

Abstract
Inducing angiogenesis is a hallmark of cancers that sustains tumor growth and metastasis. Neovascularization is a surprisingly early event during the multistage progression of cancer. Cinobufagin, an important bufadienolide originating from Chan Su, has been clinically used to treat cancer in China since the Tang dynasty. Here, we show that cinobufagin suppresses colorectal cancer (CRC) growth in vivo by downregulating angiogenesis. The hierarchized neovasculature is significantly decreased and the vascular network formation is disrupted in HUVEC by cinobufagin in a dose‐dependent way. Endothelial apoptosis is observed by inducing reactive oxygen species (ROS) accumulation and mitochondrial dysfunction which can be neutralized by N‐acetyl‐l‐cysteine (NAC). Expression of hypoxia‐inducible factor 1α (HIF‐1α) is reduced and phosphorylation of mTOR at Ser2481 and Akt at Ser473 is downregulated in HUVEC. Endothelial apoptosis is triggered by cinobufagin by stimulation of Bax and cascade activation of caspase 9 and caspase 3. Increased endothelial apoptosis rate and alterations in the HIF‐1α/mTOR pathway are recapitulated in tumor‐bearing mice in vivo. Further, the anti‐angiogenesis function of cinobufagin is consolidated based on its pro‐apoptotic effects on an EOMA‐derived hemangioendothelioma model. In conclusion, cinobufagin suppresses tumor neovascularization by disrupting the endothelial mTOR/HIF‐1α pathway to trigger ROS‐mediated vascular endothelial cell apoptosis. Cinobufagin is a promising natural anti‐angiogenetic drug that has clinical translation potential and practical application value.
Keywords: angiogenesis, apoptosis, cinobufagin, colorectal cancer, HIF‐1α
1. INTRODUCTION
Angiogenesis is a hallmark of cancer.1 Vascular endothelial cells begin to resuscitate even if the diameter of tumors is only approximately 0.2‐2.0 mm.2 This early angiogenic switch emerges in premalignant or in in situ carcinomas. Neovasculature promotes solid tumor growth and metastasis.3 Therefore, targeting angiogenesis is one of the most conventional approaches for controlling tumor progression.
In 2018, colorectal cancer (CRC) was the third main cause of estimated cancer death in the USA.4 CRC is the fifth most common cancer in China and the incidence rate has shown an upward trend from 2000 to 2011.5 Chemotherapy is the treatment of choice for postoperative CRC patients. However, the influences of chemotherapeutic agents on angiogenesis are still controversial. Chemotherapy drugs such as cisplatin, doxorubicin and vincristine increase vessel densities in neuroblastoma.6 However, the absolute counts of circulating endothelial cells, a biomarker for angiogenesis, were not changed after a 4‐week gemcitabine treatment in pancreatic carcinoma patients.7 On the contrary, low‐dose metronomic chemotherapy docetaxel treatment effectively inhibited the growth of gastric tumor by decreasing microvessel number.8 For breast cancer patients with highly angiogenic tumor subtypes, angiogenesis phenotype enhances chemotherapeutic resistance. These patients cannot derive sufficient benefit even after receiving intensified adjuvant chemotherapy.9 Therefore, seeking more effective anti‐angiogenic agents would renew our enthusiasm for this therapeutic strategy.10
Our previous work shows that intrinsic apoptosis is a feasible and efficacious pathway to eliminate transformed cells with damaged DNA and aberrant expression of oncogene. Arenobufagin isolated from skin and parotid venom glands (Chan'Su) is an active component that suppresses the progression of CRC by inducing intrinsic apoptosis.11 Cinobufagin is another cardiotonic steroid isolated from Chan'Su of the toad Bufo bufo gargarizans Cantor. It inhibits the growth of breast cancer,12 osteosarcoma13 and colorectal cancer14 by inducing apoptosis. It can also reverse multidrug resistance in colon cancer cells.15 Cinobufagin treatment significantly ameliorates nutritional status and quality of life of patients with lung cancer cachexia, suggesting that it is a well‐tolerated natural agent for cancer therapy.16 However, its function on angiogenesis has not been fully investigated.
Tumor‐hypoxia induces advanced but dysfunctional vascularization. Hypoxia‐inducible factor 1α (HIF‐1α), a transcription factor, orchestrates angiogenesis by stimulating vascular endothelial growth factor (VEGF). The PI3K/Akt/mTOR pathway is activated in most human cancers. The constitutive activation of Akt in endothelial cells results in the formation of structurally abnormal blood vessels.17 Hyperactivation of this cascade pathway and its downstream targets can facilitate tumor neovascularization by increasing VEGF secretion.18 The activation of PI3K/Akt/mTOR can, in turn, increase the transcription and stabilization of HIF‐1α which is the pivotal stimulus for angiogenesis.19 Our results provide consolidated data that cinobufagin can effectively suppress angiogenesis both in vivo and in vitro. It regulates the PI3K/Akt/mTOR pathway to trigger apoptosis in endothelial cells. Cinobufagin is a candidate natural drug that targets tumor angiogenesis with strong translation potential.
2. MATERIALS AND METHODS
2.1. Materials
Cinobufagin was purchased from Chenguang Biotechnology (Baoji, China). MTT and DMSO were purchased from Sigma‐Aldrich (St Louis, MO, USA). RPMI 1640, FBS, and antibiotics were purchased from Thermo Fisher Scientific (Boston, MA, USA). HUVEC was purchased from ATCC (Manassas, VI, America). Mice hemangioendothelioma cell EOMA was purchased from JNObio (Guangzhou, China). Hoechst staining kit, JC‐1 kit for mitochondrial membrane potential, reactive oxygen species (ROS) assay kit and ROS inhibitor N‐acetyl‐l‐cysteine (NAC) were purchased from Beyotime Biotechnology (Shanghai, China). Annexin V‐FITC/7AAD apoptosis detection kit was purchased from Becton Dickinson (BD, Franklin Lakes, NJ, USA). Antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA) and Abcam (Cambridge, UK). Rapamycin was purchased from Meilunbio (Dalian, China). Endostatin was purchased from Simcere (Shandong, China).
2.2. Tumor s.c. and orthotopic implantation model
All procedures carried out in animal experiments were done according to the ethical standards of Animal Care Ethics Committee of Southern Medical University. Male BALB/c nude mice (Laboratory Animal Center of Southern Medical University, Guangzhou, China) were maintained under controlled conditions (22°C, 12 h/12 h dark/light cycle). SW480 cells or EOMA cells (5 × 106/mice) were s.c. injected into the bilateral underarms of nude mice to establish an s.c. implantation model. To establish the orthotopic implantation model further, tumors were excised and cut into 1‐mm3 pieces when the tumor volume reached an average of 100 mm3. Tumor pieces were then used to carry out surgical orthotopic implantation in the ascending colon as previously described.20
For the SW480 cell orthotopic implantation model, animals were randomly divided into four groups (8 mice/group) 1 week after implantation: (i) control; (ii) cinobufagin 2 mg/kg; (iii) cinobufagin 4 mg/kg; and (iv) cisplatin 0.2 mg/kg. Cinobufagin and cis‐diamminedichloroplatinum (DDP) were i.p. injected every other day for 3 weeks. Weights of mice were measured with an electron balance once a week for up to 3 weeks. Thirty‐eight days after implantation, mice were killed.
For the EOMA cell s.c. implantation model, animals were randomly divided into four groups (6 mice/group) 2 weeks after injection: (i) control; (ii) cinobufagin 2 mg/kg; (iii) rapamycin 5 mg/kg; and (iv) endostatin 20 mg/kg. Cinobufagin, rapamycin and endostatin were i.p. injected every day for 2 weeks. Weights of mice were measured every 3 days. Fourteen days after implantation, mice were killed.
Organs of the mice were excised, photographed, and fixed in 4% paraformaldehyde. Tissues were then used to prepare paraffin slides for TUNEL staining and immunohistochemistry (IHC) analysis. Tumor volume (mm3) was calculated by the formula: volume = (width)2 × length/2.21
2.3. Immunohistochemistry and TUNEL
Expression of CD31 in tumors was assessed by IHC staining as described previously.22 In brief, paraffin‐embedded tumor sections were incubated with anti‐CD31 antibody (1:100) overnight at 4°C, and then incubated with secondary antibody.
Apoptosis was evaluated by TUNEL assay (Roche diagnostics, Mannheim, Germany) following the manufacturer's instructions. The image of each case was captured using a fluorescence microscope (Nikon Eclipse‐Ti; Nikon, Tokyo, Japan) and analyzed using Image‐Pro Plus 6.0 (IPP6) software.
2.4. Chicken chorioallantoic membrane assay
Chicken chorioallantoic membrane (CAM) assay was carried out as described with modification.23 Briefly, fertilized chicken eggs (South China Agricultural University) were incubated at 37°C for 10 days, and a window was then carefully created through the egg shell. Sterilized rings (5 mm × 5 mm) saturated with 0.3 mL cinobufagin (0, 0.1, 1, and 10 μM/CAM) were placed on CAM. Cinobufagin (0, 0.1, 1, and 10 μM/CAM) was added to CAM every other day and incubated at 37°C for another 7 days. Then an appropriate volume of 10% fat emulsion (Intralipose purchased from Green Cross Pharmaceutical Company [Guangzhou, China], 10%) was injected into the embryo chorioallantois for observing the density of vessels toward the CAM face. Neovascular zones were observed and photographed by a digital camera. Number of newly grown vessels was counted.
2.5. Tube formation assay
Tube formation assay was carried out as described by Nagata et al.24 HUVEC (5 × 104/well) were pretreated with different concentrations of cinobufagin for 30 minutes and seeded on the Matrigel layer (BD) in 24‐well plates. After 8 hours of incubation, tubular structures were captured and tube number was counted to evaluate tube formation.
2.6. Migration assay
For HUVEC migration assay, Transwell inserts (8 μM pore; Corning Costar Corp., Cambridge, MA, USA) were used. HUVEC (5 × 104 cells/well) were resuspended in a serum‐free RPMI 1640 medium containing 0.1% BSA and seeded on the top chambers. RPMI 1640 (700 μL) with 10% FBS was added to the bottom chambers. HUVEC were coincubated with cinobufagin (0.1, 1, and 10 μM) and allowed to invade for 48 hours. After 48 hours, the noninvasive cells on the upper surface of the filter were removed by a cotton swab. The invaded cells were stained with 0.1% crystal violet, and counted in five random fields per well.22
2.7. Annexin V and 7‐AAD staining
SW480 and HUVEC cells were seeded in 6‐well plates at a density of 1.0 × 105 cells/well overnight. After treatment with various dilutions of cinobufagin for 24 hours or 48 hours, cells were then washed with PBS twice and incubated in 300 μL binding buffer with 5 μL annexin V‐FITC and 5 μL 7‐AAD for 15 minutes in the dark at room temperature. Then, the stained cells were tested by flow cytometer (BD), and 10 000 cells were counted within 30 minutes.
2.8. Analysis of mitochondrial membrane potential
The experiment was conducted according to the protocols provided by the manufacturer (Beyotime Biotechnology). Then, both red fluorescence and green fluorescence were measured by flow cytometry.
2.9. Reactive oxygen species analysis
Cinobufagin‐treated HUVEC and control group cells were stained with 20 μM 2',7'‐Dichlorodihydrofluorescein diacetate(DCFH‐DA) in the dark for 30 minutes and DCF fluorescence intensity by flow cytometry was measured. Green fluorescence was detected in the FL1 (FITC) channel. Mean fluorescence intensity (MFI) of 10 000 cells was analyzed by WinMDI 2.8 software. MFI data were normalized to control levels and expressed as relative fluorescence intensity (DCF).
2.10. Western blotting
Total protein was extracted with RIPA buffer (Beyotime Biotechnology) and quantified using a BCA protein assay kit (Beyotime Biotechnology) following the manufacturer's instructions and then western blot was carried out as described.22 ECL solution was mixed for luminescent image development, and target protein levels were analyzed by Image J software with β‐actin as a reference protein for loading control.
2.11. Statistical analysis
All experimental data are shown as mean ± standard deviation. Experiments were repeated three times. Statistical analysis was conducted by one‐way ANOVA using statistical package SPSS 20.0 (Chicago, IL, USA). P < .05 was considered a statistically significant difference.
3. RESULTS
3.1. Cinobufagin inhibits orthotopic CRC growth by suppressing angiogenesis in vivo
The antineoplastic effects of cinobufagin were tested on mice bearing orthotopically implanted colorectal carcinomas as described previously.11, 22 The weight of tumor in situ was significantly reduced by cinobufagin and DDP (Figure 1A,C). Tumor size was also downregulated by cinobufagin in a dose‐dependent way as evaluated by vernier caliper (Figure 1D). Body weight of mice was obviously lowered by DDP (Figure 1B). However, the rapid weight loss was ameliorated in mice treated with cinobufagin compared to those treated with DDP (Figure 1B), suggesting that cinobufagin is a naturally occurring molecule with high efficiency and low toxicity.
Figure 1.
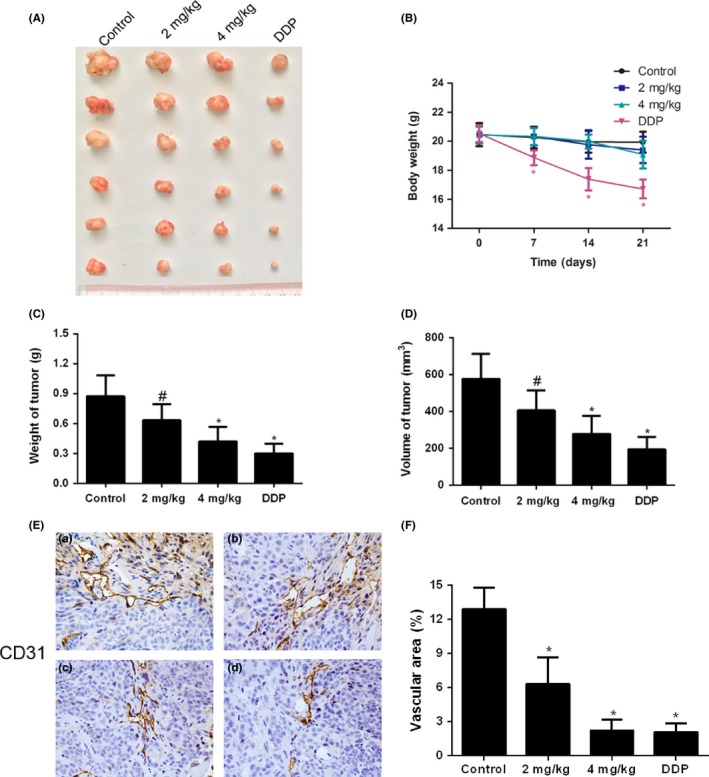
Cinobufagin inhibits tumor growth and tumor angiogenesis in orthotopically transplanted colorectal carcinoma. A, Sizes of orthotopically transplanted colorectal carcinoma. B, Body weight of mice (n = 6). C, Tumor weight of control, cinobufagin (2 mg/kg and 4 mg/kg) and cisplatin treatment groups (n = 6). D, Tumor volume of control, cinobufagin and cisplatin treatment groups (n = 6). E, Representative images of immunohistochemical staining for CD31 (100×). a, control group; b, 2 mg/kg cinobufagin group; c, 4 mg/kg cinobufagin group; d, 0.5 mg/kg cisplatin group. F, Vascular area in xenotransplanted tumor tissues. Vascular endothelial cells were stained with CD31. # P < .05; *P < .01 compared to control group
CD31 is a widely used angiogenic marker for the measurement of microvessel density (MVD).25, 26 Expression of CD31 and vascular area were dose dependently decreased in tumor tissues by cinobufagin treatment (Figure 1E,F). Collectively, these results showed that cinobufagin decreases CRC growth by downregulating angiogenesis.
3.2. Cinobufagin suppresses endothelial cell migration and angiogenesis
Neovasculature forms a hierarchized and stereotypic network that is lined with endothelial cells in normal tissues, whereas an abnormal and chaotic network forms within the tumor. CAM assay was conducted as an in vivo approach to validate the putative anti‐angiogenic effects of cinobufagin. The number of primary, secondary and tertiary blood vessels was dose‐dependently reduced by a 4‐day cinobufagin treatment in 8‐day‐old fertilized eggs (Figure 2A).
Figure 2.
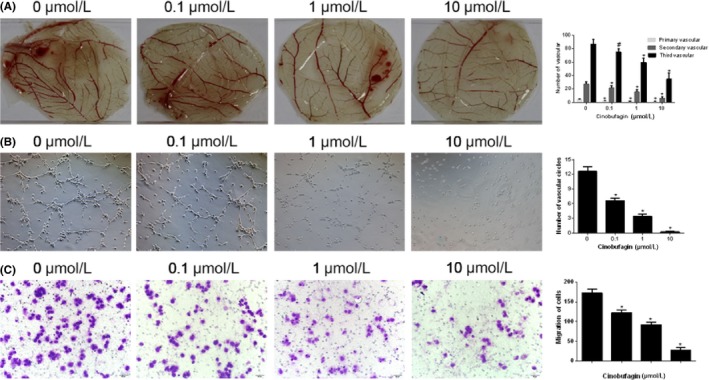
Cinobufagin suppresses endothelial cell migration and angiogenesis. A, Effects of cinobufagin on angiogenesis in chicken chorioallantoic membrane (n = 3). Concentrations of cinobufagin are shown above each part. The following figure is in accordance with this standard. B, Number of vascular circles in HUVEC treated with cinobufagin. Representative images (magnification, 100×) show tubule formation by HUVEC cells plated on Matrigel (n = 3). C, Migration of HUVEC treated with cinobufagin (n = 3). Representative images (magnification, 100×) show effect of cinobufagin on the migration of HUVEC in 24 h. # P < .05; *P < .01 compared to 0 μM
The ability of endothelial cells to engage in vascular morphogenesis was assessed by tubular formation test.27 By using cultured HUVEC, we found that vascular networks were formed spontaneously in control conditions. The formation of these structures was significantly abolished upon cinobufagin treatment (Figure 2B).
The migration of endothelial cells is an early step for the sprouting and elongation of a newly formed vessel.28 Cinobufagin significantly inhibited endothelial cell migration compared to control (Figure 2C). Together, these assays recapitulate the endothelial functions during angiogenesis. Cinobufagin suppresses neovascularization by dampening endothelial migration and tubular formation.
3.3. Cinobufagin induces endothelial cell apoptosis
The viability of HUVEC was significantly lowered by cinobufagin in a dose‐ and time‐dependent way (Figure S1). To clarify the potential anti‐angiogenic mechanism of cinobufagin, cell apoptosis was first probed with Hoechst 33342 staining. The positive ratios of HUVEC (Figure 3A) were essentially upregulated by cinobufagin. The results were further confirmed through double staining of Annexin V and 7‐AAD by flow cytometry (Figure 3B). Furthermore, ROS inhibitor NAC lowered the cell apoptosis ratio caused by cinobufagin (Figure 3C), suggesting that cinobufagin causes apoptosis and lowers cell viability through triggering oxidation reactions in HUVEC.
Figure 3.
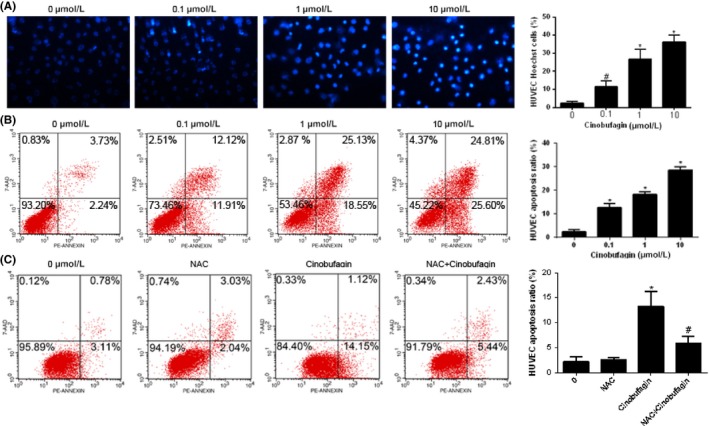
Cinobufagin induces apoptosis in HUVEC. A, Hoechst 33342 staining for HUVEC with cinobufagin treatment (n = 3). Cells were treated with different concentrations of cinobufagin (0, 0.1, 1, and 10 μM) for 24 h. Original magnification, 200×. Marked morphological changes in chromatin morphology such as crenation, condensation, and fragmentation are observed in HUVEC. B, Apoptosis rate of HUVEC cells detected by cytometry (n = 3). Cells treated with different concentrations of cinobufagin (0, 0.1, 1, and 10 μM) for 24 h were detected by flow cytometry. C, Apoptosis rate of HUVEC treated with N‐acetyl‐l‐cysteine (NAC; n = 3). l‐NAC reduced HUVEC cell apoptosis induced by cinobufagin. Cells treated with or without cinobufagin or NAC (0.1 μM) for 24 h were detected by flow cytometry. # P < .05; *P < .01 compared to 0 μM
3.4. Cinobufagin disrupts mitochondrial membrane potential and induces ROS accumulation
JC‐1 is a fluorescent, lipophilic, cationic dye that can be used to effectively distinguish between apoptotic and healthy cells. Flow cytometry showed that mitochondrial membrane potential was exhausted dose‐dependently by cinobufagin (Figure 4A). ROS production was subsequently checked by DCFH‐DA staining. ROS level was increased with dose escalation of cinobufagin (Figure 4B), indicating that cinobufagin‐induced ROS accumulation and mitochondrial dysfunction are the driving force of apoptosis.
Figure 4.
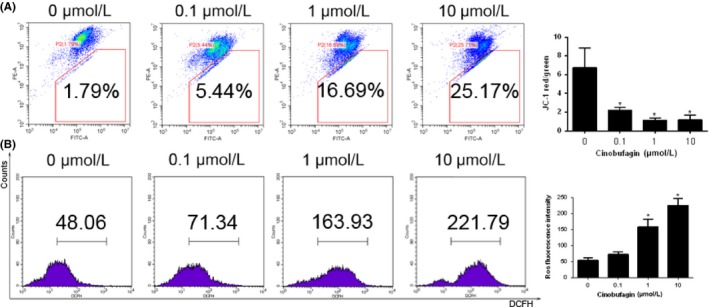
Cinobufagin lowers mitochondrial membrane potential and induces reactive oxygen species (ROS) accumulation in HUVEC. A, Mitochondrial membrane potential measured by flow cytometry (n = 3). HUVEC were treated with different concentrations of cinobufagin (0, 0.1, 1, and 10 μM) for 24 h, and then stained with JC‐1. B, ROS levels in HUVEC detected by flow cytometry. HUVEC were treated with different concentrations of cinobufagin (0, 0.1, 1, and 10 μM) for 24 h, and then stained with 2',7'‐Dichlorodihydrofluorescein diacetate(DCFH‐DA). # P < .05; *P < .01 compared to 0 μM
3.5. Cinobufagin cleaves caspase‐3 and downregulates mTOR signaling
Mitochondrial membrane potential depletion will lead to the initiation of mitochondrial outer membrane permeability (MOMP).29 Cinobufagin downregulated Bcl‐2 and activated Bax in HUVEC (Figure 5A). Mitochondrial translocation of Bax is the key event for opening the dynamic Bax/Bak‐lipid pore in the process of MOMP.11 Upon rupture of the mitochondrial outer membrane, secreted cytochrome c triggers hierarchical activation of caspase‐9 and caspase‐3. The cleaved forms of caspase‐9 and caspase‐3 were upregulated in HUVEC treated with cinobufagin (Figure 5A). Subsequently, cleavage of poly‐ADP ribose polymerase (PARP) was observed in cinobufagin‐treated HUVEC. Collectively, cinobufagin induces cascade activation of caspase to stimulate HUVEC apoptosis.
Figure 5.
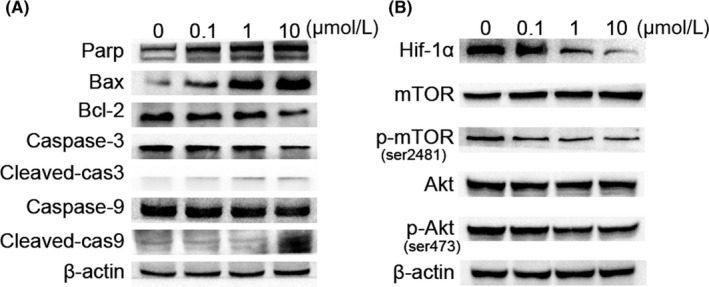
Western blot analysis of the effect of cinobufagin on caspase and mTOR signaling. A, Effect of cinobufagin on the caspase pathway (Bax, Bcl‐2, caspase‐3, cleaved‐caspase‐3, caspase‐9 and cleaved‐caspase‐9) in HUVEC cells. Western blot analysis was carried out to detect changes in the levels of Bax, Bcl‐2, caspase‐3, cleaved‐caspase‐3, caspase‐9 and cleaved‐caspase‐9 after treatment with 0, 0.1, 1, 10 μM cinobufagin for 24 h (n = 3). B, Effect of cinobufagin on the expression of hypoxia‐inducible factor 1α (HIF‐1α), mTOR, p‐mTOR (ser2481), Akt, and p‐Akt (ser473) (n = 3). HUVEC cells were treated with various concentrations of cinobufagin (0, 0.1, 1, and 10 μM) for 24 h, and then the levels of proteins were detected by western blot analysis
Members of the anti‐apoptotic Bcl‐2 subfamily normally stabilize the outer membrane by binding to pro‐apoptotic BH123 effectors, such as Bax and Bak. Hypoxia is known to modulate apoptosis sensitivity by increasing expression of anti‐apoptotic Bcl‐2 and Bcl‐xL and downregulating Bax, Bad and Bid. The serine/threonine kinase Akt is widely acknowledged as a proto‐oncogene that promotes cell survival and inhibits apoptosis. Cinobufagin decreased the expression of HIF‐1α and downregulated the phosphorylation of Akt at Ser473 dose‐dependently (Figure 5B). Hypoxia is a key stimulator of endothelial migration and angiogenesis through activating Akt.30 Reduction of HIF‐1α and p‐Akt is accompanied by downregulated survival and migration of HUVEC.
mTOR is a key nutrient and energy sensor that regulates metabolic processes. mTOR hyperactivation is the driving force for angiogenesis.31 Cinobufagin downregulated phosphorylation of mTOR at ser2481 in HUVEC, suggesting that deactivation of endothelial mTOR inhibits endothelial cell proliferation and migration. Collectively, cinobufagin suppresses the survival pathway in HUVEC through downregulation of HIF‐1α and dephosphorylation of Akt and mTOR.
3.6. Cinobufagin activates endothelial apoptosis in vivo
To verify the pro‐apoptotic effects of cinobufagin in vivo, tumor tissue slides were double stained with TUNEL and CD31 (Figure 6A). Cinobufagin downregulated the vascular area dose dependently and upregulated the apoptotic ratio of CD31‐positive cells, suggesting that cinobufagin induces the apoptosis of endothelial cells (Figure 6B,C).
Figure 6.
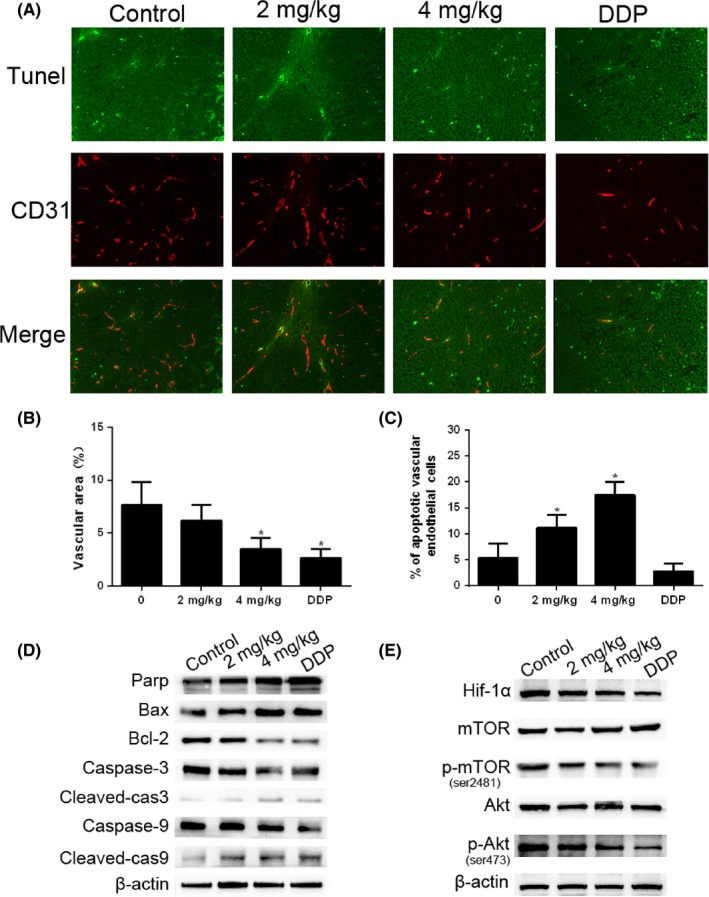
Cinobufagin induces apoptosis of endothelial cells in vivo. A, Apoptotic vascular endothelial cells were detected using TUNEL and CD31 staining in tumor tissues from BALB/c nude mice with treatment (0, 2 mg/kg, 4 mg/kg of cinobufagin and 0.5 mg/kg of cisplatin) (n = 6). Double‐stained cells indicate apoptotic endothelial vascular cells (green) in the tumor tissues (200×). Anti‐CD31 antibody was used as a marker for endothelial cells (red). B, Area of tumor endothelial cells (red) was quantified by Image‐Pro Plus 6.0 (IPP6) software. C, Percentage of apoptotic endothelial cells (double stained) in tumor issues. Immunostained plugs were assessed by microscopy at 200× magnification. *P < .01 compared with control group. D, Effect of cinobufagin on the caspase pathway (Bax, Bcl‐2, caspase‐3, cleaved‐caspase‐3, caspase‐9 and cleaved‐caspase‐9) in tumor tissues. Western blot analysis was carried out to detect changes in the levels of different groups of mice. E, Effect of cinobufagin on the expression of hypoxia‐inducible factor 1α (HIF‐1α), mTOR, p‐mTOR (ser2481), Akt, and p‐Akt (ser473). DDP, cis‐diamminedichloroplatinum
Cleavage of PARP, caspase‐3 and ‐9 was increased by cinobufagin treatment. Bax expression was upregulated, representing the triggering of a mitochondrial apoptosis process (Figure 6D). Cinobufagin also reduced the expression of HIF‐1α and significantly decreased the phosphorylation of mTOR and Akt, indicating that cinobufagin might be an mTOR inhibitor to suppress angiogenesis (Figure 6E). Taken together, cinobufagin is an efficacious angiogenesis inhibitor through triggering endothelial apoptosis. It inhibits HIF‐1α and dephosphorylates Akt and mTOR to switch the apoptosis program.
3.7. Cinobufagin suppresses hemangioendothelioma in vivo
The anti‐angiogenic effects of cinobufagin in vivo were tested on mice bearing s.c. hemangioendothelioma EOMA. Tumor weight and tumor size were significantly reduced by cinobufagin, rapamycin and the endogenous angiogenesis inhibitor endostatin, respectively (Figure 7A,C,D). Surprisingly, body weight of mice was obviously lowered by mTOR inhibitor rapamycin (Figure 7B). To further verify the pro‐apoptotic effects of cinobufagin in vivo, hemangioendothelioma tissue slides were double stained with TUNEL and CD31 (Figure 7E). Similar to rapamycin and the endogenous angiogenesis inhibitor endostatin, cinobufagin upregulated apoptotic endothelial vascular cells but with a much stronger effect (Figure 7F). Cinobufagin reduced the expression of HIF‐1α and significantly decreased the phosphorylation of mTOR and Akt similar to rapamycin (Figure 7G), and similar to the result shown in Figure 6E. Collectively, we confirmed that cinobufagin suppresses angiogenesis by regulating mTOR signaling with lower toxicity compared with DDP.
Figure 7.
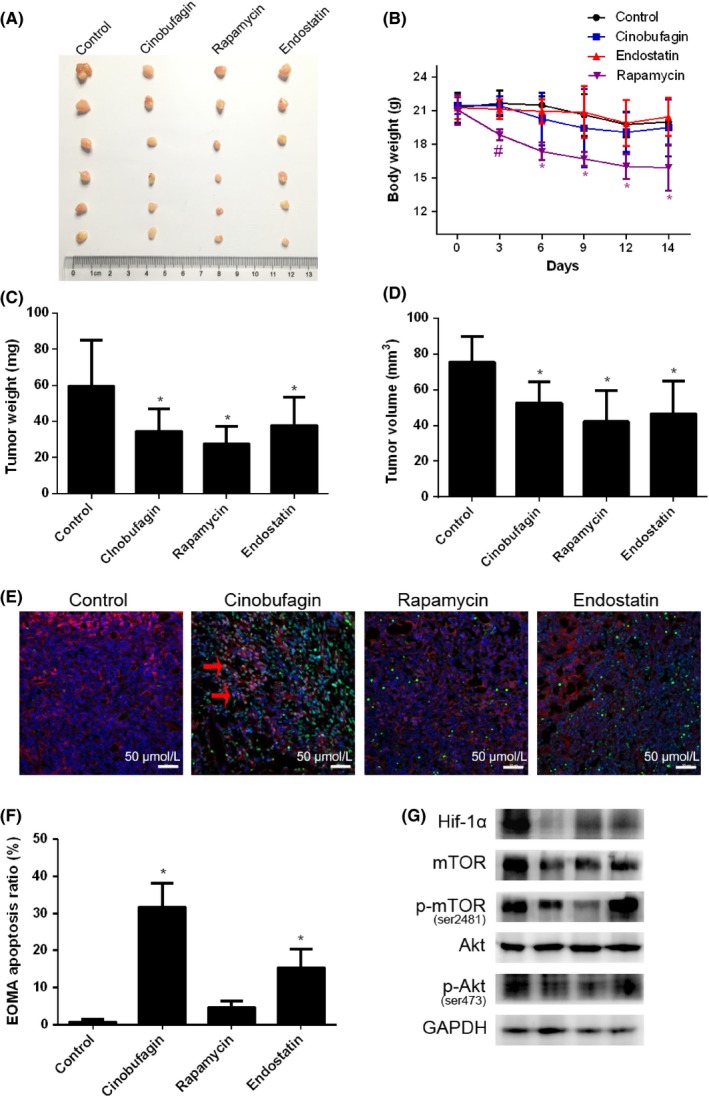
Cinobufagin suppresses hemangioendothelioma in vivo. A, Sizes of s.c. hemangioendothelioma tumor. B, Body weight of mice (n = 6). #P < .05, *P < .01 compared to control group. C, Tumor weight of control, cinobufagin, rapamycin and endostatin treatment groups (n = 6). D, Tumor volume of control, cinobufagin, rapamycin and endostatin treatment groups (n = 6). *P < .01 compared to control group. E, Apoptotic vascular endothelial cells were detected using TUNEL and CD31 staining in hemangioendothelioma tissues from BALB/c nude mice with control, cinobufagin, rapamycin and endostatin treatments (n = 6). Double‐stained cells indicate apoptotic endothelial vascular cells (green) in the tumor tissues (200×). Anti‐CD31 antibody was used as a marker for endothelial cells (red). Apoptotic vascular endothelial cells stained cyan as indicated by red arrow. F, Percentage of apoptotic endothelial cells (double stained) in tumor issues. *P < .01 compared with control group. G, Effect of control, cinobufagin rapamycin and endostatin treatment on the expression of hypoxia‐inducible factor 1α (HIF‐1α), mTOR, p‐mTOR (ser2481), Akt, and p‐Akt (ser473) by western blot analysis
4. DISCUSSION
Since the approval of bevacizumab for treating metastatic colorectal cancer in 2004, angiogenesis is an attractive target as an angiogenesis inhibitor. Blockade of angiogenesis can effectively retard tumor growth. However, angiogenesis inhibitors may also paradoxically increase metastasis by aggravating tumor hypoxia and preventing absorption of the chemotherapeutic regimen. “Vascular normalization” has been proposed as a revolutionary strategy to overcome the aforementioned shortcomings of angiogenesis inhibitors. Even so, the therapeutic window for vascular normalization is transient and unpredictable. It is too difficult to estimate and grasp the most appropriate timeframe of vascular normalization to maximize delivery of chemotherapeutic agents.32 Although still controversial, clinical studies of single bevacizumab and combination trials have shown positive outcomes that angiogenesis inhibitors can be an optional frontline therapy for metastatic CRC.33 Cinobufagin suppresses in vivo and in vitro angiogenesis and inhibits the growth of orthotopic transplanted colorectal carcinoma. It dose‐dependently downregulates tumor microvessel number, suggesting that targeting tumor neovasculature is still a valuable and effective choice for CRC management.
Tuberous sclerosis complex (TSC) patients are predisposed to highly vascularized tumors. mTORC1 is a critical driver for HIF‐1α synthesis. mTORC1 activates a 10‐fold increase of HIF‐1α transcriptional activity under hypoxic conditions.34 HIF‐1α knockdown can suppress the induction of VEGF in hypoxic cells and reduce vascular leakage.35 However, none of the HIF inhibitor compounds are currently suitable for clinical therapy. We propose a model whereby HIF‐1α is a downstream signaling molecule of mTORC1 that can induce angiogenesis for tumor progression.36 Cinobufagin significantly inhibits the phosphorylation of mTOR and subsequently downregulates HIF‐1α, which is consistent with the highlighted anti‐angiogenic effects of mTOR inhibitor.37 Hypoxic condition induces activation of Akt which is essential for HIF‐1α expression. HIF‐1α then triggers angiogenesis by upregulating the expression of VEGF in cancer cells. In turn, VEGF activates the PI3K/Akt/mTOR pathway to stimulate HUVEC proliferation and drive angiogenesis in tumors. Our results suggest that cinobufagin inhibits the Akt‐HIF‐1α signaling axis.38 Together, cinobufagin exerts its anti‐angiogenesis effects by inhibiting these survival pathways of HUVEC.
Bcl‐xL translation is regulated by mTOR through phosphorylation of its known target, 4E‐BP1.39 Rapamycin triggers apoptosis by increasing the Bax/Bcl‐xL ratio. HIF‐1 suppresses mitochondrial function and promotes the efficient use of available oxygen. Therefore, HIF‐1α reduces the generation of ROS and confers apoptosis resistance to cancer cells.40 Evasion of apoptosis may represent an important hallmark of many human cancers. Cinobufagin reduces neovascularization by increasing the ROS level which leads to mitochondrial dysfunction in endothelial cells. The ROS accumulation induced by oxidation reaction facilitates the triggering of MOMP by cinobufagin. Then, it stimulates caspases, which is a biochemical characteristic of apoptosis.41 Cinobufagin treatment increases cleavage of caspase‐9 dose‐dependently. Activated caspase‐9 in turn cleaves and activates caspase‐3 to execute intrinsic apoptosis in HUVEC.
Specific Akt/mTOR double pathway targeting agent, such as everolimus, is available and efficacious in metastatic CRC patients.42 However, everolimus treatment may induce therapy resistance by triggering a negative feedback loop which leads to ERK activation and Mcl‐1 stabilization in colon cancer cells.42 Cinobufagin dephosphorylates mTOR and Akt. Furthermore, it also disrupts the HIF‐1α‐VEGF pathway by switching the apoptosis program,43 suggesting that cinobufagin targets multiple angiogenic signaling to suppress neovascularization in CRC. In conclusion, cinobufagin suppresses CRC angiogenesis by downregulating the Akt/mTORC1/HIF‐1α pathway and triggering MOMP‐mediated apoptosis. Cinobufagin is a promising biotherapeutic agent for the treatment of CRC, particularly for patients with highly vascularized tumors.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
{kind=link}
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (81573848, 81774172), Natural Science Foundation of Guangdong Province (2014A030313323), Planned Science Technology Project of Guangzhou (201704020077, 201607010146), Guangdong Province Bureau of Traditional Chinese Medicine Scientific Research Project (No: 20151024, 20161161).
Li X, Chen C, Dai Y, et al. Cinobufagin suppresses colorectal cancer angiogenesis by disrupting the endothelial mammalian target of rapamycin/hypoxia‐inducible factor 1α axis. Cancer Sci. 2019;110:1724–1734. 10.1111/cas.13988
Li, Chen, Dai and Huang contributed equally to this study.
Contributor Information
Xuegang Sun, Email: sxg_smu@126.com.
Xueqing Yao, Email: yjb9211@21cn.com.
REFERENCES
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 2. Shahneh FZ, Baradaran B, Zamani F, Aghebati‐Maleki L. Tumor angiogenesis and anti‐angiogenic therapies. Hum Antibodies. 2013;22:15‐19. [DOI] [PubMed] [Google Scholar]
- 3. Wang LF, Liu YS, Yang B, et al. The extracellular matrix protein mindin attenuates colon cancer progression by blocking angiogenesis via Egr‐1‐mediated regulation. Oncogene. 2018;37:601‐615. [DOI] [PubMed] [Google Scholar]
- 4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7‐30. [DOI] [PubMed] [Google Scholar]
- 5. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115‐132. [DOI] [PubMed] [Google Scholar]
- 6. Michaelis M, Hinsch N, Michaelis UR, et al. Chemotherapy‐associated angiogenesis in neuroblastoma tumors. Am J Pathol. 2012;180:1370‐1377. [DOI] [PubMed] [Google Scholar]
- 7. Kondo S, Ueno H, Hashimoto J, et al. Circulating endothelial cells and other angiogenesis factors in pancreatic carcinoma patients receiving gemcitabine chemotherapy. BMC Cancer. 2012;12:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu H, Xin Y, Zhao J, et al. Metronomic docetaxel chemotherapy inhibits angiogenesis and tumor growth in a gastric cancer model. Cancer Chemother Pharmacol. 2011;68:879‐887. [DOI] [PubMed] [Google Scholar]
- 9. Gluz O, Wild P, Liedtke C, et al. Tumor angiogenesis as prognostic and predictive marker for chemotherapy dose‐intensification efficacy in high‐risk breast cancer patients within the WSG AM‐01 trial. Breast Cancer Res Treat. 2011;126:643‐651. [DOI] [PubMed] [Google Scholar]
- 10. Mi C, Ma J, Wang KS, et al. Imperatorin suppresses proliferation and angiogenesis of human colon cancer cell by targeting HIF‐1alpha via the mTOR/p70S6K/4E‐BP1 and MAPK pathways. J Ethnopharmacol. 2017;203:27‐38. [DOI] [PubMed] [Google Scholar]
- 11. Xu W, Jing L, Wang Q, et al. Bax‐PGAM5L‐Drp1 complex is required for intrinsic apoptosis execution. Oncotarget. 2015;6:30017‐30034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu L, Chen Y, Wei C, et al. Anti‐proliferative and pro‐apoptotic effects of cinobufagin on human breast cancer MCF‐7 cells and its molecular mechanism. Nat Prod Res. 2018;32:493‐497. [DOI] [PubMed] [Google Scholar]
- 13. Cao Y, Yu L, Dai G, et al. Cinobufagin induces apoptosis of osteosarcoma cells through inactivation of Notch signaling. Eur J Pharmacol. 2017;794:77‐84. [DOI] [PubMed] [Google Scholar]
- 14. Lu XS, Qiao YB, Li Y, Yang B, Chen MB, Xing CG. Preclinical study of cinobufagin as a promising anti‐colorectal cancer agent. Oncotarget. 2017;8:988‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan Z, Shi X, Qiu Y, et al. Reversal of P‐gp‐mediated multidrug resistance in colon cancer by cinobufagin. Oncol Rep. 2017;37:1815‐1825. [DOI] [PubMed] [Google Scholar]
- 16. Xie M, Chen X, Qin S, Bao Y, Bu K, Lu Y. Clinical study on thalidomide combined with cinobufagin to treat lung cancer cachexia. J Cancer Res Ther. 2018;14:226‐232. [DOI] [PubMed] [Google Scholar]
- 17. Zimna A, Kurpisz M. Hypoxia‐inducible factor‐1 in physiological and pathophysiological angiogenesis: applications and therapies. Biomed Res Int. 2015;2015:549412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karar J, Maity A. PI3K/Akt/mTOR pathway in angiogenesis. Front Mol Neurosci. 2011;4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim GD. Myricetin inhibits angiogenesis by inducing apoptosis and suppressing PI3K/Akt/mTOR signaling in endothelial cells. J Cancer Prev. 2017;22:219‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang F, Luo Y, Shao Z, et al. MicroRNA‐187, a downstream effector of TGFbeta pathway, suppresses Smad‐mediated epithelial‐mesenchymal transition in colorectal cancer. Cancer Lett. 2016;373:203‐213. [DOI] [PubMed] [Google Scholar]
- 21. Lin X, Yi Z, Diao J, et al. ShaoYao decoction ameliorates colitis‐associated colorectal cancer by downregulating proinflammatory cytokines and promoting epithelial‐mesenchymal transition. J Transl Med. 2014;12:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chunhua L, Donglan L, Xiuqiong F, et al. Apigenin up‐regulates transgelin and inhibits invasion and migration of colorectal cancer through decreased phosphorylation of AKT. J Nutr Biochem. 2013;24:1766‐1775. [DOI] [PubMed] [Google Scholar]
- 23. Cai QY, Liang GY, Zheng YF, Tan QY, Wang RW, Li K. CCR7 enhances the angiogenic capacity of esophageal squamous carcinoma cells in vitro via activation of the NF‐kappaB/VEGF signaling pathway. Am J Transl Res. 2017;9:3282‐3292. [PMC free article] [PubMed] [Google Scholar]
- 24. Nagata D, Mogi M, Walsh K. AMP‐activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem. 2003;278:31000‐31006. [DOI] [PubMed] [Google Scholar]
- 25. Koukourakis MI, Giatromanolaki A, Thorpe PE, et al. Vascular endothelial growth factor/KDR activated microvessel density versus CD31 standard microvessel density in non‐small cell lung cancer. Cancer Res. 2000;60:3088‐3095. [PubMed] [Google Scholar]
- 26. Ito K, Hamamichi S, Abe T, et al. Antitumor effects of eribulin depend on modulation of the tumor microenvironment by vascular remodeling in mouse models. Cancer Sci. 2017;108:2273‐2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Torres P, Diaz J, Arce M, et al. The salivary peptide histatin‐1 promotes endothelial cell adhesion, migration, and angiogenesis. FASEB J. 2017;31:4946‐4958. [DOI] [PubMed] [Google Scholar]
- 28. Qutub AA, Popel AS. Elongation, proliferation & migration differentiate endothelial cell phenotypes and determine capillary sprouting. BMC Syst Biol. 2009;3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schulz S, Schmitt S, Wimmer R, et al. Progressive stages of mitochondrial destruction caused by cell toxic bile salts. Biochim Biophys Acta. 2013;1828:2121‐2133. [DOI] [PubMed] [Google Scholar]
- 30. Dai T, Hu Y, Zheng H. Hypoxia increases expression of CXC chemokine receptor 4 via activation of PI3K/Akt leading to enhanced migration of endothelial progenitor cells. Eur Rev Med Pharmacol Sci. 2017;21:1820‐1827. [PubMed] [Google Scholar]
- 31. Wenes M, Shang M, Di Matteo M, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. 2016;24:701‐715. [DOI] [PubMed] [Google Scholar]
- 32. Lin Z, Zhang Q, Luo W. Angiogenesis inhibitors as therapeutic agents in cancer: challenges and future directions. Eur J Pharmacol. 2016;793:76‐81. [DOI] [PubMed] [Google Scholar]
- 33. Shih T, Lindley C. Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin Ther. 2006;28:1779‐1802. [DOI] [PubMed] [Google Scholar]
- 34. Land SC, Tee AR. Hypoxia‐inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282:20534‐20543. [DOI] [PubMed] [Google Scholar]
- 35. Zhang W, Xiong Z, Wei T, et al. Nuclear factor 90 promotes angiogenesis by regulating HIF‐1alpha/VEGF‐A expression through the PI3K/Akt signaling pathway in human cervical cancer. Cell Death Dis. 2018;9:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosner M, Pham H, Moriggl R, Hengstschlager M. Human stem cells alter the invasive properties of somatic cells via paracrine activation of mTORC1. Nat Commun. 2017;8:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pang X, Yi Z, Zhang J, et al. Celastrol suppresses angiogenesis‐mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010;70:1951‐1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomas GV, Tran C, Mellinghoff IK, et al. Hypoxia‐inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122‐127. [DOI] [PubMed] [Google Scholar]
- 39. Tirado OM, Mateo‐Lozano S, Notario V. Rapamycin induces apoptosis of JN‐DSRCT‐1 cells by increasing the Bax : Bcl‐xL ratio through concurrent mechanisms dependent and independent of its mTOR inhibitory activity. Oncogene. 2005;24:3348‐3357. [DOI] [PubMed] [Google Scholar]
- 40. Mylonis I, Kourti M, Samiotaki M, Panayotou G, Simos G. Mortalin‐mediated and ERK‐controlled targeting of HIF‐1alpha to mitochondria confers resistance to apoptosis under hypoxia. J Cell Sci. 2017;130:466‐479. [DOI] [PubMed] [Google Scholar]
- 41. Larsen BD, Sorensen CS. The caspase‐activated DNase: apoptosis and beyond. FEBS J. 2017;284:1160‐1170. [DOI] [PubMed] [Google Scholar]
- 42. He K, Chen D, Ruan H, et al. BRAFV600E‐dependent Mcl‐1 stabilization leads to everolimus resistance in colon cancer cells. Oncotarget. 2016;7:47699‐47710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cui J, Gong R, Hu S, Cai L, Chen L. Gambogic acid ameliorates diabetes‐induced proliferative retinopathy through inhibition of the HIF‐1alpha/VEGF expression via targeting PI3K/AKT pathway. Life Sci. 2018;192:293‐303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials