

Abstract
Regulatory T (Treg) cells help to maintain tolerance and prevent the development of autoimmune diseases. Retinoic acid (RA) can promote peripheral conversion of naïve T cells into Foxp3+ Treg cells. Here, we show that RA can act as an adjuvant to induce antigen‐specific type 1 Treg (Tr1) cells, which is augmented by co‐administration of IL‐2. Immunization of mice with the model antigen KLH in the presence of RA and IL‐2 induces T cells that secrete IL‐10, but not IL‐17 or IFN‐γ, and express LAG‐3, CD49b and PD‐1 but not Foxp3, a phenotype typical of Tr1 cells. Furthermore, immunization of mice with the autoantigen MOG in the presence of RA and IL‐2 induces Tr1 cells, which suppress pathogenic Th1 and Th17 cells that mediate the development of experimental autoimmune encephalomyelitis (EAE), an autoimmune disease of the CNS. Furthermore, immunization with a surrogate autoantigen, RA and IL‐2 prevents development of spontaneous autoimmune uveitis. Our findings demonstrate that the induction of autoantigen‐specific Tr1 cells can prevent the development of autoimmunity.
Keywords: autoimmune disease, immune suppression, regulatory T cell, retinoic acid, Th17 cell
Subject Categories: Immunology, Molecular Biology of Disease, Signal Transduction
Introduction
Autoimmune diseases, such as multiple sclerosis (MS) and uveitis, can develop following a breakdown in self‐tolerance and activation of autoantigen‐reactive lymphocytes, which migrate to tissues of autoantigen expression where they trigger inflammation. Current and developing treatments against autoimmune diseases mainly target the mediators of inflammation. These include antibodies and other biological drugs that neutralize the inflammatory cytokines TNF, IL‐17 and IL‐23, or block the function (CD52, CD20) or migration (VLA‐4) of pathogenic lymphocytes 1, 2, 3. While these treatments reduce the symptoms of certain autoimmune diseases, they can be associated with drug unresponsiveness and side effects due to complete suppression of cytokines that play key roles in host defence against infection. Alternatively, re‐balancing the immune response in favour of immune regulation may influence the development of autoimmunity and may hold the key to safer and more effective treatment approaches. Indeed, the development of autoantigen‐specific regulatory T (Treg) cells that specifically target and inhibit autoreactive inflammatory lymphocytes without impairing other functions of the immune system is considered the “Holy Grail” in the prevention or treatment of autoimmune diseases 4, 5. However, isolating and expanding autoantigen‐specific Treg cells in vivo has proven to be challenging and has restricted the development of effective Treg‐based therapy. Nevertheless, studies in animal models and humans have shown that nasal or parenteral administration of peptides corresponding to sequences from self‐antigens either in solution, trapped in nanoparticles or presented by tolerogenic dendritic cells (DC) can attenuate autoimmune diseases through induction of tolerance and Treg cells 6, 7, 8, 9, 10. Conventional or natural Treg cells that develop in the thymus constitutively express Foxp3, a lineage‐defining transcription factor 11. Inducible Foxp3+ Treg cells can also develop in the periphery following conversion from naïve conventional Foxp3− T cells, especially under the influence of TGF‐β. Adaptive Treg or Tr1 cells can also be generated in the periphery in response to antigen stimulation 12, 13. These Treg cells do not express Foxp3 but secrete the immunosuppressive cytokine IL‐10 and are characterized by surface expression of CD49b and LAG‐3 14.
Treg cells play a central role in the protection against autoimmune diseases by maintaining self‐tolerance through continuous inhibition of autoreactive immune cells in the periphery. They can suppress the development of inflammation through a variety of mechanisms including the secretion of the immunosuppressive cytokine IL‐10, as well as their surface expression of the immune checkpoint CTLA‐4, PD‐1 and LAG‐3, which inhibit a broad range of immune cells, including antigen presenting cells (APCs), B cells, NK cells, CD4 and CD8 T cells 15.
Environmental factors can have a powerful influence on susceptibility to autoimmune diseases, and these can operate through modulation of T‐cell function. Retinoic acid (RA), the active metabolite of vitamin A, is a key regulator of CD4 T‐cell homeostasis, particularly in the gut, where maintenance of self‐tolerance is essential to homeostasis 16. RA is an important cofactor for the induction of extra‐thymic Treg cells; it greatly enhances Treg cell conversion induced by TCR activation and TGF‐β in vitro and facilitates the differentiation of inducible Treg cells following oral administration of antigens 17, 18, 19. Moreover, RA plays a critical role in maintaining natural Treg cells in inflammatory conditions 20, 21. RA can also modulate other immune cells, such as DCs and γδ T cells, and has been assessed as a potential treatment for autoimmune disease 22, 23, while inhibitors of RA are effective in a model of cancer 24.
Recent studies have shown that treatment with low‐dose IL‐2 induces the expansion of Treg cells without inducing the activation of effector T cells. This approach has shown high efficacy in mouse models of type 1 diabetes, food allergy and Alzheimer's disease 25, 26, 27. These studies, coupled with encouraging effects of low‐dose IL‐2 in clinical trials for type 1 diabetes, systemic lupus erythematosus and chronic graft‐vs.‐host disease, suggest that IL‐2 may be a promising first‐line treatment against autoimmunity 28, 29, 30, 31, 32, 33.
In this study, we examined the hypothesis that RA and IL‐2 could act as adjuvants to promote the induction of autoantigen‐specific Treg cells in vivo and that this could attenuate the development of autoimmune disease. We found that immunization of mice with foreign or self‐antigens in combination with RA and IL‐2 induced Tr1‐type antigen‐specific T cells that express the immune checkpoints LAG‐3, PD‐1 and CTLA‐4, but not Foxp3, and produce the anti‐inflammatory cytokine IL‐10. Furthermore, in vivo expansion of these antigen‐specific Tr1 cells impaired the induction and migration of Th1 and Th17 cells that mediate inflammation in experimental autoimmune encephalomyelitis (EAE), experimental autoimmune uveitis (EAU) and spontaneous uveitis models.
Results and Discussion
RA and IL‐2 act as adjuvants for induction of Tr1 cells in vivo
RA plays a role in maintenance of tolerance and can enhance TGF‐β‐induced conversion of naïve T cells into Treg cells in vitro 19, 24. Treg cells express the receptor for IL‐2 and it has been shown that treatment with low‐dose IL‐2 can expand Foxp3+ Treg cells in vitro and in vivo and that these Treg cells suppress inflammatory responses 34. Here, we assessed the capacity of RA to act as an adjuvant to induce antigen‐specific Treg cells in vivo in response to immunization of mice with the model foreign antigen KLH and if this could be augmented by addition of IL‐2. Flow cytometric analysis (gating strategy shown in Fig EV1) revealed that immunization of mice with KLH and RA promoted induction of a population of CD49b+ LAG‐3+ CD4 T cells that was further enhanced by co‐administration of IL‐2 (Fig 1A and B). In contrast, immunization with KLH alone or KLH and IL‐2 did not expand CD49b+ LAG‐3+ CD4 T cells in vivo (Fig 1A and B). The frequency and absolute number of Foxp3+ T cells were not enhanced by any immunization protocol examined (Fig 1A and B). Co‐expression of CD49b and LAG‐3, without Foxp3, on CD4 T cells is consistent with the induction of Tr1‐type Treg cells 14. Assessment of other markers expressed by Tr1 cells revealed that the induced Tr cells expressed CTLA‐4, PD‐1, CD226 and ICOS, as well as CD25 (Fig 1C).
Figure EV1. Gating strategy for flow cytometry analysis of Tr1 cells.
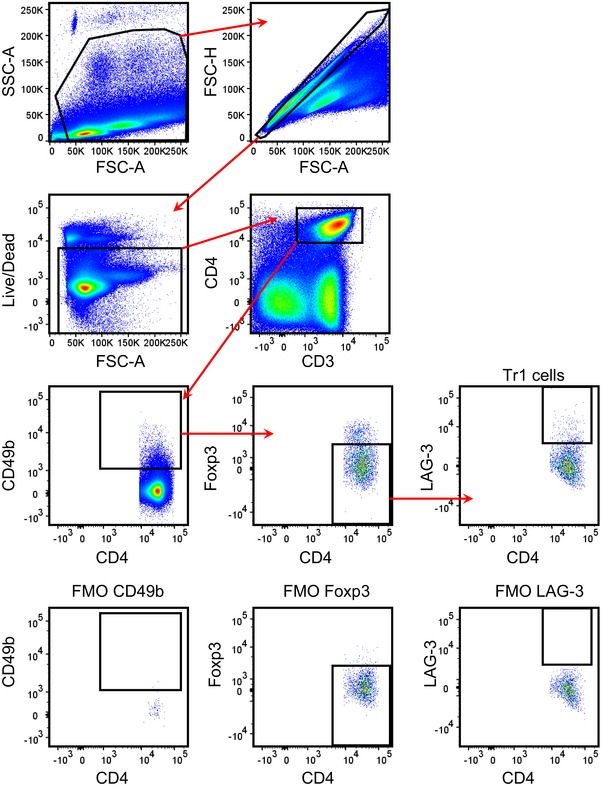
Figure 1. Immunization with antigen, RA and IL‐2 induces antigen‐specific IL‐10‐secreting LAG‐3+ CD4 T cells.
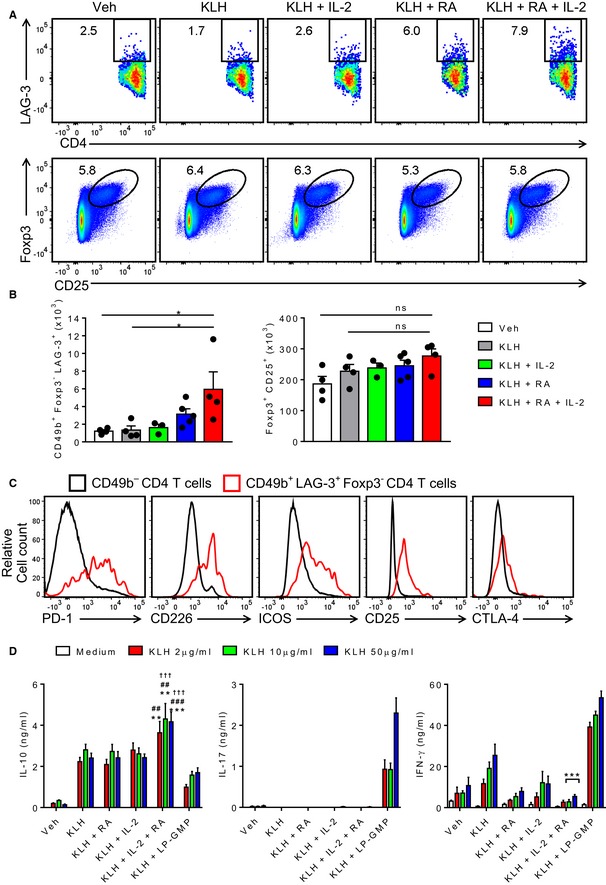
- Ex vivo flow cytometry on LN cells of mice 7 days after two s.c. immunizations (14 days apart) with PBS + DMSO (Veh), KLH + DMSO (KLH), KLH + IL‐2, KLH + RA, KLH + RA + IL‐2. Results are representative FACS plots of LAG3+CD4+ or CD25+Foxp3+ cells after gating on live single CD3+CD4+CD49b+Foxp3− and live single CD3+CD4+ lymphocytes, respectively.
- Mean absolute numbers of LAG‐3+CD49b+Foxp3− or CD25+Foxp3+ CD4 T cells in LNs as described in (A). Bars are mean + SEM (n = 3, 4 or 5). *P < 0.05 by one‐way ANOVA with Tukey post hoc test.
- Expression of Tr1 markers PD‐1, CD226, ICOS, CD25 and CTLA‐4 on CD49b+ LAG‐3+ Foxp3−CD4+ T cells or on CD49b−CD4+ T cells.
- IL‐10, IL‐17 and IFN‐γ production by ELISA on supernatants of spleen cells from mice immunized with PBS + DMSO (Veh), KLH + DMSO (KLH), KLH + RA, KLH + IL‐2, KLH + RA + IL‐2 or KLH + LP‐GMP (LP1569 + c‐di‐GMP) and cultured for 5 days with increasing concentrations of KLH. Bars are mean + SEM (n = 4 or 5). **P < 0.01, ***P < 0.001, KLH + IL‐2 + RA vs. KLH, ## P < 0.01, ### P < 0.001 KLH + IL‐2 + RA vs. KLH + RA, ††† P < 0.001 KLH + IL‐2 + RA vs. KLH + IL‐2 by two‐way ANOVA and Tukey post hoc test.
Assessment of the antigen specificity and cytokine production of the induced T cells showed that immunization of mice with KLH, RA and IL‐2 induced KLH‐specific T cells in the spleens that produced large amounts of IL‐10, but did not produce IFN‐γ or IL‐17 (Fig 1D). In contrast, immunization with KLH combined with the TLR2 agonist LP1569 and the STING agonist cyclic di‐GMP (c‐di‐GMP), an adjuvant combination that we have used to promote effector T‐cell responses in a vaccine for an infectious disease 35, strongly promoted antigen‐specific IFN‐γ and IL‐17 production, with low concentrations of IL‐10 (Fig 1D). Immunization with KLH alone, KLH and RA or KLH and IL‐2 induced antigen‐specific IL‐10 production, but this was significantly stronger in mice immunized with KLH, RA and IL‐2 (Fig 1D). Furthermore, immunization with KLH alone also induced IFN‐γ, whereas IFN‐γ was not induced by immunization with KLH, RA and IL‐2. LAG3+ Foxp3− CD4 T cells from mice immunized with KLH, RA and IL‐2 proliferated and expressed high levels of CTLA‐4 in response to re‐stimulation with KLH (Fig EV2). These findings demonstrate that immunization of mice with a foreign antigen with RA and IL‐2 as adjuvants promotes induction of antigen‐specific Tr1 cells that secrete the anti‐inflammatory cytokine IL‐10 and express the immune checkpoints CTLA‐4, PD‐1 and LAG‐3.
Figure EV2. T cells from mice immunized with KLH, RA and IL‐2 proliferate in response to KLH and express CTLA4.
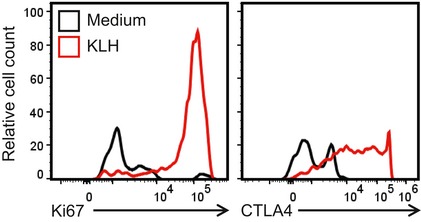
Representative histograms of Ki67 and CTLA‐4 expression by CD4 T cells from LNs of mice immunized twice with KLH, RA and IL‐2 and cultivated for 3 days with KLH or medium only. Cells were gated as single live CD3+ CD4+ Foxp3− LAG3+.
Mice immunized with MOG, RA and IL‐2 are resistant to the induction of EAE
Having shown that immunization with a model antigen in the presence of RA and IL‐2 induces the development of antigen‐specific Tr1 cells that produce IL‐10 but not inflammatory cytokines, we examined the possibility that RA and IL‐2 could also act as Tr1‐inducing adjuvants for an autoantigen and thereby prevent the induction of autoimmune disease. We first demonstrated that immunization of mice with the myelin peptide MOG33–55 (MOG) in the presence of RA and IL‐2 induced CD4 T cells that secreted IL‐10 (Fig 2A and B). The CD4 T cells generated in the presence of MOG, RA and IL‐2 expressed LAG‐3 and PD‐1, markers of Tr1 cells (Fig 2B). Interestingly, the frequency of Foxp3+CD25+ Treg cells was unchanged (Fig 2B). Immunization with MOG only did not induce Tr1 cells (Fig 2B). Immunization of mice with MOG in the presence of RA or IL‐2, 21 and 7 days before induction of EAE, delayed the onset of disease (Fig 2C). However, the best protection was observed in mice immunized with a combination of MOG, RA and IL‐2; these mice had mild or no clinical signs of EAE and those that did had delayed disease onset when compared with mice immunized with MOG and RA or MOG and IL‐2 (Fig 2C). In contrast, immunization of mice with MOG, RA (Fig EV3A) or IL‐2 (Fig EV3B) only did not prevent the development of EAE.
Figure 2. Induction of Tr1 cells by immunization of mice with MOG, RA and IL‐2 protects against the development of EAE.
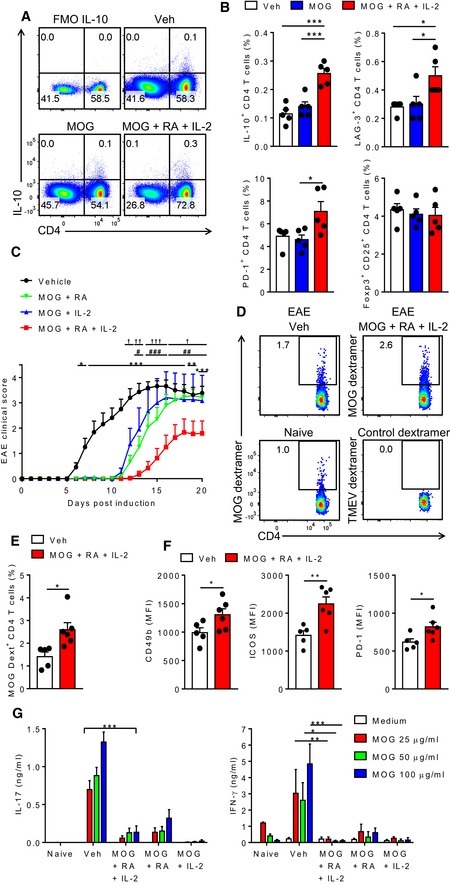
- Flow cytometry on LN cells of mice 7 days after two s.c. immunizations (14 days apart) with PBS + DMSO (Veh), MOG + DMSO (MOG), MOG + RA + IL‐2. Cells were gated on live single CD3+ lymphocytes. FMO: Fluorescence Minus One.
- Mean percentages of IL‐10+, LAG‐3+, PD1+ or Foxp3+ CD25+ CD4 T cells as described in A. Cells were gated on live single CD3+ lymphocytes (top left panel) or on live single CD3+ CD4+ lymphocytes. Bars are mean + SEM (n = 5). *P < 0.05, ***P < 0.001 by one‐way ANOVA and Tukey post hoc test.
- EAE clinical scores in mice immunized s.c. with MOG + RA, MOG + IL‐2, MOG + RA + IL‐2, or PBS + DMSO (Veh), 21 and 7 days before induction of EAE. Results are mean + SEM (n = 12, except for MOG + IL‐2, where n = 6; combined from two experiments). *P < 0.05, **P < 0.01, ***P < 0.001, MOG + IL‐2 + RA vs. Veh, # P < 0.05, ## P < 0.01, ### P < 0.001, MOG + IL‐2 + RA vs. MOG + RA, † P < 0.05, †† P < 0.01, ††† P < 0.001, MOG + IL‐2 + RA vs. MOG + IL‐2 by two‐way ANOVA and Tukey post hoc test.
- Representative FACS plots of MOG‐dextramer+ CD4 T cells in LNs of naïve mice or 3 days after induction of EAE. Cells were gated as single live F4/80− CD8− B220− CD4+ CD44+.
- Mean percentages of MOG‐dextramer+ CD4 T cells in LNs 3 days after induction of EAE. Cells were gated as single live F4/80− CD8− B220− CD4+ CD44+. Bars are mean + SEM (n = 5 or 6). *P < 0.05 by unpaired two‐tailed t‐test.
- Mean fluorescence intensity (MFI) of CD49b, ICOS and PD‐1 by MOG‐dextramer+ CD4 T cells in LNs 3 days after induction of EAE. Cells were gated as single live F4/80− CD8− B220− CD4+ CD44+ dextramer+. Bars are mean + SEM (n = 5 or 6). *P < 0.05, **P < 0.01 by unpaired two‐tailed t‐test.
- MOG‐specific IL‐17 and IFN‐γ production by spleen cells from day 7 of EAE, quantified in supernatants by ELISA after 5 d culture with increasing concentrations of MOG. Bars are mean + SEM (n = 3 or 4). *P < 0.05, **P < 0.01 and ***P < 0.001 by two‐ANOVA and Tukey post hoc test.
Figure EV3. Immunization with either MOG, RA or IL‐2 alone does not prevent the development of EAE .
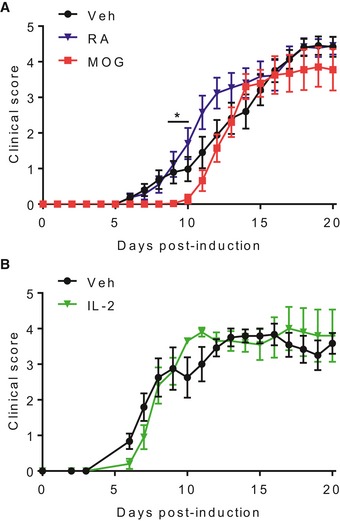
- EAE clinical scores of mice injected s.c. with MOG, RA or PBS + DMSO (Veh), 21 and 7 days before induction of EAE. Results are mean + SEM (n = 14, 15 or 17, combined from three experiments). *P < 0.05, MOG vs. Veh by mixed effects model with Tukey post hoc test.
- EAE clinical scores of mice injected s.c. with IL‐2 or PBS + DMSO (Veh), 21 and 7 days before induction of EAE. Results are mean + SEM (n = 5 or 6). Statistics were performed by two‐way ANOVA and Bonferroni post hoc test.
MOG‐dextramer staining of LN cells 3 days after induction of EAE revealed a significantly increased proportion of MOG‐specific CD4 T cells in mice immunized with MOG, RA and IL‐2 when compared with vehicle‐immunized control or naïve mice (Fig 2D and E). These cells had a higher expression of CD49b, ICOS and PD‐1, markers of Tr1 cells (Fig 2F). Seven days after the induction of EAE, spleen cells from vehicle‐treated mice secreted IL‐17 and IFN‐γ following ex vivo stimulation with MOG (Fig 2G). In contrast, spleen cells from mice immunized with MOG and RA, MOG and IL‐2 or with MOG, RA and IL‐2 produced significantly lower concentrations of MOG‐specific IL‐17 and IFN‐γ.
The development of EAE is dependent on migration of encephalitogenic T cells into the CNS. Therefore, we assessed the effect of inducing MOG‐specific Tr1 cells on the migration of pathogenic T cells. CD4 T cells in the spleens of mice immunized with MOG, RA and IL‐2 had significantly reduced cell surface expression of CD11a, an integrin involved in the migration of T cells into the CNS (Fig 3A and B). Consistent with this, flow cytometric analysis of the spinal cords on day 12 of EAE showed that mice immunized with MOG, RA and IL‐2 had significantly reduced numbers of infiltrating lymphocytes in the CNS (Fig 3C and D). Furthermore, the frequency and absolute numbers of IL‐17‐ and IFN‐γ‐secreting CD4 T cells infiltrating the CNS were significantly lower in mice immunized with MOG, RA and IL‐2 when compared with mice immunized with vehicle only (Fig 3E and F). Interestingly, we found an increased expression of LAG‐3 CD4 T cells in the CNS of mice immunized with MOG, RA and IL‐2 (Fig 3G). Moreover, we found a highly significant inverse correlation between the frequency of LAG‐3‐expressing CD4 T cells and the frequency of IL‐17‐ or IFN‐γ‐expressing CD4 T cells in the spinal cords of the mice (Fig 3H). Our findings suggest that immunization of mice with MOG, RA and IL‐2 induces a population of Tr1‐like T cells that suppress the induction and migration of pathogenic Th1 and Th17 cells that mediate EAE.
Figure 3. Suppressed pathogenic Th17 responses correlate with LAG‐3+ CD4 T cells.
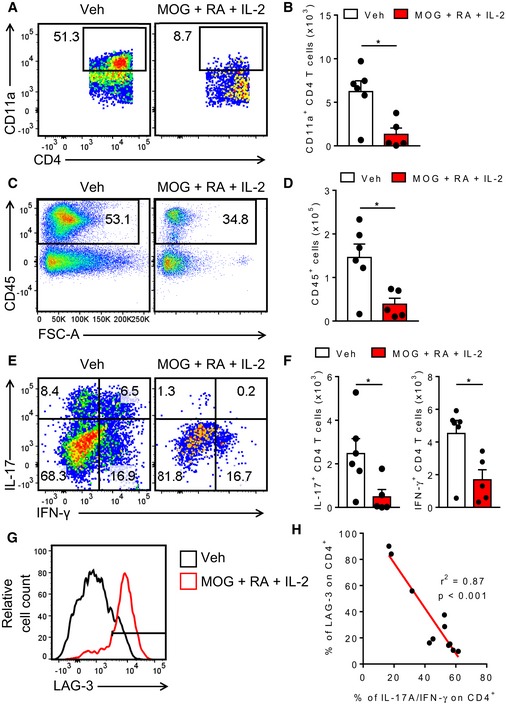
- Representative FACS plots of CD11a+ CD4 T cells in the spleen on day 7 of EAE in mice immunized with MOG, RA and IL‐2 or PBS + DMSO (Veh) only. Cells were gated as single live CD45+ CD3+ CD4+.
- Mean absolute numbers of CD11a+ CD4 T cells in the spleen on day 7 of EAE in mice immunized with MOG, RA and IL‐2 or PBS + DMSO (Veh). Bars are mean + SEM (n = 4 or 5). *P < 0.05 by unpaired two‐tailed t‐test.
- Representative FACS plots of CD45+ T cells in the spinal cords of mice on day 12 of EAE. Cells were gated as single live CD45+.
- Mean absolute numbers of infiltrating CD45+ cells in the spinal cords of mice on day 12 of EAE. Bars are mean + SEM (n = 5 or 6). *P < 0.05 by unpaired two‐tailed t‐test.
- Representative FACS plots of infiltrating IL‐17+ and IFN‐γ+ CD4 T cells in the spinal cords on day 12 of EAE. Cells were gated as single live CD45+ CD3+ CD4+.
- Mean absolute numbers of infiltrating IL‐17+ and IFN‐γ+ CD4 T cells in the spinal cords on day 12 of EAE. Bars are mean + SEM (n = 5 or 6). *P < 0.05 by unpaired two‐tailed t‐test.
- Representative histogram of LAG‐3 expression on CD4 T cells in the spinal cords on day 12 of EAE. Cells were gated as single live CD45+ CD3+ CD4+.
- Inverse correlation between LAG‐3 and IL‐17 and IFN‐γ expression on CD4 T cells in the spinal cords on day 12 of EAE. *P < 0.001 by unpaired two‐tailed t‐test.
Tr1 cells generated with RA and IL‐2 suppress encephalitogenic CD4 T cells
We assessed the capacity of Tr1 cells induced by immunization with MOG, RA and IL‐2 to suppress pathogenic Th1 and Th17 cells in vitro and in vivo. We expanded T cells from the LNs of mice immunized with MOG, RA and IL‐2 by culture with antigen in the presence of RA and IL‐2. This short‐term MOG‐specific CD4 T‐cell line expressed the memory marker CD44 as well as LAG‐3, PD‐1, CD226, ICOS and CD25, while expression of the naïve T‐cell marker CD62L was decreased (Fig EV4A–C). Interestingly, cells from mice immunized with MOG alone and cultured in vitro with MOG or MOG, RA and IL‐2 did not exhibit the same level of expression of CD49b, ICOS and PD‐1 (Fig EV4C). This demonstrates that immunization with MOG, RA and IL‐2 is required to generate Tr1 cells, which can be expanded in vitro by re‐stimulation with MOG in the presence of RA and IL‐2. These MOG‐specific Treg cells suppressed antigen‐induced IL‐17 and IFN‐γ production by Th1/Th17 cell from mice with EAE in a dose‐dependent manner (Fig 4A).
Figure EV4. Immunization with MOG, RA and IL‐2 induce T cells that express markers of Tr1 cells.
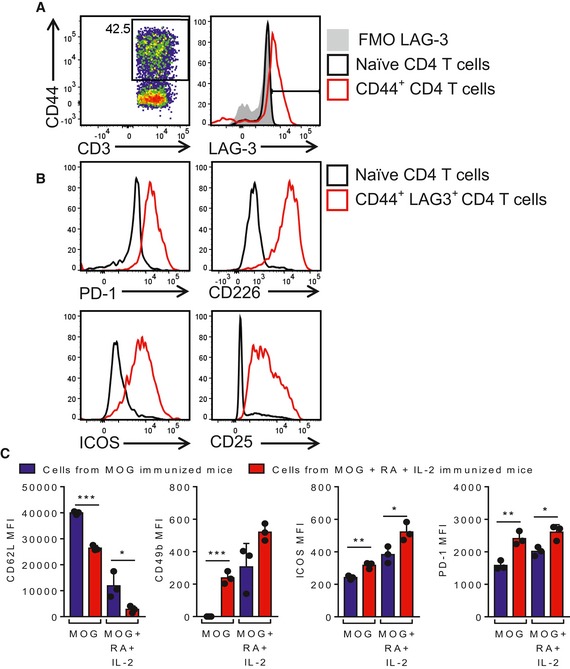
- Representative FACS plots of CD44 expression in CD4 T cells and representative histogram of LAG‐3 expression by total CD4 and CD44+ CD4 T cells in naïve and amplified CD4 T cells, respectively. Amplified cells are LN cells of mice immunized twice with MOG, RA and IL‐2 and amplified in vitro with MOG, RA and IL‐2 for 8 days.
- Representative histograms of PD‐1, CD226, ICOS and CD25 expression on naïve CD4 T cells and amplified CD44+ CD4 T cells from mice immunized with MOG, RA and IL‐2.
- Mean fluorescence intensity (MFI) of CD62L, CD49b, ICOS and PD‐1 expression on naïve CD4 T cells and amplified CD44+ CD4 T cells from LNs of mice immunized twice with MOG (blue bars) or MOG, RA and IL‐2 (red bars) and amplified in vitro with MOG or MOG, RA and IL‐2 for 8 days. Results are mean + SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, by unpaired two‐tailed t‐test.
Figure 4. MOG‐specific Tr1 cells suppress pathogenic Th1 and Th17 cells.
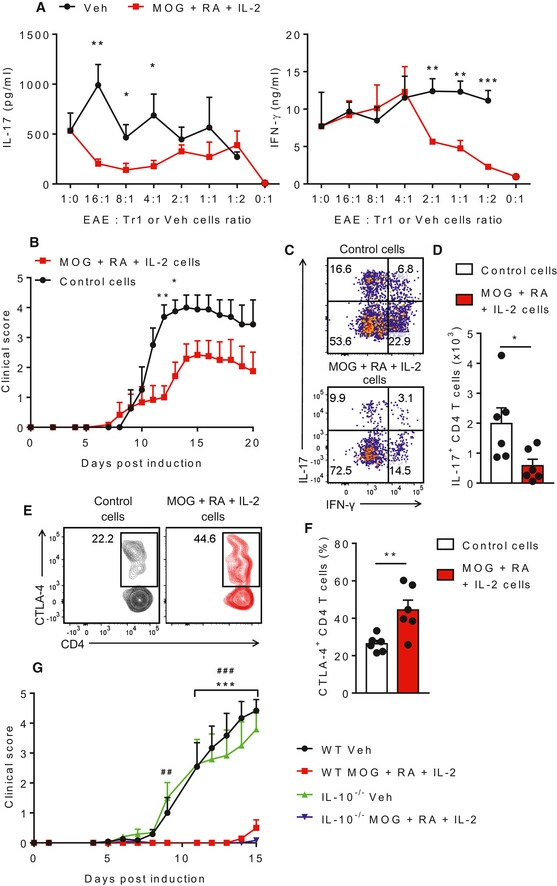
- IL‐17 and IFN‐γ production by ELISA on supernatants of spleen cells from mice with EAE (EAE) co‐cultured for 3 days with MOG‐specific Tr1 cells (from mice immunized twice with MOG, RA and IL‐2 and amplified in vitro with MOG, RA and IL‐2 for 8 days) or control T cells from LNs of mice injected with PBS + DMSO (Veh). Bars are mean + SD of one experiment. *P < 0.05, **P < 0.01 and ***P < 0.001 by unpaired two‐tailed t‐test.
- EAE clinical scores of mice transferred 1 day before induction of EAE with MOG‐specific Tr1 cells amplified in vitro with MOG, RA and IL‐2. Results are mean + SEM (n = 4 or 6). *P < 0.05 and **P < 0.01 by two‐way ANOVA with Bonferroni post hoc test.
- Representative FACS plots of IL‐17+ and IFN‐γ+ CD4 T cells in the spinal cords on day 20 of EAE. Cells were gated as live single CD45.2+CD3+CD4+ cells.
- Mean absolute numbers of IL‐17+ CD4 T cells in the spinal cords on day 20 of EAE. Results are mean + SEM (n = 6). *P < 0.05 by unpaired two‐tailed t‐test.
- Representative FACS plots of CTLA‐4+ CD4 T cells in the spleens of mice with EAE. Cells were gated as live single CD45.1+CD3+CD4+.
- Mean percentages of CTLA‐4+ CD4 T cells in the spleens of mice with EAE. Results are mean + SEM (n = 6). **P < 0.01 by unpaired two‐tailed t‐test.
- EAE clinical scores of WT and IL‐10−/− mice immunized s.c. with MOG + RA + IL‐2, or PBS + DMSO (Veh), 7 and 21 days before induction of EAE. Results are mean + SEM (n = 6 or 7). ***P < 0.001, WT MOG + IL‐2 + RA vs. WT Veh, ## P < 0.01, ### P < 0.001, IL‐10−/− MOG + IL‐2 + RA vs. IL‐10−/− Veh by two‐way ANOVA and Tukey post hoc test.
We next provided definitive evidence that Tr1 cells mediated the protective effect of immunization with MOG, RA and IL‐2 on the development of EAE. We transferred lymphocytes from naïve mice (control) or T cells from CD45.1+ mice immunized with MOG, RA and IL‐2 and amplified in vitro with MOG, RA and IL‐2 into naïve CD45.2+ recipient mice 1 day before inducing EAE. Amplifying the cells in vitro with antigen not only enhanced the number of T cells for transfer but also ensured that the majority of these cells were antigen‐specific Tr1 cells. Mice that received the Tr1 cells had a delayed onset of EAE associated with a reduction in the severity of clinical signs when compared to mice that received control cells (Fig 4B). Flow cytometric analysis of the spinal cords of the mice on day 20 of EAE showed that mice injected with Tr1 cells had a significant reduction in IL‐17+CD4+CD45.2+ T cells (Fig 4C and D). Interestingly, the spleens of those mice contained a significant number of donor CD4+CD45.1+ T cells that expressed high levels of CTLA‐4 (Fig 4E and F). These findings demonstrate that MOG‐specific Tr1 cells induced by immunization of mice with MOG, RA and IL‐2 suppress encephalitogenic T cells that mediate EAE. Since the Treg cells induced by immunization with MOG, RA and IL‐2 secrete IL‐10, we examined the possibility that this cytokine mediated protection in the EAE model. Consistent with the data in Fig 2C, immunization with MOG, RA and IL‐2 attenuated the induction of EAE in wild‐type mice (Fig 4G). Attenuation of EAE was also observed in IL‐10−/−, suggesting that the Treg‐induced protection was independent of IL‐10. To assess the antigen specificity of suppression by the Tr1 cells and the possibility of bystander suppression of EAE, we immunized mice with OVA323–339 peptide (OVA) or MOG together with RA and IL‐2 before induction of EAE. While immunization with MOG, RA and IL‐2 attenuated the symptoms of EAE, the disease developed fully in mice immunized with OVA, RA and IL‐2, with disease scores similar to those seen in non‐immunized control mice (Fig EV5). These data demonstrate that the protection is antigen‐specific.
Figure EV5. Protection against EAE mediated by induction of Tr1 cells is antigen‐specific.
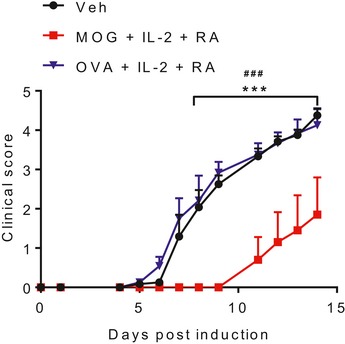
EAE clinical scores of mice immunized s.c. with MOG + IL‐2 + RA, OVA + IL‐2 + RA, or PBS + DMSO (Veh), 21 and 7 days before induction of EAE. Results are mean + SEM (n = 5 or 6). ***P < 0.001, MOG + IL‐2 + RA vs. Veh, ### P < 0.001, MOG + IL‐2 + RA vs. OVA + IL‐2 + RA by two‐way ANOVA with Tukey post hoc test.
Generation of Tr1 cells protects against development of induced or spontaneous uveitis
Having shown that Tr1 cells induced by immunization with MOG, RA and IL‐2 can inhibit the development of EAE by suppressing encephalitogenic T‐cell activity, we assessed whether this immunization approach could confer protection in another autoimmune disease, uveitis, using a distinct autoantigen. Immunization of mice with the autoantigen IRBP1–20 (IRBP) together with RA and IL‐2 (21 and 7 days prior to immunization) significantly delayed the onset of EAU and strongly reduced retinal inflammation (vasculitis and vitreous haze) when compared with mice immunized with IRBP alone or vehicle control mice (Fig 5A and B).
Figure 5. Induction of Tr1 cells by immunization of mice with autoantigen, RA and IL‐2 protects against EAU and spontaneous uveitis.
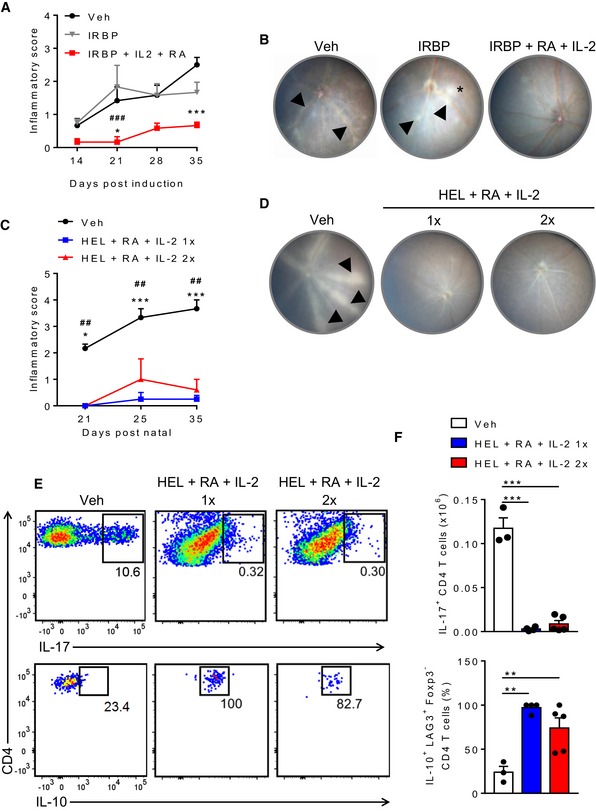
- Retinal inflammatory scores of mice immunized s.c. with PBS + DMSO (Veh), IRBP + DMSO (IRBP) or IRBP + RA + IL‐2 21 and 7 days before induction of EAU by s.c. injection of IRBP emulsified in CFA. Bars are mean + SEM (n = 6), *P < 0.05 and ***P < 0.001, IRBP + IL‐2 + RA vs. Veh, ### P < 0.001 IRBP + IL‐2 + RA vs. IRBP by two‐way ANOVA with Tukey post hoc test.
- Representative fundus images of each group of mice 35 days after induction of EAU. Arrowheads indicate area with retinal vasculitis and the asterisk indicates a region of vitreous haze, where the image appears blurred.
- Retinal inflammation scores in HEL‐TCR transgenic mice s.c. injected with PBS + DMSO (Veh) or HEL + RA + IL‐2 on pn 18 (1×) or on pn 18 and 25 (2×). Results are mean + SEM (n = 3, 4 or 5). *P < 0.05 and ***P < 0.001, HEL + IL‐2 + RA 1× vs. Veh, ## P < 0.01 HEL + IL‐2 + RA 2× vs. Veh by two‐way ANOVA with Tukey post hoc test.
- Representative photographs of each group of retinal fundus of HEL‐TCR transgenic mice at pn 35. Arrowheads indicate representative areas with retinal vasculitis.
- Representative FACS plots of IL‐17+ CD4 or IL‐10+ CD4 T cells in the retina on day 36 of HEL‐TCR transgenic mice. Cells were gated on live single CD3+CD4+ and CD3+CD4+LAG‐3+Foxp3−, respectively.
- Mean absolute number of IL‐17+ CD4 T cells in the retina (upper panels) or mean percentages of IL‐10+ LAG‐3+ Foxp3− CD4 T cells in the spleens of HEL‐TCR transgenic mice with vasculitis (lower panels). Results are mean + SEM (n = 3, 4 or 5). **P < 0.01 and ***P < 0.001 by one‐way ANOVA with Bonferroni post hoc test.
We next assessed if the same immunization approach could protect against uveitis that developed spontaneously. We used the double transgenic TCR‐HEL mice, which start developing signs of uveitis resembling the human features of posterior uveitis by post‐natal day (pn) 21 as previously described 36. Mice immunized with HEL, RA and IL‐2 on pn 18 only (1×) or on pn 18 and pn 25 (2×) were completely protected against the development of spontaneous autoimmune uveitis (Fig 5C and D). This dramatic reduction in clinical signs of uveitis was associated with a reduced number of IL‐17‐producing CD4 T cells in the retina and increased IL‐10 production by LAG3+ Foxp3− T cells in the spleen as assessed by flow cytometry (Fig 5E and F).
Collectively, our findings demonstrate that immunization with an antigen and RA in combination with IL‐2 as adjuvants can protect against development of autoimmune diseases through the induction of antigen‐specific Tr1 cells that suppress the function of pathogenic Th1 and Th17 cells. Our demonstration that the induced Tr1 cells can suppress retinal inflammation in the double transgenic TCR‐HEL mice where autoimmune uveitis develops without the need for immunization with autoantigen, suggests that attenuation of autoimmunity is not due to unresponsiveness to the self‐antigens. Furthermore, our demonstration that transfer of T cells from mice immunized with MOG, RA and IL‐2 reduced the severity of EAE in recipient mice suggests that the attenuation of autoimmune disease is mediated by Tr1 cells. Finally, co‐culture experiments confirmed that induced Tr1 cells actively suppress pathogenic T cells that mediate autoimmunity.
In addition to genetic factors, environmental influences, including the microbiome and exposure to helminth pathogens, can influence the development of autoimmunity by enhancing immune regulation 37, 38. In this study, we have exploited a natural metabolite of vitamin A, found in many foods, and a host protein, the cytokine IL‐2, together with a self‐antigen for expanding the pool of Tr1 cells from the natural repertoire in vivo. We show for the first time that RA and IL‐2 in combination with an antigen selectively promotes antigen‐specific IL‐10‐producing Tr1 cells that suppress pathogenic Th1 and Th17 cells. Furthermore, we demonstrate that this immunization approach is effective in preventing the development of autoimmunity in models of MS and uveitis. It has previously been reported that chronic treatment of mice with RA protects against DSS‐induced colitis in mice by promoting IL‐22 production by γδ T cells and ILC2 22 and attenuates collagen‐induced arthritis through induction of Foxp3+ Treg cells 39. In the present study, rather than treating mice with RA, we used it as an adjuvant, together with IL‐2 that binds to a receptor expressed on most Treg cells, and an autoantigen to promote induction of autoantigen‐specific Treg cells. These Treg cells were not conventional Foxp3+ Treg cells, but expressed PD‐1, ICOS, LAG‐3, CTLA‐4 and CD49b, markers of Tr1 cells and also secreted IL‐10, though the mechanism of protection was not mediated by IL‐10. However, IL‐10‐secreting Tr1 can also suppress T‐cell responses by killing APCs 40. Furthermore, antigen‐specific induced Treg cells can suppress naïve T cells with identical antigen specificity, independently of IL‐10, by removing MHC class II‐peptide complexes from the surface of APCs 41.
Induction of a tolerogenic environment in the host through the induction of antigen‐specific Treg cells has considerable potential in the treatment of autoimmune diseases. Indeed, adoptive cell therapy with Treg cells is already showing promising results in early clinical studies in type I diabetes and Crohn's disease patients 42, 43, 44. However, ex vivo protocols for expansion of Foxp3+ Treg cells have proven challenging and the stability and long‐term survival of these cells can be compromised after transfer. Approaches to induce and expand antigen‐specific Tr1, including direct in vivo administration of peptides from self‐antigens 45, 46, 47, 48, have progressed to clinical trials 10, 49. Interestingly the method of administration, such as auto‐antigens trapped in nanoparticles or presented by tolerogenic DCs, influences the efficacy of Treg‐mediated suppression 8, 9, 10. Our findings suggest that the addition of Treg cell‐inducing adjuvants, such as IL‐2 and RA, to autoantigen‐loaded nanoparticles or APCs could further improve the efficacy of Treg cell‐inducing approaches for treatment of autoimmune diseases. In addition, our findings shed new light on the role of autoantigen‐specific Treg cells in suppressing pathogenic T cells and demonstrate that shifting the T‐cell balance in favour of immune regulation can prevent the development of autoimmune diseases.
Material and Methods
Mice
C57BL/6 mice (8–10 week old females) were housed in the Comparative Medicine Unit of Trinity College Dublin, or the Medical Research Facility at the University of Aberdeen. IL10−/− mice were provided by the Comparative Medicine Unit of Trinity College Dublin. TCR‐HEL double transgenic mice were provided by Medical Research Facility at the University of Aberdeen 36. All animals were bred under SPF conditions and maintained according to the regulations of the European Union. All EAU experiments were performed according to the regulations of the Animal License Act (UK) and to the Association for Research in Vision and Ophthalmology statement for The Use of Animals in Ophthalmic and Vision Research. All other experiments were performed under licence from the Health Products Regulatory Authority in Ireland and with approval from the Trinity College Dublin Animal Research Ethics Committee. All animals were randomly allocated into experimental groups 1 week before starting each experiment.
EAE
EAE was induced in C57BL/6 wild‐type or IL‐10−/− mice by immunization s.c. with 100 μg of MOG emulsified in CFA containing 4 mg/ml (0.4 mg/mouse) heat killed Mycobacterium tuberculosis (Chondrex) and i.p. injection of pertussis toxin (250 ng, Kaketsuken) on days 0 and 2. Mice were weighed and monitored for signs of EAE daily. The disease severity was assessed as follows: 0, no clinical sign; 1, limp tail; 2, ataxic gait; 3, hind limb weakness; 4, hind limb paralysis; 5, tetraplegia/moribund.
EAU and spontaneous autoimmune uveitis
For EAU model, mice were immunized s.c. with 500 μg of IRBP emulsified in CFA containing additional 3.5 mg/ml M. tuberculosis H37Ra (Difco) and were also injected i.p. with pertussis toxin (1 μg, Health Protection Agency). Spontaneous autoimmune uveitis developed in TCR‐HEL double transgenic mice at pn 21 with the peak at pn 28 as previously described 36. Clinical images of the retinal fundus were acquired using an endoscopic imaging system as described previously 50.
Induction of antigen‐specific Treg cells
C57BL/6 mice were injected s.c. with PBS, KLH (1 μg, Calbiochem), MOG (100 μg, GenScript), IRBP (100 μg, New England Peptide), OVA (100 μg, Sigma‐Aldrich) together with DMSO (Sigma‐Aldrich), RA (375 μg, Enzo), IL‐2 (2,500 IU, Immunotools) or LP1569 (50 μg, EMC Microcollections) and c‐di‐GMP (10 μg, Invivogen). Mice were immunized twice, 21 and 7 days before induction of EAE or EAU. TCR‐HEL double transgenic mice were injected s.c. with PBS and DMSO or HEL (100 μg, Sigma‐Aldrich) with RA and IL‐2 on day pn 18 or on day pn 18 and pn 25.
Isolation of mononuclear cells from the spinal cord and retina
For spinal cord cell isolation, mice were euthanized and perfused with phosphate‐buffered saline (PBS), and their spinal cords were isolated. Mononuclear cells were isolated using a tissue lyser (Qiagen) and stimulated for 2–4 h with PMA (10 ng/ml, Sigma‐Aldrich) and ionomycin (1 μg/ml, Sigma‐Aldrich) in the presence of brefeldin A (5 μg/ml, Sigma‐Aldrich), and then stained for intracellular cytokines and analysed by FACS. For analysis of eye‐infiltrating cells, retinal tissue was separated from the choroid and vitreous and digested for 40 min at 37°C with mixture of Liberase (10 μg/ml, Roche) and DNase I (10 μg/ml, Roche). Cells were stimulated for 5 h with PMA (50 ng/ml), ionomycin (1 μM) and GolgiSTOP (BD Bioscience) and then stained for intracellular cytokines and analysed by FACS.
Flow cytometry
For IL‐10 detection in LNs cells of mice immunized twice with MOG, RA and IL‐2, the cells were stimulated with PMA (50 ng/ml), and ionomycin (1 μg/ml) in the presence of brefeldin A (10 μg/ml) for 3 h prior to staining. Cells were incubated with a live/dead stain (Invitrogen) and then surface stained with antibodies specific for CD3 (1/400, clone 17A2), CD4 (1/200, clone RM4‐5), LAG‐3 (1/200, clone C9B7W), CD49b (1/200, clone DX‐5) from eBioscience, CD25 (1/200, clone PC61), CD45.1 (1/400, clone A20), CD45.2 (1/400, clone 104), CD226 (1/200, clone TX42.), PD‐1 (1/200, clone 29F.1A12) from Biolegend and ICOS (1/200, clone 7E.17G9), CD62L (1/200, clone MEL‐14) and CD11a (1/200, clone M17/4) from BD Biosciences. The cells were washed, fixed and permeabilized using 1–2% PFA (Pierce) or a Foxp3/Transcription Factor Staining Buffer Set (eBioscience) for staining of intranuclear proteins. The cells were stained in 0.5% saponin (Sigma‐Aldrich) or permeabilization buffer (eBioscience) containing antibodies specific for Foxp3 (1/200, clone FJK‐16s), Ki67 (1/200, clone SolA15), CTLA‐4 (1/200, clone UC10‐4B9) from eBioscience, IL‐17A (1/200, clone TC11‐18H10.1, Biolegend), IFN‐γ (1/200, clone XMG1.2, BD Biosciences) and IL‐10 (1/200 or 1/20, clone JES5‐16E3, BD Biosciences). Cells were analysed using a flow cytometer LSR Fortessa (BD) or LSR II analyser (BD), and the data were analysed with FlowJo software.
Dextramer staining
MOG‐specific CD4 T cells were stained using an APC‐conjugated IAb/MOG35–55 dextramer as previously described 51. LNs cells from naïve mice or from mice on day 3 of EAE were isolated and plated in 50 μl of RPMI (Sigma‐Aldrich) containing 2.5% FCS (Sigma‐Aldrich) and stained with 5 μl specific (IAb/MOG35‐55) or negative control (IAs/TMEV70–86) dextramer for 2.5 h at 37°C. Cells were washed and incubated with a live/dead stain (Invitrogen) and then surface stained with antibodies specific for F4/80 (1/400, clone BM8, Biolegend), CD8 (1/400, clone 53‐6.7, eBioscience), B220 (1/400, clone HIS24, eBioscience), CD4 (1/200), CD44 (1/400, clone IM7, Biolegend), ICOS (1/200), PD‐1 (1/200) and CD49b (1/200). Cells were washed and analysed immediately using a flow cytometer LSR Fortessa.
Antigen‐specific Tr1 cell amplification, transfer and suppression assay
For MOG‐specific Tr1 cells, LN cells from mice immunized twice with MOG or MOG, RA and IL‐2 were cultured at 2 × 106 cells/ml with MOG (100 μg/ml) alone or with MOG, RA (10−6 M) and IL‐2 (10 ng/ml) for 8 days. After 4 days of culture, more IL‐2 (10 ng/ml) was added to the culture. For Tr1 cell transfer, mice were injected i.v. with 5 × 106 amplified MOG‐specific T cells or with 5 × 106 spleen cells from mice injected with PBS + DMSO the day before active induction of EAE. For MOG‐specific Tr1 suppression assay, spleen cells from mice on day 3 of EAE were cultured with MOG (50 μg/ml) and increasing concentrations of in vitro expanded MOG‐specific Tr1 cells or LN cells from mice injected with PBS + DMSO. After 3 days, supernatants were collected and cytokine concentrations were determined by ELISA.
Antigen‐specific cytokine production and ELISA
Spleen cells (2 × 106 cells/ml) from mice immunized with KLH or MOG and adjuvants were stimulated with medium, KLH (2–50 μg/ml) or MOG (25–100 μg/ml), respectively. IL‐10, IL‐17 (DuoSet, R&D Systems) and IFN‐γ (BD Biosciences) concentrations were quantified in the supernatants by ELISA.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 and the R software. A mixed effects model accounting for multiple observations within mice and treatment groups was used for analysis of data from pooled experiments shown in Fig EV3. This allowed for ANOVA and Tukey post hoc test to be used to identify statistical differences of means between groups. This model was fit using the packages lme4, lmerTest and emmeans within the R environment 52, 53. For other experiments, ANOVA with Tukey post hoc test, Bonferroni post hoc test or Student's t‐test was used to determine statistical differences of means between groups.
Author contributions
MR, KHGM, AMa and JVF conceived and planned the experiments. MR, MC, AMa, LK, MMW, AMi, TY, SMQ and AMM carried out the experiments. CM and JR produced and provided MOG‐dextramer reagent. MR and KHGM wrote the manuscript. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Barry Moran for assistance with flow cytometry, Nicolas Jourdan for assistance with statistics and Dr Yihsia Liu for help with retina isolation. This work was supported by Science Foundation Ireland (grants #11/PI/1036 and #16/IA/4468) to KHGM and Marie Curie Intra‐European Fellowship (#629498) to MR and KHGM. Work in Aberdeen was supported by internal grant from Development Trust of University of Aberdeen (UoA) (#RG14251).
EMBO Reports (2019) 20: e47121
References
- 1. Feldmann M, Steinman L (2005) Design of effective immunotherapy for human autoimmunity. Nature 435: 612–619 [DOI] [PubMed] [Google Scholar]
- 2. Garber K (2012) Anti‐IL‐17 mAbs herald new options in psoriasis. Nat Biotechnol 30: 475–477 [DOI] [PubMed] [Google Scholar]
- 3. Dolgin E (2016) New anti‐IL‐23 drugs raise hopes for psoriasis plaque clearance. Nat Biotechnol 34: 1218–1219 [DOI] [PubMed] [Google Scholar]
- 4. Wraith DC (2017) The future of immunotherapy: a 20‐year perspective. Front Immunol 8: 1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T (2006) Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self‐tolerance and autoimmune disease. Immunol Rev 212: 8–27 [DOI] [PubMed] [Google Scholar]
- 6. Streeter HB, Rigden R, Martin KF, Scolding NJ, Wraith DC (2015) Preclinical development and first‐in‐human study of ATX‐MS‐1467 for immunotherapy of MS. Neurol Neuroimmunol Neuroinflamm 2: e93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alhadj Ali M, Liu YF, Arif S, Tatovic D, Shariff H, Gibson VB, Yusuf N, Baptista R, Eichmann M, Petrov N et al (2017) Metabolic and immune effects of immunotherapy with proinsulin peptide in human new‐onset type 1 diabetes. Sci Transl Med 9: eaaf7779 [DOI] [PubMed] [Google Scholar]
- 8. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, Pahau H, Lee BT, Ng J, Brunck ME et al (2015) Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype‐positive rheumatoid arthritis patients. Sci Transl Med 7: 290ra87 [DOI] [PubMed] [Google Scholar]
- 9. Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ et al (2012) Microparticles bearing encephalitogenic peptides induce T‐cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol 30: 1217–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clemente‐Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C et al (2016) Expanding antigen‐specific regulatory networks to treat autoimmunity. Nature 530: 434–440 [DOI] [PubMed] [Google Scholar]
- 11. Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4: 330–336 [DOI] [PubMed] [Google Scholar]
- 12. Mills KH (2004) Regulatory T cells: friend or foe in immunity to infection? Nat Rev Immunol 4: 841–855 [DOI] [PubMed] [Google Scholar]
- 13. Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG (1997) A CD4+ T‐cell subset inhibits antigen‐specific T‐cell responses and prevents colitis. Nature 389: 737–742 [DOI] [PubMed] [Google Scholar]
- 14. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona‐Limon P, Guo B, Herbert DR, Bulfone A, Trentini F et al (2013) Coexpression of CD49b and LAG‐3 identifies human and mouse T regulatory type 1 cells. Nat Med 19: 739–746 [DOI] [PubMed] [Google Scholar]
- 15. Schmidt A, Oberle N, Krammer PH (2012) Molecular mechanisms of treg‐mediated T cell suppression. Front Immunol 3: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hall JA, Grainger JR, Spencer SP, Belkaid Y (2011) The role of retinoic acid in tolerance and immunity. Immunity 35: 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y (2007) Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 Treg cells via retinoic acid. J Exp Med 204: 1775–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M et al (2011) Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 34: 435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coombes JL, Siddiqui KR, Arancibia‐Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F (2007) A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF‐beta and retinoic acid‐dependent mechanism. J Exp Med 204: 1757–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou X, Kong N, Wang J, Fan H, Zou H, Horwitz D, Brand D, Liu Z, Zheng SG (2010) Cutting edge: all‐trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol 185: 2675–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, Wang J, Chen M, Liu Y, Shen Y et al (2014) Critical role of all‐trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci USA 111: E3432–E3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, Misiak A, Dungan LS, Sutton CE, Streubel G et al (2013) Retinoic acid expression associates with enhanced IL‐22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med 210: 1117–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raverdeau M, Breen CJ, Misiak A, Mills KH (2016) Retinoic acid suppresses IL‐17 production and pathogenic activity of gammadelta T cells in CNS autoimmunity. Immunol Cell Biol 94: 763–773 [DOI] [PubMed] [Google Scholar]
- 24. Dyck L, Wilk MM, Raverdeau M, Misiak A, Boon L, Mills KH (2016) Anti‐PD‐1 inhibits Foxp3+ Treg cell conversion and unleashes intratumoural effector T cells thereby enhancing the efficacy of a cancer vaccine in a mouse model. Cancer Immunol Immunother 65: 1491–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dansokho C, Ait Ahmed D, Aid S, Toly‐Ndour C, Chaigneau T, Calle V, Cagnard N, Holzenberger M, Piaggio E, Aucouturier P et al (2016) Regulatory T cells delay disease progression in Alzheimer‐like pathology. Brain 139: 1237–1251 [DOI] [PubMed] [Google Scholar]
- 26. Grinberg‐Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, Cagnard N, Carpentier W, Tang Q, Bluestone J et al (2010) IL‐2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 207: 1871–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bonnet B, Vigneron J, Levacher B, Vazquez T, Pitoiset F, Brimaud F, Churlaud G, Klatzmann D, Bellier B (2016) Low‐dose IL‐2 induces regulatory t cell‐mediated control of experimental food allergy. J Immunol 197: 188–198 [DOI] [PubMed] [Google Scholar]
- 28. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, Fonfrede M, Rosenzwajg M, Bernard C, Klatzmann D (2013) Low‐dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double‐blind, placebo‐controlled trial. Lancet Diabetes Endocrinol 1: 295–305 [DOI] [PubMed] [Google Scholar]
- 29. Rosenzwajg M, Churlaud G, Mallone R, Six A, Derian N, Chaara W, Lorenzon R, Long SA, Buckner JH, Afonso G et al (2015) Low‐dose interleukin‐2 fosters a dose‐dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun 58: 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP III, Armand P, Cutler C, Ho VT, Treister NS et al (2011) Interleukin‐2 and regulatory T cells in graft‐versus‐host disease. N Engl J Med 365: 2055–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Humrich JY, von Spee‐Mayer C, Siegert E, Alexander T, Hiepe F, Radbruch A, Burmester GR, Riemekasten G (2015) Rapid induction of clinical remission by low‐dose interleukin‐2 in a patient with refractory SLE. Ann Rheum Dis 74: 791–792 [DOI] [PubMed] [Google Scholar]
- 32. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, Jin Y, Gan Y, Hu X, Jia R et al (2016) Low‐dose interleukin‐2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med 22: 991–993 [DOI] [PubMed] [Google Scholar]
- 33. Klatzmann D, Abbas AK (2015) The promise of low‐dose interleukin‐2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 15: 283–294 [DOI] [PubMed] [Google Scholar]
- 34. Ye C, Brand D, Zheng SG (2018) Targeting IL‐2: an unexpected effect in treating immunological diseases. Signal Transduct Target Ther 3: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Allen AC, Wilk MM, Misiak A, Borkner L, Murphy D, Mills KHG (2018) Sustained protective immunity against Bordetella pertussis nasal colonization by intranasal immunization with a vaccine‐adjuvant combination that induces IL‐17‐secreting TRM cells. Mucosal Immunol 11: 1763–1776 [DOI] [PubMed] [Google Scholar]
- 36. Lambe T, Leung JC, Ferry H, Bouriez‐Jones T, Makinen K, Crockford TL, Jiang HR, Nickerson JM, Peltonen L, Forrester JV et al (2007) Limited peripheral T cell anergy predisposes to retinal autoimmunity. J Immunol 178: 4276–4283 [DOI] [PubMed] [Google Scholar]
- 37. Finlay CM, Walsh KP, Mills KH (2014) Induction of regulatory cells by helminth parasites: exploitation for the treatment of inflammatory diseases. Immunol Rev 259: 206–230 [DOI] [PubMed] [Google Scholar]
- 38. Mills KH (2011) TLR‐dependent T cell activation in autoimmunity. Nat Rev Immunol 11: 807–822 [DOI] [PubMed] [Google Scholar]
- 39. Kwok SK, Park MK, Cho ML, Oh HJ, Park EM, Lee DG, Lee J, Kim HY, Park SH (2012) Retinoic acid attenuates rheumatoid inflammation in mice. J Immunol 189: 1062–1071 [DOI] [PubMed] [Google Scholar]
- 40. Magnani CF, Alberigo G, Bacchetta R, Serafini G, Andreani M, Roncarolo MG, Gregori S (2011) Killing of myeloid APCs via HLA class I, CD2 and CD226 defines a novel mechanism of suppression by human Tr1 cells. Eur J Immunol 41: 1652–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, Kabat J, Matsumura R, Dorward DW, Glass DD et al (2019) Regulatory T cells mediate specific suppression by depleting peptide‐MHC class II from dendritic cells. Nat Immunol 20: 218–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hebuterne X, Bouhnik Y, Nachury M, Brun V, Bastian H, Belmonte N et al (2012) Safety and efficacy of antigen‐specific regulatory T‐cell therapy for patients with refractory Crohn's disease. Gastroenterology 143: 1207–1217 e2 [DOI] [PubMed] [Google Scholar]
- 43. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K et al (2015) Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 7: 315ra189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marek‐Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, Wujtewicz MA, Witkowski P, Mlynarski W, Balcerska A et al (2012) Administration of CD4+CD25highCD127‐ regulatory T cells preserves beta‐cell function in type 1 diabetes in children. Diabetes Care 35: 1817–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou X, Bailey‐Bucktrout SL, Jeker LT, Penaranda C, Martinez‐Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA (2009) Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo . Nat Immunol 10: 1000–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gregori S, Roncarolo MG (2018) Engineered T regulatory type 1 cells for clinical application. Front Immunol 9: 233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh‐hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H (2014) Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20: 62–68 [DOI] [PubMed] [Google Scholar]
- 48. Bailey‐Bucktrout SL, Martinez‐Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, Fehling HJ, Bluestone JA (2013) Self‐antigen‐driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 39: 949–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C et al (2013) Antigen‐specific tolerance by autologous myelin peptide‐coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med 5: 188ra75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Klaska IP, Muckersie E, Martin‐Granados C, Christofi M, Forrester JV (2017) Lipopolysaccharide‐primed heterotolerant dendritic cells suppress experimental autoimmune uveoretinitis by multiple mechanisms. Immunology 150: 364–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Massilamany C, Upadhyaya B, Gangaplara A, Kuszynski C, Reddy J (2011) Detection of autoreactive CD4 T cells using major histocompatibility complex class II dextramers. BMC Immunol 12: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed‐effects models using lme4. J Stat Softw 67: 48 [Google Scholar]
- 53. Kuznetsova A, Brockhoff PB, Christensen RHB (2017) lmerTest package: tests in linear mixed effects models. J Stat Softw 82: 26 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File