

Abstract
Osteosarcoma is the most common primary malignant bone tumor. Raddeanin A (RA) is an active oleanane‐type triterpenoid saponin extracted from the traditional Chinese herb Anemone raddeana Regel that exerts antitumor activity against several cancer types. However, the effect of RA on osteosarcoma remains unclear. In the present study, we showed that RA inhibited proliferation and induced apoptosis of osteosarcoma cells in a dose‐ and time‐dependent way in vitro and in vivo. RA treatment resulted in excessive reactive oxygen species (ROS) generation and JNK and ERK1/2 activation. Apoptosis induction was evaluated by the activation of caspase‐3, caspase‐8, and caspase‐9 and poly‐ADP ribose polymerase (PARP) cleavage. RA‐induced cell death was significantly restored by the ROS scavenger glutathione (GSH), the pharmacological inhibitor of JNK SP600125, or specific JNK knockdown by shRNA. Additionally, signal transducer and activator of transcription 3 (STAT3) activation was suppressed by RA in human osteosarcoma, and this suppression was restored by GSH, SP600125, and JNK‐shRNA. Further investigation showed that STAT3 phosphorylation was increased after JNK knockdown. In a tibial xenograft tumor model, RA induced osteosarcoma apoptosis and notably inhibited tumor growth. Taken together, our results show that RA suppresses proliferation and induces apoptosis by modulating the JNK/c‐Jun and STAT3 signaling pathways in human osteosarcoma. Therefore, RA may be a promising candidate antitumor drug for osteosarcoma intervention.
Keywords: Jun amino‐terminal kinase, osteosarcoma, Raddeanin A, signal transducer and activator of transcription 3, SP600125
1. INTRODUCTION
Osteosarcoma is the most common nonhematological malignancy of bone in children aged 15‐19 years and typically arises around the growth plate of long bones, with highly aggressive and early systemic metastases.1 With surgical advances and the development of dose‐intensive, multiagent chemotherapy, the 5‐year overall survival rate for osteosarcoma has increased to approximately 50‐70%.1, 2 Despite clinical and therapeutic improvements, patients with metastatic or recurrent disease have a poor prognosis, with a 5‐year overall survival rate of approximately 20% in the past three decades.1, 3 Therefore, novel therapeutic approaches with a lower working dose and improved targeting are urgently needed.
Raddeanin A (RA) is an active oleanane‐type triterpenoid saponin extracted from a commonly used traditional Chinese medicinal herb, Anemone raddeana Regel, which exerts antitumor activity through inhibiting proliferation and angiogenesis and inducing apoptosis in multiple cancer cell types, such as human colorectal cells, gastric cancer cells, and human hepatocellular carcinoma cells.4, 5, 6, 7 RA mainly functions to reduce inflammation and tumor activity. Furthermore, recent research has shown that RA can suppress the progression of human colorectal carcinoma.4, 5, 6, 7 However, the effects of RA on human osteosarcoma cells are unknown.
Reactive oxygen species (ROS) function as second messengers in signal transduction and gene regulation in a variety of cell types and under several biological conditions, such as in the presence of cytokines, growth factors, and hormone treatments, and are involved in ion transport, transcription, neuromodulation, and apoptosis.8, 9 As a heterogeneous group of diatomic oxygen molecules from free and nonfree radical species, ROS trigger apoptosis by causing various cellular stresses and regulate several apoptotic effectors, such as caspases, Bcl‐2, and cytochrome c.8 Broadly speaking, ROS are capable of activating p38 MAPK, JNK, ERK, and Akt pathways, depending on the context.8, 10, 11, 12
Jun amino‐terminal kinase, a stress‐activated protein kinase of the MAPK family, regulates a variety of cellular events, including autophagy and apoptosis, which are complex events depending on cell type, nature of the death stimulus and activity of other signaling pathways.13 A major target of the JNK signaling pathway is the activator protein‐1 (AP‐1) transcription factor, which is activated in part by the phosphorylation of c‐Jun and related molecules.9, 10, 14, 15 Some reports have indicated that there is an interaction between a region within c‐Jun and specific sites within signal transducer and activator of transcription 3 (STAT3) in vitro and in vivo.16, 17, 18, 19 Additionally, JNK is an upstream kinase responsible for the phosphorylation of STAT3,16, 20, 21, 22 and both are known to play critical roles in the regulation of apoptosis signal transduction in cancer.
Signal transducer and activator of transcription 3 is one of the six members of a family of transcription factors that regulates gene expression related to the cell cycle, cell survival, and the immune response.23 Compared with the transient activation of STAT3 in normal cells, STAT3 remains abnormally active in tumors and promotes the induction and survival of cancer.24 Our previous results showed that selective inactivation of STAT3 blocks tumorigenesis in human osteosarcoma.25, 26, 27 Our most recent studies have shown that a natural compound, alternol, suppresses cell proliferation and migration and induces apoptosis and cell cycle arrest by modulating ROS‐dependent MAPK and STAT3 signaling pathways in human osteosarcoma cells.27 These studies have demonstrated the key status of STAT3 in human osteosarcoma development and provide compelling evidence that STAT3 inactivation might become a novel therapeutic approach for drug discovery in human osteosarcoma.
In the present study, we investigated the antitumor activity of RA on human osteosarcoma and explored the possible mechanisms of RA induction. We found that RA induces apoptosis in human osteosarcoma cells by modulating ROS‐dependent JNK/c‐Jun and STAT3 signaling pathways. Collectively, we show the effects of RA in human osteosarcoma and its possible mechanisms.
2. MATERIALS AND METHODS
2.1. Cells and cell culture
Human osteosarcoma cell lines 143B and SJSA were obtained from ATCC (Manassas, VA, USA). The cells were cultured in high‐glucose DMEM (Cyclone, Logan, UT, USA) with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific) at 37°C in an atmosphere of 5% CO2.
2.2. Materials and chemicals
Raddeanin A was purchased from Shanghai Yuanye Biotechnology, Ltd (Shanghai, China). Stock solution (20 mmol/L) was prepared by dissolution in DMSO and stored in the dark at −20°C. GSH (glutathione, γ‐glutamyl cysteinyl glycine), cycloheximide (CHX), SP600125 (JNK inhibitor), and NSC74859 (STAT3 inhibitor, S3I‐201) were purchased from Sigma‐Aldrich (St Louis, MO, USA). LY3214996 (Erk1/2 inhibitor) and Q‐VD‐OPh (caspase inhibitor) were purchased from MCE (Monmouth Junction, NJ, USA). Antibodies against STAT3, phospho‐STAT3S727, Bcl‐2, Bcl‐xl, Bax, total‐ and cleaved PARP, cleaved caspase‐3, cleaved caspase‐8, cleaved caspase‐9, c‐Jun, phospho‐c‐Jun, phospho‐JNK, JNK, ERK1/2, phospho‐ERK1/2 and actin were purchased from Cell Signaling Technology (Beverly, MA, USA).
2.3. Cell viability assay
Cell proliferation was measured using the CCK‐8 assay (Dojindo, Kumamoto, Japan). Briefly, cells were subcultured in a 96‐well plate at a density of 5 × 103 cells/mL and incubated overnight and then treated with various concentrations of RA with or without GSH or SP600125. After 24 or 48 hours of incubation, the cells were incubated with CCK‐8 working solution for 1 hour at 37°C following the manufacturer's protocol. The resulting absorbance at 450 nm was measured using a microplate reader iMark (Molecular Devices, Sunnyvale, CA, USA).
2.4. Clone formation assay
Cells were seeded evenly at a density of 200 cells/well in six‐well plates. After incubation overnight, the cells were treated with various concentrations of RA with or without GSH for approximately 14 days until the cells formed visible colonies. The medium was then discarded, and the cells were washed with PBS three times. After fixation with 4% paraformaldehyde, the colonies were stained with 0.1% crystal violet for 15 minutes. Colony number was counted manually.
2.5. Flow cytometric analysis of apoptosis
Osteosarcoma cell lines were seeded in six‐well plates at a density of 5 × 105 cells/well and treated at the indicated concentration for 24 hours with RA in the presence or absence of GSH or SP600125 or other treatment. Then, the cells were collected, washed with ice‐cold PBS, and resuspended in 1× binding buffer containing Annexin V‐FITC/PI (BD Biosciences, Mountain View, CA, USA). After incubation for 15 minutes at room temperature in the dark, the samples were analyzed by flow cytometry (BD Biosciences, Mountain View, CA, USA).
2.6. Measurement of intracellular ROS generation
Intracellular ROS generation was detected using a Reactive Oxygen Species Assay Kit (Beyotime, Beijing, China). Briefly, the cells were seeded in six‐well plates at a density of 5 × 105 cells/well and exposed to RA (2 μmol/L) with or without GSH for 12 hours. Cells were stained with 10 μmol/L 2′‐7′‐dichlorodihydrofluorescein diacetate (DCFH‐DA) at 37°C in the dark for 30 minutes. Then, the cells were washed with serum‐free DMEM three times, and ROS levels were measured by fluorescence microscopy and flow cytometry (BD Biosciences).
2.7. JNK knockdown in human osteosarcoma cell lines
Four JNK‐targeted shRNA sequences were designed under the guidelines of the WI siRNA Selection Program (http://sirna.wi.mit.edu/). The JNK‐shRNA sequence was 5′‐CCTGAAACGATATCAGAATTT‐3′; the control sequence was 5′‐CAACAAGATGAAGAGCACCAA‐3′, and the shRNA vector was pLKO.1. The protocol for the construction of JNK‐shRNA lentiviruses was as previously described.28 The plasmids were carefully transfected into 293T cells. After 48 hours of transfection, the supernatant was harvested and filtered using a 0.45‐μm filter. The same procedure was used to construct the empty lentiviral vector as the control group. Stable JNK‐shRNA‐transfected cells were screened with 2 μg/mL puromycin to improve transfection efficiency.
2.8. siRNA‐mediated STAT3 knockdown
Cells were seeded in a six‐well plate. After 24 hours, siRNA duplex targeting STAT3 was transfected using Lipofectamine 2000 (Invitrogen Life Technologies, Los Gatos, CA, USA) according to the instructions. The siRNA‐STAT3 sequence was 5′‐CGTCATTAGCAGAATCTCA‐3′.
2.9. Animal model
All animal care and experimental studies were carried out according to the guidelines and approval of the Animal Investigation Committee of the Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine. Male BALB/c athymic nude mice (4 weeks) were bred and maintained at the animal experimental center in Shanghai General Hospital. 143B (1 × 106) was injected into the medullary cavity of the right tibia to establish a primary orthotopic model. One week after tibial injection, mice were randomly allocated to the 5 mg/kg RA group (n = 5), 10 mg/kg RA group (n = 5), and vehicle (DMSO) group (n = 5). Each mouse in the RA groups received a weight‐based dose of drug by i.p. injection every 3 days.
2.10. TUNEL assay
Apoptosis in tumor samples was identified using a TUNEL Assay Kit (Beyotime) according to the manufacturer's instructions. Briefly, paraffin‐embedded slides were deparaffinized in xylene, rehydrated in gradient ethanol and incubated in proteinase K for antigen retrieval. After washing with PBS three times, the sections were stained with TUNEL detection mixture for 1 hour at 37°C in a humidified chamber. The cell nuclei were stained with DAPI (Beyotime). Apoptotic tumor cells in randomly chosen fields on the slides were observed using a fluorescence microscope (Leica, Wetzler, Germany).
2.11. Statistical analysis
All data are presented as the mean ± SD from at least three independent experiments. Fisher's exact test, unpaired Student's t test, and one‐way ANOVA were used to compare differences between the control and treatment groups. All statistical analyses were carried out using SPSS version 18.0 software (IBM Corporation, Chicago, IL, USA). P‐values <0.05 (*) and <0.01 (**) were considered significant.
3. RESULTS
3.1. Raddeanin A activates ROS generation in human osteosarcoma cells
Reactive oxygen species are important regulators in various pathways, including apoptosis, and they promote sustained JNK activation.8 Mitochondria are the major intracellular source of ROS.8, 13, 29, 30, 31, 32, 33, 34 Glutathione is present in the cells in both reduced (GSH) and oxidized (GSSG) states, and the harmful effect of ROS is counterbalanced by antioxidants such as GSH. To investigate whether ROS levels were increased because of RA treatment, DCFH‐DA was used in a fluorescence microplate experiment. As shown in Figure 1A, cells treated with RA showed a dramatic enhancement in the DCFH‐DA fluorescence signal compared with control cells, where ROS were scavenged by the antioxidant GSH. Consistent with the microscopic data, as shown in Figure 1B, the DCFH‐DA flow cytometry assay showed increased ROS levels in cells treated with RA, and this increase could be strongly inhibited by GSH. For example, ROS production resulting from treatment with 2 μmol/L RA for 12 hours was 1000‐fold higher (Figure 1C) than that of the blank control, whereas the same ROS level was sixfold higher (Figure 1C) than that after pretreatment with 1.5 mmol/L GSH for 2 hours, which confirmed the efficacy of GSH pretreatment. These results show that GSH significantly attenuates ROS induction and that RA activates ROS generation in human osteosarcoma cells.
Figure 1.
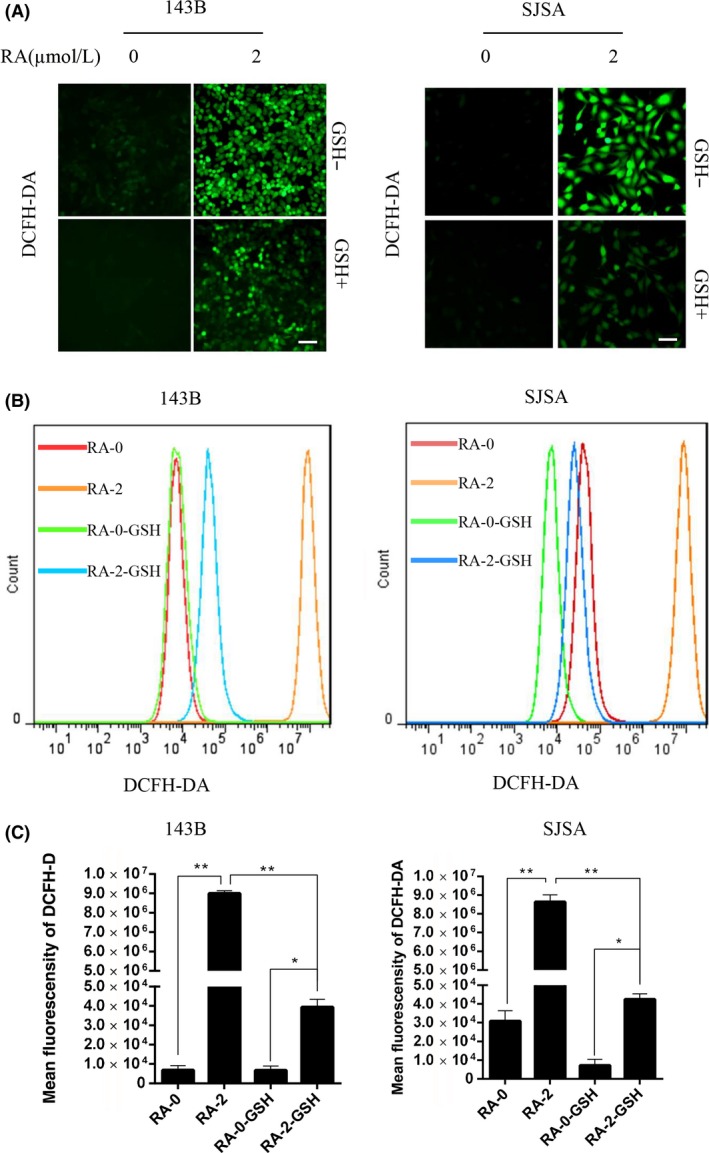
Raddeanin A (RA) induced intracellular reactive oxygen species (ROS) generation. A, Human osteosarcoma cells were preincubated with glutathione (GSH) for 2 h and then treated with 2 μmol/L RA for 24 h. Cells were stained with 2′‐7′‐dichlorodihydrofluorescein diacetate (DCFH‐DA) at 37°C in the dark for 30 min, and ROS levels were determined by fluorescence microscopy. Scale bar, 50 μm. B, Cells were stained with DCFH‐DA, and ROS levels were determined by flow cytometry. C, Mean fluorescent intensity is shown as histograms. Data are presented as the means ± SD (n = 3). *P < 0.05, **P < 0.01, significantly different compared with the untreated control group
3.2. Raddeanin A inhibits cell proliferation and induces apoptosis by ROS generation
To investigate the antiproliferative activity of RA, 143B and SJSA were treated with RA for 48 hours, and cell viability was then measured by CCK‐8 assays (Figure 2A). After RA treatment, cell viability was significantly decreased in a dose‐dependent method. RA treatment also reduced the number of colonies, as determined using a clone formation assay (Figure 2B,C). Importantly, GSH pretreatment alleviated RA cytotoxicity, as demonstrated by recovery of both clone formation (Figure 2B) and cell viability (Figure 2A) in GSH‐pretreated samples. Clone formation rate increased from approximately 30% (29.97 ± 5.20) to approximately 50% (49.31 ± 5.47) with GSH pretreatment, which was a significant difference. These results show that GSH partially rescued osteosarcoma cells from ROS damage stemming from RA treatment, confirming that RA cytotoxicity was mediated by ROS generation.
