

Abstract
Notch signaling plays a key role in many cell fate decisions during development by directing different gene expression programs via the transcription factor CSL, known as Su(H) in Drosophila. Which target genes are responsive to Notch signaling is influenced by the chromatin state of enhancers, yet how this is regulated is not fully known. Detecting a specific increase in the histone variant H3.3 in response to Notch signaling, we tested which chromatin remodelers or histone chaperones are required for the changes in enhancer accessibility to Su(H) binding. We show a crucial role for the Brahma SWI/SNF chromatin remodeling complex, including the actin‐related BAP55 subunit, in conferring enhancer accessibility and enabling the transcriptional response to Notch activity. The Notch‐responsive regions have high levels of nucleosome turnover which depend on the Brahma complex, increase in magnitude with Notch signaling, and primarily involve histone H3.3. Together these results highlight the importance of SWI/SNF‐mediated nucleosome turnover in rendering enhancers responsive to Notch.
Keywords: Drosophila, histone H3.3, Notch, nucleosome turnover, SWI/SNF
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Signal Transduction
Introduction
Many cell fate decisions during development are directed by Notch signaling between neighboring cells, and misregulation of the pathway results in a variety of complex diseases 1, 2. Notch, the receptor, becomes cleaved upon binding to cell‐surface ligands, freeing the Notch intracellular domain (NICD) to travel directly to the nucleus and activate target gene expression. Depending on the context, different genes are targeted by the Notch transcription complex due to differential enhancer accessibility 3, 4, 5. Furthermore, successful activation involves large‐scale changes in histone modifications and chromatin accessibility across the target enhancers 4, 6, 7, 8. How these changes in chromatin structure are brought about remains to be determined.
The conserved DNA binding partner of NICD is known as Suppressor of Hairless (Su(H)) in Drosophila melanogaster or CSL more generally. In the absence of Notch activity, Su(H) partners with co‐repressive proteins to prevent transcription, often acting on the same genes which are induced upon Notch signal activation 5. The switch from genes being repressed (Notch‐OFF) to activated (Notch‐ON) involves a change in the dynamics of Su(H) binding, so that it acquires a longer residence time when participating in the activating complex, as well as increased accessibility of the DNA 8. Several histone acetyltransferases and methyltransferases contribute to this switch 8, 9, 10, and their actions could explain some of the changes in histone post‐translational modifications that have been observed 4, 6, 7. However, the histone modifiers that have been identified do not explain how target enhancer accessibility is regulated, making it likely that other factors contribute.
One way that chromatin structure can be altered is by a change in the density or dynamics of the nucleosomes, coordinated by chromatin remodeling complexes which fall into four categories based on the classification of their ATPase domains: Imitation Switch (ISWI), Chromodomain helicase DNA‐binding (CHD), Inositol‐Requiring Protein 80 (INO80), and Switching‐Defective/Sucrose Non‐Fermenting (SWI/SNF) complexes 11, 12, 13. Their common property of DNA translocation provides the force needed to dislodge histone–DNA contacts and is tailored to achieve nucleosome repositioning, sliding, ejection, or editing depending on the complex 14. Chromatin remodeling has been shown to facilitate gene expression in a variety of contexts. For example, INO80 is recruited by Oct4 at pluripotency genes to maintain their accessibility in ES cells 15, and by reducing nucleosome occupancy, it facilitates oncogene transcription in melanoma 16. Similarly, SWI/SNF remodelers establish accessible enhancers in fibroblasts following their recruitment by lineage‐specific transcription factors and FOS/JUN 17 and function to shift nucleosomes away from GATA1 sites in hematopoietic stem cells allowing TAL1‐dependent transcription 18. Conversely, in some cases chromatin remodeling can be inhibitory, such as at the MMLV promoter where SWI/SNF recruitment by the glucocorticoid receptor (GR) subsequently inhibits GR binding 19, 20. These diverse roles are highlighted by the fact that mutations affecting remodeling complexes can both promote and suppress tumor progression 11, 21.
The contribution made by chromatin remodeling to Notch‐dependent transcription is also unclear, as conflicting models have been proposed depending on the system and the type of analysis. For example, while two developmental studies based on genetics and phenotypic analysis argued that SWI/SNF complexes contribute positively to Notch‐dependent transcription 22, 23, another proposed an inhibitory effect, detecting increased expression of a key target gene when SWI/SNF components were depleted 24. Confounding the situation further, a non‐nuclear effect on Notch trafficking was reported following depletion of Snr1, a member of SWI/SNF complexes in Drosophila 25. It is possible that histone variants also play a role in regulating enhancer accessibility, and a recent study suggests that the acetylation of histone H2A.Z by Tip60 supports Notch‐dependent gene expression 10. However, none of these studies have analyzed the effects on chromatin dynamics directly, making it important to investigate how depletion of chromatin remodelers can bring about these effects on Notch‐dependent transcription mechanistically.
Given that enhancer accessibility appears to play a key role in Notch‐mediated transcription, we set out to investigate the nucleosome dynamics at target enhancers and to distinguish which chromatin remodelers are critical for enabling target gene activation by Notch. Firstly, we find that Notch signaling regulates nucleosome turnover at target enhancers and promotes incorporation of the histone variant H3.3. Secondly, by testing several classes of chromatin remodelers, we find that the BRM SWI/SNF complex, including the actin‐related BAP55 subunit, is required for nucleosome turnover and the enhancer accessibility required for the Notch response. Thus, SWI/SNF complexes are vital for the Notch response, and we propose a model whereby dynamic chromatin remodeling poises Notch‐responsive genes and facilitates their rapid activation.
Results
Ectopic Notch signaling increases the local concentration of histone H3.3 at the E(spl)‐C
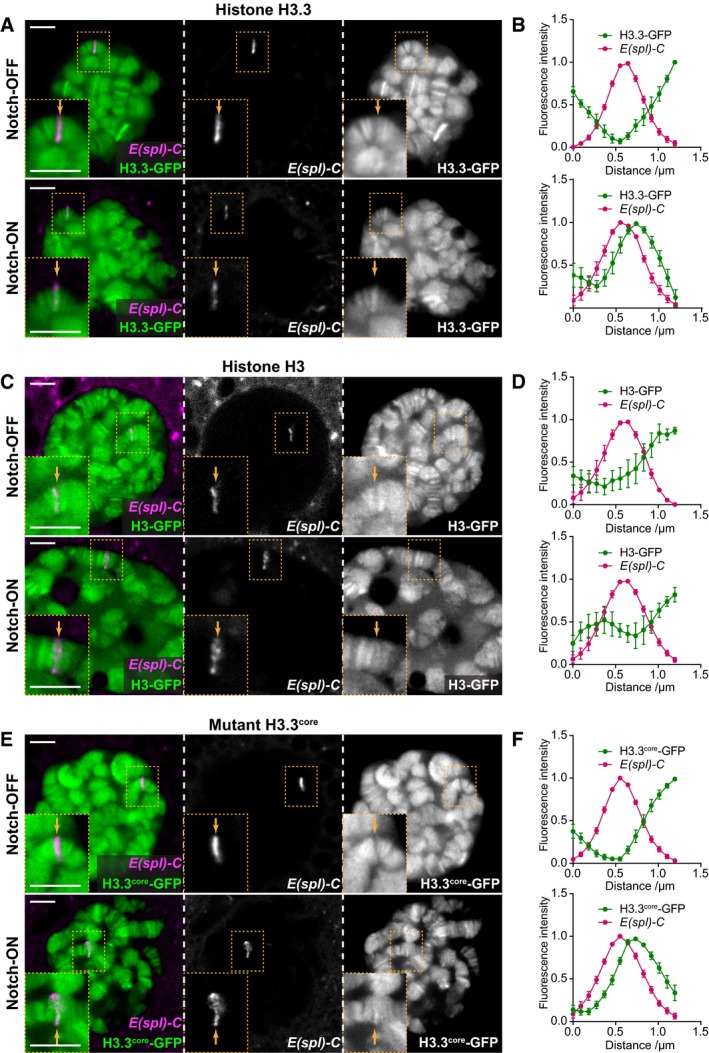
Previous work has shown that Notch signaling promotes large‐scale changes in chromatin structure, including rapid increases in histone acetylation and increased chromatin accessibility 4, 6, 7, 8. In Drosophila, these changes have been most clearly observed at the Enhancer of split‐Complex (E(spl)‐C), a 60‐kb region where 11 highly Notch‐responsive genes are concentrated 26, 27, 28, 29. We therefore chose to investigate whether there are any large‐scale changes in the histone variant H3.3 or canonical histone H3 occupancy at this region following Notch activation, making use of a live tag marking the E(spl)‐C in Drosophila larval salivary glands 8. Without Notch signaling, both histones H3.3‐GFP and H3‐GFP were present at low levels at the E(spl)‐C compared to surrounding regions. However, when we activated Notch signaling in this tissue by expressing a constitutively active form of the Notch receptor, NΔECD 30, 31, under control of the GAL4/UAS system, the levels of H3.3‐GFP were strongly increased compared to surrounding regions (Fig 1A, Notch‐ON). This pattern was found to be reproducible when the relative fluorescence intensity was quantified across the locus in images taken from live salivary glands (Fig 1B). No such change was detected when we examined the effects on histone H3 in a similar manner (Fig 1C and D).
Figure 1. H3.3 levels increase at the E(spl)‐C in Notch‐ON nuclei.
-
A–F(A, C, E) Live imaging of histone‐GFP (green) and ParB‐mCherry (magenta) expressed in larval salivary gland nuclei using 1151‐Gal4. H3.3‐GFP levels are increased at the E(spl)‐C in the presence of constitutively active Notch, NΔECD (Notch‐ON), compared to control Notch‐OFF nuclei expressing LacZ (A). The same is seen with H3.3core‐GFP (E), but there is little change in H3‐GFP between Notch‐OFF and Notch‐ON nuclei (C). ParB‐mCherry binds to its cognate int DNA sequence inserted within the E(spl)‐C 8, 64. Yellow dotted box contains E(spl)‐C, and yellow arrow indicates position of E(spl)‐C on chromosome. Scale bars (white) = 5 μm. (B, D, F) Quantifications of relative fluorescence intensity of histone‐GFP and ParB‐mCherry across the E(spl)‐C in Notch‐OFF (upper) and Notch‐ON (lower) conditions. Mean ± SEM; n nuclei = 7, 6, 5, 8, 9, and 11 and n glands = 5, 5, 5, 5, 5, and 4 where each gland represents a biological replicate (from top to bottom).
Histone H3.3 has been associated with actively transcribed genes and can be incorporated into the chromatin independently of DNA replication 32, 33. To verify that the Notch‐dependent increase in H3.3‐GFP was replication‐independent, we used a mutant form of histone H3.3, H3.3core‐GFP, which is only incorporated in a replication‐independent manner 32. Indeed, with H3.3core‐GFP we saw the same pattern as with H3.3‐GFP (Fig 1E and F), suggesting that the local increase in histone H3.3 concentration at the E(spl)‐C is not due to an increased level of endoreplication at this locus, and thus likely represents changes associated with Notch‐induced transcription. Furthermore, when histone H3.3 tagged with another fluorophore, mKO, was expressed on a much shorter timescale under Notch‐OFF and Notch‐ON conditions, making use of a heat‐shock‐inducible FRT construct 34, the same pattern of incorporation was observed (Fig EV1). This showed that the incorporation of histone H3.3 under Notch‐ON conditions is dynamic and ongoing during the period of Notch activity.
Figure EV1. Histone H3.3 is incorporated dynamically at the E(spl)‐C in Notch‐ON nuclei.
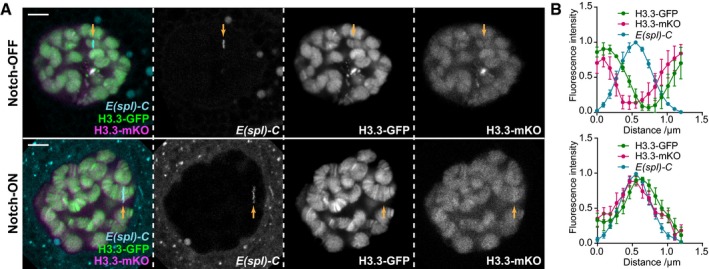
- Live imaging of H3.3‐GFP (green), H3.3‐mKO (magenta), and ParB‐mCherry (cyan) expressed in larval salivary gland nuclei using 1151‐Gal4 with the UAS‐FRT‐H3.3‐GFP‐PolyA‐FRT‐H3.3‐mKO‐PolyA transgene 34. H3.3‐mKO expression was initiated by heat‐shock‐inducible flippase expression approximately 24 h before imaging (see Materials and Methods for details), and ParB‐mCherry indicates the E(spl)‐C as in Fig 1. H3.3‐mKO incorporation shows a similar pattern to H3.3‐GFP in Notch‐OFF (LacZ expression) and Notch‐ON (NΔECD expression) nuclei, as higher levels are present relative to surrounding regions in the Notch‐ON condition. Yellow arrow indicates position of E(spl)‐C on chromosome. Scale bars (white) = 5 μm.
- Quantifications of relative fluorescence intensity of H3.3‐GFP, H3.3‐mKO, and ParB‐mCherry across the E(spl)‐C in Notch‐OFF (upper) and Notch‐ON (lower) conditions. Mean ± SEM; n nuclei = 3 and 5 and n glands = 3 and 3 where each gland represents a biological replicate (from top to bottom).
The BRM chromatin remodeling complex is required for Notch‐responsive accessibility
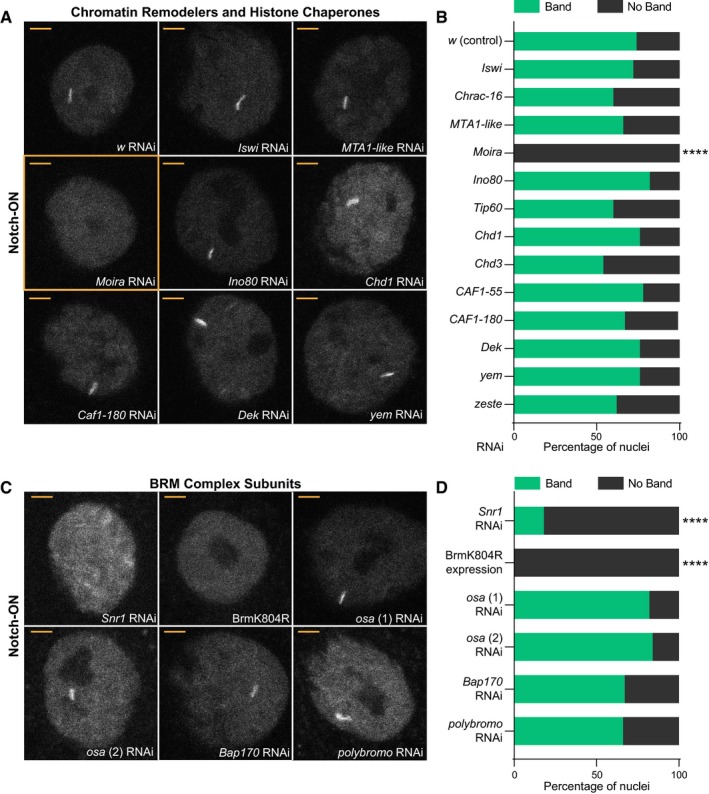
The incorporation of histone H3.3 at Notch‐regulated genes, along with the previously detected changes in accessibility 8, suggests an involvement of chromatin remodeling complexes and/or histone chaperones. In order to ascertain which of these are needed, we tested if any remodelers or chaperones are required for the recruitment of Su(H) to the E(spl)‐C. We have previously shown that Notch activity promotes robust recruitment of Su(H)‐GFP, detectable as a band of fluorescence when salivary gland nuclei are imaged live under conditions of UAS‐NΔECD expression (Fig 2A) 8. We therefore performed RNAi knockdown of different chromatin remodelers and histone chaperones and assessed the impact on Su(H) recruitment under these Notch‐ON conditions. To verify that the RNAi lines were effective, RNA was extracted from the salivary glands and expression levels quantified by reverse transcription–qPCR (Fig EV2A). Only those chromatin remodelers and chaperones where a reduced expression was detected were analyzed further. In the majority of cases, we detected little or no change in Su(H) recruitment (Fig 2A and B). For example, knockdown of components in the ISWI, NuRD, and INO80 complexes failed to perturb Su(H) recruitment (for a review of chromatin remodelers in Drosophila, see 35). Likewise, knockdown of chromatin assembly factors or H3.3‐specific chaperones such as DEK 36 or Yemanuclein (YEM) 37 had no effect. In contrast, depletion of core components of the BRM (BAF/PBAF) SWI/SNF chromatin remodeling complex 38, 39 had a striking effect. Knockdown of Moira (SMARCC1/2) eliminated visible recruitment of Su(H)‐GFP in all nuclei, while knockdown of Snr1 (SMARCB1) prevented the formation of a single clear band of recruitment in most nuclei (Fig 2A–D).
Figure 2. The BRM complex is required for Su(H) recruitment in Notch‐ON nuclei.
-
A–D(A, C) Effects from depleting chromatin remodelers and histone chaperones, as indicated (wide range shown in A, BRM complex components shown in C), on recruitment of Su(H)‐GFP in Notch‐ON nuclei (expressing NΔECD) of larval salivary glands. w RNAi is a control and BrmK804R is expression of dominant‐negative Brm. Different OSA RNAi stocks used in (C) are denoted by (1) and (2). In all conditions except Moira RNAi (A), Snr1 RNAi, and BrmK804R expression (C), nuclei exhibit a bright accumulation of Su(H)‐GFP at a single locus when imaged live. Scale bars (yellow) = 5 μm. (B, D) Percentage of Notch‐ON nuclei retaining a single clear band of Su(H)‐GFP when the indicated RNAi is co‐expressed with NΔECD. For each genotype, 5 nuclei from each of the 10 glands were scored (50 nuclei total). **** A significant fraction of nuclei lost the fluorescent band when core components of the BRM complex were perturbed; P < 0.0001, two‐tailed Fisher's exact test calculated using the raw (non‐percentage) scoring data.
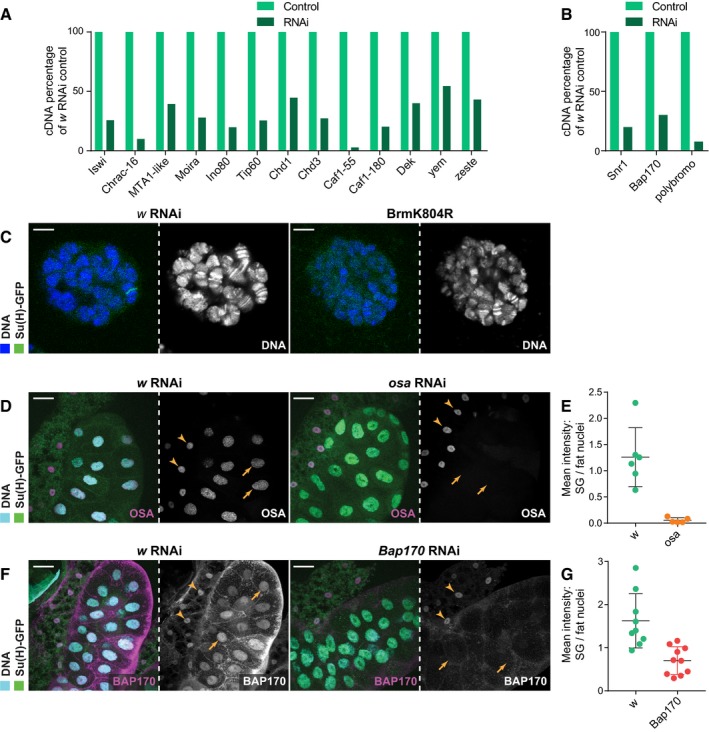
Figure EV2. The RNAi lines used successfully reduce RNA and protein levels.
-
A, BEffects of the indicated RNAi expression in salivary glands on cDNA levels, measured by reverse transcription–qPCR; percentage cDNA compared to w RNAi control after normalizing to an internal control for each sample (RpL32 for most but RpII215 for Moira as Moira RNAi appeared to affect RpL32 levels). All reduce their respective cDNA levels, with polybromo RNAi causing a greater reduction than Snr1, despite not having an effect on Su(H) recruitment (B). Note that samples included RNA extracted from adjoining fat cells as well as salivary glands, and thus, some variability in the knockdowns is attributed to residual expression of chromatin remodelers and chaperones in the fat tissue where the RNAi was not expressed. Mean, n = 2.
-
CLive Hoechst 33342 staining (blue) of salivary glands expressing either w RNAi or BrmK804R. Chromosomes are observed with distinctive banding patterns under both conditions despite chromosomes being slightly smaller with BrmK804R expression. Scale bars (white) = 5 μm.
-
D–G(D, F) Immunofluorescence staining of OSA (C; magenta) and BAP170 (E; magenta) in salivary glands expressing osa (stock (2) in Fig 2) and Bap170 RNAi, respectively, compared to w RNAi control glands. osa RNAi depletes all detectable OSA protein and Bap170 RNAi removes most BAP170 protein. Yellow arrows indicate salivary gland nuclei and yellow arrowheads indicate fat cell nuclei for comparison where RNAi is not expressed. Scale bars (white) = 50 μm. (E, G) Quantifications of OSA (n = 5) (E) and BAP170 (n = 10) (G) nuclear levels from maximum projection images with salivary gland nuclei normalized to fat cell nuclei. Mean ± SEM.
Expression of a commonly used dominant‐negative form of the Brahma ATPase BrmK804R 40 had the same effect as Moira knockdown, preventing formation of the Su(H)‐GFP band (Fig 2C and D). This demonstrates that the ATPase activity of the BRM complex is required for Su(H) recruitment. Furthermore, DNA staining of salivary gland nuclei showed that the chromosomes retained their characteristic DNA banding patterns, although the chromosomes were somewhat reduced in size (Fig EV2C). Thus, there was no global disruption to the nuclear architecture when BrmK804R was expressed, suggesting that the effects on Su(H) recruitment were specific.
Two different BRM complexes have been reported, BAP and PBAP (BAF and PBAF), which are distinguished by specific subunits OSA (ARIID1A/B) in BAP or BAP170 (ARID2) and Polybromo (PBRM1) in PBAP 38, 39. Surprisingly, these subunits do not appear to be essential for Notch‐dependent Su(H) recruitment. A robust band of Su(H)‐GFP was still detectable in nuclei depleted for OSA, BAP170, or Polybromo (Fig 2C and D), even though little or no detectable RNA or protein remained (Fig EV2B and D–G). This suggests either that the two complexes can compensate for each other or that the specialized subunits are not necessary for the Notch‐mediated effects on chromatin.
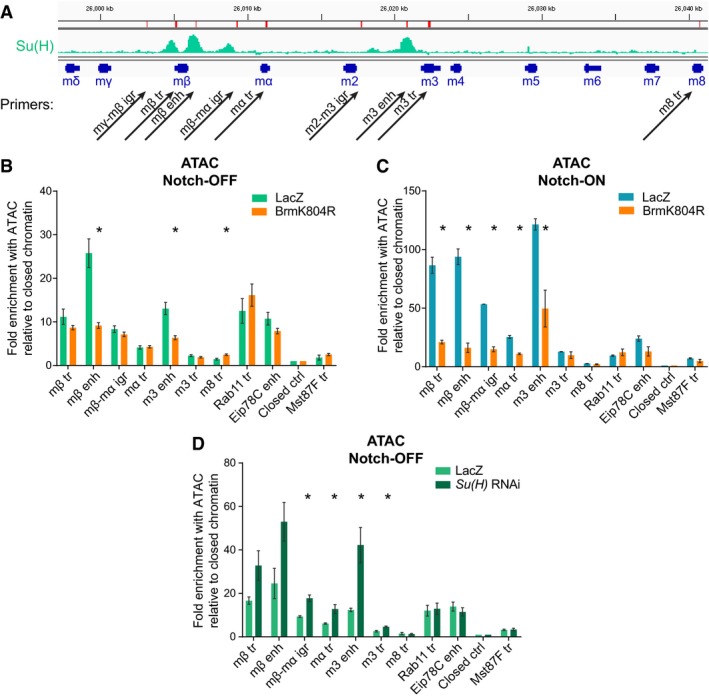
Su(H) recruitment in the Notch‐ON condition correlates with increased chromatin accessibility 8. We therefore used the assay for transposase‐accessible chromatin (ATAC) 41 to determine whether the BRM complex is required for this Notch‐induced change, using qPCR to analyze different regions of the E(spl)‐C (chosen regions illustrated in Fig 3A). Expression of BrmK804R had a very localized effect on accessibility measured with ATAC, causing a strong reduction at the E(spl)mβ‐HLH and E(spl)m3‐HLH enhancer regions in both Notch‐OFF (Fig 3B) and Notch‐ON (Fig 3C) conditions. The effects in the Notch‐ON condition were the most dramatic, with BrmK804R largely abolishing the increases in accessibility induced by Notch across the E(spl)‐C so that the locus resembled that in the Notch‐OFF condition.
Figure 3. The BRM complex is required for chromatin accessibility at Notch‐responsive regions.
-
AGenomic region encompassing the E(spl)‐C; green graphs indicate ChIP enrichment for Su(H) in Kc167 cells (log2 scale is −0.5 to 2.9, data published previously in 4); gene models are depicted in dark blue. Positions of primer pairs used in qPCR experiments are indicated with black arrows. Abbreviations are as follows: “igr”, intergenic region; “tr”, transcribed region; and “enh”, enhancer.
-
B, CChromatin accessibility in Notch‐OFF (B) and Notch‐ON (NΔECD expression, C) salivary gland nuclei measured by ATAC‐qPCR; fold enrichment at the indicated regions compared to a “closed ctrl” region. Expression of dominant‐negative Brm, BrmK804R, led to reduced accessibility of E(spl)mβ‐HLH and E(spl)m3‐HLH enhancer regions in Notch‐OFF conditions, and to a more widespread reduction in accessibility in Notch‐ON conditions. “Eip78C enh” corresponds to the ecdysone receptor‐binding region of the Eip78C enhancer, which is highly accessible but not Notch‐responsive; “Rab11 tr” and “Mst87F tr” represent highly and lowly expressed control genes, respectively. Mean ± SEM; n = 3; *P < 0.05 with two‐tailed Welch's t‐test comparing LacZ and BrmK804R samples.
-
DChromatin accessibility in salivary gland nuclei depleted for Su(H) by RNAi, measured by ATAC‐qPCR; fold enrichment at the indicated regions compared to a “closed ctrl” region. Accessibility is increased across most of the E(spl)‐C compared to controls expressing LacZ. Control primer regions are as in (B) and (C). Mean ± SEM; n = 3; *P < 0.05 with two‐tailed Welch's t‐test compared to LacZ controls.
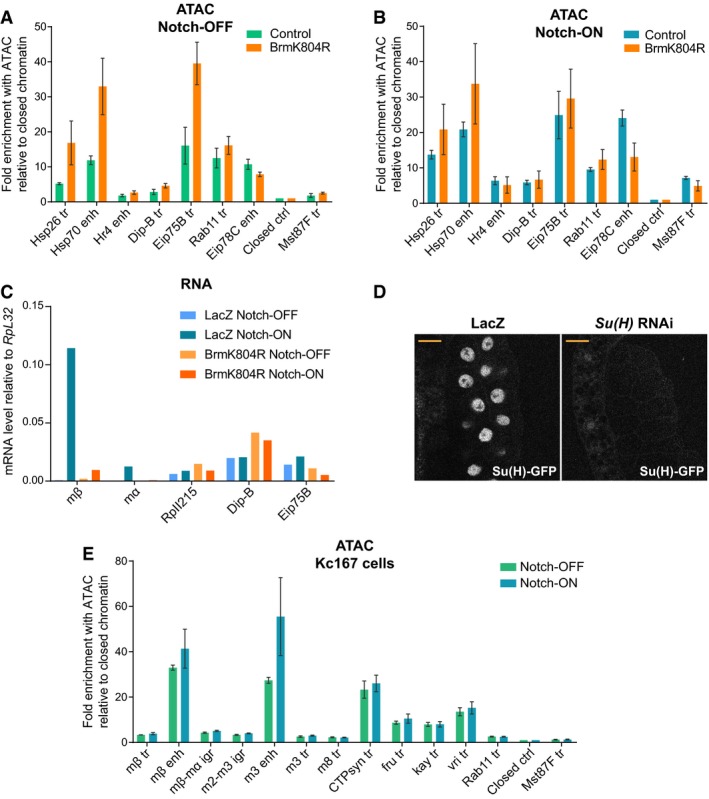
To determine whether the BRM complex plays the same role at other inducible enhancers, several additional regions were analyzed, including heat‐shock and ecdysone‐responsive regions. In contrast to the Notch‐responsive regions in the E(spl)‐C, the ecdysone‐responsive regions of Hr4, Dip‐B, and Eip75B showed no decrease in accessibility in the presence of BrmK804R (Fig EV3A and B) and, unlike E(spl)mβ‐HLH and E(spl)mα‐HLH, there was no decrease in transcription of these genes (Fig EV3C). Surprisingly, two heat‐shock promoters underwent an increase in accessibility when BrmK804R was expressed (Fig EV3A and B). These data suggest that SWI/SNF chromatin remodeling complexes are differentially deployed depending on the regulatory mechanisms operating, and demonstrate that there is some specificity in the role that BRM complexes play at Notch‐regulated loci.
Figure EV3. The effects of BrmK804R on the accessibility and expression of Notch‐inducible genes are not widespread.
-
A, BChromatin accessibility in Notch‐OFF (A) and Notch‐ON (NΔECD expression, B) salivary gland nuclei measured by ATAC‐qPCR; fold enrichment at the indicated regions compared to a “closed ctrl” region. Expression of dominant‐negative Brm, BrmK804R, had little effect or increased the accessibility of some regions. “Hsp26 tr” and “Hsp70 enh” are heat‐shock‐responsive regions, while “Hr4 enh”, “Dip‐B tr”, and “Eip75B” are ecdysone‐responsive. “Rab11 tr”, “Eip78C”, “Closed ctrl”, and “Mst87F tr” are the same as shown in Fig 3B and C. Mean ± SEM; n = 3.
-
CEffect of Notch activation (NΔECD versus LacZ expression) and BrmK804R expression on gene expression in salivary glands measured by reverse transcription–qPCR. Genes shown are Notch‐responsive E(spl)mβ‐HLH (mβ) and E(spl)mα‐HLH (mα), housekeeping gene RpII215, and ecdysone‐responsive Dip‐B and Eip75B. Out of those shown, BrmK804R expression only reduces the Notch‐responsive expression of E(spl)mβ‐HLH and E(spl)mα‐HLH. Mean, n = 2.
-
DEffect of Su(H) RNAi expression on Su(H)‐GFP levels detected with live imaging compared to LacZ expression control. No Su(H)‐GFP was left detectable, and this genotype was used for the ATAC experiment in Fig 3D. Scale bars (yellow) = 50 μm.
-
EAn acute Notch response in Kc167 cells involves increased enhancer accessibility. Chromatin accessibility across the E(spl)‐C in Notch‐ON (EGTA‐treated) and Notch‐OFF (PBS control) Kc167 cells detected by ATAC‐qPCR. Fold enrichment of the indicated regions compared to a “closed ctrl” region; positions of E(spl)‐C primers in the genome are shown in Fig 3A. “CTPsyn tr”, “fru tr”, “kay tr”, and “vri tr” are highly accessible control regions which do not respond to Notch. Mean ± SEM; n = 3.
To rule out the possibility that the effects of BrmK804R on accessibility at the E(spl)‐C were indirect, resulting from a reduced Su(H) recruitment, we knocked down Su(H) with RNAi in the Notch‐OFF condition and performed ATAC. The knockdown was effective in removing all detectable Su(H)‐GFP when salivary glands were imaged live (Fig EV3D), and these conditions resulted in an increased accessibility across the E(spl)‐C (Fig 3D). This increase in accessibility is consistent with the known role of Su(H) as a repressor of target genes in the absence of Notch signaling and contrasts with the consequences of inhibiting the BRM complex.
Together these results demonstrate that the BRM complex is necessary to maintain a degree of accessibility at enhancers, even before the cells experience Notch signaling, and is then essential for the Notch activity‐dependent increase in accessibility of the E(spl)‐C.
The BRM complex is required for acute Notch responses in Kc167 cells
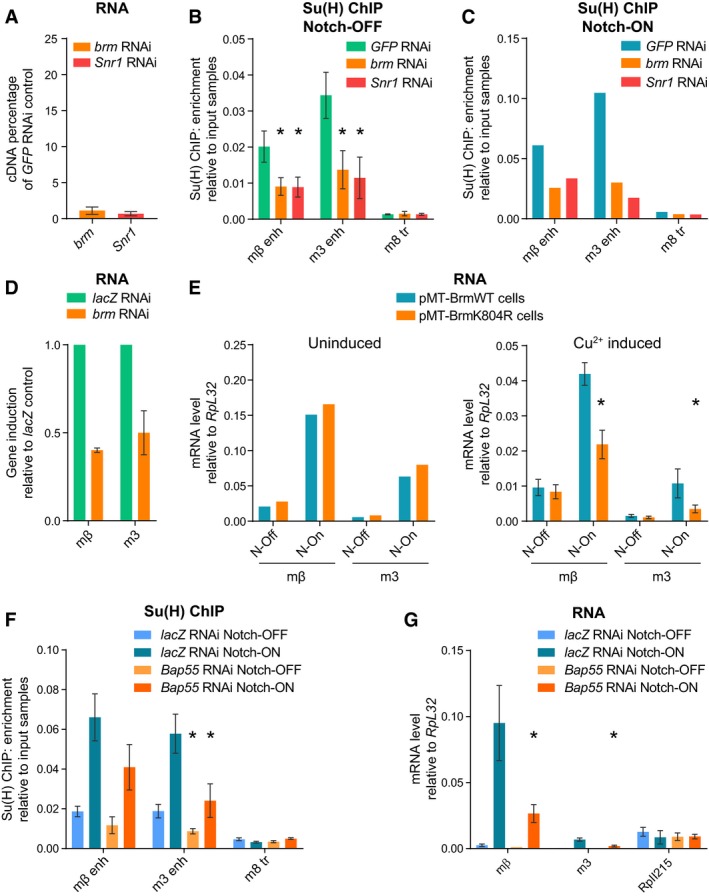
To test the role of the BRM complex in a system where we could acutely manipulate Notch activity, we turned to Drosophila Kc167 cells. In these cells, Notch signaling is rapidly activated by the addition of the calcium chelator EGTA, which, by destabilizing the negative regulatory region, elicits the rapid cleavage of the Notch receptor and activates target genes within 30 min 4, 28, 42. As in the salivary gland, gene activation is accompanied by an increase in Su(H) recruitment, detectable by chromatin immunoprecipitation (ChIP) 4. To test the involvement of the BRM complex in this context, we performed RNAi for two core components of the BRM complex, Brm and Snr1 (Fig 4A), and analyzed the effects on Su(H) recruitment by ChIP with qPCR. In both Notch‐OFF (Fig 4B) and Notch‐ON (EGTA treatment; Fig 4C) cells, the level of Su(H) recruitment was decreased when Brm or Snr1 was depleted by RNAi, showing that the BRM complex is essential for Su(H) recruitment. The transcription of the target genes E(spl)mβ‐HLH and E(spl)m3‐HLH, which are usually strongly induced following Notch activation, was also decreased by brm RNAi (Fig 4D).
Figure 4. The BRM complex is required for Su(H) recruitment and Notch‐dependent transcription in Kc167 cells.
-
AEffect of brm and Snr1 RNAi on brm and Snr1 cDNA levels, respectively, measured by reverse transcription–qPCR in Kc167 cells; percentage cDNA compared to GFP RNAi. The knockdowns are highly effective, with only 1–2% of brm and Snr1 cDNA remaining detectable. Mean ± SEM; n = 3.
-
B, CKnockdown of components of the BRM complex reduces Su(H) recruitment in both Notch‐OFF (B) and Notch‐ON (C) conditions. Fold enrichment of Su(H) occupancy at the indicated positions detected by ChIP, relative to input, in Kc167 cells treated with brm, Snr1, or GFP RNAi as a control. Notch‐ON conditions (C) were induced by 30 min of EGTA treatment. Mean ± SEM, n = 3 (B); mean, n = 2 (C); *P < 0.05 with one‐tailed Student's t‐test compared to GFP RNAi control.
-
DEffect of brm RNAi on E(spl)mβ‐HLH (mβ) and E(spl)m3‐HLH (m3) induction by Notch activation (EGTA treatment) measured by reverse transcription–qPCR; shown as fold difference to lacZ RNAi control. Mean ± SEM; n = 3.
-
EEffect of Brm dominant‐negative on expression of E(spl)mβ‐HLH (mβ) and E(spl)m3‐HLH (m3) measured by reverse transcription–qPCR. Expression was analyzed in stable cell lines containing pMT‐inducible BrmWT or BrmK804R in the absence (left, uninduced) or presence of copper sulfate (right, Cu2+ induced). The response of E(spl)mβ‐HLH and E(spl)m3‐HLH to Notch activation (“N‐On” = EGTA treatment versus “N‐Off” = PBS control) was reduced in the BrmK804R‐expressing cells compared to BrmWT‐expressing cells, only when induced with copper (right graph). Mean, n = 2 (left); mean ± SEM, n = 3 (right); *P < 0.05 with one‐tailed Student's t‐test comparing BrmWT and BrmK804R.
-
FKnockdown of actin‐related subunit, BAP55, reduces Su(H) recruitment in both Notch‐OFF (PBS treatment) and Notch‐ON (EGTA treatment) conditions. Fold enrichment of Su(H) occupancy at the indicated positions detected by ChIP, relative to input, in Kc167 cells treated with Bap55 or lacZ RNAi as a control. Mean ± SEM; n = 3; *P < 0.05 with one‐tailed Student's t‐test compared to lacZ RNAi control.
-
GEffect of Bap55 RNAi on E(spl)mβ‐HLH (mβ) and E(spl)m3‐HLH (m3) expression levels measured by reverse transcription–qPCR in Notch‐OFF (PBS treatment) and Notch‐ON (EGTA treatment) conditions. Expression level of RpII215 is shown as a control gene. Mean ± SEM; n = 3; *P < 0.05 with one‐tailed Student's t‐test compared to lacZ RNAi control.
In order to confirm that the ATPase activity of the BRM complex was essential, we made stable cell lines expressing BrmK804R or the wild‐type form, BrmWT, as a control (under control of the copper‐inducible pMT promoter) 43. Expression of E(spl)mβ‐HLH and E(spl)m3‐HLH was rapidly upregulated by EGTA‐induced Notch activation in control conditions (Fig 4E, left). However, following copper‐induced expression of BrmK804R for 24 h, cells had a significantly reduced upregulation of E(spl)mβ‐HLH and E(spl)m3‐HLH compared to cells expressing BrmWT (Fig 4E, right). This shows that the ATPase function of the BRM complex is key to the Notch response in these cells.
Another sub‐complex associated with SWI/SNF chromatin remodelers contains actin‐related proteins (ARPs) and is proposed to facilitate sliding and ejection of nucleosomes 44, 45, 46. To test whether this sub‐complex is important for the activity of Notch‐responsive enhancers, we analyzed the effects from knocking down the Drosophila ARP homolog, BAP55, in Kc167 cells. Strikingly, this had similar consequences to depletion of the core Brm and Snr1 subunits. Firstly, Su(H) recruitment was decreased in both Notch‐OFF and Notch‐ON conditions (Fig 4F), and secondly, Notch‐induced RNA levels were reduced (Fig 4G). These results demonstrate an essential role for BAP55 in the Notch response and, given the data that ARPs are required for histone ejection 46, suggest that this aspect of SWI/SNF function is important mechanistically for Notch enhancer activation.
Nucleosome turnover increases with Notch signaling and is dependent on the BRM complex
Chromatin remodelers are thought to slide, replace, or eject nucleosomes 13. Even the short pulse of activity in Kc167 cells was sufficient to bring about a change in chromatin accessibility measured with ATAC (Fig EV3E), suggesting that the BRM complex could be moving or depleting histones at the Notch‐regulated enhancers. Additionally, the histone variant H3.3 has been associated with nucleosome turnover 47, 48. Given the results showing changes in accessibility and histone H3.3 levels, and the involvement of BAP55, we were prompted to measure whether nucleosome turnover was occurring. To do this, we used the CATCH‐IT technique, which relies on the incorporation of a methionine analog called azidohomoalanine into newly synthesized proteins 49, 50. Click chemistry is used to biotinylate this residue so that any chromatin containing newly synthesized proteins can therefore be isolated, and a wash with high salt and urea leaves only the histone H3/H4 tetramers bound to the DNA such that histone turnover is distinguished from the incorporation of other DNA‐binding proteins. We performed CATCH‐IT in Kc167 cells, incubating them with media containing azidohomoalanine for 4 h after a 1‐h period of methionine starvation, in the presence or absence of NICD. To achieve this, we used a cell line where NICD was expressed from the copper‐inducible pMT promoter 4 for 1 h prior to and during both the methionine starvation and azidohomoalanine labeling. Using this approach, we detected differential levels of histone turnover, with active enhancer regions showing approximately fivefold higher levels of turnover than the surrounding less active regions (Fig 5A). Notably, the enriched turnover detected at Su(H)‐binding enhancer regions increased by two‐ to threefold in the presence of NICD.
Figure 5. Nucleosome turnover at Su(H)‐bound enhancers is increased in Notch‐ON cells and is dependent on the BRM complex.
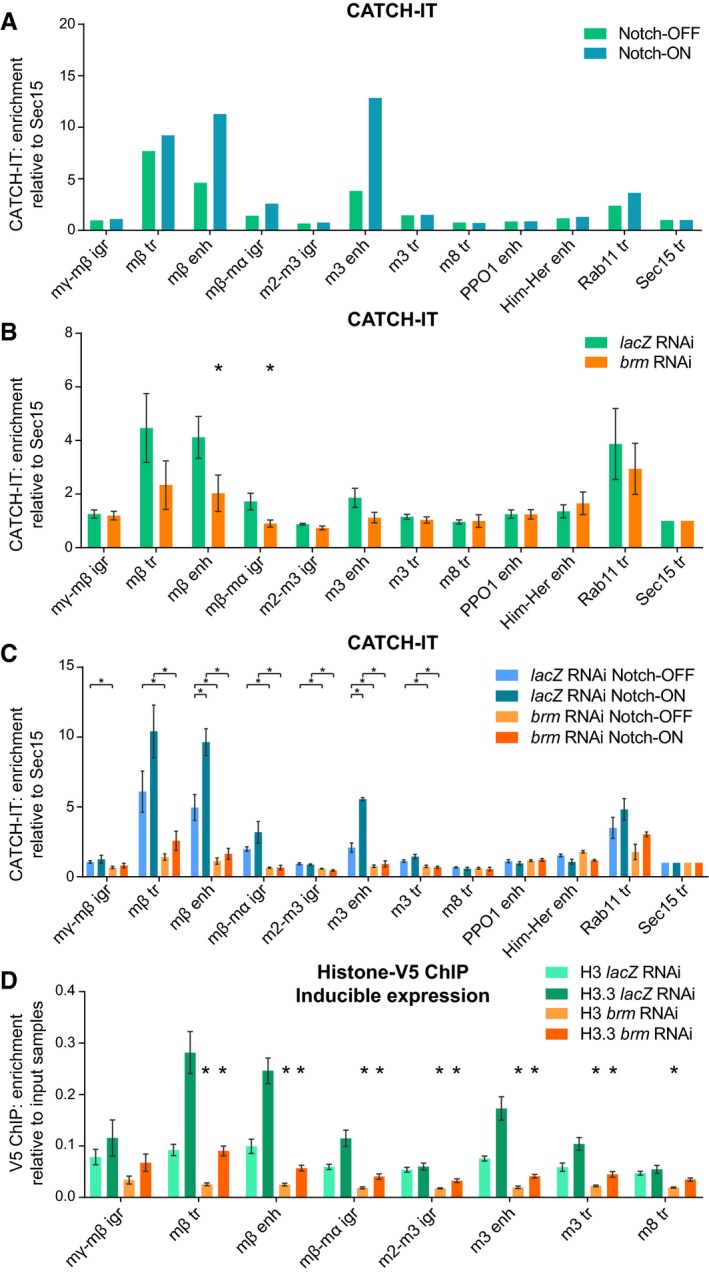
- Nucleosome turnover measured by CATCH‐IT‐qPCR; fold enrichment over input samples compared to Sec15 tr control region. Su(H)‐bound enhancers show increased nucleosome turnover in response to Notch signaling. Notch signaling is activated in Kc167 cells by 6 h of copper induction of pMT‐NICD with copper excluded in the control. Positions of E(spl)‐C primers are shown in Fig 3A; the remaining primers are control non‐Notch‐responsive regions. Mean, n = 2.
- brm RNAi reduces nucleosome turnover at Notch‐responsive regions. CATCH‐IT‐qPCR results as in A after brm or lacZ RNAi as a control. Mean ± SEM; n = 5; *P < 0.05 with one‐tailed Student's t‐test comparing brm and lacZ RNAi.
- Brm is required for Notch‐responsive nucleosome turnover. CATCH‐IT‐qPCR results after brm or lacZ RNAi as in (B) and pMT‐NICD expression as in (A). Mean ± SEM; n = 3; *P < 0.05 with two‐tailed Student's t‐test compared to control (lacZ RNAi Notch‐ON bars are compared to lacZ RNAi Notch‐OFF bars, and brm RNAi bars are compared to their respective lacZ RNAi control bars).
- brm RNAi reduces incorporation of histone H3.3. V5 ChIP–qPCR in Kc167 cells after lacZ or brm RNAi treatment in cells with H3‐V5 or H3.3‐V5 expression induced from the pMT promoter by 3 h of copper treatment, shown as fold enrichment over input samples. Mean ± SEM; n = 3. *P < 0.05 with two‐tailed Welch's t‐test compared to lacZ RNAi control.
We then tested whether knockdown of the BRM complex would affect the levels of nucleosome turnover measured with CATCH‐IT. Depletion of Brm by RNAi resulted in a localized decrease in histone turnover at the Notch‐responsive regions with relatively little change at control regions (Fig 5B), strengthening the evidence that the BRM complex has a critical role in Notch signaling and providing a specific mechanism by which this may occur. Furthermore, Brm depletion had an even greater effect in the Notch‐ON condition when brm RNAi was combined with copper‐inducible NICD expression (Fig 5C), illustrating the importance of the BRM complex for the Notch response.
Given that we observed increased histone H3.3 recruitment in Notch‐ON cells in vivo, we next sought to measure the effects on the histone variant H3.3 by expressing V5‐tagged histone proteins and performing ChIP 51. When H3‐V5 and H3.3‐V5 were expressed from a constitutive promoter, it was evident that H3.3 predominated over H3 throughout the Notch‐responsive regions of the E(spl)‐C, while the distal region at the E(spl)m8‐HLH gene had similar levels of both variants (Fig EV4). Neither Notch activity (Fig EV4D and E) nor depletion of Brm (Fig EV4F) had a large impact on these distributions of H3 or H3.3, arguing that there is no gross change in the overall levels of histone H3 or H3.3 during an acute Notch response and that the BRM complex is not essential for the incorporation of H3.3 per se.
Figure EV4. Notch activation does not affect the distribution of histones H3 and H3.3.
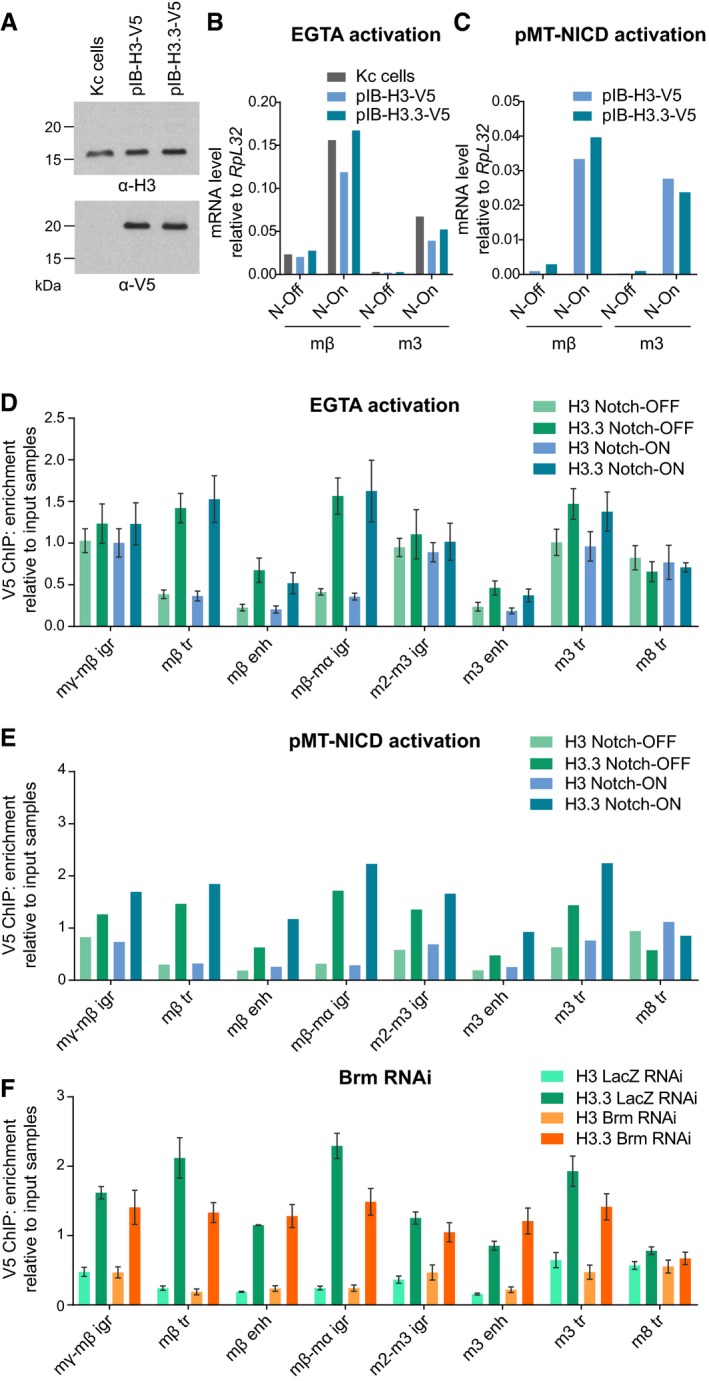
-
AH3‐V5 and H3.3‐V5 expression in stable cell lines compared to un‐transfected “Kc cells”, demonstrated by Western blots probed with H3 and V5 antibodies. V5‐tagged histones have a larger molecular weight and are not detectable in the H3 blot due to low levels of expression in comparison with endogenous H3.
-
B, CEffect of Notch activation by EGTA (B) or copper‐inducible NICD expression (C) on expression of E(spl)mβ‐HLH and E(spl)m3‐HLH in stable cell lines expressing H3‐V5 and H3.3‐V5, measured by reverse transcription–qPCR. Both methods of activation strongly induce both genes. “N‐On” denotes EGTA or copper treatment, and “N‐Off” denotes PBS alone or no copper.
-
D, ENotch activation does not affect H3 and H3.3 levels across the E(spl)‐C. V5 ChIP–qPCR in Kc cells expressing H3‐V5 or H3.3‐V5 from a ubiquitous promoter with Notch signaling activated by EGTA (D) or 6 h of copper‐inducible NICD expression (E), shown as fold enrichment over input samples. H3‐V5 and H3.3‐V5 show a differential pattern across the E(spl)‐C, but Notch activation causes no detectable change in levels compared to controls treated with PBS (D) or no copper (E). Mean ± SEM, n = 3 (D); mean, n = 2 (E).
-
FV5 ChIP–qPCR in Kc cells expressing H3‐V5 or H3.3‐V5 from a ubiquitous promoter after lacZ or brm RNAi treatment, shown as fold enrichment over input samples. The changes caused by brm RNAi are minimal and do not occur at enhancer regions. Mean ± SEM; n = 3.
To test the dynamics of the two histone variants in this system, we then expressed H3‐V5 and H3.3‐V5 from the pMT promoter so that we could monitor their incorporation over a relatively short timescale. By approximately 90 min after their induction, the labeled histones had started to be incorporated into the chromatin (Fig EV5). By 3 h after induction, differential incorporation of H3.3‐V5 could be observed at specific regions, replicating the pattern seen across the E(spl)‐C with CATCH‐IT, while the levels of H3‐V5 incorporation were lower and largely uniform (Fig 5D). Crucially, the incorporation of H3.3‐V5 was greatly reduced following depletion of Brm, in agreement with the BRM complex having a key role in nucleosome turnover.
Figure EV5. Time‐course of copper‐inducible histone‐V5 expression.
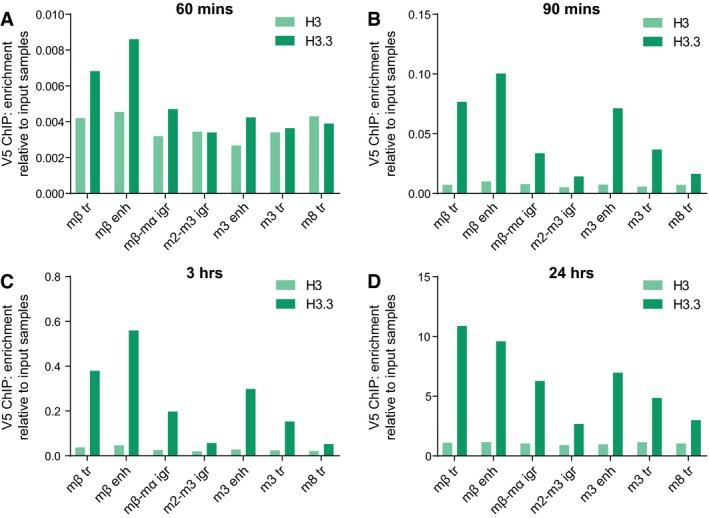
-
A–DV5 ChIP–qPCR in Kc cells with H3‐V5 and H3.3‐V5 expression induced by copper from the pMT promoter for 60 min (A), 90 min (B), 3 h (C), and 24 h (D), shown as fold enrichment over input samples. Differential incorporation of H3.3 across the E(spl)‐C is clear after 90 min. n = 1 (A and B); mean, n = 2 (C and D).
Discussion
In summary, we have shown that the BRM chromatin remodeling complex is essential for Notch‐responsive gene activation. We propose that the BRM complex is required to maintain high levels of accessibility at Notch‐responsive enhancers where it promotes rapid nucleosome turnover. Techniques that measure the nucleosome dynamics were critical in uncovering the role of the BRM complex, since the steady state levels of histones bound to the DNA do not change. Furthermore, the requirement for the ARP BAP55, whose homolog has been implicated in histone eviction 46, lends further support for our model that the BRM complex promotes nucleosome turnover. This has two implications for Notch signaling. Firstly, in the absence of Notch signaling, the BRM complex brings about local turnover of nucleosomes that enables Su(H) to access enhancers with its co‐repressors, poising the enhancers for activation (Fig 6A). Secondly, BRM is responsible for the dramatic increase in chromatin accessibility at responsive genes following Notch activation (Fig 6B). It is possible that the BRM complex also plays a key role in switching off the Notch response upon cessation of signaling, since the continual turnover of nucleosomes provides a mechanism for the rapid resetting of chromatin states. In future, more fine‐grained studies will be needed to determine precisely which nucleosomes are targeted by BRM complexes and what the dynamics of these interactions are.
Figure 6. Model of BRM complex action.
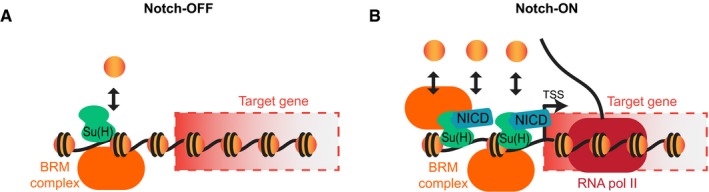
- In the absence of Notch signaling, the BRM complex maintains the accessibility of Notch‐responsive enhancers to allow Su(H) recruitment by promoting nucleosome turnover.
- When Notch signaling is activated, the nucleosome turnover at Notch‐responsive enhancers increases, increasing the accessibility of the chromatin and allowing more Su(H) to bind. The BRM complex is essential for the process to occur. Target genes are activated via co‐activators.
Our evidence for the involvement of SWI/SNF chromatin remodeling in Notch signaling responses, even before signaling takes place, is fully consistent with a previous observation that BRM was recruited to Hes1 and Hes5 in mouse myoblasts before Notch induction 52. It also fits with genetic data that hinted at a role for the BRM complex in regulating Notch target genes 22. The fact that an antagonistic effect of Brm on Notch activity has been detected in some contexts 24 may be attributed to this role in enabling initial recruitment of CSL, as it would bring associated co‐repressors 53, 54, 55. Furthermore, since we observe consistent effects from inhibiting the BRM complex on Notch‐regulated transcription in two different contexts, it is likely that the recruitment of SWI/SNF complexes will be a key step in selecting enhancer repertoires in different cell types. The SWI/SNF‐dependent nucleosome turnover is therefore likely to have an integral role in generating accessible enhancer landscapes critical for the specificity of signaling pathway responses. To achieve this, BRM complexes must be recruited to target enhancers by the cell‐type transcription factors that confer specificity. Candidates in the case of Notch include Runx and GATA factors which are associated with regulated enhancers in blood cell lineages (both in flies and in mammals) 3, 7, 56, 57. Similarly, it has been suggested that the pioneer factors GATA3 and FOXA1 are required to recruit SWI/SNF complexes to enable binding of the glucocorticoid receptor at responsive sites 58.
The subsequent role of the BRM complex in bringing about a dramatic change in chromatin accessibility at responsive genes following Notch activation is critical for their upregulation (Fig 6B). A striking feature of activated enhancers is that they exhibit a substantial increase in certain histone modifications 4, 59, 60, which could be facilitated by the high level of histone turnover that we detect. Indeed, a recent study has found that nucleosome turnover is predictive of the propagation of histone modifications 61. Furthermore, the recruitment of CSL complexes is greatly enhanced following Notch activation 6, 7, 62 and may be an essential step for engaging sufficient low‐affinity interactions with Mediator to promote active initiation of transcription. We have previously shown that this enhanced recruitment involves changes in chromatin accessibility 8, and we now demonstrate that the BRM complex is required to enact this change. A similar model has been proposed for serum stimulation acting via FOS/JUN, where serum‐induced changes in chromatin accessibility relied on BRM recruitment 17. As several studies have shown direct interactions between NICD complexes and SWI/SNF subunits in a range of contexts 23, 24, 52, it is plausible that this interaction is responsible for SWI/SNF complex recruitment in Notch‐ON conditions. Certainly, the BRM complex facilitates the recruitment of Su(H) in Notch‐ON conditions, as evident from the depleted band of fluorescence in the presence of BrmK804R (Fig 2C), and most likely does so by promoting nucleosome turnover. The final outcome therefore differs in Notch‐ON versus and Notch‐OFF conditions, but it remains unclear how this is brought about. For example, does BRM target different nucleosomes or interact with more prolonged dynamics in the Notch‐ON state?
Our results argue therefore that SWI/SNF complexes are likely to play an integral role in Notch target enhancer activation. How general this effect of the BRM complex will be is an open question—to what extent is SWI/SNF remodeling critical for all types of enhancer activity? In the basal state, this complex does not appear to be required in every case, as neither heat‐shock nor ecdysone‐regulated loci exhibited a similar loss of accessibility when the BRM complex was perturbed. However, the mammalian SWI/SNF subunit Brg1 is required for a robust transcriptional response to glucocorticoid and was found to occupy many glucocorticoid response elements prior to hormone treatment 58. Thus, it has been proposed that the selection and activation of hormone‐responsive enhancers is reliant on the pre‐patterning of specialized chromatin environments through the actions of SWI/SNF complexes in a similar manner to that proposed here. Elucidating how widely the SWI/SNF regulation of nucleosome turnover that we have detected in Notch‐responsive regions underpins other classes of enhancer activation will be important, especially in the context of the frequency of mutations affecting SWI/SNF subunits in a broad spectrum of cancers.
Materials and Methods
Fly stocks
For expression of all UAS constructs in the salivary gland, 1151‐Gal4 was used (L S Shashidhara, Centre for Cellular and Molecular Biology, Hyderabad, India) 63. Notch signaling was activated by UAS‐NΔECD 30, 31. The E(spl)‐C was imaged using the ParB‐INT DNA tagging system where UAS‐ParB1‐mCherry was expressed in the presence of the INT sequence inserted between E(spl)m7‐HLH and E(spl)m8‐HLH 8, 64. Histone‐GFP imaging made use of UAS‐H3‐GFP, UAS‐H3.3‐GFP, and UAS‐H3.3core‐GFP constructs (flies kindly provided by Kami Ahmad) 32, 65. Dynamic H3.3‐mKO imaging made use of the UAS‐FRT‐H3.3‐GFP‐PolyA‐FRT‐H3.3‐mKO‐PolyA construct crossed with hs‐FLP (flies kindly provided by Xin Chen) 34. Su(H) recruitment was monitored using Su(H)‐GFP 8. Dominant‐negative Brm was expressed from UAS‐BrmK804R 40. RNAi lines used are listed in Table 1.
Table 1.
RNAi lines used
| RNAi | Sourcea | Phenotype shown in published work |
|---|---|---|
| w | BL‐35573 | Used as a control in 8, 68 |
| Iswi | BL‐32845 | 69 |
| Chrac‐16 | BL‐51155 | 69 |
| MTA1‐like | BL‐33745 | 68 |
| Ino80 | BL‐33708 | 70 |
| Tip60 | BL‐28563 | 71 |
| Chd1 | BL‐34665 | 70 |
| Chd3 | V‐13636 | Not used previously |
| Snr1 | BL‐32372 | 72 |
| Moira | BL‐34919 | 73 |
| osa (1) | BL‐31266 | 74 |
| osa (2) | V‐7810 | 75 |
| Bap170 | BL‐26308 | 76 |
| polybromo | BL‐32840 | 76 |
| Caf1‐55 | V‐26455 | 77 (the same construct used, inserted in a different chromosome) |
| Caf1‐180 | BL‐28918 | 77 |
| Dek | BL‐28696 | Not used previously |
| yem | V‐26808 | 78 |
| zeste | BL‐31615 | Not used previously |
Bloomington Drosophila Stock Center is abbreviated to BL, and Vienna Drosophila RNAi Center is abbreviated to V.
Live imaging of salivary gland nuclei
Salivary glands were dissected and mounted as described previously 8, using Shields and Sang M3 Insect Medium (Sigma S3652) supplemented with 5% FBS (Sigma F9665) and 1× Antibiotic‐Antimycotic (Gibco 15240062) for dissection and the same medium with the addition of 2.5% methyl‐cellulose (Sigma) for mounting. For DNA stains, salivary glands were incubated in dissecting media containing 200 μg/ml Hoechst 33342 (ThermoFisher) for 10 min at room temperature before washing with PBS and mounting.
Image acquisition was performed with Nikon D‐Eclipse C1 confocal microscope using lasers at 405, 488, and 543 nm. Images captured of nuclei used the 60× oil objective with a 4.5× zoom level, and images of glands used the 40× oil objective. To monitor Su(H)‐GFP recruitment, nuclei were scanned slowly through the Z‐stack using a 2× zoom level while looking for accumulations of fluorescence which were scored as bands. The scoring was not conducted blind to genotype. However, strict criteria were used to reduce subjective bias: The band of fluorescence must be bright, occupy a volume in the z‐axis, and most importantly, persist during microscope scanning. Ten glands and five nuclei per gland were analyzed and scored per condition, with the five nuclei closest to the coverslip chosen each time.
For dynamic H3.3‐mKO imaging, heat shocks at 37°C were performed on larvae for 1 h approximately 24 h before imaging. Twenty‐four hours was chosen as a time point shortly after the mKO signal was first detectable. Image acquisition was performed with an Olympus FV1000 confocal microscope using a 60×/1.35 NA objective with spectral detectors. GFP was imaged using a laser at 488 nm, and subsequently, mKO and mCherry were imaged simultaneously using a laser at 543 nm to excite both fluorophores. The spectral detectors were set to detect wavelengths of 555–585 nm for mKO and 680–750 nm for mCherry. These wavelengths were chosen to ensure that the signal from mCherry was not observed in the mKO channel. To ensure that the two signals were fully distinguished, the Spectral_Unmixing plugin (Joachim Walter) was used in Fiji 66 to process the images before quantification.
For quantifications of histone‐GFP and mKO, representative images where the E(spl)‐C could be clearly observed were used with the Fiji software 66 as follows. The images were rotated such that the E(spl)‐C was vertical and a rectangle 1.29 μm by 2.58 μm was placed over it, centered on the peak fluorescence of the ParB‐mCherry marker. The “plot profile” function was used to obtain mean fluorescence intensity across the rectangle in each channel. Arbitrary fluorescence values were adjusted such that the highest value obtained was set to 1 and the lowest to 0, and the mean values were taken from several nuclei (n numbers given in figure legend).
Immunofluorescence staining
Staining of salivary glands was performed as described 8 except for the following changes. Glands were permeabilized with 1% Triton X‐100 in PBS for 30 min. Antibodies against OSA and BAP170 were gifts from Peter Verrijzer 67 and were used at dilutions of 1:200 and 1:100, respectively.
Kc167 cell culture, Notch activation, and generation of stable lines
Kc167 cells (Drosophila Genomics Resource Center) were cultured in Schneider's Drosophila medium (Gibco 21720024) supplemented with 5% FBS (Sigma F9665) and 1× Antibiotic‐Antimycotic (Gibco 15240062) at 25°C. Notch was activated either by NICD expression from the pMT vector (cell line described below) or by EGTA treatment where the medium was replaced with 4 mM EGTA (Bioworld) in PBS for 30 min.
Stable cell lines were generated by transfection followed by antibiotic selection. 18 μg of the relevant plasmid was mixed with 925 μl Opti‐MEM (Gibco 31985070) and 54 μl FuGENE HD Transfection Reagent (Promega E2311) at room temperature for 30 min before adding dropwise to cells plated in 10‐cm plates. After 24–48 h, the medium was replaced to contain antibiotic for selection. Cells were grown in the presence of antibiotic and experiments were performed after significant cell death and recovery had taken place to indicate selection (usually after approximately 3 weeks).
CATCH‐IT was performed in the pMT‐NICD cell line generated previously 4, where cells were maintained with 2 μg/ml puromycin (Sigma).
Cell lines expressing BrmWT and BrmK804R were generated using plasmids kindly provided by Neus Visa 43. BrmK804R was re‐made by mutagenesis to ensure homogeneity between the two constructs using Pfu polymerase with the primers listed in Table 2. The BrmWT and BrmK804R sequences were then cloned into the pMT‐puro vector (Addgene 17923) by digestion with SpeI and PmeI (NEB) and ligation (T4 ligase; Promega). After transfection of pMT‐BrmWT and pMT‐BrmK804R, cells were selected with 5 μg/ml and maintained with 2 μg/ml puromycin.
Table 2.
All primers used
| Name | Sequence |
|---|---|
| BrmK804R mutagenesis | |
| K804R_forward | CCGATGAAATGGGTTTGGGTCGAACCATTCAAACCATTTCGC |
| K804R_reverse | GCGAAATGGTTTGAATGGTTCGACCCAAACCCATTTCATCGG |
| Histone‐V5 cloning into pMT vector | |
| SpeI_his_forward | CCTACTAGTCATGGCTCGTACCAAGCAAACTGC |
| XhoI_his_reverse | CACCTCGAGGCGGCCGCCACTGTGCTGGATA |
| T7 primers for making double‐stranded RNA | |
| T7_GFP_forward | TAGTAATACGACTCACTATAGGGAAAGGGCAGATTGTGTGGAC |
| T7_GFP_reverse | TAGTAATACGACTCACTATAGGGTCAAGGACGACGGGAACTAC |
| T7_LacZ_forward | CCGAATTAATACGACTCACTATAGGGCCGCTGGATAACGACATTGG |
| T7_LacZ_reverse | CCGAATTAATACGACTCACTATAGGGGACTGTAGCGGCTGATGTTG |
| T7_Brm_forward | TAGTAATACGACTCACTATAGGGCGATCATAAACCCAAGGTGG |
| T7_Brm_reverse | TAGTAATACGACTCACTATAGGGCAGTGATGGTTCTTCATGCG |
| T7_Snr1_forward | TAGTAATACGACTCACTATAGGGCTGGACATGGAGCTAGAGGG |
| T7_Snr1_reverse | TAGTAATACGACTCACTATAGGGCGTCGGTAAGCGTCTCTAGG |
| T7_Bap55_forward | TAGTAATACGACTCACTATAGGGACGGCATGATTGACAACTGG |
| T7_Bap55_reverse | TAGTAATACGACTCACTATAGGGTGCCACGACTGAGTTAGGTT |
| qPCR: E(spl)‐C | |
| mγ‐mβ igr_forward | GGAGTTGAGGAGTTGGTCG |
| mγ‐mβ igr_reverse | ATAAGTGTGGTTGGGTGCCT |
| mβ tr_forward | AGAAGTGAGCAGCAGCCATC |
| mβ tr_reverse | GCTGGACTTGAAACCGCACC |
| mβ enh_forward | AGAGGTCTGTGCGACTTGG |
| mβ enh_reverse | GGATGGAAGGCATGTGCT |
| mβ‐mα igr_forward | AAGCCAGTGGACTCTGCTCT |
| mβ‐mα igr_reverse | TGATCTCCAAGCGGAGTATG |
| mα tr_forward | GCAGGAGGACGAGGAGGATG |
| mα tr_reverse | GATCCTGGAATTGCATGGAG |
| m2‐m3 igr_forward | GCGCGTATTTCCCAAATAAA |
| m2‐m3 igr_reverse | GATTGTACGTGCATGGGAAA |
| m3 enh_forward | ACACACACAAACACCCATCC |
| m3 enh_reverse | CGAGGCAGTAGCCTATGTGA |
| m3 tr_forward | CGTCTGCAGCTCAATTAGTC |
| m3 tr_reverse | AGCCCACCCACCTCAACCAG |
| m8 tr_forward | CAATTCCACGAAGCACAGTC |
| m8 tr_reverse | GAGGAGCAGTCCATCGAGTT |
| qPCR: additional controls for ATAC | |
| Rab11 tr_forward | ACTGAAAATGGGCCGTTTCG |
| Rab11 tr_reverse | AGGAGTGGTAATCGACGGTC |
| Eip78C enh_forward | AGAAGTAGGGGCCGTCAAGT |
| Eip78C enh_reverse | GTGTAAGACCCGTCGCATTT |
| Closed ctrl_forward | GCATTTTTGTGGCAGAGGCA |
| Closed ctrl_reverse | CTCTTTCGGTGTCGCCTTCT |
| Mst87F tr_forward | ATCCTTTGCCTCTTCAGTCC |
| Mst87F tr_reverse | AATAATGATACAAAATCTGGTTACGC |
| Hsp26 tr_forward | TTGAATTCGATCTGTGCTCTGT |
| Hsp26 tr_reverse | CGGGTATAAAAGCAGCGTCG |
| Hsp70 enh_forward | TCGTTTTGTGACTCTCCCTCT |
| Hsp70 enh_reverse | TGTGACAGAGTGAGAGAGCA |
| Hr4 enh_forward | GGCACCTGACGGTTGATAGT |
| Hr4 enh_reverse | CAGCCCGAAGAATCTACCAG |
| Dip‐B tr_forward | TCAACTGCAACCGGATGATA |
| Dip‐B tr_reverse | ATAACCTCATCGGCCACGTA |
| Eip75B tr_forward | AGCAACTTGGCCAGGAACT |
| Eip75B tr_reverse | AACCTGGAGCTGATCGAGAA |
| CTPsyn tr_forward | TCGATTGTTGTTGGCTGAGC |
| CTPsyn tr_reverse | TTCCTTCGCTCTTCCTGTCC |
| fru tr_forward | CTCTTTCGCACACTTGGCAT |
| fru tr_reverse | CCGTTCGTTGCCCATCTAAG |
| kay tr_forward | CTCTCTCATTGGCTCTCCCC |
| kay tr_reverse | TGAAGCGGAGACCACACAAT |
| vri tr_forward | TGTGTGTTTGTGTCTGCGAG |
| vri tr_reverse | TCACTCACCCTCACCATGAC |
| qPCR: additional controls for RT–qPCR | |
| RpL32_forward | ACGTTGTGCACCAGGAACTT |
| RpL32_reverse | CCGCTTCAAGGGACAGTATC |
| RpII215_forward | GACTCGACTGGAATTGCACC |
| RpII215_reverse | TCTTCATCGGGATACTCGCC |
| qPCR: additional controls for CATCH‐IT | |
| PPO1 enh_forward | AAGTCCCAACCGCAAAACTG |
| PPO1 enh_reverse | GCTATCGACTAAACCACAACGT |
| Him‐Her enh_forward | CGAACCGAGTTGTGGGAAAT |
| Him‐Her enh_reverse | CCCTTGGAGTGACAATTAGCTG |
| Rab11 tr_forward | ACTGAAAATGGGCCGTTTCG |
| Rab11 tr_reverse | AGGAGTGGTAATCGACGGTC |
| Sec15 tr_forward | GGTAGCGGTTCTCTTGCTTG |
| Sec15 tr_reverse | GTAACCGTCAGCTGTTGGAC |
Constitutive expression of histone‐V5 proteins made use of pIB‐H3‐V5 and pIB‐H3.3‐V5 plasmids kindly provided by Dirk Schübeler and used as described 51. Cells were selected with 50 μg/ml and maintained with 20 μg/ml blasticidin (ThermoFisher R21001). For Notch activation in these cells, they were further transfected with pMT‐NICD, and selected and then maintained with 5 and 2 μg/ml puromycin, respectively.
For inducible expression of histone‐V5 proteins, H3 and H3.3 sequences were cloned from pIB‐H3‐V5 and pIB‐H3.3‐V5 into the pMT‐puro vector using SpeI and XhoI sites (NEB) with the primers listed in Table 2. After transfection of pMT‐H3‐V5 and pMT‐H3.3‐V5, cells were selected with 5 μg/ml and maintained with 2 μg/ml puromycin.
To induce expression from all pMT constructs, 5 mM CuSO4 was added to normal culture media. Induction was performed for 24 h for experiments with pMT‐BrmWT and pMT‐BrmK804R, and for the lengths of time specified for other experiments (see CATCH‐IT method for details in this experiment).
Assay for transposase‐accessible chromatin
ATAC using salivary glands was performed exactly as described previously with no changes 8. ATAC was performed in Kc167 cells in a similar manner with the following changes. After a 30‐min EGTA treatment in 10‐cm culture plates containing approximately 40 million cells, cells were immediately harvested taking a quarter of the cells for the experiment (roughly 10 million). Cells were pelleted at 500 × g at 4°C for 5 min, washed in 10 ml of cold PBS, and pelleted again. The cells were then lysed by resuspending in 50 μl lysis buffer (10 mM Tris–HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.3% NP‐40), vortexing for 10 s, keeping on ice for 3 min, and vortexing again. Nuclei were pelleted at 400 × g at 4°C for 5 min and resuspended in 30 μl TD buffer (Illumina FC‐121‐1030). 25 μl was used for the tagmentation reaction and the rest of the protocol performed exactly as described previously for salivary glands 8.
RNAi in Kc167 cells
300‐ to 800‐base pair regions of brm, Snr1, and Bap55 DNA were amplified from genomic DNA, with GFP or lacZ sequences amplified from plasmids as controls, using either Q5 or Phusion High‐Fidelity DNA Polymerases (NEB M0491 and M0530, respectively) and overhanging primers containing the T7 promoter sequence listed in Table 2. In vitro transcription was performed using the MEGAscript T7 Transcription Kit (Invitrogen AM1334). RNA was purified by phenol–chloroform extraction and then annealed to form double‐stranded RNA by heating to 75°C and cooling slowly. 100 μg double‐stranded RNA was mixed with 3.5 ml Opti‐MEM (Gibco 31985070) and added to approximately 10 million cells in a 10‐cm plate for 30 min before topping up to 10 ml with normal culture medium. Volumes were scaled down for some smaller experiments.
RNA extraction and reverse transcription
To extract RNA from Kc167 cells, TRI reagent solution (Invitrogen AM6738) was used followed by phenol–chloroform extraction and isopropanol precipitation at −20°C overnight. For reverse transcription, RNA was resuspended in water and first DNase‐treated with the DNA‐free DNA Removal Kit (Invitrogen AM1906), before reverse‐transcribing with M‐MLV Reverse Transcriptase (Promega M1705) using Oligo(dT)15 Primers (Promega C1101). cDNA was diluted fivefold before analysis with qPCR.
The same protocol was used to extract RNA from salivary glands, exactly as described previously 8.
Western blot
Approximately 20 million Kc167 cells were lysed in 100 μl lysis buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 10% glycerol, 0.5% Triton X‐100) on ice for 30 min before debris was removed by centrifugation at 13,000 × g at 4°C for 30 min. Samples were then combined with 2× loading buffer (10 mM Tris–HCl, pH 6.8, 20% glycerol, 4% SDS, 0.025% bromophenol blue, 2% mercaptoethanol) and boiled. Proteins were resolved using standard protocols with 15% SDS–PAGE and transferred to nitrocellulose. Blots were probed with antibodies against histone H3 (Abcam ab1791) and V5 (Invitrogen R960‐25) at dilutions of 1:1,000 and 1:4,000, respectively. Horseradish peroxidase‐conjugated secondary antibodies were used and detected with the ECL system (GE Life Sciences).
Chromatin immunoprecipitation
Su(H) and V5 ChIP were performed largely as described previously 4, 62, using 2.5 μg goat Su(H) antibody (Santa Cruz Biotechnology, no longer available) and 1‐2 μg V5 antibody (Invitrogen R960‐25). Briefly, cells were cross‐linked with 1% formaldehyde (Sigma F8775) in PBS for 10 min at 25°C. After lysis, chromatin was diluted twofold for sonication and then a further fivefold for pre‐clearing with goat or mouse IgG and 40 μl protein G or protein A/G PLUS‐Agarose (Santa Cruz Biotechnology sc‐2002 and sc‐2003) for Su(H) and V5 ChIP, respectively. Immunoprecipitation was performed with 40 μl of the same beads at 4°C overnight, followed by washes, elution by vortexing, and de‐cross‐linking with 0.3 M NaCl, 0.1 mg/ml RNase A and 0.1 mg/ml proteinase K treatment. DNA was purified with the QIAquick PCR purification kit (Qiagen) and eluted in 100 μl water for analysis with qPCR.
CATCH‐IT
Schneider's Drosophila medium without methionine (PAN Biotech), supplemented with 5% FBS and 1× Antibiotic‐Antimycotic was added to cells for 1 h, followed by adding either 4 mM azidohomoalanine (Aha; AnaSpec AS‐63669) or 4 mM methionine (Sigma) as a control for 4 h. To activate Notch, pMT‐NICD cells were induced with 5 mM CuSO4 for 1 h before the medium was substituted for methionine‐free medium, also containing 5 mM CuSO4, so that cells were incubated with CuSO4 for a total of 6 h.
CATCH‐IT was performed as previously described 50, except where stated otherwise. Briefly, cells were harvested and nuclei were extracted with 30 μl of 10% NP‐40. Nuclei were resuspended in 180 μl of HB125 buffer, and the following were added: 5 μl of 2 nM biotin‐alkyne (Invitrogen B10185), 10 μl of 100 mM THPTA (Sigma 762342) premixed with 2 μl of 100 mM copper sulfate (Jena Bioscience CLK‐M1004), and 6 μl of freshly prepared 500 mM sodium ascorbate (Jena Bioscience CLK‐M1005). Cycloaddition reaction was performed for 30 min at room temperature on a rotor. Reaction with MNase (Sigma N3755) was performed at 37°C for 3 min. After capture with Dynabeads M‐280 Streptavidin (Invitrogen 11205) as described, captured chromatin and input chromatin samples were treated with 0.25 mg/ml RNase A (Roche) and 0.25 mg/ml proteinase K (ThermoFisher). DNA was purified with the QIAquick PCR purification kit (Qiagen) and analyzed by qPCR.
qPCR
All qPCR was performed using LightCycler 480 SYBR Green I Mastermix (Roche 04707516001) as described previously 8. For all experiments, two technical replicate qPCRs were performed per sample and the mean taken for analysis. Replicate numbers given in figure legends do not count these technical replicates and instead refer only to repeats of the full experimental protocol from start to finish with different cells or animals (biological replicates). For reverse transcription experiments, relative amounts of the genes of interest were normalized to the control gene RpL32. For ChIP, immunoprecipitated samples were normalized to input samples. For CATCH‐IT, pulldown samples were normalized to input samples and then to the Sec15 transcribed region. All primers used are shown in Table 2.
Author contributions
SJB and ZP conceived the project and wrote the manuscript, SJB supervised the work, and ZP performed the experiments and analyzed the data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Jin Li, Damiano Porcelli, and members of the Bray Lab for technical advice and helpful discussions. This work was supported by a Programme Grant from the Medical Research Council to SJB (MR/L007177/1) and by a studentship from BBSRC for ZP (1502069).
EMBO Reports (2019) 20: e46944
References
- 1. Penton AL, Leonard LD, Spinner NB (2012) Notch signaling in human development and disease. Semin Cell Dev Biol 23: 450–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bray SJ (2016) Notch signalling in context. Nat Rev Mol Cell Biol 17: 722–735 [DOI] [PubMed] [Google Scholar]
- 3. Terriente‐Felix A, Li J, Collins S, Mulligan A, Reekie I, Bernard F, Krejci A, Bray S (2013) Notch cooperates with Lozenge/Runx to lock haemocytes into a differentiation programme. Development 140: 926–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skalska L, Stojnic R, Li J, Fischer B, Cerda‐Moya G, Sakai H, Tajbakhsh S, Russell S, Adryan B, Bray SJ (2015) Chromatin signatures at Notch‐regulated enhancers reveal large‐scale changes in H3K56ac upon activation. EMBO J 34: 1889–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bray SJ, Gomez‐Lamarca M (2018) Notch after cleavage. Curr Opin Cell Biol 51: 103–109 [DOI] [PubMed] [Google Scholar]
- 6. Castel D, Mourikis P, Bartels SJJ, Brinkman AB, Tajbakhsh S, Stunnenberg HG (2013) Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev 27: 1059–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, Blacklow SC, Liu XS, Aster JC (2014) NOTCH1‐RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci USA 111: 705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gomez‐Lamarca MJ, Falo‐Sanjuan J, Stojnic R, Abdul Rehman S, Muresan L, Jones ML, Pillidge Z, Cerda‐Moya G, Yuan Z, Baloul S et al (2018) Activation of the Notch signaling pathway in vivo elicits changes in CSL nuclear dynamics. Dev Cell 44: 611–623 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oswald F, Rodriguez P, Giaimo BD, Antonello ZA, Mira L, Mittler G, Thiel VN, Collins KJ, Tabaja N, Cizelsky W et al (2016) A phospho‐dependent mechanism involving NCoR and KMT2D controls a permissive chromatin state at Notch target genes. Nucleic Acids Res 44: 4703–4720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Giaimo BD, Ferrante F, Vallejo DM, Hein K, Gutierrez‐Perez I, Nist A, Stiewe T, Mittler G, Herold S, Zimmermann T et al (2018) Histone variant H2A.Z deposition and acetylation directs the canonical Notch signaling response. Nucleic Acids Res 46: 8197–8215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nair SS, Kumar R (2012) Chromatin remodeling in cancer: a gateway to regulate gene transcription. Mol Oncol 6: 611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Venkatesh S, Workman JL (2015) Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16: 178–189 [DOI] [PubMed] [Google Scholar]
- 13. Längst G, Manelyte L (2015) Chromatin remodelers: from function to dysfunction. Genes (Basel) 6: 299–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clapier CR, Iwasa J, Cairns BR, Peterson CL (2017) Mechanisms of action and regulation of ATP‐dependent chromatin‐remodelling complexes. Nat Rev Mol Cell Biol 18: 407–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang L, Du Y, Ward JM, Shimbo T, Lackford B, Zheng X, Miao Y, Zhou B, Han L, Fargo DC et al (2014) INO80 facilitates pluripotency gene activation in embryonic stem cell self‐renewal, reprogramming, and blastocyst development. Cell Stem Cell 14: 575–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou B, Wang L, Zhang S, Bennett BD, He F, Zhang Y, Xiong C, Han L, Diao L, Li P et al (2016) INO80 governs superenhancer‐mediated oncogenic transcription and tumor growth in melanoma. Genes Dev 30: 1440–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, Greenberg ME (2017) AP‐1 transcription factors and the BAF complex mediate signal‐dependent enhancer selection. Mol Cell 68: 1067–1082 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu G, Schones DE, Cui K, Ybarra R, Northrup D, Tang Q, Gattinoni L, Restifo NP, Huang S, Zhao K (2011) Regulation of nucleosome landscape and transcription factor targeting at tissue‐specific enhancers by BRG1. Genome Res 21: 1650–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fryer CJ, Archer TK (1998) Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature 393: 88–91 [DOI] [PubMed] [Google Scholar]
- 20. Nagaich AK, Walker DA, Wolford R, Hager GL (2004) Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol Cell 14: 163–174 [DOI] [PubMed] [Google Scholar]
- 21. Kadoch C, Crabtree GR (2015) Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv 1: e1500447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Armstrong JA, Sperling AS, Deuring R, Manning L, Moseley SL, Papoulas O, Piatek CI, Doe CQ, Tamkun JW (2005) Genetic screens for enhancers of brahma reveal functional interactions between the BRM chromatin‐remodeling complex and the Delta‐Notch signal transduction pathway in Drosophila . Genetics 170: 1761–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takeuchi JK, Lickert H, Bisgrove BW, Sun X, Yamamoto M, Chawengsaksophak K, Hamada H, Yost HJ, Rossant J, Bruneau BG (2007) Baf60c is a nuclear Notch signaling component required for the establishment of left–right asymmetry. Proc Natl Acad Sci USA 104: 846–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Das AV, James J, Bhattacharya S, Imbalzano AN, Antony ML, Hegde G, Zhao X, Mallya K, Ahmad F, Knudsen E et al (2007) SWI/SNF chromatin remodeling ATPase Brm regulates the differentiation of early retinal stem cells/progenitors by influencing Brn3b expression and Notch signaling. J Biol Chem 282: 35187–35201 [DOI] [PubMed] [Google Scholar]
- 25. Xie G, Chen H, Jia D, Shu Z, Palmer WH, Huang Y‐C, Zeng X, Hou SX, Jiao R, Deng W‐M (2017) The SWI/SNF complex protein Snr1 is a tumor suppressor in Drosophila imaginal tissues. Cancer Res 77: 862–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bailey AM, Posakony JW (1995) Suppressor of hairless directly activates transcription of enhancer of split complex genes in response to Notch receptor activity. Genes Dev 9: 2609–2622 [DOI] [PubMed] [Google Scholar]
- 27. Cooper MTD, Tyler DM, Furriols M, Chalkiadaki A, Delidakis C, Bray S (2000) Spatially restricted factors cooperate with Notch in the regulation of enhancer of split genes. Dev Biol 221: 390–403 [DOI] [PubMed] [Google Scholar]
- 28. Housden BE, Fu AQ, Krejci A, Bernard F, Fischer B, Tavaré S, Russell S, Bray SJ (2013) Transcriptional dynamics elicited by a short pulse of Notch activation involves feed‐forward regulation by E(spl)/Hes genes. PLoS Genet 9: e1003162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schaaf CA, Misulovin Z, Gause M, Koenig A, Dorsett D (2013) The Drosophila enhancer of split gene complex: architecture and coordinate regulation by notch, cohesin, and polycomb group proteins. G3 (Bethesda) 3: 1785–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fortini ME, Rebay I, Caron LA, Artavanis‐Tsakonas S (1993) An activated Notch receptor blocks cell‐fate commitment in the developing Drosophila eye. Nature 365: 555–557 [DOI] [PubMed] [Google Scholar]
- 31. Rebay I, Fehon RG, Artavanis‐Tsakonas S (1993) Specific truncations of Drosophila Notch define dominant activated and dominant negative forms of the receptor. Cell 74: 319–329 [DOI] [PubMed] [Google Scholar]
- 32. Ahmad K, Henikoff S (2002) The histone variant H3.3 marks active chromatin by replication‐independent nucleosome assembly. Mol Cell 9: 1191–1200 [DOI] [PubMed] [Google Scholar]
- 33. Mito Y, Henikoff JG, Henikoff S (2005) Genome‐scale profiling of histone H3.3 replacement patterns. Nat Genet 37: 1090–1097 [DOI] [PubMed] [Google Scholar]
- 34. Tran V, Lim C, Xie J, Chen X (2012) Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science 338: 679–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bouazoune K, Brehm A (2006) ATP‐dependent chromatin remodeling complexes in Drosophila . Chromosom Res 14: 433–449 [DOI] [PubMed] [Google Scholar]
- 36. Sawatsubashi S, Murata T, Lim J, Fujiki R, Ito S, Suzuki E, Tanabe M, Zhao Y, Kimura S, Fujiyama S et al (2010) A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev 24: 159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orsi GA, Algazeery A, Meyer RE, Capri M, Sapey‐Triomphe LM, Horard B, Gruffat H, Couble P, Aït‐Ahmed O, Loppin B (2013) Drosophila Yemanuclein and HIRA cooperate for de novo assembly of H3.3‐containing nucleosomes in the male pronucleus. PLoS Genet 9: e1003285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mohrmann L, Verrijzer CP (2005) Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochim Biophys Acta Gene Struct Expr 1681: 59–73 [DOI] [PubMed] [Google Scholar]
- 39. Hodges C, Kirkland JG, Crabtree GR (2016) The many roles of BAF (mSWI/SNF) and PBAF complexes in cancer. Cold Spring Harb Perspect Med 6: a026930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elfring LK, Daniel C, Papoulas O, Deuring R, Sarte M, Moseley S, Beek SJ, Waldrip WR, Daubresse G, DePace A et al (1998) Genetic analysis of brahma: the Drosophila homolog of the yeast chromatin remodeling factor SWI2/SNF2. Genetics 148: 251–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (2015) ATAC‐seq: a method for assaying chromatin accessibility genome‐wide Curr Protoc Mol Biol 109: 21.29.1–21.29.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rand MD, Grimm LM, Artavanis‐Tsakonas S, Patriub V, Blacklow SC, Sklar J, Aster JC (2000) Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol 20: 1825–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu S, Waldholm J, Böhm S, Visa N (2014) Brahma regulates a specific trans‐splicing event at the mod(mdg4) locus of Drosophila melanogaster . RNA Biol 11: 134–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cairns BR, Erdjument‐Bromage H, Tempst P, Winston F, Kornberg RD (1998) Two actin‐related proteins are shared functional components of the chromatin‐remodeling complexes RSC and SWI/SNF. Mol Cell 2: 639–651 [DOI] [PubMed] [Google Scholar]
- 45. Schubert HL, Wittmeyer J, Kasten MM, Hinata K, Rawling DC, Héroux A, Cairns BR, Hill CP (2013) Structure of an actin‐related subcomplex of the SWI/SNF chromatin remodeler. Proc Natl Acad Sci USA 110: 3345–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clapier CR, Kasten MM, Parnell TJ, Viswanathan R, Szerlong H, Sirinakis G, Zhang Y, Cairns BR (2016) Regulation of DNA translocation efficiency within the chromatin remodeler RSC/Sth1 potentiates nucleosome sliding and ejection. Mol Cell 62: 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang C, Zhu B (2014) H3.3 turnover: a mechanism to poise chromatin for transcription, or a response to open chromatin? BioEssays 36: 579–584 [DOI] [PubMed] [Google Scholar]
- 48. Deaton AM, Gómez‐Rodríguez M, Mieczkowski J, Tolstorukov MY, Kundu S, Sadreyev RI, Jansen LE, Kingston RE (2016) Enhancer regions show high histone H3.3 turnover that changes during differentiation. Elife 5: e15316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deal RB, Henikoff JG, Henikoff S (2010) Genome‐wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328: 1161–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Teves SS, Deal RB, Henikoff S (2012) Measuring genome‐wide nucleosome turnover using CATCH‐IT. Methods Enzymol 513: 169–184 [DOI] [PubMed] [Google Scholar]
- 51. Wirbelauer C, Bell O, Schübeler D (2005) Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter‐proximal bias. Genes Dev 19: 1761–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kadam S, Emerson BM (2003) Transcriptional specificity of human SWI/SNF BRG1 and BRM chromatin remodeling complexes. Mol Cell 11: 377–389 [DOI] [PubMed] [Google Scholar]
- 53. Kao HY, Ordentlich P, Koyano‐Nakagawa N, Tang Z, Downes M, Kintner CR, Evans RM, Kadesch T (1998) A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev 12: 2269–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hsieh JJ, Zhou S, Chen L, Young DB, Hayward SD (1999) CIR, a corepressor linking the DNA binding factor CBF1 to the histone deacetylase complex. Proc Natl Acad Sci USA 96: 23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Collins KJ, Yuan Z, Kovall RA (2014) Structure and function of the CSL‐KyoT2 corepressor complex: a negative regulator of Notch signaling. Structure 22: 70–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Muratoglu S, Hough B, Mon ST, Fossett N (2007) The GATA factor Serpent cross‐regulates lozenge and u‐shaped expression during Drosophila blood cell development. Dev Biol 311: 636–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mehta C, Johnson KD, Gao X, Ong IM, Katsumura KR, McIver SC, Ranheim EA, Bresnick EH (2017) Integrating enhancer mechanisms to establish a hierarchical blood development program. Cell Rep 20: 2966–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hoffman JA, Trotter KW, Ward JM, Archer TK (2018) BRG1 governs glucocorticoid receptor interactions with chromatin and pioneer factors across the genome. Elife 7: e35073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T‐K, Koche RP et al (2007) Genome‐wide maps of chromatin state in pluripotent and lineage‐committed cells. Nature 448: 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA et al (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 107: 21931–21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chory EJ, Calarco JP, Hathaway NA, Bell O, Neel DS, Crabtree GR (2019) Nucleosome turnover regulates histone methylation patterns over the genome. Mol Cell 73: 61–72 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Krejci A, Bray S (2007) Notch activation stimulates transient and selective binding of Su(H)/CSL to target enhancers. Genes Dev 21: 1322–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Roy S, VijayRaghavan K (1997) Homeotic genes and the regulation of myoblast migration, fusion, and fibre‐specific gene expression during adult myogenesis in Drosophila . Development 124: 3333–3341 [DOI] [PubMed] [Google Scholar]
- 64. Saad H, Gallardo F, Dalvai M, Tanguy‐le‐Gac N, Lane D, Bystricky K (2014) DNA dynamics during early double‐strand break processing revealed by non‐intrusive imaging of living cells. PLoS Genet 10: e1004187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Henikoff S, Ahmad K, Platero JS, van Steensel B (2000) Heterochromatic deposition of centromeric histone H3‐like proteins. Proc Natl Acad Sci USA 97: 716–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moshkin YM, Mohrmann L, van Ijcken WFJ, Verrijzer CP (2007) Functional differentiation of SWI/SNF remodelers in transcription and cell cycle control. Mol Cell Biol 27: 651–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nikalayevich E, Ohkura H (2015) The NuRD nucleosome remodelling complex and NHK‐1 kinase are required for chromosome condensation in oocytes. J Cell Sci 128: 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Börner K, Jain D, Vazquez‐Pianzola P, Vengadasalam S, Steffen N, Fyodorov DV, Tomancak P, Konev A, Suter B, Becker PB (2016) A role for tuned levels of nucleosome remodeler subunit ACF1 during Drosophila oogenesis. Dev Biol 411: 217–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ellis K, Friedman C, Yedvobnick B (2015) Drosophila domino exhibits genetic interactions with a wide spectrum of chromatin protein‐encoding loci. PLoS One 10: e0142635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kwon MH, Callaway H, Zhong J, Yedvobnick B (2013) A targeted genetic modifier screen links the SWI2/SNF2 protein domino to growth and autophagy genes in Drosophila melanogaster . G3 (Bethesda) 3: 815–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Berson A, Sartoris A, Nativio R, Van Deerlin V, Toledo JB, Porta S, Liu S, Chung C‐Y, Garcia BA, Lee VM‐Y et al (2017) TDP‐43 promotes neurodegeneration by impairing chromatin remodeling. Curr Biol 27: 3579–3590 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oh H, Slattery M, Ma L, Crofts A, White KP, Mann RS, Irvine KD (2013) Genome‐wide association of Yorkie with chromatin and chromatin‐remodeling complexes. Cell Rep 3: 309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Anderson AE, Galko MJ (2014) Rapid clearance of epigenetic protein reporters from wound edge cells in Drosophila larvae does not depend on the JNK or PDGFR/VEGFR signaling pathways. Regeneration (Oxf) 1: 11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zeng X, Lin X, Hou SX (2013) The Osa‐containing SWI/SNF chromatin‐remodeling complex regulates stem cell commitment in the adult Drosophila intestine. Development 140: 3532–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. He J, Xuan T, Xin T, An H, Wang J, Zhao G, Li M (2014) Evidence for chromatin‐remodeling complex PBAP‐controlled maintenance of the Drosophila ovarian germline stem cells. PLoS One 9: e103473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yu Z, Wu H, Chen H, Wang R, Liang X, Liu J, Li C, Deng W‐M, Jiao R (2013) CAF‐1 promotes Notch signaling through epigenetic control of target gene expression during Drosophila development. Development 140: 3635–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chen W‐Y, Shih H‐T, Liu K‐Y, Shih Z‐S, Chen L‐K, Tsai T‐H, Chen M‐J, Liu H, Tan BC‐M, Chen C‐Y et al (2015) Intellectual disability‐associated dBRWD3 regulates gene expression through inhibition of HIRA/YEM‐mediated chromatin deposition of histone H3.3. EMBO Rep 16: 528–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File