

Abstract
We evaluated the safety, efficacy, pharmacokinetics, pharmacodynamics and predictive biomarkers of tirabrutinib, a second‐generation, enhanced‐selectivity Bruton's tyrosine kinase inhibitor in Japanese patients with relapsed/refractory B‐cell non−Hodgkin lymphoma (B‐cell NHL) and chronic lymphocytic leukemia (CLL). This was an open‐label, multicenter, phase I study. Seventeen patients (male N = 8) with a median age of 70 years were enrolled in 4 dose cohorts (160 mg once daily [N = 3], 320 mg once daily [N = 3], 480 mg once daily [N = 4] and 300 mg twice daily [N = 7]); 4 patients had continued tirabrutinib administration as of 4 January 2018. The maximum tolerated dose was not reached. Pneumonitis (N = 1) was the dose‐limiting toxicity for 300 mg twice daily. Common adverse events (AEs) were rash (35.3%) and vomiting (29.4%). Eight patients (47.1%) developed grade ≥3 AEs: neutropenia (23.5%), anemia (11.8%) and leukopenia (11.8%) were frequent. The overall response rate (≥PR) was 76.5% (13/17 patients), including 4 DLBCL patients with no CD79A/B or MYD88 mutations, and 1 CLL patient with a TP53 mutation, providing promising data for future developments. Of 16 patients with measurable lesions during the screening period, 12 showed ≥50% reductions in tumor diameter. In many patients, the tumor size decreased soon after beginning treatment. The maximum serum concentration for tirabrutinib was 611, 1220, 1280 and 886 ng/mL on Day 1 and 484, 971 1940, and 961 ng/mL on Day 28 for Cohorts 1‐4, respectively. Tirabrutinib pharmacokinetics were linear, with little accumulation following multiple doses. Tirabrutinib was well tolerated and showed promising efficacy for B‐cell NHL/CLL.
Keywords: B‐cell malignancy, B‐cell non−Hodgkin lymphoma, chronic lymphocytic leukemia, safety, tirabrutinib
1. INTRODUCTION
Bruton's tyrosine kinase (BTK) has an important role in B‐cell signaling, cell proliferation and survival,1 and is, therefore, an attractive target for therapeutic intervention in B‐cell malignancies and autoimmune disorders. Ibrutinib, the first‐in‐class irreversible BTK inhibitor, is approved in the United States for treatment of mantle cell lymphoma (MCL), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), Waldenström's macroglobulinemia (WM) and marginal zone lymphomas. In contrast, ibrutinib is approved in Japan only for the treatment of MCL and CLL/SLL.2, 3, 4 Although the overall response rate (ORR) with ibrutinib is high, complete response (CR) is not frequently achieved. Ibrutinib can also lead to clinically significant toxicities in some patients, including bleeding and atrial fibrillation. It is likely that these adverse events (AEs) are partly due to the low selectivity of ibrutinib.5 Thus, an unmet need remains for novel, more specific BTK inhibitors that might improve efficacy, with less toxicity and shorter therapy duration. More specific second‐generation BTK inhibitors, including acalabrutinib (ACP‐196), tirabrutinib (ONO‐4059/GS‐4059) and BGB‐3111, have been developed to overcome the limitations of first‐generation BTK inhibitors, such as “off‐target” AEs or the development of resistance.
Tirabrutinib (ONO‐4059/GS‐4059) is a covalent and selective oral inhibitor of BTK, currently under clinical development for the treatment of CLL, B‐cell non−Hodgkin lymphoma (B‐cell NHL) and rheumatoid arthritis. Tirabrutinib has demonstrated cytostatic properties in vitro (against the activated B‐cell‐like diffuse large B‐cell lymphoma [ABC‐DLBCL] cell line, TMD8) and in vivo (TMD8 transplanted mouse tumor model).6 Furthermore, promising efficacy data have been reported, and tolerability was observed up to 480 mg once daily (QD) in B‐cell NHL and 600 mg QD in CLL in a European phase I study (ONO‐4059POE001, #NCT01659255).7 However, this European phase I trial did not include Asian patients and, therefore, the tolerability, safety, efficacy, pharmacokinetics (PK) and pharmacodynamics (PD) of tirabrutinib in Japanese patients with B‐cell NHL and CLL are unknown.
The present open‐label, multicenter, non−randomized phase I study aimed to evaluate the maximum tolerated dose (MTD), safety, efficacy, PK, PD and predictive biomarkers of tirabrutinib monotherapy in Japanese patients with relapsed or refractory B‐cell NHL and CLL.
2. PATIENTS AND METHODS
2.1. Patients
Patients with relapsed/refractory B‐cell NHL and CLL were enrolled in this study.
The main inclusion criteria were: age ≥20 years; confirmed histopathological diagnosis and documented history of relapsed or refractory B‐cell non−Hodgkin lymphoma (DLBCL, MCL, follicular lymphoma [FL], marginal zone lymphoma or WM/lymphoplasmacytic lymphoma [LPL]) and CLL/SLL for which no curative or high‐priority therapy exists; Eastern Cooperative Oncology Group performance status of ≤2; bidimensionally measurable disease >1.5 cm in its largest diameter (except for patients with CLL and WM); and life expectancy ≥90 days.
Exclusion criteria included: uncontrolled autoimmune hemolytic anemia (CLL only); pancreatitis; cerebellar disorders; disease significantly affecting gastrointestinal function; history of severe allergic or anaphylactic reactions; known active infection, including positive result for HIV, HBV (patients who are HBs antigen negative are to be excluded if they are positive for HBs antibody or HBc antibody and HBV‐DNA has been detected by quantitative analysis) or HCV; significant uncontrolled concomitant diseases; prior use of standard anti–lymphoma/leukemia therapy or radiation therapy within 28 days of receiving the first dose of tirabrutinib; prior tirabrutinib treatment; pregnancy; and abnormal laboratory values (creatine clearance < 50 mL/min [mL/min/1.73 m2]; aspartate aminotransferase or alanine aminotransferase ≥2.5 times the upper limit of normal; platelet count < 50 × 109/L; neutrophils < 1.0 × 109/L; hemoglobin < 8.0 g/dL; and total bilirubin ≥1.5 times the upper limit of normal).
Informed consent was obtained from all patients prior to enrollment. The study protocol and all amendments were reviewed by ethics committees at each participating center. The study was conducted according to the protocol, Good Clinical Practice guidelines, applicable local regulations and the Declaration of Helsinki.
2.2. Study design
Patients were enrolled from January 2015 to February 2016 in 4 dose cohorts of 160 mg QD, 320 mg QD, 480 mg QD and 300 mg BID (Cohorts 1‐4). The study design was a 3 + 3 dose escalation scheme where patients were observed for a 28‐day period to identify dose‐limiting toxicities (DLT), which are defined as drug‐related AEs that occurred in the first 28 days after the first dose. Further information on the definition of DLT is described in Data S1.
Tirabrutinib administration was continued until progressive disease, unacceptable AEs or consent withdrawal. PK was assessed on Day 1 and Day 28 in Cycle 1. Criteria for the discontinuation or interruption, resumption and dose modifications included AEs meeting DLT criteria, AEs not related to tirabrutinib, a patient's wish to discontinue, not meeting inclusion criteria and meeting exclusion criteria. The full criteria for discontinuation or interruption, resumption, and dose modification are described in Data S1.
2.3. Assessments
2.3.1. Safety
Adverse events were evaluated using the Common Terminology Criteria for Adverse Events (Ver. 4.0). Laboratory tests, vital signs and electrocardiograms were also assessed using conventional methods.
2.3.2. Efficacy
The assessment of best clinical response was based on International Workshop Criteria for B‐cell NHL patients,8 WM patients9 and CLL patients.10 Drug efficacy evaluation was based on the following: overall response rate (ORR), CR (CR unconfirmed or CR with incomplete blood count recovery), partial response rate (PR or very good PR), progression‐free survival, overall survival, duration of response and event‐free survival.
2.3.3. Pharmacokinetics
Blood for the PK analysis was drawn on Days 1 and 28 of Cycle 1 (pre−dose and at 30 minutes, 1, 2, 3, 4, 6, 8 and 12 hours post–dose) and on Days 2, 8 and 15 of Cycle 1 (pre−dose and at 4 hours post–dose). The maximum plasma concentration (C max), time to maximum plasma concentration (T max), area under the concentration–time curve (AUC) and T 1/2 were assessed (Phoenix WinNonlin Ver.6.2.1).
2.3.4. Pharmacodynamics
Bruton's tyrosine kinase phosphorylation inhibition in peripheral blood mononuclear cells was assessed using western blotting analysis at Day 1 of Cycle 1, from a blood sample taken pre−dose and at 2 hours post–dose. Primary antibodies included anti–phosphorylated BTK (pBTK) (Abcam, Cambridge, MA, USA), anti–BTK (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti–beta actin (Sigma‐Aldrich). Additional details are provided in Data S1.
2.3.5. Biomarker analysis
For chemokine analysis, blood was drawn: during the screening period (14 days to 1 day before start of administration); on Days 1, 2, 8, 15 and 28 of Cycle 1; and on Day 1 (all before administration of the study drug) of Cycles 3, 4, 5 and 6. Blood samples were analyzed at a central testing facility (including CCL‐3, CCL‐4, CXCL‐4, CXCL‐7, CXCL‐10, CXCL‐12, CXCL‐13 and BIM).
For CLL and non−germinal center B‐cell‐like (non−GCB) DLBCL patients, cell‐free DNA from blood samples was used for central analysis of genetic predictive markers (including 17p deletion and mutation, 11q, 13q, trisomy 12 and TP53 mutational status in CLL, and CD79A, CD79B, MYD88 and CARD11 mutational status in DLBCL). Cell‐free DNA was extracted using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA), and mutations were determined by next‐generation sequencing. Determination of DLBCL subtype was performed locally by immunohistochemistry according to the Hans criteria.11
2.4. Statistical analysis
Safety and efficacy analyses were based on all patients enrolled in this study who received at least 1 tirabrutinib dose. Best overall response was used for response analysis. ORR (CR plus PR, including PR with residual lymphocytosis) was calculated for the entire study population, and for each treatment cohort and histology.
3. RESULTS
3.1. Patients and treatment
Overall, 17 patients with a median age of 70 years (range, 37‐80) were enrolled (N = 3, 3, 4 and 7 patients in cohorts 1‐4, respectively), and 4 patients had continued tirabrutinib administration as of 4 January 2018. The histological subtypes were FL (N = 5), MCL (N = 4), non−GCB DLBCL (N = 4), WM (N = 2), DLBCL (N = 1) and CLL (N = 1). The median number of prior therapies was 3 (range, 1‐11), with 94.1% (16/17 patients) of patients having had prior exposure to a rituximab‐containing regimen. Patient characteristics are shown in Table 1. The median duration of study drug administration was 161 days (range, 5‐1037 days), and the median number of cycles administered was 7 (range, 1‐38).
Table 1.
Baseline demographics and disease characteristics of patients (N = 17)
| Characteristic | 160 mg QD, N = 3 | 320 mg QD, N = 3 | 480 mg QD, N = 4 | 300 mg BID, N = 7 | Total N = 17 |
|---|---|---|---|---|---|
| Age | |||||
| Median, years (range) | 70.0 (62‐70) | 72.0 (68‐72) | 74.5 (68‐79) | 71.0 (37‐80) | 70.0 (37‐80) |
| <65 y, n (%) | 1 (33.3) | 0 | 0 | 1 (14.3) | 2 (11.8) |
| ≥65 y, n (%) | 2 (66.7) | 3 (100.0) | 4 (100.0) | 6 (85.7) | 15 (88.2) |
| Sex | |||||
| Male, n (%) | 3 (100.0) | 1 (33.3) | 1 (25.0) | 3 (42.9) | 8 (47.1) |
| Female, n (%) | 0 | 2 (66.7) | 3 (75.0) | 4 (57.1) | 9 (52.9) |
| Histologic subtype, n (%) | |||||
| Non−GCB DLBCL | 0 | 0 | 2 (50.0) | 2 (28.6) | 4 (23.5) |
| DLBCL | 0 | 0 | 0 | 1 (14.3) | 1 (5.9) |
| MCL | 1 (33.3) | 2 (66.7) | 1 (25.0) | 0 | 4 (23.5) |
| FL | 0 | 1 (33.3) | 1 (25.0) | 3 (42.9) | 5 (29.4) |
| WM | 1 (33.3) | 0 | 0 | 1 (14.3) | 2 (11.8) |
| CLL | 1 (33.3) | 0 | 0 | 0 | 1 (5.9) |
| Relapsed or refractory after the latest treatment, n (%) | |||||
| Relapsed | 0 | 2 (66.7) | 2 (50.0) | 4 (57.1) | 8 (47.1) |
| Refractory | 3 (100.0) | 1 (33.3) | 2 (50.0) | 3 (42.9) | 9 (52.9) |
| Prior therapies, n (%) | |||||
| Prior rituximab | 2 (66.7) | 3 (100.0) | 4 (100.0) | 7 (100.0) | 16 (94.1) |
CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B‐cell lymphoma; FL, follicular lymphoma; MCL, mantle cell lymphoma; non−GCB DLBCL, non−germinal center B‐cell‐like diffuse large B‐cell lymphoma; WM, Waldenström's macroglobulinemia.
3.2. Safety and tolerability
Tirabrutinib was generally well tolerated (Table 2). The MTD was not reached. No DLT were observed in Cohorts 1‐3. One DLT (grade 3 pneumonitis) was observed in 1 patient with WM in Cohort 4. After a 3‐week drug‐free period, the patient recovered with the administration of steroids and oxygen. The patient then continued treatment with tirabrutinib 160 mg QD and was undergoing treatment at the time of data cut‐off (4 January 2018).
Table 2.
AEs reported for ≥10% of total population in all cohorts
| 160 mg QD N = 3, n (%) | 320 mg QD N = 3, n (%) | 480 mg QD N = 4, n (%) | 300 mg BID N = 7, n (%) | Total (all cohorts) N = 17, n (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | Any grade | Grade ≥3 | Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| All AEs | 3 (100.0) | 1 (33.3) | 3 (100.0) | 2 (66.7) | 4 (100.0) | 2 (50.0) | 7 (100.0) | 3 (42.9) | 17 (100.0) | 8 (47.1) |
| Rash | 1 (33.3) | 0 | 2 (66.7) | 0 | 2 (50.0) | 0 | 1 (14.3) | 0 | 6 (35.3) | 0 |
| Vomiting | 0 | 0 | 1 (33.3) | 0 | 2 (50.0) | 0 | 2 (28.6) | 0 | 5 (29.4) | 0 |
| Neutropenia | 1 (33.3) | 1 (33.3) | 2 (66.7) | 2 (66.7) | 1 (25.0) | 1 (25.0) | 0 | 0 | 4 (23.5) | 4 (23.5) |
| Anemia | 0 | 0 | 0 | 0 | 2 (50.0) | 1 (25.0) | 1 (14.3) | 1 (14.3) | 3 (17.6) | 2 (11.8) |
| Constipation | 0 | 0 | 1 (33.3) | 0 | 1 (25.0) | 0 | 1 (14.3) | 0 | 3 (17.6) | 0 |
| Diarrhea | 0 | 0 | 1 (33.3) | 0 | 0 | 0 | 2 (28.6) | 0 | 3 (17.6) | 0 |
| Nausea | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 2 (28.6) | 0 | 3 (17.6) | 0 |
| Malaise | 2 (66.7) | 0 | 0 | 0 | 1 (25.0) | 0 | 0 | 0 | 3 (17.6) | 0 |
| Arthralgia | 1 (33.3) | 0 | 1 (33.3) | 0 | 0 | 0 | 1 (14.3) | 0 | 3 (17.6) | 0 |
| Insomnia | 0 | 0 | 0 | 0 | 0 | 0 | 3 (42.9) | 0 | 3 (17.6) | 0 |
| Leukopenia | 0 | 0 | 2 (66.7) | 2 (66.7) | 0 | 0 | 0 | 0 | 2 (11.8) | 2 (11.8) |
| Lymphopenia | 0 | 0 | 1 (33.3) | 1 (33.3) | 1 (25.0) | 0 | 0 | 0 | 2 (11.8) | 1 (5.9) |
| Hypophosphatemia | 0 | 0 | 0 | 0 | 2 (50.0) | 1 (25.0) | 0 | 0 | 2 (11.8) | 1 (5.9) |
| Pyrexia | 0 | 0 | 0 | 0 | 0 | 0 | 2 (28.6) | 0 | 2 (11.8) | 0 |
| Cystitis | 0 | 0 | 1 (33.3) | 0 | 0 | 0 | 1 (14.3) | 0 | 2 (11.8) | 0 |
| Nasopharyngitis | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 1 (14.3) | 0 | 2 (11.8) | 0 |
| Pharyngitis | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 1 (14.3) | 0 | 2 (11.8) | 0 |
| Aspartate aminotransferase increased | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 1 (14.3) | 0 | 2 (11.8) | 0 |
| Gamma‐glutamyltransferase increased | 0 | 0 | 1 (33.3) | 0 | 0 | 0 | 1 (14.3) | 0 | 2 (11.8) | 0 |
| Thrombocytopenia | 0 | 0 | 1 (33.3) | 0 | 1 (25.0) | 0 | 0 | 0 | 2 (11.8) | 0 |
| Hypokalemia | 0 | 0 | 0 | 0 | 2 (50.0) | 0 | 0 | 0 | 2 (11.8) | 0 |
| Myalgia | 2 (66.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (11.8) | 0 |
| Dysgeusia | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 1 (14.3) | 0 | 2 (11.8) | 0 |
| Headache | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 1 (14.3) | 0 | 2 (11.8) | 0 |
| Upper respiratory tract inflammation | 1 (33.3) | 0 | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 2 (11.8) | 0 |
MedDRA version 20.1J.
“Neutropenia” includes “neutropenia” and “neutrophil count decrease.” AE, adverse events.
The most common AEs were rash (35.3%), vomiting (29.4%) and neutropenia (23.5%). Other AEs reported, for ≥15% of the population, included anemia, constipation, diarrhea, nausea, malaise, arthralgia and insomnia (17.6% each). Grade 3 or 4 AEs were mainly hematologic toxicities, with the most common being neutropenia (23.5%), anemia (11.8%) and leukopenia (11.8%). No AEs leading to death were reported.
Nine serious AEs (SAEs) were reported for 7 patients, of which 4 were reported as study drug‐related, including Mallory‐Weiss syndrome, de novo reactivation of HBV, acute myeloid leukemia and pneumonitis (1 patient each). One patient in the 160‐mg QD cohort had de novo reactivation of HBV during Cycle 13; after starting entecavir, study drug treatment was continued. At screening, HBs antigen and HBV DNA tests were negative, and HBs and HBc antibody tests were positive. Careful monitoring of liver function and HBV DNA was helpful to initiate the antivirus drug and continue the treatment of tirabrutinib. Mallory‐Weiss syndrome was observed during Cycle 1 in 1 patient in the 480‐mg cohort. Nausea and vomiting persisted in this patient after starting the study drug. No obvious hemorrhagic lesions were observed in the stomach or duodenum, but signs of Mallory‐Weiss syndrome were observed in the esophagus. The patient received nothing by mouth and famotidine treatment was initiated. There was no tirabrutinib‐induced lymphocytosis observed in this study.
3.3. Efficacy
The ORR for all patients was 76.5% (13/17 patients) (Table 3), and the subgroup analysis showed that the ORR for non−GCB DLBCL was 75.0% (3/4 patients) (Table 4). Median progression‐free survival in Cohort 3 was 109 days and was not estimable in Cohorts 1, 2 and 4. ORR was 80.0% (4/5 patients including 2 CR) for DLBCL, 40.0% (2/5 patients including 1 CR) for FL, 100% (4/4 patients including 3 CRs) for MCL, 100% (2/2 patients) for WM and 100% (1/1 patient) for CLL. The best overall response in DLBCL patients was CR in 2 patients and stable disease in 1 patient, among 3 patients who received 300 mg BID; in the 2 patients who received 480 mg QD, the best overall response was PR. Four patients with DLBCL (3/4 classified as non−GCB DLBCL) who achieved objective responses had no CD79A/B, MYD88 or CARD11 mutations. Of note, a CARD11 mutation was identified in 1 non−GCB DLBCL patient who did not achieve PR or a more response. At the time of the data cut‐off, 1 CLL patient with a TP53 mutation (del 13q and del 17p) had achieved PR and was still receiving tirabrutinib therapy. Twelve of 16 patients with measurable lesions during the screening period showed ≥50% reductions in tumor diameter (Figure 1A). Of 13 patients who had achieved PR or more, 8 showed early response within 1 month after the beginning of the study treatment (Figure 1B).
Table 3.
Response rate by dosage of tirabrutinib
| 160 mg QD N = 3 | 320 mg QD N = 3 | 480 mg QD N = 4 | 300 mg BID N = 7 | Total N = 17 | |
|---|---|---|---|---|---|
| Overall response (CR, CRu, CRi, VGPR or PRa), | 3 (100.0) | 3 (100.0) | 3 (75.0) | 4 (57.1) | 13 (76.5) |
| n (%) [95% CI] | [29.2, 100.0] | [29.2, 100.0] | [19.4, 99.4] | [18.4, 90.1] | [50.1, 93.2] |
| Complete response (CR, CRu, CRi), | 1 (33.3) | 2 (66.7) | 0 (.0) | 3 (42.9) | 6 (35.3) |
| n (%) [95% CI] | [.8, 90.6] | [9.4, 99.2] | [.0, 60.2] | [9.9, 81.6] | [14.2, 61.7] |
| Partial response (VGPR or PRa), | 2 (66.7) | 1 (33.3) | 3 (75.0) | 1 (14.3) | 7 (41.2) |
| n (%) [95% CI] | [9.4, 99.2] | [.8, 90.6] | [19.4, 99.4] | [.4, 57.9] | [18.4, 67.1] |
The response rate and its 95% CI were estimated using the Clopper–Pearson method.
CI, confidence interval; CR, complete response; CRi, CR with incomplete blood count recovery; CRu, CR unconfirmed; PR, partial response; VGPR, very good PR.
Includes patients with lymphocytosis assessed as modified PR.
Table 4.
Subgroup analysis of overall response rate
| Diagnosis (subtype) | n/N (%) [95% confidence interval] |
|---|---|
| N = 17 | |
| Non−GCB DLBCL | 3/4 (75.0) [19.4, 99.4] |
| DLBCL | 1/1 (100.0) [2.5, 100.0] |
| Mantle cell lymphoma | 4/4 (100.0) [39.8, 100.0] |
| Follicular lymphoma | 2/5 (40.0) [5.3, 85.3] |
| Waldenström's macroglobulinemia | 2/2 (100.0) [15.8, 100.0] |
| Chronic lymphocytic leukemia | 1/1 (100.0) [2.5, 100.0] |
The rates and their 95% confidence intervals were calculated using the Clopper‐Pearson method.
DLBCL, diffuse large B‐cell lymphoma; non−GCB DLBCL, non−germinal center B‐cell‐like diffuse large B‐cell lymphoma.
Figure 1.
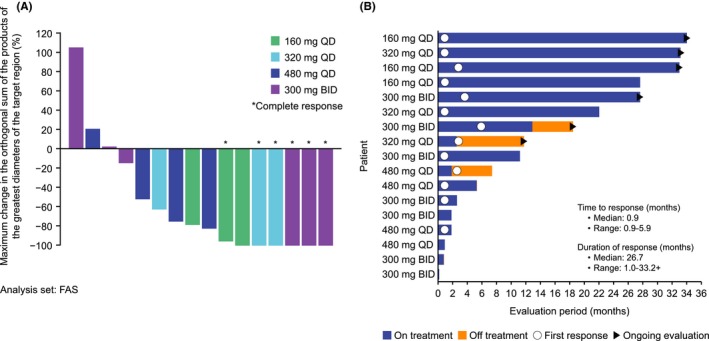
Maximum change in the orthogonal sum of the products of the greatest diameters of the target region (A) and the time to response and duration of response (B). +, censor; FAS, full analysis set
CXCL‐10 levels in responders tended to be lower on Day 8 of Cycle 1 (C1D8) than baseline and tended to be higher on Day 28 of Cycle 1 (C1D28) than on C1D8 (Figure 2). Among 13 responders, C1D28/C1D8 > 1 was reported in 8 (61.5%). Among 4 non−responders, C1D28/C1D8 > 1 was reported in 1 (50.0%) of the 2 with measurements available for C1D28. When patients with CXCL‐10 > 1000 pg/mL at baseline were excluded to eliminate the potential effect of inflammatory diseases, C1D28/C1D8 > 1 was reported in 7 patients out of 9 responders (77.8%). Among the 4 non−responders, 3 patients had baseline CXCL‐10 levels > 1000 pg/mL, and in the remaining patient, CXCL‐10 could not be measured on C1D28. There were no observable differences in the levels of other chemokines between responders and non−responders, including CCL‐3, CCL‐4, CXCL‐4, CXCL‐7, CXCL‐12, CXCL‐13 and BIM.
Figure 2.
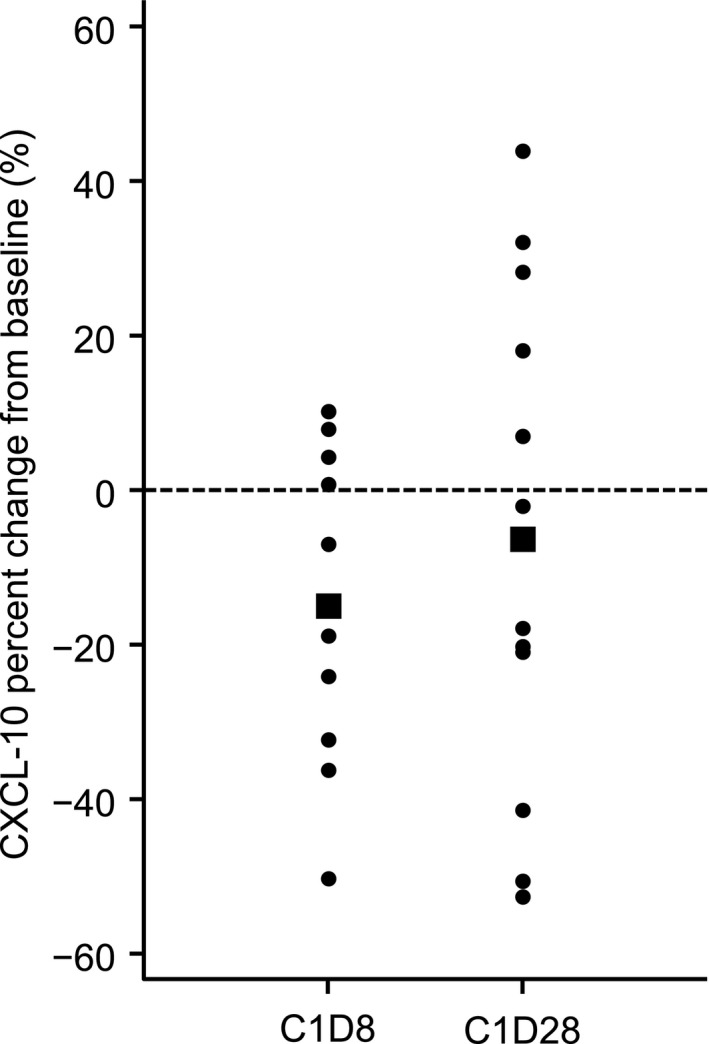
CXCL‐10 percent change from baseline in all 13 responders (partial response or more) on C1D8 and C1D28. Bold squares represent the average value
3.4. Pharmacokinetics
The plasma concentrations of tirabrutinib for all cohorts are shown in Figure 3A,B. The PK parameters C max, T max, AUC12 h, AUC24 h, AUCinf and T 1/2 on C1D1 and C1D28 are shown in Table S1.
Figure 3.
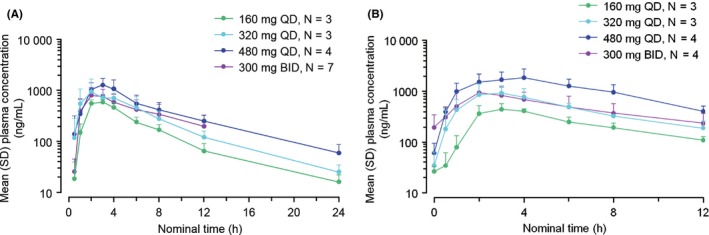
Mean plasma concentration of tirabrutinib on Cycle 1, Day 1 (A) and on Cycle 1, Day 28 (B) in the fasting state (once or twice a day for 28 d) (Cohorts 1, 2, 3 and 4, Cycle 1, Day 1). The error bars indicate standard deviation (SD). BID, twice daily; QD, once daily
3.5. Pharmacodynamics
Treatment with tirabrutinib suppressed the expression level of pBTK in all 11 patients with confirmed pBTK expression at the basal level. pBTK on C1D1 was rapidly eliminated in these 11 patients (data not shown). Expression was suppressed in the CLL patient in a time‐dependent manner. Complete suppression was observed in the other patients at 2 hours post–dose on Day 1 of Cycle 1 (Figure 4). Basal expression of pBTK was below the detection limit in 6 of 17 patients.
Figure 4.
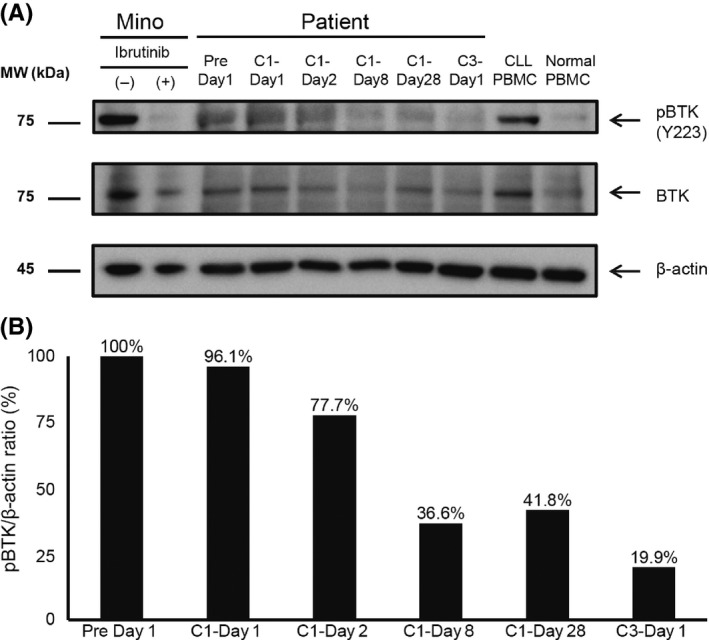
Phosphorylated Bruton's tyrosine kinase (pBTK) levels, analyzed by western blot in samples from patients with CLL. (A) Western blot analysis, (B) relative amount of pBTK (Pre Day 1 = 100%). Levels of pBTK were determined by imaging analyzer and normalized to those of β‐actin. Mino, TP53 mutation marker as standard; CLL, chronic lymphocytic leukemia; PBMC, peripheral blood mononuclear cells
4. DISCUSSION
This study evaluated the MTD, safety, efficacy, PK and PD of tirabrutinib monotherapy in 17 Japanese patients with relapsed or refractory B‐cell NHL and CLL. Furthermore, we conducted the first biomarker analysis of tirabrutinib in humans. We found that tirabrutinib was well tolerated and demonstrated promising efficacy in a group of Japanese B‐cell NHL and CLL patients. Tolerability up to 300 mg BID was confirmed, and the MTD was not reached. Therefore, the recommended dosing schedule of tirabrutinib in Japanese patients with B‐cell NHL and CLL is either 480 mg QD or 300 mg BID. However, it is still necessary to consider which dosage is recommended for each histological subtype.
No DLT was identified in Cohorts 1‐3. A DLT (grade 3 pneumonitis) was observed in 1 WM patient in Cohort 4 (300 mg BID), but it resolved with supportive measures, and study treatment was resumed at a lower dose. Organized pneumonia was present before the patient entered the study. All AEs were manageable and resolved with temporary dose interruption or the administration of granulocyte colony‐stimulating factor or other agents. There were no clinically significant AEs in this group of Japanese patients, and the safety profile and most common AEs (neutropenia and anemia) were similar to those reported in the European phase I study.7
A response was seen beginning at 160 mg QD. No correlation was found between response rate and dose, possibly because the dose was gradually increased from an initial dose at which an adequate response was observed in a previous phase I study and the number of enrolled patients was small.7 Histological subtypes may have also influenced the response.
This study has some limitations, including the small sample size of each subtype. However, a notable result was that the ORR was 80% (4/5 patients) in patients with DLBCL. CR was seen in 2 patients in the 300 mg BID cohort, while the best response in the 480 mg QD cohort was PR. This trend toward improved response in the 300‐mg BID cohort may be because the arithmetic mean trough tirabrutinib plasma concentrations on C1D28 were 3.2‐fold higher with 300 mg BID than with 480 mg QD. This result is supported by pre−clinical pharmacological studies in the TMD8 model.12 Therefore, BID dosing may improve the efficacy of tirabrutinib in patients with DLBCL. Of note, 4 patients with DLBCL who achieved objective responses had no CD79A/B, MYD88 or CARD11 mutations. A study of DLBCL patients treated with ibrutinib reported that 55.5% of patients with ABC tumors with B‐cell receptor mutations responded to treatment, and this rate increased to 80% for those who also had MYD88 mutations.13
In the current study, 1 CLL patient (160 mg QD) had a TP53 mutation but achieved PR, and treatment is currently ongoing, despite the poor prognosis with TP53 mutation reported for this type of case treated by conventional chemotherapy.14, 15, 16 The efficacy of tirabrutinib against CLL with TP53 mutation may be comparable with that of ibrutinib.
For patients with CXCL‐10 levels > 1000 pg/mL, it was difficult to evaluate the effect of the study drug on CXCL‐10 levels because an increase in CXCL‐10 levels to >1000 pg/mL can be related to other complications (autoimmune disease or infection). Therefore, we only evaluated the change in CXCL‐10 levels in patients with CXCL‐10 ≤ 1000 pg/mL at baseline. CXCL‐10 levels in responders with CXCL‐10 ≤ 1000 pg/mL at baseline tended to be higher on C1D28 vs C1D8. This suggests that the administration of tirabrutinib suppresses CXCL‐10 production from tumor cells over a short period; therefore, a fast‐acting antitumor effect rapidly lowers CXCL‐10 levels, and B‐cell functions (including regulatory B‐cells) are inhibited for a long period, accelerating Th1 functions.17
Pharmacokinetics analysis demonstrated that the area under the concentration‐time curve and maximum serum concentration were increased in a dose‐dependent manner. Treatment with tirabrutinib at doses of ≥160 mg QD fully suppressed the expression of pBTK in all patients with confirmed basal expression of pBTK. In addition, pBTK in peripheral mononuclear cells was suppressed at 160 mg QD, indicating tumor shrinkage. However, although suppression of phosphorylation was seen in most patients, there were some non−responders. Therefore, further investigation is needed to validate pBTK as a biomarker.
In conclusion, in Japanese patients with B‐cell NHL and CLL, tirabrutinib was well tolerated and showed promising efficacy with an acceptable PK profile for future development. Overall responses (≥PR) were observed in 76.5% of patients, including 4 DLBCL patients with no CD79A/B or MYD88 mutations, and 1 CLL patient with a TP53 mutation, providing promising data for future developments. Based on the results of this phase I study, phase II studies of tirabrutinib monotherapy have already been initiated in Japanese patients with specific subtypes of B‐cell NHL. In addition, large‐scale clinical trials of tirabrutinib are required to elucidate the differences between tirabrutinib and other BTK inhibitors, such as ibrutinib and acalabrutinib.
CONFLICT OF INTEREST
W. Munakata has received grants from Ono Pharmaceutical Co., Ltd. K. Ando has received grants from Ono Pharmaceutical Co., Ltd. K. Hatake has received grants from Ono Pharmaceutical Co., Ltd. and Chugai Pharmaceutical Co., Ltd. N. Fukuhara has received grants from Ono Pharmaceutical Co., Ltd. T. Kinoshita has received grants and/or personal fees from Chugai Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Solasia Pharma K.K., Ono Pharmaceutical Co., Ltd., Gilead Sciences, Inc., MSD, Zenyaku Kogyo, Bristol‐Myers Squibb, Kyowa Hakko Kirin Co., Ltd., Eisai Co., Ltd., and Janssen Pharmaceutica. S. Fukuhara has received grants from Ono Pharmaceutical Co., Ltd. Y. Shirasugi has received personal fees from Novartis. M. Yokoyama has received grants from Ono Pharmaceutical Co., Ltd. and personal fees from Chugai Pharmaceutical Co., Ltd. S. Ichikawa has received grants from Ono Pharmaceutical Co., Ltd. K. Ohmachi has received grants from Ono Pharmaceutical Co., Ltd. and personal fees from Ono Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Eisai Co., Ltd., Chugai Pharmaceutical Co., Ltd., Pfizer Inc., Takeda Pharmaceutical Co., Ltd., and Meiji Seika Pharma Co., Ltd. N. Gion is an employee of Ono Pharmaceutical Co., Ltd., and has received personal fees. A. Aoi is an employee of Ono Pharmaceutical Co., Ltd., and has received personal fees. K. Tobinai has received grants and/or personal fees from Ono Pharmaceutical Co., Ltd., Celgene, Zenyaku Kogyo, HUYA Bioscience International LLC, Eisai Co., Ltd., Takeda Pharmaceutical Co., Ltd., Mundipharma, Janssen Pharmaceutical, Kyowa Hakko Kirin Co., Ltd., Chugai Pharmaceutical Co., Ltd., GlaxoSmithKline, and AbbVie Inc.
Supporting information
ACKNOWLEDGMENTS
This work was supported by Ono Pharmaceutical Co., Ltd. The study sponsor participated in the study design, collection, analysis and interpretation of data, in the writing of the report, and in the decision to submit the paper for publication. The authors thank all participating patients and their families who made this study possible. The authors would like to thank Dr Kazuo Tamura (General Medical Research Center, Fukuoka University, Fukuoka), Dr Kunihiro Tsukasaki (International Medical Center, Saitama Medical University, Saitama) and Dr Hirokazu Nagai (Nagoya Medical Center, Nagoya) for their strict review of the clinical data as members of the Efficacy and Safety Monitoring Committee. The authors would like to thank J. Ludovic Croxford, PhD, of Edanz Medical Writing for providing medical writing services, which were funded by Ono Pharmaceutical Co., Ltd.
Munakata W, Ando K, Hatake K, et al. Phase I study of tirabrutinib (ONO‐4059/GS‐4059) in patients with relapsed or refractory B‐cell malignancies in Japan. Cancer Sci. 2019;110:1686–1694. 10.1111/cas.13983
Clinical trial register: JapicCTI‐142682 (http://www.clinicaltrials.jp/).
REFERENCES
- 1. Niiro H, Clark EA. Regulation of B‐cell fate by antigen‐receptor signals. Nat Rev Immunol. 2002;2:945‐956. [DOI] [PubMed] [Google Scholar]
- 2. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle‐cell lymphoma. N Engl J Med. 2013;369:507‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med. 2015;372:1430‐1440. [DOI] [PubMed] [Google Scholar]
- 5. Walter HS, Jayne S, Rule SA, et al. Long‐term follow‐up of patients with CLL treated with the selective Bruton's tyrosine kinase inhibitor ONO/GS‐4059. Blood. 2017;129:2808‐2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kozaki R, Hutchinson C, Sandrine J, Dyer M. Kinome reprogramming in DLBCL by the BTK‐specific inhibitor ONO‐4059 highlights synergistic combinations for clinical application; Abstract P431. Haematologica. 2014;99:137‐138. [Google Scholar]
- 7. Walter HS, Rule SA, Dyer MJ, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS‐4059 in relapsed and refractory mature B‐cell malignancies. Blood. 2016;127:411‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non−Hodgkin's lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244. [DOI] [PubMed] [Google Scholar]
- 9. Owen RG, Kyle RA, Stone MJ, et al. Response assessment in Waldenstrom macroglobulinaemia: update from the VIth International Workshop. Br J Haematol. 2013;160:171‐176. [DOI] [PubMed] [Google Scholar]
- 10. Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute‐Working Group 1996 guidelines. Blood. 2008;111:5446‐5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B‐cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275‐282. [DOI] [PubMed] [Google Scholar]
- 12. Yoshizawa T, Yasuhiro T, Honda H, Kawabata K. ONO‐4059—a Potent and Selective Reversible Bruton's Tyrosine Kinase (Btk) Inhibitor: Single Agent, Twice Daily (BD) Dosing and Dosing with Food Results in Sustained, High Trough Levels of ONO‐4059, Translating into 100% Tumour Remission in a TMD‐8 Xenograft Model. Blood. 2014;124:4502. [Google Scholar]
- 13. Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fornecker LM, Aurran‐Schleinitz T, Michallet AS, et al. Salvage outcomes in patients with first relapse after fludarabine, cyclophosphamide, and rituximab for chronic lymphocytic leukemia: the French intergroup experience. Am J Hematol. 2015;90:511‐514. [DOI] [PubMed] [Google Scholar]
- 15. Tam CS, O'Brien S, Plunkett W, et al. Long‐term results of first salvage treatment in CLL patients treated initially with FCR (fludarabine, cyclophosphamide, rituximab). Blood. 2014;124:3059‐3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cramer P, Fink AM, Busch R, et al. Second‐line therapies of patients initially treated with fludarabine and cyclophosphamide or fludarabine, cyclophosphamide and rituximab for chronic lymphocytic leukemia within the CLL8 protocol of the German CLL Study Group. Leuk Lymphoma. 2013;54:1821‐1822. [DOI] [PubMed] [Google Scholar]
- 17. Schmidt NW, Thieu VT, Mann BA, Ahyi AN, Kaplan MH. Bruton's tyrosine kinase is required for TLR‐induced IL‐10 production. J Immunol. 2006;177:7203‐7210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials