Liu J, He Z, Zhang Y, You H, Huang J, Lin X, Feng XH (2017) Tumor suppressor bromodomain‐containing protein 7 cooperates with Smads to promote transforming growth factor‐beta responses. Oncogene 36: 362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Drost J, Mantovani F, Tocco F, Elkon R, Comel A, Holstege H, Kerkhoven R, Jonkers J, Voorhoeve PM, Agami R et al (2010) BRD7 is a candidate tumour suppressor gene required for p53 function. Nat Cell Biol 12: 380–389 [DOI] [PubMed] [Google Scholar]
- 35. Burrows AE, Smogorzewska A, Elledge SJ (2010) Polybromo‐associated BRG1‐associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc Natl Acad Sci USA 107: 14280–14285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaeser MD, Aslanian A, Dong MQ, Yates JR III, Emerson BM (2008) BRD7, a novel PBAF‐specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J Biol Chem 283: 32254–32263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harte MT, O'Brien GJ, Ryan NM, Gorski JJ, Savage KI, Crawford NT, Mullan PB, Harkin DP (2010) BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1‐dependent transcription. Cancer Res 70: 2538–2547 [DOI] [PubMed] [Google Scholar]
- 38. Tae S, Karkhanis V, Velasco K, Yaneva M, Erdjument‐Bromage H, Tempst P, Sif S (2011) Bromodomain protein 7 interacts with PRMT5 and PRC2, and is involved in transcriptional repression of their target genes. Nucleic Acids Res 39: 5424–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Park SW, Herrema H, Salazar M, Cakir I, Cabi S, Basibuyuk Sahin F, Chiu YH, Cantley LC, Ozcan U (2014) BRD7 regulates XBP1s' activity and glucose homeostasis through its interaction with the regulatory subunits of PI3K. Cell Metab 20: 73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chiu YH, Lee JY, Cantley LC (2014) BRD7, a tumor suppressor, interacts with p85alpha and regulates PI3K activity. Mol Cell 54: 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma A, Tsoucas D, Qiu X, Lim K, Rao P et al (2018) A major chromatin regulator determines resistance of tumor cells to T cell‐mediated killing. Science 359: 770–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Satoh MS, Lindahl T (1992) Role of poly(ADP‐ribose) formation in DNA repair. Nature 356: 356–358 [DOI] [PubMed] [Google Scholar]
- 43. Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL, Dawson TM, Poirier GG (2008) Proteome‐wide identification of poly(ADP‐ribose) binding proteins and poly(ADP‐ribose)‐associated protein complexes. Nucleic Acids Res 36: 6959–6976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kraus WL, Lis JT (2003) PARP goes transcription. Cell 113: 677–683 [DOI] [PubMed] [Google Scholar]
- 45. Gibson BA, Kraus WL (2012) New insights into the molecular and cellular functions of poly(ADP‐ribose) and PARPs. Nat Rev Mol Cell Biol 13: 411–424 [DOI] [PubMed] [Google Scholar]
- 46. James DI, Smith KM, Jordan AM, Fairweather EE, Griffiths LA, Hamilton NS, Hitchin JR, Hutton CP, Jones S, Kelly P et al (2016) First‐in‐class chemical probes against poly(ADP‐ribose) glycohydrolase (PARG) inhibit DNA repair with differential pharmacology to olaparib. ACS Chem Biol 11: 3179–3190 [DOI] [PubMed] [Google Scholar]
- 47. Li N, Zhang Y, Han X, Liang K, Wang J, Feng L, Wang W, Songyang Z, Lin C, Yang L et al (2015) Poly‐ADP ribosylation of PTEN by tankyrases promotes PTEN degradation and tumor growth. Genes Dev 29: 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S et al (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461: 614–620 [DOI] [PubMed] [Google Scholar]
- 49. Kim MK, Dudognon C, Smith S (2012) Tankyrase 1 regulates centrosome function by controlling CPAP stability. EMBO Rep 13: 724–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Levaot N, Voytyuk O, Dimitriou I, Sircoulomb F, Chandrakumar A, Deckert M, Krzyzanowski PM, Scotter A, Gu S, Janmohamed S et al (2011) Loss of Tankyrase‐mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 147: 1324–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin Y, Kang T, Zhou BP (2014) Doxorubicin enhances Snail/LSD1‐mediated PTEN suppression in a PARP1‐dependent manner. Cell Cycle 13: 1708–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bassermann F, Eichner R, Pagano M (2014) The ubiquitin proteasome system ‐ implications for cell cycle control and the targeted treatment of cancer. Biochim Biophys Acta 1843: 150–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A et al (2011) RNF146 is a poly(ADP‐ribose)‐directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 13: 623–629 [DOI] [PubMed] [Google Scholar]
- 54. Wang Z, Michaud GA, Cheng Z, Zhang Y, Hinds TR, Fan E, Cong F, Xu W (2012) Recognition of the iso‐ADP‐ribose moiety in poly(ADP‐ribose) by WWE domains suggests a general mechanism for poly(ADP‐ribosyl)ation‐dependent ubiquitination. Genes Dev 26: 235–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. DaRosa PA, Wang Z, Jiang X, Pruneda JN, Cong F, Klevit RE, Xu W (2015) Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP‐ribosyl)ation signal. Nature 517: 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kang HC, Lee YI, Shin JH, Andrabi SA, Chi Z, Gagne JP, Lee Y, Ko HS, Lee BD, Poirier GG et al (2011) Iduna is a poly(ADP‐ribose) (PAR)‐dependent E3 ubiquitin ligase that regulates DNA damage. Proc Natl Acad Sci USA 108: 14103–14108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xue Z, Zhao J, Niu L, An G, Guo Y, Ni L (2015) Up‐regulation of MiR‐300 promotes proliferation and invasion of osteosarcoma by targeting BRD7. PLoS ONE 10: e0127682 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58. Zhou J, Ma J, Zhang BC, Li XL, Shen SR, Zhu SG, Xiong W, Liu HY, Huang H, Zhou M et al (2004) BRD7, a novel bromodomain gene, inhibits G1‐S progression by transcriptionally regulating some important molecules involved in ras/MEK/ERK and Rb/E2F pathways. J Cell Physiol 200: 89–98 [DOI] [PubMed] [Google Scholar]
- 59. Liu Y, Zhao R, Wei Y, Li M, Wang H, Niu W, Zhou Y, Qiu Y, Fan S, Zhan Y et al (2018) BRD7 expression and c‐Myc activation forms a double‐negative feedback loop that controls the cell proliferation and tumor growth of nasopharyngeal carcinoma by targeting oncogenic miR‐141. J Exp Clin Cancer Res 37: 64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu Y, Zhao R, Wang H, Luo Y, Wang X, Niu W, Zhou Y, Wen Q, Fan S, Li X et al (2016) miR‐141 is involved in BRD7‐mediated cell proliferation and tumor formation through suppression of the PTEN/AKT pathway in nasopharyngeal carcinoma. Cell Death Dis 7: e2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Peng C, Liu HY, Zhou M, Zhang LM, Li XL, Shen SR, Li GY (2007) BRD7 suppresses the growth of nasopharyngeal carcinoma cells (HNE1) through negatively regulating beta‐catenin and ERK pathways. Mol Cell Biochem 303: 141–149 [DOI] [PubMed] [Google Scholar]
- 62. Kikuchi M, Okumura F, Tsukiyama T, Watanabe M, Miyajima N, Tanaka J, Imamura M, Hatakeyama S (2009) TRIM24 mediates ligand‐dependent activation of androgen receptor and is repressed by a bromodomain‐containing protein, BRD7, in prostate cancer cells. Biochim Biophys Acta 1793: 1828–1836 [DOI] [PubMed] [Google Scholar]
- 63. Park YA, Lee JW, Kim HS, Lee YY, Kim TJ, Choi CH, Choi JJ, Jeon HK, Cho YJ, Ryu JY et al (2014) Tumor suppressive effects of bromodomain‐containing protein 7 (BRD7) in epithelial ovarian carcinoma. Clin Cancer Res 20: 565–575 [DOI] [PubMed] [Google Scholar]
- 64. Wu WJ, Hu KS, Chen DL, Zeng ZL, Luo HY, Wang F, Wang DS, Wang ZQ, He F, Xu RH (2013) Prognostic relevance of BRD7 expression in colorectal carcinoma. Eur J Clin Invest 43: 131–140 [DOI] [PubMed] [Google Scholar]
- 65. Ye JZ, de Lange T (2004) TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat Genet 36: 618–623 [DOI] [PubMed] [Google Scholar]
- 66. Ozaki Y, Matsui H, Asou H, Nagamachi A, Aki D, Honda H, Yasunaga S, Takihara Y, Yamamoto T, Izumi S et al (2012) Poly‐ADP ribosylation of Miki by tankyrase‐1 promotes centrosome maturation. Mol Cell 47: 694–706 [DOI] [PubMed] [Google Scholar]
- 67. Xu J, Lamouille S, Derynck R (2009) TGF‐beta‐induced epithelial to mesenchymal transition. Cell Res 19: 156–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lonn P, van der Heide LP, Dahl M, Hellman U, Heldin CH, Moustakas A (2010) PARP‐1 attenuates Smad‐mediated transcription. Mol Cell 40: 521–532 [DOI] [PubMed] [Google Scholar]
- 69. Miki Y, Swensen J, Shattuck‐Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266: 66–71 [DOI] [PubMed] [Google Scholar]
- 70. Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378: 789–792 [DOI] [PubMed] [Google Scholar]
- 71. Wu JN, Roberts CW (2013) ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov 3: 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B et al (2011) Olaparib in patients with recurrent high‐grade serous or poorly differentiated ovarian carcinoma or triple‐negative breast cancer: a phase 2, multicentre, open‐label, non‐randomised study. Lancet Oncol 12: 852–861 [DOI] [PubMed] [Google Scholar]
- 73. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT (2017) The PI3K pathway in human disease. Cell 170: 605–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu WJ, Zhang Y, Zeng ZL, Li XB, Hu KS, Luo HY, Yang J, Huang P, Xu RH (2013) beta‐phenylethyl isothiocyanate reverses platinum resistance by a GSH‐dependent mechanism in cancer cells with epithelial‐mesenchymal transition phenotype. Biochem Pharmacol 85: 486–496 [DOI] [PubMed] [Google Scholar]
- 75. Goldberg MS, Xing D, Ren Y, Orsulic S, Bhatia SN, Sharp PA (2011) Nanoparticle‐mediated delivery of siRNA targeting Parp1 extends survival of mice bearing tumors derived from Brca1‐deficient ovarian cancer cells. Proc Natl Acad Sci USA 108: 745–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang F, Wang Y, Wang L, Luo X, Huang K, Wang C, Du M, Liu F, Luo T, Huang D et al (2013) Poly(ADP‐ribose) polymerase 1 is a key regulator of estrogen receptor alpha‐dependent gene transcription. J Biol Chem 288: 11348–11357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bai P, Houten SM, Huber A, Schreiber V, Watanabe M, Kiss B, de Murcia G, Auwerx J, Menissier‐de Murcia J (2007) Poly(ADP‐ribose) polymerase‐2 [corrected] controls adipocyte differentiation and adipose tissue function through the regulation of the activity of the retinoid X receptor/peroxisome proliferator‐activated receptor‐gamma [corrected] heterodimer. J Biol Chem 282: 37738–37746 [DOI] [PubMed] [Google Scholar]
- 78. Hu K, Liao D, Wu W, Han AJ, Shi HJ, Wang F, Wang X, Zhong L, Duan T, Wu Y et al (2014) Targeting the anaphase‐promoting complex/cyclosome (APC/C)‐ bromodomain containing 7 (BRD7) pathway for human osteosarcoma. Oncotarget 5: 3088–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hong J, Hu K, Yuan Y, Sang Y, Bu Q, Chen G, Yang L, Li B, Huang P, Chen D et al (2012) CHK1 targets spleen tyrosine kinase (L) for proteolysis in hepatocellular carcinoma. J Clin Invest 122: 2165–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7