Figure 2.
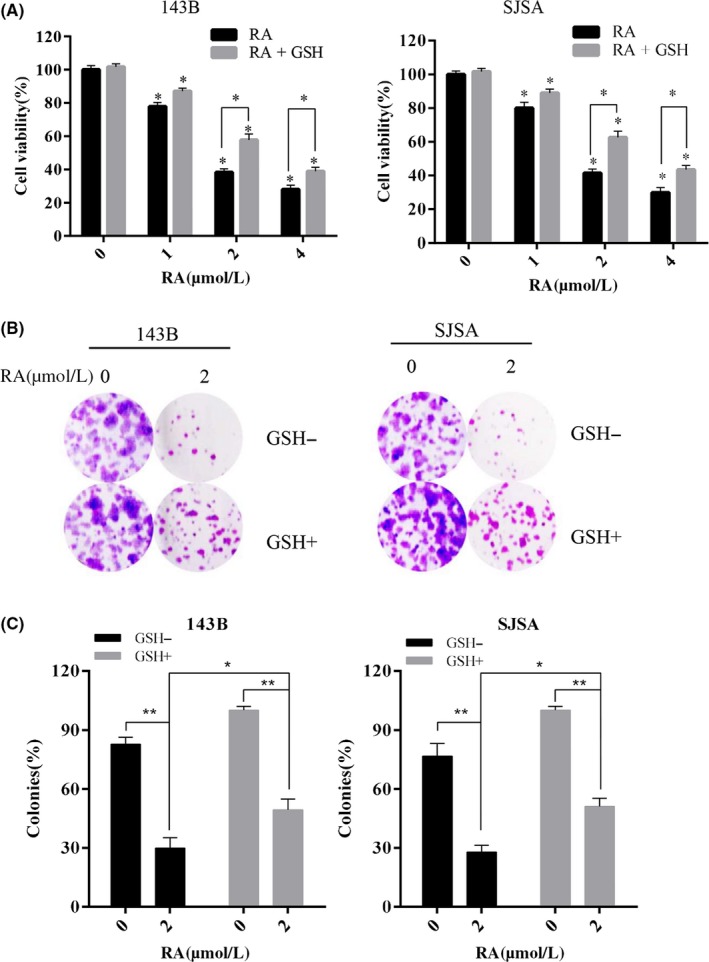
Raddeanin A (RA) inhibited cell viability through reactive oxygen species (ROS) production. A,B, Cells were treated with 2 μmol/L RA in the presence or absence of 1.5 mmol/L glutathione (GSH). Cell clone formation was evaluated by clonogenic assay. C, Human osteosarcoma cell lines were treated with RA for 24 h in the presence or absence of 1.5 mmol/L GSH. Cell viability was measured by CCK‐8. *P < 0.05, **P < 0.01, significantly different compared with the untreated control group
To quantify cell apoptosis, we used Annexin V‐FITC/propidium iodide (PI) double staining. The results showed that the treatment of osteosarcoma cells with RA resulted in an increase in both early and late apoptotic cells (Figure 3A). Following pretreatment with 1.5 mmol/L GSH for 2 hours, the percentage of apoptotic cells was significantly reduced, as determined by flow cytometric analysis of apoptosis (Figure 3A).
Figure 3.
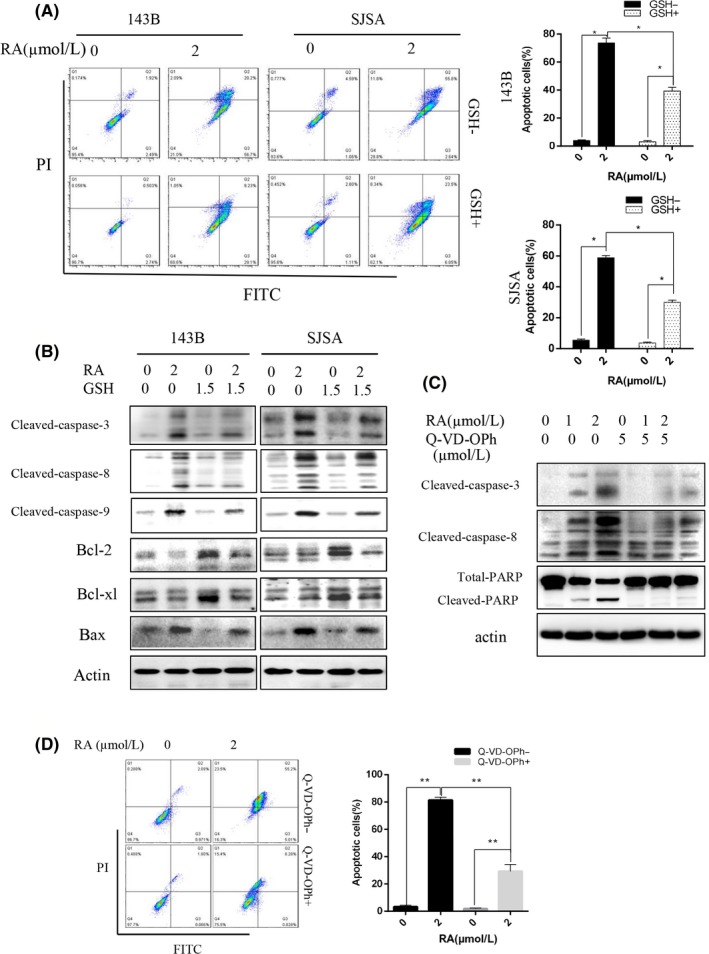
Raddeanin A (RA) induced apoptosis in human osteosarcoma cells. A, Cells were treated with RA for 48 h and analyzed using Annexin V‐FITC/propidium iodide (PI) flow cytometry. Histograms indicate the levels of apoptosis from three separate experiments. B, Cells were treated with RA for 48 h in the presence or absence of glutathione (GSH). Apoptosis‐related proteins were analyzed by western blotting. C, Cells were treated with RA in the presence or absence of the caspase inhibitor Q‐VD‐OPh, and cleaved caspase‐3 and caspase‐8 and total and cleaved PARP were analyzed. D, RA‐induced apoptosis in osteosarcoma cells pretreated with Q‐VD‐OPh was detected by Annexin V‐PE/FITC flow cytometry. *P < 0.05, **P < 0.01, significantly different compared with the untreated control group
We also investigated the expression of important signaling proteins involved in DNA repair and apoptosis by western blotting. As shown in Figure 3C, protein expression levels of cleaved PARP, cleaved caspase‐8, cleaved caspase‐9, cleaved caspase‐3, and Bax were all remarkably increased, and the protein expression levels of the antiapoptotic proteins Bcl‐2 and Bcl‐xl were decreased in osteosarcoma cells after RA treatment. The reduction in cleaved PARP, cleaved caspase‐8, cleaved caspase‐9, cleaved caspase‐3, and Bax protein was mitigated after RA treatment in osteosarcoma cells pretreated with 1.5 mmol/L GSH for 2 hours (Figure 3B). Additionally, the increase in Bcl‐2 and Bcl‐xl protein was promoted after pretreatment with GSH (Figure 3B). The caspase inhibitor Q‐VD‐OPh sharply reduced RA‐induced apoptosis as determined by western blotting and flow cytometry (Figure 3C,D) These results all indicate that RA rescues caspase‐dependent apoptosis and morphological apoptosis dependent on ROS generation.
3.3. Raddeanin A activates the JNK/c‐Jun signaling pathway dependent on ROS production
To evaluate the molecular mechanism of the antitumor activity of RA in osteosarcoma, activation of cell signaling molecules was evaluated. In particular, JNK and STAT3 activities were investigated because both are known to play critical roles in the regulation of apoptosis signal transduction.9, 10, 23, 35 RA induced the phosphorylation of ERK1/2, JNK, and c‐Jun (Figure 4A,B) proteins in 143B and SJSA osteosarcoma cells in a concentration‐ and time‐dependent method.
Figure 4.
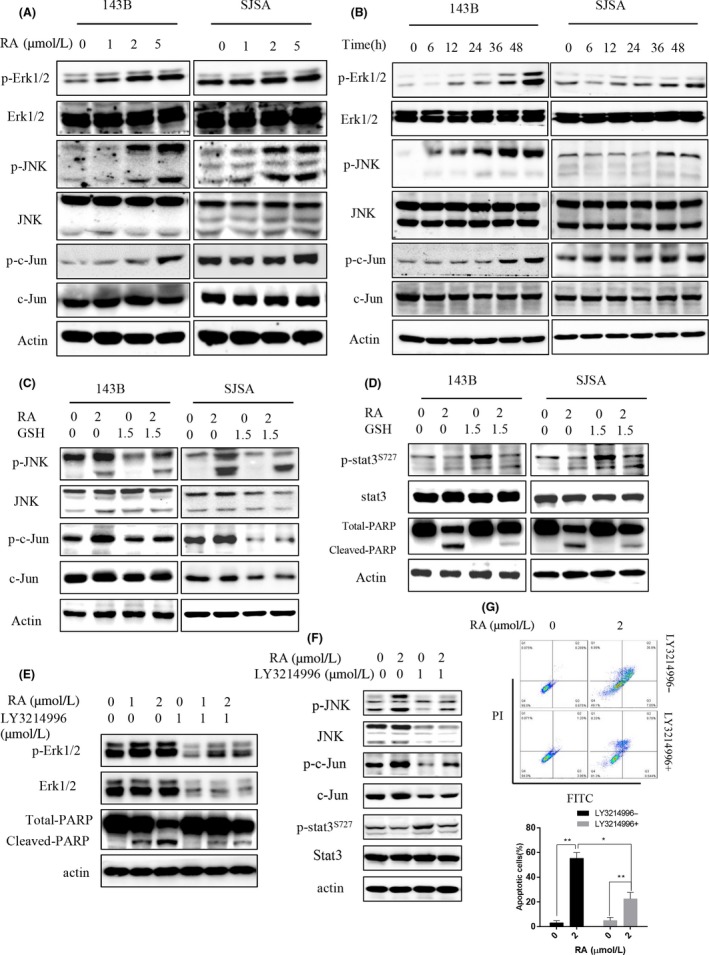
Raddeanin A (RA) activated the JNK signal pathway and increased reactive oxygen species (ROS) generation. A, Cells were treated with various concentrations of RA for 48 h. B, Cells were incubated with 2 μmol/L RA for different lengths of time. Expression of phospho‐Erk1/2, Erk1/2, phospho‐JNK, JNK, phospho‐c‐Jun, and c‐Jun was analyzed by western blotting. C,D, Cells were preincubated with glutathione (GSH) for 2 h and then treated with RA for 48 h. Expression of phospho‐JNK, JNK, phospho‐c‐Jun, c‐Jun, phospho‐STAT3, STAT3, and total and cleaved PARP was analyzed by western blotting. E,F, Cells were treated with RA in the presence or absence of the Erk1/2 inhibitor LY3214996, phospho‐Erk1/2, Erk1/2, phospho‐JNK, JNK, phospho‐c‐Jun, c‐Jun, total and cleaved PARP, phospho‐STAT3S727, and STAT3 were analyzed. G, RA‐induced apoptosis in osteosarcoma cells pretreated with LY3214996 was detected by flow cytometry. *P < 0.05, and **P < 0.01, significantly different compared with the untreated control group. PARP, poly‐ADP ribose polymerase; STAT3, signal transducer and activator of transcription 3
ROS generation is an early event in apoptosis and directly relates to caspase and MAPK activation, especially JNK signaling pathway activation.8, 15 We investigated whether ROS generated by RA induced apoptosis through JNK signal transduction. 143B and SJSA cells were subjected to pretreatment with the ROS scavenger GSH at 1.5 mmol/L for 2 hours, followed by RA treatment. Expression of phosphorylated JNK and c‐Jun was measured by western blot analysis. RA‐induced JNK and c‐Jun phosphorylation was obviously inhibited by pretreatment with GSH, suggesting that RA‐induced ROS‐mediated osteosarcoma cell apoptosis is JNK dependent (Figure 4C). In addition, increased expression of cleaved PARP induced by RA was attenuated in osteosarcoma cells pretreated with GSH (Figure 4D). The Erk1/2 inhibitor LY3214996 was used to assess the effect of Erk1/2 on osteosarcoma cells after RA treatment as shown in Figure 4E,F and G. The inhibitor inhibited RA‐induced apoptosis in osteosarcoma (Figure 4E,G). Interestingly, LY3214996 also inhibited JNK expression and marginally increased phospho‐STAT3S727 (Figure 4F). These results suggest that ROS generated by RA trigger JNK‐ and c‐Jun‐mediated osteosarcoma cell apoptosis.
3.4. Raddeanin A induces apoptosis by the JNK/c‐Jun pathway in human osteosarcoma cells
To further investigate the molecular mechanism by which RA induces apoptosis in human osteosarcoma, we used the specific JNK inhibitor SP600125 (Figure 5A) and shRNA targeting JNK (Figure 5B,E). As shown in Figure 5A and E, the expression of phospho‐c‐Jun and total c‐Jun was remarkably downregulated in SP600125‐treated cells. Meanwhile, the accumulation of cleaved PARP induced by RA was decreased in human osteosarcoma cells pretreated with SP600125 and JNK‐shRNA. The percentage of apoptotic cells was notably reduced in osteosarcoma cells treated with RA in the presence of SP600125 and JNK‐shRNA (Figure 5C,F). Although some reports have indicated that SP600125 elevates ROS generation SP600125 abolished the RA‐induced inhibition of proliferation in 143B and SJSA (Figure 5D), and the percent viability was significantly increased by approximately 15% in the presence of SP600125. RA induced apoptosis in a concentration‐dependent way in 143B and 143B‐JNK‐shRNA cells (Figure 5E). Western blot assay results indicated that phospho‐JNK was activated and that cleaved PARP accumulated in 143B‐JNK‐shRNA cells treated with RA. Compared with 143B cells treated with RA, cleaved PARP accumulation was reduced in 143B‐JNK‐shRNA cells (Figure 5E). Furthermore, pretreatment of cells with GSH and/or SP600125 remarkably reduced the RA‐induced activation of phospho‐JNK, phospho‐c‐Jun and cleaved PARP (Figure 6H). Collectively, these results show that RA induced apoptosis through the JNK/c‐Jun pathway.
Figure 5.
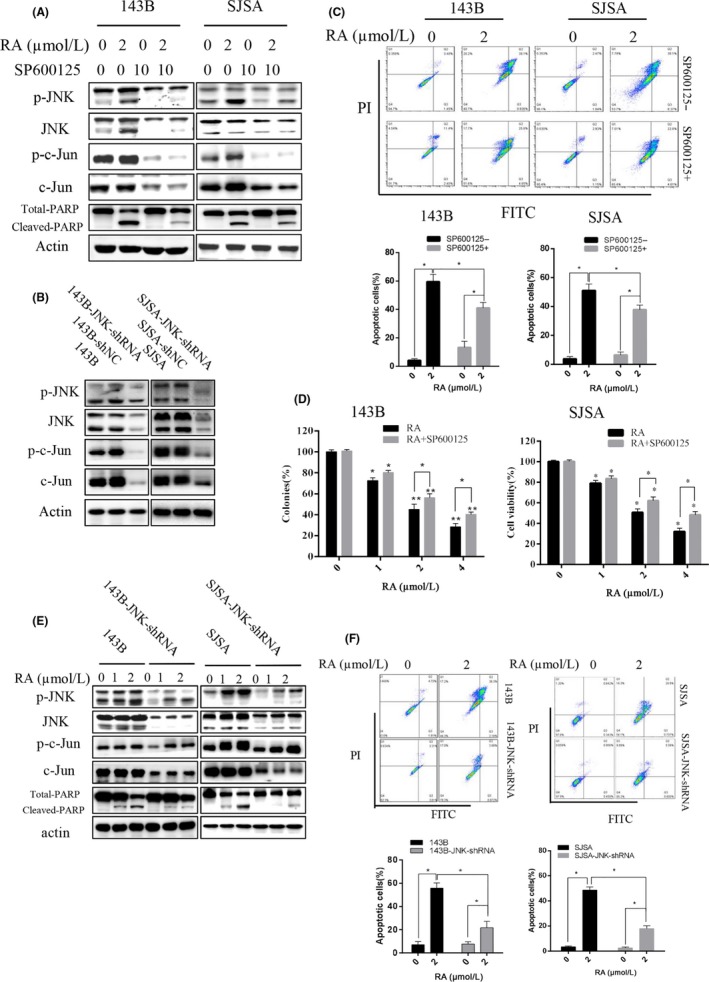
Role of JNK in apoptosis induced by Raddeanin A (RA) in human osteosarcoma cells. A, Expression of phospho‐JNK, JNK, phospho‐c‐Jun, c‐Jun, and total and cleaved PARP was analyzed by western blotting using the JNK inhibitor SP600125. B, JNK was knocked down in osteosarcoma cells using shRNA. C, Cells were analyzed using Annexin V‐ FITC/propidium iodide (PI) flow cytometry pretreated with SP600125. Histograms indicate degrees of apoptosis from three separate experiments. D, Cell viability was measured by CCK‐8. E, Expression of phospho‐JNK, JNK, phospho‐c‐Jun, c‐Jun, and total and cleaved PARP was analyzed by western blotting using shRNA targeting JNK. F, Cells were analyzed using Annexin V‐PE/FITC flow cytometry pretreated with JNK knockdown. *P < 0.05, **P < 0.01, significantly different compared with the untreated control group. PARP, poly‐ADP ribose polymerase
Figure 6.
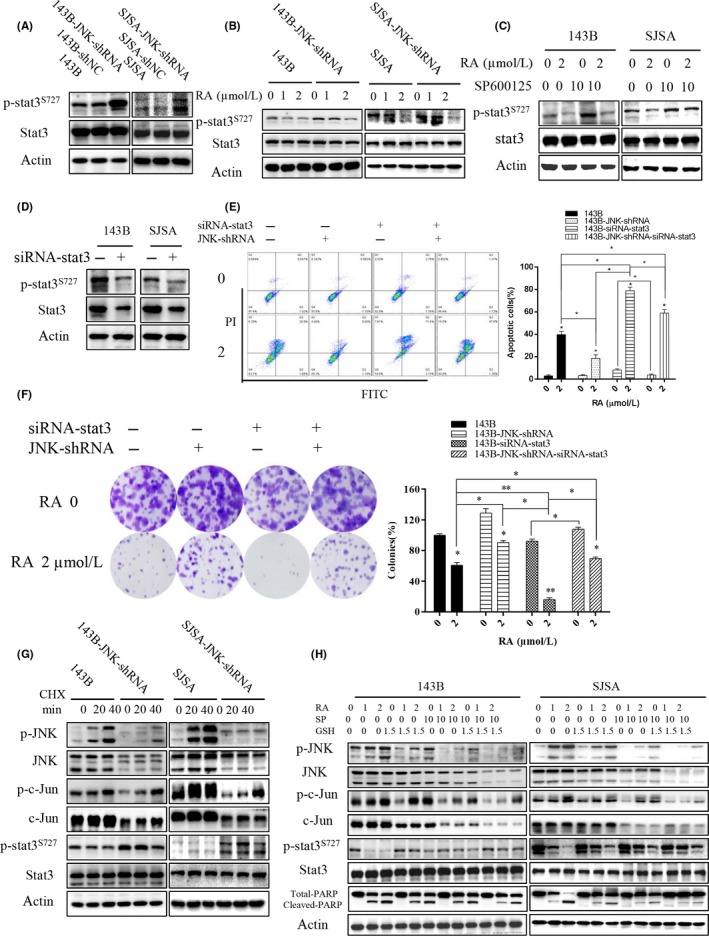
ROS/JNK/STAT3 pathway mediated Raddeanin A (RA)‐induced apoptosis in human osteosarcoma cells. A‐C, Expression of phospho‐STAT3S727 and STAT3 was analyzed by western blotting. A, Cells treated with shRNA targeting JNK in 143B and SJSA cells. B, Cells treated with RA after JNK knockdown compared with control. C, Cells treated with RA in the presence or absence of SP600125. D‐F, Cells treated with siRNA against STAT3. D, Expression of phospho‐STAT3S727 and STAT3 is detected. E, RA‐induced apoptosis detected in the presence or absence of siRNA‐STAT3 and/or JNK knockdown by flow cytometry. Histograms indicate degrees of apoptosis from three separate experiments. F, Clone formation is detected. Histograms indicate the colony‐formation rate from three separate experiments. G, Cells were incubated in 10 mg/mL CHX (cycloheximide) for various times (0, 20, and 40 min). H, Cells were pretreated with or without GSH/SP600125 for 2 h and then incubated in various concentrations of RA for 48 h. Expression of phospho‐JNK, JNK, phospho‐c‐Jun, c‐Jun, phospho‐STAT3S727, STAT3, and total and cleaved PARP was analyzed by western blotting. GSH, glutathione; PARP, poly‐ADP ribose polymerase; ROS, reactive oxygen species; STAT3, signal transducer and activator of transcription 3
3.5. Raddeanin A‐induced JNK pathway activation inhibits STAT3 phosphorylation
Our team previously reported that STAT3 is a critical regulator of osteosarcoma development and progression.25, 26, 27 The effect of RA on the JNK and STAT3 signaling pathways was assessed to investigate the molecular mechanism of the antitumor activity of RA using pharmacological inhibitors and shRNA. As shown in Figure 6A, knockdown of JNK with shRNA remarkably increased the formation of phospho‐STAT3 compared with that noted in control cells. Analogously, the reduction in phospho‐STAT3 was attenuated in cells pretreated with SP600125 (Figure 6B). The reduction in phospho‐STAT3 was attenuated with the activation of phospho‐JNK and phospho‐c‐Jun in osteosarcoma cells subjected to JNK knockdown or SP600125 combined with RA treatment (Figure 6B,C). Similarly, in human osteosarcoma cells pretreated with GSH and/or SP600125, the reduction in phospho‐STAT3 was attenuated by RA treatment (Figure 6H).
To address the effect of RA‐induced JNK/c‐Jun activation on STAT3 phosphorylation, an siRNA against STAT3 was used (Figure 6D‐F). As shown in Figure 6E, RA‐induced apoptosis was weakened by JNK‐shRNA, but the apoptosis was enhanced by siRNA‐STAT3. Although JNK‐shRNA rescued RA‐induced apoptosis, siRNA‐STAT3 counteracted this effect. The apoptotic difference of siRNA‐STAT3 and JNK‐shRNA‐siRNA‐STAT3 on osteosarcoma cells was statistically significant (Figure 6E), and the difference of colony formation was similarly statistically significant (Figure 6F). The effect of RA on colony formation was partly weakened by JNK‐shRNA or SP600125, but the weakened effect was cancelled by siRNA‐STAT3 or a specific pharmacological STAT3 inhibitor (NSC 74859, S3I‐201) (Figures 6F and S1A). Furthermore, RA‐induced apoptosis was weakened by SP600125, and the effect was cancelled by NSC74859 (Figure S1B). In short, RA induced JNK/c‐Jun activation and phospho‐STAT3 inhibition in human osteosarcoma cells.
When JNK‐shRNA cells were treated with the protein synthesis inhibitor CHX, phospho‐STAT3 was obviously increased compared with that in control cells (Figure 6G). CHX also resulted in high levels of active JNK, which are necessary for tumor necrosis factor (TNF)‐induced apoptosis.9 With the activation of CHX‐induced phospho‐JNK, levels of phospho‐STAT3 were slightly decreased in two groups treated with CHX from 20 to 40 minutes; however, STAT3 phosphorylation was increased in the groups after JNK knockdown (Figure 6G). This result indicated that phospho‐STAT3 was a likely downstream protein of JNK in the cascade reaction induced by RA. CHX completely inhibited protein synthesis, confirming the inducible nature of phosphorylated STAT3 with downregulation of JNK expression. Furthermore, pretreatment of cells with GSH and/or SP600125 partly rescued the RA‐induced inhibition of phospho‐STAT3 (Figure 6H). Taken together, these results indicate that RA‐induced activation of the JNK pathway inhibits phospho‐STAT3 in human osteosarcoma cells.
3.6. Raddeanin A inhibits tumor growth and induces apoptosis in osteosarcoma xenografts
To determine whether RA inhibits osteosarcoma growth and induces apoptosis in vivo, we established osteosarcoma xenografts in the medullary cavity of tibia for each mouse injected with 143B. When the tumors were established, the mice were injected with either vehicle (DMSO) or RA every 3 days. As shown in Figure 7A, RA given at 5 or 10 mg/kg significantly inhibited tumor growth, and tumor weights were markedly reduced in the RA‐injection groups compared with the vehicle group. In Figure 7B, RA treatment at 5 and 10 mg/kg resulted in significant decreases in the weights of the tumor‐bearing leg by 33.8% and 58%, respectively, after eight doses. Moreover, we detected apoptotic cells in osteosarcoma tissue sections using the TUNEL assay. As shown in Figure 7C, 5 and 10 mg/kg RA notably induced apoptosis in a dose‐dependent way in the tumor tissues. To address the mechanism of RA‐induced apoptosis in vivo, p‐JNK, JNK, p‐c‐Jun, c‐Jun, p‐STAT3S727, and STAT3 proteins were detected in tumor tissues by western blot (Figure 7D). In vivo, RA (10 mg/kg) plus SP600125 (1 mg/kg) weakened the effect of RA treatment alone, but the weakened effect was partially rescued by NSC73859 (2 mg/kg; Figure S2). The results showed that RA induced JNK/c‐Jun activation and inhibited STAT3 activation in vivo. Taken together, these data showed that RA inhibits tumor growth and promotes the apoptosis of osteosarcoma cells in a preclinical osteosarcoma xenograft model.
Figure 7.
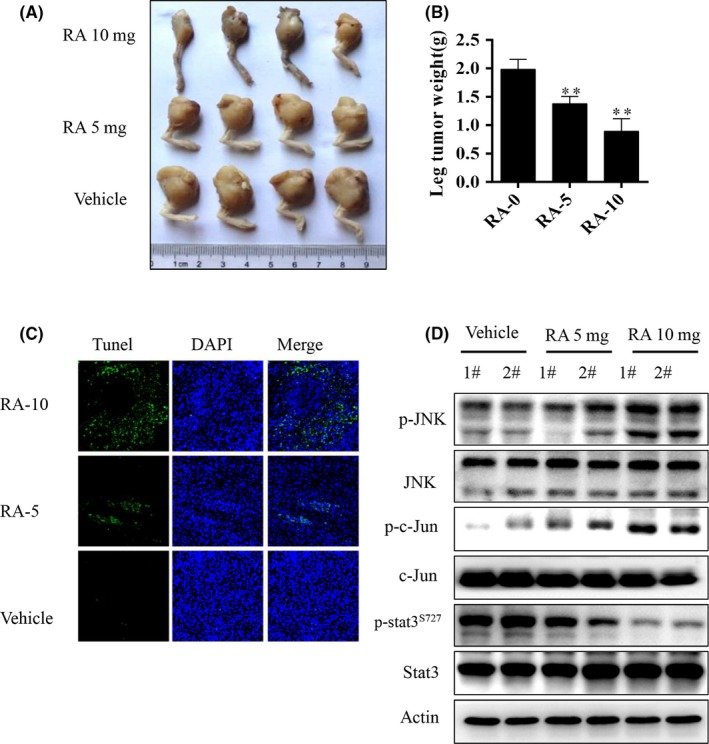
Raddeanin A (RA) inhibited tumor growth and induced apoptosis in an orthotopic osteosarcoma mouse model. 143B was injected into the medullary cavity of the tibia of each mouse. A, Macroscopic appearance of primary osteosarcoma tumors in the tibia and B, tumor weight quantification in BALB/c nude mice after treatment with RA or vehicle. C, TUNEL assay of tumor samples from legs in different treatment groups. D, Expression of phospho‐JNK, JNK, phospho‐c‐Jun, c‐Jun, phospho‐STAT3S727, and STAT3 was analyzed in tumor tissues by western blotting. **P < 0.01 significantly different compared with vehicle control. STAT3, signal transducer and activator of transcription 3
4. DISCUSSION
In the present study, we investigated the antiproliferative and apoptotic effects of RA on human osteosarcoma cell lines. We found that RA significantly reduced cell proliferation, blocked clone formation and induced apoptosis of human osteosarcoma cells in vitro and in vivo. In addition, RA‐induced apoptosis was mediated by mitochondrial‐dependent pathways through ROS generation, apoptotic proteins, JNK/c‐Jun pathway activation and STAT3 dephosphorylation. As shown in our research, RA, a promising therapeutic agent, might suppress osteosarcoma development through the activation of integrated intracellular signaling pathways.
Natural bioactive compounds derived from traditional Chinese medicinal herbs have been widely investigated because of their antiproliferative, antiangiogenic, and antitumor effects on various diseases, as well as their safety, efficacy and immediate availability. The effects of natural compounds on the inhibition of osteosarcoma have previously been reported by our research team.25, 26, 27, 36 For instance, we found that pectolinarigenin significantly suppressed osteosarcoma cell proliferation, induced apoptosis and inhibited migration and invasion in osteosarcoma cells.26 Alternol, a novel compound purified from microbial fermentation products, not only inhibited osteosarcoma cell proliferation and migration and induced caspase‐dependent apoptosis and G2/M cell cycle arrest in vitro, but also suppressed tumor growth in an orthotopic tibial osteosarcoma model.27 In addition, our team has demonstrated that two natural compounds, toosendanin and erianin, suppress human osteosarcoma growth and metastasis.25, 36 The results of the present research indicate that RA, a natural bioactive compound, significantly suppressed cell proliferation and induced apoptosis in a dose‐ and time‐dependent method. Wang et al7 verified that RA could suppress osteoclast formation and bone resorption, possibly through inhibition of the SRC/AKT signaling pathway in human breast cancer cells and in a mouse calvarial model. RA suppresses the angiogenesis and growth of human colorectal tumors by inhibiting the VEGFR2 pathway.4 These reports support our results, which indicate that RA has potential as a therapeutic agent for osteosarcoma. RA has been shown to induce ROS generation in other cell lines, such as the prostate cancer cell line RM‐1 (Figure S3). However, those reports did not investigate the effects of RA‐induced JNK/c‐Jun activation on STAT3 deactivation in human osteosarcoma. Our investigation demonstrated that the imbalance of GSSG/GSH, as a result of the RA inhibition of glutathione reductase (GR), may result in excessive ROS production (Figure S4) in human osteosarcoma.
In the present research, RA was found to induce ROS generation, leading to activation of the proapoptotic protein Bax and suppression of the antiapoptotic proteins Bcl‐2 and Bcl‐xl. Generally, caspase family proteases can be activated through two pathways: the death receptor‐mediated pathway and the mitochondria‐dependent pathway.8, 13 Cleavage of caspases leads to cleavage of the specific substrate PARP, thus inducing apoptosis in cancer cells. ROS production in response to RA can be attenuated using the peroxide scavenger, along with inhibition of cleaved caspase‐3, cleaved caspase‐8, and cleaved caspase‐9, leading to inhibition of cell apoptosis. The ROS scavenger GSH reduced the level of ROS induced by RA, leading to increased cell proliferation and inhibition of apoptosis. Moreover, clone formation was increased in the presence of GSH. Furthermore, we found that RA caused ROS accumulation in human osteosarcoma cells by activating the JNK pathway to induce apoptosis. Decreased ROS generation inhibited activation of the JNK pathway and suppressed cleavage of the proapoptotic protein PARP. These data indicate that ROS accumulation contributes to RA‐induced apoptosis of human osteosarcoma cells and activates the JNK/c‐Jun pathway, which is known to induce apoptosis.
MAPK pathways are one of the numerous downstream cascades of the ROS signaling pathway closely associated with cell proliferation, differentiation, mitosis, survival, and apoptosis.10 JNK are extremely important in the process of apoptotic cell death as one subgroup of the MAPK family. JNK and ERK, as stress‐activated protein kinases, are activated by various stress stimuli, such as bioactive compounds, to induce apoptosis.9, 12, 13, 15, 29, 32, 34, 37, 38, 39 Consistent with these results, we observed that JNK activation and ERK1/2 activation were involved in RA‐induced apoptosis in a drug concentration‐ and time‐dependent method. In the present study, JNK activation was mediated by ROS, as demonstrated by the phenomenon that treatment with the ROS scavenger GSH attenuated the RA‐induced increase in phosphorylated JNK. Additionally, when JNK activity was inhibited by the specific pharmacological inhibitor SP600125 and the specific JNK‐shRNA, cell proliferation and apoptosis and PARP cleavage decreased. JNK inhibition could also mitigate RA‐mediated apoptosis without affecting upstream ROS production by RA (Figure S5). Indeed, blockage of JNK activation attenuated the anticancer activity of RA, further supporting a central role of JNK in mediating the proapoptotic effect of RA.
STAT3 has been implicated in the development of different human malignancies.23 Previous studies have confirmed that aberrant constitutive activation of STAT3 is strongly associated with the initiation, maintenance, and progression of various malignancies.23, 40 Our research team has reported that inhibition of STAT3 phosphorylation has beneficial clinical therapeutic effects on human osteosarcoma. Persistently active STAT3 has been identified in many human cancers and appears to be required for the continued growth or resistance to apoptosis of cultured human cancer cells.20, 25, 41, 42 In this study, phospho‐JNK activation induced significant phospho‐STAT3 degradation. Disruption of the JNK signaling pathway by treatment with the JNK inhibitor SP600125 or shRNA knockdown of JNK abrogated STAT3 phosphorylation in RA‐induced malignant transformation of the cells. Lim and Cao showed that STAT3 is a target of JNK, which may regulate STAT3 activity through both Ser‐727 phosphorylation‐dependent and phosphorylation‐independent mechanisms.16 Additionally, several reports have demonstrated that there are interacting regions in STAT3 and c‐Jun that participate in cooperative transcriptional activation.14, 18, 19, 43 These concepts support our finding that phospho‐STAT3 was suppressed by the activation of phospho‐JNK and phospho‐c‐Jun.
Additionally, expression profiling and functional studies in vitro and in vivo have shown that STAT3 activation is mediated by the combined action of JAK, SRC, c‐ABL, and JNK.22, 23 In the present research, JNK knockdown using shRNA led to the accumulation of phospho‐STAT3, while the total STAT3 level remained steady. However, the transcription factor c‐Jun, a significant downstream effector of JNK, was abolished in terms of both the phosphorylated form and total protein after JNK knockdown in human osteosarcoma cells. The protein synthesis inhibitor CHX immediately activates JNK.9 However, within 40 minutes of the treatment of 143‐JNK‐shRNA cells with CHX, compared with 143B cells, phospho‐STAT3 accumulated after JNK inhibition, which demonstrates the inhibitory effect of JNK on STAT3 activation in human osteosarcoma. Based on these results, it could be concluded that JNK activation, reduced STAT3 phosphorylation, and ROS generation are involved in RA‐induced human osteosarcoma cell apoptosis. Additionally, when JNK activity was blocked by SP600125, and ROS were scavenged by GSH, respectively or in combination, PARP cleavage decreased, and phospho‐STAT3 accumulated in RA‐induced apoptosis. From these data, we propose that RA‐induced activation of c‐Jun and JNK may have negative regulatory effects on STAT3 transcriptional activity in that they downregulate phosphorylation in a concerted contributory way. Therefore, intracellular ROS generation by RA treatment induces osteosarcoma cell apoptosis by JNK/c‐Jun pathway activation and STAT3 inhibition.
In conclusion, our research shows that RA has the potential to inhibit osteosarcoma growth by JNK/c‐Jun activation and STAT3 inhibition and to induce osteosarcoma apoptosis in a mouse primary tibia model. Therefore, RA is a potential bioactive compound for treating osteosarcoma and could be used as a potent clinically therapeutic agent. However, it is possible that RA exerts its activity against osteosarcoma by impairing/activating other signaling pathways. To understand the function of RA in osteosarcoma, the molecular mechanism requires comprehensive investigation.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGEMENTS
This project was supported by the NSFC (81502604, 81501584, and 81702973), The Shanghai Science and Technology Commission (14140904000), the Doctoral Innovation Fund of Shanghai Jiaotong University School of Medicine (No. BXJ201732), the Shanghai Municipal Commission of Health and Family Planning (No. 20164Y0270), and a Research Grant from the Shanghai Hospital Development Center (SHDC12013107).
Wang Z, Shen J, Sun W, et al. Antitumor activity of Raddeanin A is mediated by Jun amino‐terminal kinase activation and signal transducer and activator of transcription 3 inhibition in human osteosarcoma. Cancer Sci. 2019;110:1746–1759. 10.1111/cas.14008
Wang and Shen contributed equally to this paper.
REFERENCES
- 1. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: current treatment and a collaborative pathway to success. J Clin Oncol. 2015;33(27):3029‐3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer. 2014;14(11):722‐735. [DOI] [PubMed] [Google Scholar]
- 3. Tang N, Song WX, Luo J, Haydon RC, He TC. Osteosarcoma development and stem cell differentiation. Clin Orthop Relat Res. 2008;466(9):2114‐2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guan YY, Liu HJ, Luan X, et al. Raddeanin A, a triterpenoid saponin isolated from Anemone raddeana, suppresses the angiogenesis and growth of human colorectal tumor by inhibiting VEGFR2 signaling. Phytomedicine. 2015;22(1):103‐110. [DOI] [PubMed] [Google Scholar]
- 5. Xue G, Zou X, Zhou JY, et al. Raddeanin A induces human gastric cancer cells apoptosis and inhibits their invasion in vitro. Biochem Biophys Res Commun. 2013;439(2):196‐202. [DOI] [PubMed] [Google Scholar]
- 6. Li JN, Yu Y, Zhang YF, Li ZM, Cai GZ, Gong JY. Synergy of Raddeanin A and cisplatin induced therapeutic effect enhancement in human hepatocellular carcinoma. Biochem Biophys Res Commun. 2017;485(2):335‐341. [DOI] [PubMed] [Google Scholar]
- 7. Wang Q, Mo J, Zhao C, et al. Raddeanin A suppresses breast cancer‐associated osteolysis through inhibiting osteoclasts and breast cancer cells. Cell Death Dis. 2018;9(3):376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. 2013;19(6):546‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ray RM, Jin S, Bavaria MN, Johnson LR. Regulation of JNK activity in the apoptotic response of intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;300(5):G761‐G770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000;103:14. [DOI] [PubMed] [Google Scholar]
- 11. Wan‐Wan L, Hsu YW. Cycloheximide‐induced cPLA2 activation is via the MKP‐1 down‐regulation and ERK activation. Cell Signal. 2000;12(7):457‐461. [DOI] [PubMed] [Google Scholar]
- 12. Zhang Z, Ren Z, Chen S, et al. ROS generation and JNK activation contribute to 4‐methoxy‐TEMPO‐induced cytotoxicity, autophagy, and DNA damage in HepG2 cells. Arch Toxicol. 2018;92(2):717‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci. 2005;118(Pt 2):265‐267. [DOI] [PubMed] [Google Scholar]
- 14. Ivanov VN, Krasilnikov M, Ronai Z. Regulation of Fas expression by STAT3 and c‐Jun is mediated by phosphatidylinositol 3‐kinase‐AKT signaling. J Biol Chem. 2002;277(7):4932‐4944. [DOI] [PubMed] [Google Scholar]
- 15. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19(2):142‐149. [DOI] [PubMed] [Google Scholar]
- 16. Lim CP, Cao X. Serine phosphorylation and negative regulation of Stat3 by JNK. J Biol Chem. 1999;274(43):8. [DOI] [PubMed] [Google Scholar]
- 17. Zhang X, Wrzeszczynska MH, Horvath CM, Darnell JE Jr. Interacting regions in Stat3 and c‐Jun that participate in cooperative transcriptional activation. Mol Cell Biol 1999;19(10):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ivanov VN, Bhoumik A, Krasilnikov M, et al. Cooperation between STAT3 and c‐Jun suppresses FAS transcription. Mol Cell. 2001;7:12. [DOI] [PubMed] [Google Scholar]
- 19. Li Y, Kundu P, Seow SW, et al. Gut microbiota accelerate tumor growth via c‐jun and STAT3 phosphorylation in APC Min/+ mice. Carcinogenesis. 2012;33(6):1231‐1238. [DOI] [PubMed] [Google Scholar]
- 20. Tarafder S, Chen E, Jun Y, et al. Tendon stem/progenitor cells regulate inflammation in tendon healing via JNK and STAT3 signaling. FASEB J. 2017;31(9):3991‐3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei ZZ, Yu SP, Lee JH, et al. Regulatory role of the JNK‐STAT1/3 signaling in neuronal differentiation of cultured mouse embryonic stem cells. Cell Mol Neurobiol. 2014;34(6):881‐893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu J, Chen B, Lu Y, Guan Y, Chen F. JNK‐dependent Stat3 phosphorylation contributes to Akt activation in response to arsenic exposure. Toxicol Sci. 2012;129(2):363‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol. 2016;11(2):308‐318. [DOI] [PubMed] [Google Scholar]
- 24. Chakraborty D, Sumova B, Mallano T, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun. 2017;8(1):1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang T, Li J, Yin F, et al. Toosendanin demonstrates promising antitumor efficacy in osteosarcoma by targeting STAT3. Oncogene. 2017;36(47):6627‐6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang T, Li S, Li J, et al. Natural product pectolinarigenin inhibits osteosarcoma growth and metastasis via SHP‐1‐mediated STAT3 signaling inhibition. Cell Death Dis. 2016;7(10):e2421. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27. Zuo D, Zhou Z, Wang H, et al. Alternol, a natural compound, exerts an anti‐tumour effect on osteosarcoma by modulating of STAT3 and ROS/MAPK signalling pathways. J Cell Mol Med. 2017;21(2):208‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De A1, Lewis XZ, Gambhir SS. Noninvasive imaging of lentiviral‐mediated reporter gene expression in living mice. Mol Ther 2003;7(1):681‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahn J, Chung YW, Park JB, Yang KM. Omega‐hydroxyundec‐9‐enoic acid induces apoptosis by ROS mediated JNK and p38 phosphorylation in breast cancer cell lines. J Cell Biochem. 2018;119(1):998‐1007. [DOI] [PubMed] [Google Scholar]
- 30. Xie X, Zhao Y, Ma CY, et al. Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br J Pharmacol. 2015;172(15):3929‐3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang T, Li Y, Park KA, et al. Cucurbitacin induces autophagy through mitochondrial ROS production which counteracts to limit caspase‐dependent apoptosis. Autophagy. 2012;8(4):559‐576. [DOI] [PubMed] [Google Scholar]
- 32. Li HY, Zhang J, Sun LL, et al. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: an in vitro and in vivo study. Cell Death Dis. 2015;6:e1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang G, Zhang T, Sun W, et al. Arsenic sulfide induces apoptosis and autophagy through the activation of ROS/JNK and suppression of Akt/mTOR signaling pathways in osteosarcoma. Free Radic Biol Med. 2017;106:24‐37. [DOI] [PubMed] [Google Scholar]
- 34. Park S, Lim W, Bazer FW, Song G. Apigenin induces ROS‐dependent apoptosis and ER stress in human endometriosis cells. J Cell Physiol. 2018;233(4):3055‐3065. [DOI] [PubMed] [Google Scholar]
- 35. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang H, Zhang T, Sun W, et al. Erianin induces G2/M‐phase arrest, apoptosis, and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2016;7(6):e2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chambers JW, LoGrasso PV. Mitochondrial c‐Jun N‐terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J Biol Chem. 2011;286(18):16052‐16062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim JH, Shim JW, Eum DY, et al. Downregulation of UHRF1 increases tumor malignancy by activating the CXCR4/AKT‐JNK/IL‐6/Snail signaling axis in hepatocellular carcinoma cells. Sci Rep. 2017;7(1):2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shen K, Xie J, Wang H, et al. Cambogin induces caspase‐independent apoptosis through the ROS/JNK pathway and epigenetic regulation in breast cancer cells. Mol Cancer Ther. 2015;14(7):1738‐1749. [DOI] [PubMed] [Google Scholar]
- 40. Mahendrarajah N, Borisova ME, Reichardt S, et al. HSP90 is necessary for the ACK1‐dependent phosphorylation of STAT1 and STAT3. Cell Signal. 2017;39:9‐17. [DOI] [PubMed] [Google Scholar]
- 41. Peng Z, Zhang C, Zhou W, Wu C, Zhang Y. The STAT3/NFIL3 signaling axis‐mediated chemotherapy resistance is reversed by Raddeanin A via inducing apoptosis in choriocarcinoma cells. J Cell Physiol. 2018;233(7):5370‐5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Q, Yang Z, Jia Z, et al. ISL‐1 is overexpressed in non‐Hodgkin lymphoma and promotes lymphoma cell proliferation by forming a p‐STAT3 p‐c‐Jun ISL‐1 complex. Mol Cancer 2014;29:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou Z, Lu X, Wang J, Xiao J, Liu J, Xing F. microRNA let‐7c is essential for the anisomycin‐elicited apoptosis in Jurkat T cells by linking JNK1/2 to AP‐1/STAT1/STAT3 signaling. Sci Rep. 2016;6:24434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials