

Abstract
Delta‐like protein 3 (DLL3) is a ligand of Notch signaling, which mediates cell‐fate decisions and is tumor‐suppressive or oncogenic depending on the cellular context. Previous studies show that DLL3 is highly expressed in small cell lung cancer (SCLC) but not in normal lung tissue, suggesting that DLL3 might be associated with neuroendocrine tumorigenesis. However, its role in SCLC remains unclear. To investigate the role of DLL3 in tumorigenesis in SCLC, we performed loss‐of‐function and gain‐of‐function assays using SCLC cell lines. In vitro analysis of cell migration and invasion by transwell assay showed that DLL3 knockdown reduced migration and invasion of SCLC cells, whereas DLL3 overexpression increased these activities. In addition, DLL3 positively regulated SNAI1 expression and knockdown of SNAI1 attenuated the migration and invasion ability of SCLC cells. Moreover, upregulated DLL3 expression induced subcutaneous tumor growth in mouse models. These results indicate that DLL3 promoted tumor growth, migration and invasion in an SCLC model by modulating SNAI1/Snail.
Keywords: delta‐like protein 3, migration, NOTCH, small cell lung cancer, SNAI1
1. INTRODUCTION
Lung cancer is the leading cause of cancer‐related death worldwide, with small cell lung cancer (SCLC) accounting for approximately 15% of lung cancer cases.1, 2 Most SCLC patients initially respond to chemotherapy and radiotherapy but usually relapse and acquire resistant disease. The prognosis of patients with SCLC remains poor, with these patients frequently requiring multiple types of treatment.3
The Notch signaling pathway regulates many fundamental processes essential for normal development, including control of cell differentiation, survival, proliferation and angiogenesis. In mammals, there are 4 Notch receptors (NOTCH1 to NOTCH4) and 2 families of ligands (Jagged [JAG1 and JAG2] and delta‐like ligands [DLL1, DLL3 and DLL4]).4 Notch signaling in tumorigenesis plays an oncogenic or tumor‐suppressive role depending on the cellular context.5, 6 In SCLC, activation of Notch signaling reduces tumor growth, with previous studies identifying inactivating mutations in NOTCH 7, 8, 9, 10 and suggesting their roles as tumor suppressors. However, Lim et al11 reported that Notch‐active SCLC cells were more chemoresistant and promoted neuroendocrine cell growth, indicating that Notch signaling also plays a pro‐tumorigenic role in SCLC.
Delta‐like protein 3 (DLL3) is a ligand that reportedly inhibits Notch signaling.12 A recent report showed that DLL3 is highly expressed in SCLC but not in normal lung tissue.13 Moreover, DLL3 is a downstream target of achaete‐scute homologue 1 (ASCL1),14, 15 which plays a pivotal role in neuroendocrine cell differentiation and SCLC growth.16, 17 These findings suggest a potentially pivotal role for DLL3 in SCLC; however, little is known about DLL3 functions in SCLC. Here, we investigated the effect of DLL3 in SCLC tumorigenesis.
2. MATERIALS AND METHODS
2.1. Cell lines
We used 9 SCLC cell lines (SBC‐3, SBC‐5, MS‐1, RERF‐LC‐MA, H69, H82, H209, H592 and H1688), with SBC‐3, SBC‐5, MS‐1 and RERF‐LC‐MA obtained from the Japanese Collection of Research Bioresources Cell Bank (Osaka. Japan), and H69, H82, H209 and H1688 obtained from the American Type Culture Collection (Manassas, VA, USA). H592 was a gift from Dr Hirotoshi Dosaka‐Akita (Department of Medical Oncology, Faculty of Medicine and Graduate School of Medicine, Hokkaido University, Hokkaido, Japan). SBC‐3, SBC‐5 and RERF‐LC‐MA were maintained in minimum essential medium, and MS‐1, H82, H209, H592 and H1688 were maintained in RPMI 1640 medium supplemented with 10% FBS at 37°C and 5% CO2.
2.2. Antibodies and western blot analysis
We used primary antibodies targeting DLL3 (1:750; ab103102; Abcam, Cambridge, UK), NOTCH1 intracellular domain (NICD1; 1:500; #3608; Cell Signaling Technology, Danvers, MA, USA), NICD2 (1:5000; #5732; Cell Signaling Technology), NICD3 (1:1000; 55114‐1‐AP; Proteintech, Rosemont, IL, USA), NICD4 (1:1500; #2423; Cell Signaling Technology), E‐cadherin (1:200; sc‐8426; Santa Cruz Biotechnology, Dallas, TX, USA), Vimentin (VIM; 1:200; V6630; Sigma‐Aldrich, St. Louis, MO, USA), Snail (1:1000; #3879; Cell Signaling Technology), phospho‐Smad2/Smad3 (1:1000; #8828; Cell Signaling Technology), Smad2/Smad3 (1:1000; #8685; Cell Signaling Technology), Smad4 (1:1000; #38454; Cell Signaling Technology) and ASCL1 (1:250; #556604; BD Pharmingen, Franklin Lakes, NJ, USA). All analyses included staining with Ponceau S, which confirmed equivalent protein loading. Standardization was performed by measuring actin with the anti–actin antibody (1:1500; A2066; Sigma‐Aldrich).
2.3. Quantitative RT‐PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. RNA was reverse‐transcribed into cDNA using TaqMan reverse transcription reagents with random hexamers (Applied Biosystems, Foster City, CA, USA). Expression of DLL3, NOTCH1, NOTCH2, NOTCH3, NOTCH4, CDH1 (ie, E‐cadherin), VIM, SNAI1, ASCL1 and GAPDH mRNA was determined by quantitative RT‐PCR (qRT‐PCR) using the ABI Prism 7900HT system (Applied Biosystems) according to the manufacturer's instructions. TaqMan universal PCR master mix with DLL3, NOTCH1, NOTCH2, NOTCH3, NOTCH4 and GAPDH reagents (Applied Biosystems) or SYBR Green PCR master mix (Applied Biosystems) was used along with the following primers: CDH1 forward, 5′‐CACGGTAACCGATCAGAATG‐3′ and reverse, 5′‐ACCTCCATCACAGAGGTTCC‐3′; VIM forward, 5′‐AATTGCAGGAGGAGATGCTT‐3′ and reverse, 5′‐GAGACGCATTGTCAACATCC‐3′; SNAI1 forward, 5′‐AGGTTGGAGCGGTCAGC‐3′ and reverse, 5′‐CCTTCTCTAGGCCCTGGCT‐3′; ASCL1 forward, 5′‐CAAACGCCGGCTCAACTTC‐3′ and reverse, 5′‐TTGACCAACTTGACGCGGTT‐3′ and GAPDH forward, 5′‐CTGACTTCAACAGCGACACC‐3′ and reverse, 5′‐TGCTGTAGCCAAATTCGTTG‐3′. The mean relative expression of each gene was determined against that of GAPDH. All PCR amplifications were performed using a MicroAmp optical 96‐well reaction plate (Thermo Fisher Scientific, Waltham, MA, USA).
2.4. siRNA transfection
H69, H82, MS‐1 and H592 cells were seeded at a density of 6 × 105/well, 4 × 105/well, 5 × 105/well and 5 × 105/well, respectively, into 6‐well plates the day before transfection. The DLL3‐siRNA, SNAI1‐siRNA and NOTCH1‐siRNA along with ON‐TARGET plus SMART reagents were obtained from Thermo Fisher Scientific. Cells were transfected with 100 pmol siRNA in Opti‐MEM medium (Invitrogen, Carlsbad, CA, USA) using 50 μL Lipofectamine RNAiMAX (Invitrogen). To confirm the efficiency of siRNA transfection, DLL3, SNAI1/Snail and NOTCH1 levels were measured by qRT‐PCR and western blot at 72‐hour post–transfection. Nonspecific siRNA against the target sequence (ON‐TARGET plus non–targeting pool; Thermo Fisher Scientific) were used as controls.
2.5. DLL3 overexpression
The human cDNA‐ORF clone of the DLL3 gene (DLL3‐ORF plasmid), blank‐vector (pCMV6‐entry) and the transfection reagent TurboFectin 8.0 were purchased from OriGene Technologies (Rockville, MD, USA). SBC‐5 cells were divided equally into 2 groups: DLL3‐overexpressing (transfected with the DLL3‐ORF plasmid) and control (transfected with pCMV6‐entry) cells. The day prior to transfection, cells were seeded at a density of 2 × 105/well into 6‐well plates, followed by transfection with 2 μg DLL3‐ORF plasmid or vector in serum‐free Opti‐MEM I (Thermo Fisher Scientific) using 12 μL TurboFectin 8.0. After 24 hours, the transfected cells were diluted at 1:10 into 10‐cm dishes, and the culture medium was replaced with complete medium containing G418 (600 μg/mL). Stable positive clones were obtained after screening with G418 (Thermo Fisher Scientific).
2.6. Cell‐proliferation assays
Anchorage‐dependent and anchorage‐independent cell growth were measured by MTT assay using 96‐well plates with or without poly‐HEMA coating at 72 hours after DLL3‐siRNA transfection or at 72 hours after seeding DLL3‐overexpressing cells. The MTT assay was performed according to the manufacturer's instructions (Thermo Fisher Scientific), and the light absorption was determined at 560 nm using a microplate reader (Varioskan Flash; Thermo Fisher Scientific).
2.7. Migration and invasion assay
Cell migration and invasion assays were performed using 24‐well Transwell plates precoated with or without Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). Cells were then plated in the upper chamber with culture medium containing .1% FBS, with the lower chamber containing culture medium supplemented with 20% FBS. After incubation for 4 hours (MS‐1 cells; migration assay), 6 hours (H69, H82 and SBC‐5 cells; migration assay), 24 hours (H82 cells; invasion assay) or 48 hours (H69 and MS‐1 cells; invasion assay), the membrane was stained with Diff‐Quik (Sysmex, Hyogo, Japan). The number of migrated or invaded cells was counted in 5 random fields of view using a BZ‐9000 microscope (KEYENCE, Osaka, Japan).
2.8. In vivo tumorigenicity
All animal husbandry procedures and experiments were performed under protocols approved by the Institutional Animal Care Committee at Hokkaido University (Approval number 16‐0009). SBC‐5 cells transfected with the scrambled or DLL3‐overexpressing vector (3.0 × 106 cells) were diluted in 200‐μL PBS and injected subcutaneously into the right posterior leg of athymic, 5‐week‐old female nude mice (nu+/nu+; n = 5/group). The tumors were then measured twice weekly using digital calipers, and tumor volume (TV) was determined using the formula TV = (length) × (width) × (height)/2.18 Control or DLL3‐overexpressing cells were injected into 2 other mice, respectively, followed by resection on day 20 after injection for western blot and immunohistochemical analyses.
2.9. Immunohistochemical staining
Dissected xenograft tumors were fixed in 10% formalin for 24 hours at room temperature, placed in 70% ethanol, embedded in paraffin, and then sectioned at a thickness of 5 μm. The sections were deparaffinized using xylene and rehydrated using graded concentrations of ethanol. For antigen retrieval, sections were placed in 10 mmol/L citrate buffer (pH 6.0) and heated in a pressure cooker. The sections were then immersed in methanol containing 3% H2O2 for 10 minutes to block endogenous peroxidase activity followed by incubation with normal goat serum to block nonspecific antibody binding. The sections were reacted consecutively with each primary antibody targeting E‐cadherin (1:500; #3195; Cell Signaling Technology), VIM (1:200; #5741; Cell Signaling Technology) and Snail (1:100; #3879; Cell Signaling Technology) at 4°C overnight. Immunostaining was performed using the biotin‐streptavidin immunoperoxidase method, with 3,3‐diaminobenzidine used as the chromogen. Hematoxylin solution was used for counterstaining.
2.10. Statistical analysis
Significant differences in means between 2 samples were analyzed using Student's t test, with the level of significance set at P < 0.05 were performed. Statistical analyses were performed using JMP software (JMP Pro v11.0.0; SAS Institute, Cary, NC, USA).
3. RESULTS
3.1. DLL3 is expressed in human small cell lung cancer cell lines
We first explored the expression levels of DLL3 and its receptor NOTCH1 in 9 human SCLC cell lines. As shown in Figure 1A, DLL3 mRNA and protein were detected in all SCLC cell lines, whereas NOTCH1 protein was found only in SBC‐3, SBC‐5, MS‐1 and H82 cells (Figure 1B). Because previous studies reported that NOTCH1 negatively regulates ASCL1, and that DLL3 is a target of ASCL1,19, 20 we also determined ASCL1 levels in SCLC cell lines. ASCL1 protein was detected in H1688, H592 and H209 cells, all of which were lacking the NOTCH1 protein (Figure 1C).
Figure 1.
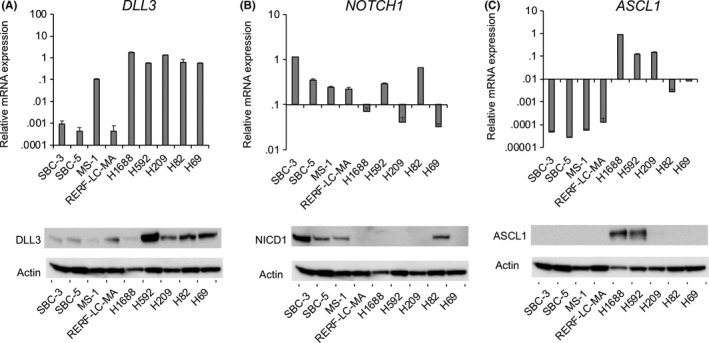
DLL3, NOTCH1 and ASCL1 expression in small cell lung cancer (SCLC) cell lines. The mRNA and protein expression of (A) DLL3, (B) NOTCH1 and (C) ASCL1 were measured by quantitative RT‐PCR and western blot. The mRNA data were normalized to GAPDH expression (n = 3; mean ± SD)
3.2. DLL3 downregulation attenuates the migration and invasion of small cell lung cancer cells
To investigate the functional role of DLL3 in SCLC, H69, H82, MS‐1 and H592 cells that showed DLL3 expression were transfected with DLL3‐siRNA followed by confirmation of downregulation of DLL3 mRNA and protein levels in both cell lines (Figure 2A). Comparison of cell proliferation between control and DLL3‐siRNA transfected cells indicated that DLL3 downregulation slightly suppressed the anchorage‐dependent proliferation of H82 cells, although no differences were observed between H69, MS‐1 or H592 control and DLL3‐siRNA‐transfected cells (Figure 2B). Next, we evaluated the ability of migration and invasion in H69, H82 and MS‐1 when DLL3 was inhibited. DLL3 downregulation significantly reduced the number of migrated cells for H69, H82 and MS‐1 and the number of invaded cells for MS‐1 (Figures 2C,D and S1). Moreover, we observed a lower number of invaded cells following DLL3‐siRNA transfection as compared with control cells for H69 and H82 (Figures 2D and S1).
Figure 2.
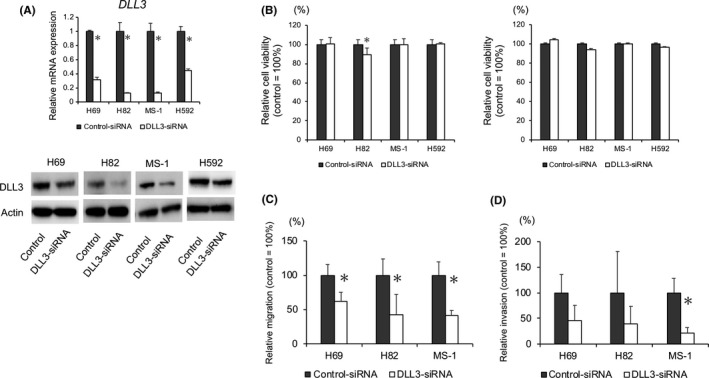
Effect of DLL3 downregulation on small cell lung cancer (SCLC) cell proliferation, migration and invasion. A, DLL3 mRNA and protein levels in H69, H82, MS‐1 and H592 cells transfected with control or DLL3‐siRNA and measured by qRT‐PCR and western blot at 72 h post–transfection. B, Anchorage‐dependent (left) and anchorage‐independent (right) cell growth measured by MTT assays using 96‐well plates with or without poly‐HEMA coating at 72 h after control or DLL3‐siRNA transfection (n = 3; mean ± SD). C, H69, H82 or MS‐1 cells were plated in the upper chamber 48 h after transfection with control or DLL3‐siRNA. After incubation for 4 h in MS‐1 or 6 h in H69 and H82, the number of migrated cells was counted in 5 random fields of view (n = 3; mean ± SD). D, H69, H82 or MS‐1 cells were plated in the upper chamber pre–coated with Matrigel at 48 h after transfection with control or DLL3‐siRNA. After incubation for 24 h in H69 cells or 48 h in H82 and MS‐1 cells, the number of invaded cells was counted in 5 random fields of view (n = 3; mean ± SD). *P < 0.05
3.3. DLL3 downregulation attenuates NOTCH1 expression
We then investigated whether DLL3 downregulation affects Notch signaling by evaluating the expression of Notch receptors in H69, H82, MS‐1 and H592 cells. Suppression of DLL3 levels by siRNA downregulated NOTCH1 mRNA levels in H69, H82 and MS‐1 cells (Figure 3A), with protein levels of NICD1 also reduced by DLL3 downregulation in H82 and MS‐1 cells, although no differences of NICD1 protein levels were observed in H69 and H592 cells (Figure 3B). We then evaluated the expression of the Notch target genes, hairy/enhancer‐of‐split related with YRPW motif protein 1 (HEY1) and Hes family BHLH transcription factor 1 (HES1) in H69, finding that DLL3 downregulation attenuated HES1 mRNA expression and significantly inhibited HEY1 expression in H69 cells (Figure 3C). NOTCH2/NICD2, NOTCH3/NICD3 and NOTCH4/NICD4 expression were unaffected by DLL3 downregulation, except for NOTCH3 mRNA levels in MS‐1 cells or in other cell lines transfected with DLL3‐siRNA (Figure 3A,B). DLL3 downregulation had no impact on ASCL1 levels in H592 cells or in other cell lines transfected with DLL3‐siRNA (Figure 3D).
Figure 3.
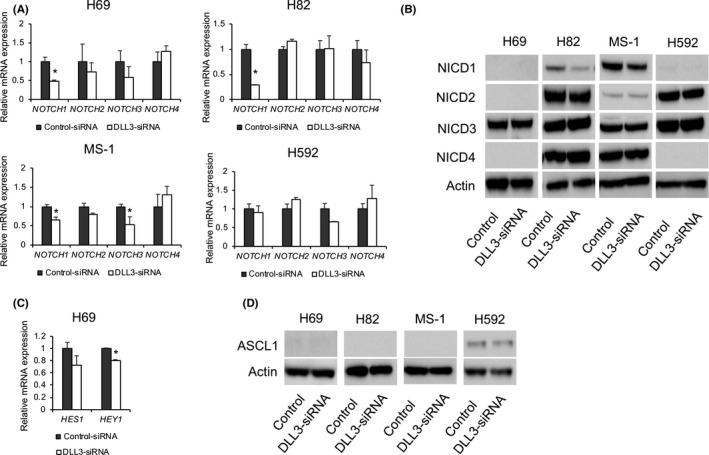
Effect of DLL3 downregulation on Notch signaling in small cell lung cancer (SCLC)‐cell. A, mRNA and B, protein levels of Notch receptors in H69, H82, MS‐1 and H592 cells transfected with control or DLL3‐siRNA and measured by quantitative RT‐PCR and western blot at 72‐h post–transfection (n = 3; mean ± SD). C, mRNA levels of HES1 and HEY1 in cells transfected with control or DLL3‐siRNA and measured by quantitative qRT‐PCR at 72‐h post–transfection (n = 3; mean ± SD). D, Protein levels of ASCL1 in H69, H82, MS‐1 and H592 cells transfected with control or DLL3‐siRNA and measured by western blot at 72‐h post–transfection. *P < 0.05
3.4. Snail plays a key role in DLL3‐mediated small cell lung cancer‐cell migration and invasion
Because DLL3 downregulation reduced the migration and invasion of SCLC cells relative to control cells, we investigated the mechanisms associated with this change in phenotype. Because the Notch signaling pathway reportedly regulates the epithelial‐mesenchymal transition (EMT),21, 22 we evaluated levels of the EMT markers E‐cadherin, VIM and Snail following DLL3 downregulation in SCLC cells. DLL3 downregulation attenuated SNAI1 mRNA expression in H69 cells and significantly inhibited SNAI1 mRNA level in H82 and MS‐1 cells (Figure 4A). Interestingly, Snail protein levels were also attenuated in H82 and MS‐1 cells, but changes in these levels relative to controls were not observed in H69 cells (Figure 4B). In addition, VIM mRNA level was upregulated by DLL3 downregulation in H82 cells, although VIM protein levels exhibited only marginal changes relative to controls (Figure 4A,B). Moreover, we found minimal differences in the mRNA and protein levels of other EMT markers between DLL3‐downregulated cell and controls.
Figure 4.
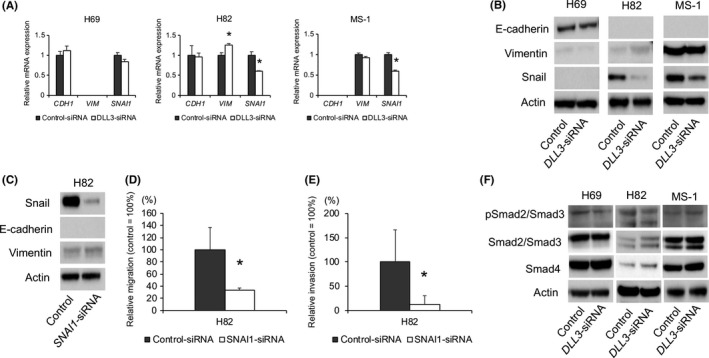
Effect of DLL3 or Snail downregulation on epithelial‐mesenchymal transition (EMT)‐marker levels in small cell lung cancer (SCLC)‐cells. (A) mRNA and (B) protein levels of EMT markers in H69, H82 and MS‐1 cells transfected with control or DLL3‐siRNA and measured by quantitative RT‐PCR and western blot at 72‐h post–transfection (n = 3; mean ± SD). C, Snail, E‐cadherin and VIM protein levels in H82 cells transfected with control or SNAI1‐siRNA and measured by western blot at 72‐h post–transfection. D, Cells were plated in the upper chamber 48 h after transfection with control or SNAI1‐siRNA and after incubation for 6 h, the number of migrated cells was counted in 5 random fields of view (n = 3; mean ± SD). E, Cells were plated in the upper chamber pre–coated with Matrigel at 48 h after transfection with control or SNAI1‐siRNA. After incubation for 48 h in H82 cells, the number of invaded cells was counted in 5 random fields of view (n = 3; mean ± SD). F, Protein levels of phospho‐Smad2/Smad3, Smad2/Smad3 and Smad4 in H69, H82 and MS‐1 cells transfected with control or DLL3‐siRNA and measured by western blot at 72‐h post–transfection. *P < 0.05
To explore whether DLL3 exerts its function through Snail, we transfected H82 cells with SNAI1‐siRNA and verified downregulation of Snail protein levels (Figure 4C). We observed that Snail downregulation markedly inhibited cell migration and invasion (Figures 4D,E and S2). Because NOTCH1 levels were attenuated following DLL3 downregulation, we evaluated H82 cell proliferation, migration and invasion following NOTCH1‐siRNA transfection. We found that NOTCH1 downregulation did not affect the levels of other Notch receptors and had no impact on cell proliferation, migration and invasion (Figure S3A‐D). In addition, NOTCH1 downregulation had no effect on Snail levels (Figure S1E). Because transforming growth factor‐β (TGF‐β)/Smad signaling plays a key role in EMT and is also related to Notch signaling and Snail,21, 23, 24, 25 we examined the expression of phospho‐Smad2, phospho‐Smad3, Smad2, Smad3 and Smad4, finding that DLL3 downregulation had no effect on the protein levels in the 3 cell lines (Figure 4F).
3.5. DLL3 overexpression induces small cell lung cancer‐cell proliferation and migration
To confirm the tumorigenic role of DLL3 in SCLC, SBC‐5 cells exhibiting low expression of DLL3 were transfected with the DLL3‐expression vector, followed by verification of upregulated DLL3 mRNA and protein levels in the transfected cells (Figure 5A). DLL3 overexpression significantly promoted cell growth based on both anchorage‐dependent and anchorage‐independent proliferation observed relative to control SBC‐5 cells (Figure 5B). In addition, cell‐migration assays showed that DLL3 overexpression significantly upregulated SBC‐5‐cell migration (Figure 5C). We could not assess SBC‐5 invasion, because neither the control and the DLL3‐overexpressing cells stably invaded the transwell.
Figure 5.
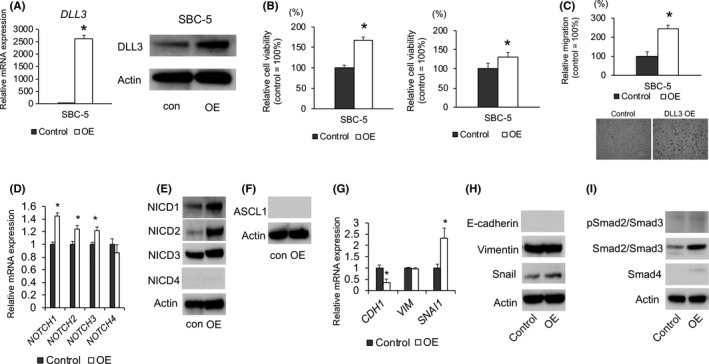
Effect of DLL3 overexpression on the proliferation, migration, NOTCH signaling and epithelial‐mesenchymal transitionmarker levels in SBC‐5 cells. A, quantitative RT‐PCR (left) and western blot (right) confirmation of elevated DLL3 mRNA and protein levels in SBC‐5 cells transfected with a DLL3‐expression vector (n = 3; mean ± SD). B, Anchorage‐dependent (left) and anchorage‐independent (right) cell growth measured by MTT assays using 96‐well plates with or without poly‐HEMA coating at 72 h after seeding of SBC‐5 cells transfected with an empty vector or the DLL3‐expression vector (n = 3; mean ± SD). C, SBC‐5 cells transfected with an empty vector or the DLL3‐expression vector were plated in the upper chamber and after incubation for 6 h, the number of migrated cells was counted in 5 random fields of view (n = 3; mean ± SD). Quantitative RT‐PCR and western blot analyses of (D) mRNA and (E) protein levels of Notch receptors in SBC‐5 cells transfected with an empty vector or DLL3‐ expression vector (n = 3; mean ± SD). F, Protein levels of ASCL1 in SBC‐5 cells transfected with an empty vector or DLL3‐expression vector. (G) mRNA and (H) protein levels of epithelial‐mesenchymal transition markers in SBC‐5 cells transfected with an empty vector or the DLL3‐expression vector (n = 3; mean ± SD). I, Protein levels of phospho‐Smad2/Smad3, Smad2/Smad3 and Smad4 in SBC‐5 cells transfected with a control or DLL3‐expression vector and measured by western blot. *P < 0.05. con, control; OE:DLL3 overexpression
3.6. DLL3 overexpression upregulates Snail expression
We then investigated whether DLL3 overexpression affects Notch signaling and EMT‐marker levels. DLL3 overexpression increased NOTCH1/2/3 mRNA and protein levels and no difference was observed in ASCL1 protein levels (Figure 5D,E,F). DLL3 overexpression increased Snail mRNA and protein levels (Figure 5G,H). In addition, DLL3 overexpression downregulated CDH1 mRNA levels relative to those in control cells, and E‐cadherin protein levels were undetected in SBC‐5 cells (Figure 5G,H). Although the expression of Smad2/Smad3 was elevated in DLL3‐overexpressing SBC‐5 cells as compared with control cells, phospho‐Smad2/Smad3 levels were unaffected by DLL3 overexpression (Figure 5I).
3.7. DLL3 overexpression promotes subcutaneous tumor growth of small cell lung cancer cells in vivo
We then investigated whether DLL3 overexpression promotes SCLC tumor growth in vivo. Tumor volumes in nude mice implanted with DLL3‐ overexpressing SBC‐5 cells were significantly larger than those observed in control mice (Figure 6A,B), and we verified sustained upregulation of DLL3 protein levels in the tumors at 20‐days post–implantation with DLL3‐overexpressing SBC‐5 cells (Figure 6C). NICD3 protein levels were upregulated in the DLL3‐overexpressing group (Figure 6D). Furthermore, we observed upregulated Snail levels in these tumors as compared with levels in control mice (Figure 6E,F). We found no change in morphology following DLL3 overexpression (Figure 6F) and there was no significant difference in VIM and E‐cadherin levels between control cells and DLL3‐overexpressing cells (Figure 6E,F).
Figure 6.
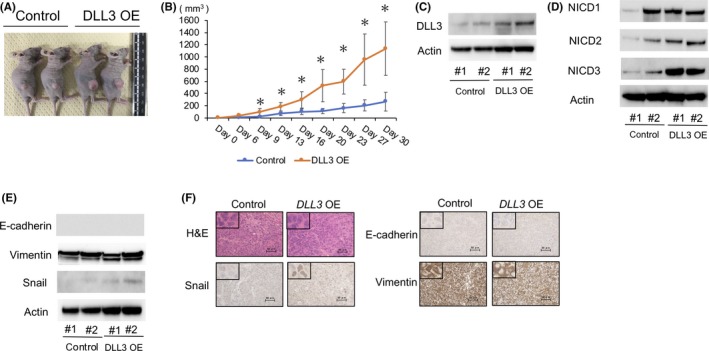
Effect of DLL3 overexpression on SBC‐5 subcutaneous tumor formation in vivo. SBC‐5 cells transfected with an empty vector or the DLL3‐expression vector were implanted into nude mice. A, Photograph of representative tumors on day 20 after implantation. B, Tumor‐growth curves following injection of DLL3 overexpressing or control cells into nude mice. Data indicate the average tumor volume (n = 5/group; mean ± SD). C, Western blot verification of DLL3 levels in DLL3‐overexpressing tumors, relative to those in control tumors on day 20 after implantation. D, Western blot analysis of protein levels of NOTCH1 intracellular domain (NICD1), NICD2 and NICD3 in DLL3‐overexpressing tumors, relative to those in control tumors on day 20 after implantation. E, Western blot analysis of protein levels of epithelial‐mesenchymal transition markers in DLL3‐overexpressing tumors, relative to those in control tumors on day 20 after implantation. F, Representative low‐magnification and high‐magnification images of xenograft tumors subjected to H&E staining and immunohistochemical staining for Snail, E‐cadherin and VIM. Scale bars, 50 μm. *P < 0.05. DLL3 OE,DLL3 overexpression
4. DISCUSSION
In this study, we demonstrated that DLL3 regulates the proliferation, migration and invasion of SCLC cells, suggesting its role as an oncogene in SCLC. Moreover, our findings suggested a potential role for Snail in DLL3‐mediated SCLC‐cell migration and invasion. To the best of our knowledge, this represents the first study reporting an oncogenic function associated with DLL3 in SCLC.
We detected DLL3 mRNA and protein in all 9 SCLC cell lines to varying degrees in the present study. Immunohistochemistry to evaluate DLL3 levels in resected SCLC tissue demonstrated that 83% of SCLC patients were positive for DLL3 protein (unpublished data from our institute), which was similar to the 88% rate determined in a phase I trial of rovalpituzumab tesirine (Rova‐T)26 and 84% in a study from Japan.27 Our results and others indicate that DLL3 is highly expressed in SCLC.
Although DLL3 is reportedly associated with NOTCH and/or ASCL1,12, 14, 15 its role in tumorigenesis remains unknown. In the present study, we demonstrated that DLL3 overexpression promoted the growth of SCLC cells in vitro and in vivo and that DLL3 downregulation suppressed this proliferation phenotype. By contrast, a previous study reported methylation of the DLL3 promoter region in association with upregulated apoptosis in hepatocellular carcinoma cells.23 In addition, Notch proteins reportedly play both tumor‐promoting and tumor‐suppressive roles depending on the tumor type.5, 6 Therefore, it is possible that DLL3 might also exhibit a context‐dependent role according to cancer type.
We observed that DLL3 overexpression increased the migration and invasion of SCLC cells. This agreed with a recent finding of elevated DLL3 levels evaluated immunohistochemically and associated with lymph node metastasis and advanced clinical disease stage according to surgically resected tissue from SCLC patients (unpublished data provided by MF, JSK and HK). In addition, a study reported that Rova‐T, a DLL3‐targeted antibody‐drug conjugate, comprised of a humanized anti–DLL3 monoclonal antibody conjugated to a DNA‐damaging pyrrolobenzodiazepine (PBD) dimer toxin, was developed and showed anti–tumor efficacy in vivo.13 The recent phase I clinical study of Rova‐T found that relapsed SCLC patients achieved clinical outcomes.26 Therefore, our data will aid in the further development of DLL3‐targeted treatment, including Rova‐T, especially in metastatic or recurrent cases.
Unlike other activating DLL ligands, DLL3 does not bind or activate Notch receptors when presented in trans, but instead inhibits Notch signaling in cis.12 Moreover, previous studies reported that DLL3 localized in the Golgi apparatus and in vesicles of late‐endosome and lysosomes promoted the degradation of full‐length NOTCH1 and reduced NOTCH1 expression at the cell surface, thereby preventing its activation by other ligands.28, 29 These findings and those of other studies suggesting tumor suppressive roles associated with Notch signaling in SCLC8, 19, 20 led us to hypothesize that DLL3 promotes SCLC tumorigenesis by inhibiting Notch signaling; however, our results showed that DLL3 positively regulated NOTCH1 expression. In addition, we found no difference in ASCL1 levels between DLL3‐siRNA transfected or overexpressing and control cells. Furthermore, NOTCH1 downregulation in H82 cells did not result in a loss of migration and invasion capabilities, suggesting that the oncogenic function of DLL3 in SCLC is promoted through a Notch‐independent pathway. Consistent with our results, a previous report showed that DLL3 overexpression showed no differences in levels of cleaved NOTCH1 in a human hepatocellular cell line.30 Furthermore, Hes5, a downstream target of Notch signaling, was downregulated in mice harboring a loss‐of‐function mutation in Dll3.31 These findings, as well as our results, suggest that DLL3 is not necessarily an inhibitor of Notch signaling.
In our study, we found that DLL3 promoted SCLC‐cell migration and invasion by modulating Snail expression, despite our observation of no significant alterations in E‐cadherin or VIM levels. The zinc‐finger transcription factor Snail is upregulated in some solid malignancies, such as SCLC, and is associated with risks of metastasis and poor survival.25, 32, 33 Snail is involved in EMT by downregulating the levels of epithelial molecules, such as E‐cadherin, Occludin and Claudins.25 However, Snail1 is a weaker mesenchymal promoter relative to PRRX1 and TWIST, which are transcription factors associated with EMT.34, 35 Moreover, Barrallo‐Gimeno et al36 indicated that Snail acts primarily as an inducer of cell movement rather than an inducer of EMT. A previous study reported that TGF‐β stimulated N‐cadherin expression and the migration of ovarian cancer cells without downregulation of E‐cadherin expression and a complete EMT.37 In breast cancer cells, Snail induction could conversely result in a migratory phenotype despite retention of the VIM and E‐cadherin levels.38 These findings suggest that Snail associated with DLL3 regulation itself might induce migration or invasion independent of EMT induction and without affecting the expression of EMT markers, including E‐cadherin and VIM.
Snail is regulated by various complex signals, such as those associated with TGF‐β signaling.21, 24, 25 Although the expression of Smad2/Smad3 was elevated in DLL3‐overexpressing SBC‐5 cells as compared with control cells, phospho‐Smad2/Smad3 levels were unaffected by DLL3 overexpression. Moreover, DLL3 downregulation had no effect on the Smad protein levels in H69, H82 and MS‐1 cells, indicating that TGF‐β signaling might have little impact on DLL3‐mediated regulation. In addition to TGF‐β signaling, Notch reportedly involved in crosstalk with pathways associated with mitogen‐activated protein kinases (MAPK), phosphatidylinositol 3‐kinase (PI3‐K) and/or NF‐κB pathway, which also regulate Snail.21, 24, 25, 39 Therefore, these other Notch‐related pathways might also affect Snail regulation. Further investigation is necessary to elucidate the precise mechanism associated with DLL3‐Snail signaling in SCLC.
In conclusion, we identified DLL3 as a regulator of SCLC‐cell proliferation, migration and invasion and an oncogene associated with modulating Snail expression. Our findings suggested that DLL3 might represent a therapeutic target for SCLC, especially in metastatic or recurrent cases.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We thank M. Maeda (Department of Respiratory Medicine, Faculty of Medicine and Graduate School of Medicine, Hokkaido University) for providing special support and experimental assistance during this study.
Furuta M, Kikuchi H, Shoji T, et al. DLL3 regulates the migration and invasion of small cell lung cancer by modulating Snail. Cancer Sci. 2019;110:1599–1608. 10.1111/cas.13997
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Kalemkerian GA, Akerley W, Bogner P, et al. Small cell lung cancer. J Natl Compr Canc Netw. 2013;11:78‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Puglisi M, Dolly S, Faria A, Myerson JS, Popat S, O'Brien ME. Treatment options for small cell lung cancer – do we have more choice? Br J Cancer. 2010;102:629‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fiuza UM, Arias AM. Cell and molecular biology of Notch. J Endocrinol. 2007;194:459‐474. [DOI] [PubMed] [Google Scholar]
- 5. Lobry C, Oh P, Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: it's NOTCH what you think. J Exp Med. 2011;208:1931‐1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Previs RA, Coleman RL, Harris AL, Sood AK. Molecular pathways: translational and therapeutic implications of the Notch signaling pathway in cancer. Clin Cancer Res. 2015;21:955‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sriuranpong V, Borges MW, Ravi RK, et al. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001;61:3200‐3205. [PubMed] [Google Scholar]
- 9. Kunnimalaiyaan M, Chen H. Tumor suppressor role of Notch‐1 signaling in neuroendocrine tumors. Oncologist. 2007;12:535‐542. [DOI] [PubMed] [Google Scholar]
- 10. Wael H, Yoshida R, Kudoh S, Hasegawa K, Niimori‐Kita K, Ito T. Notch1 signaling controls cell proliferation, apoptosis and differentiation in lung carcinoma. Lung Cancer. 2014;85:131‐140. [DOI] [PubMed] [Google Scholar]
- 11. Lim JS, Ibaseta A, Fischer MM, et al. Intratumoural heterogeneity generated by Notch signalling promotes small‐cell lung cancer. Nature. 2017;545:360‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ladi E, Nichols JT, Ge W, et al. The divergent DSL ligand Dll3 does not activate Notch signaling but cell autonomously attenuates signaling induced by other DSL ligands. J Cell Biol. 2005;170:983‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saunders LR, Bankovich AJ, Anderson WC, et al. A DLL3‐targeted antibody‐drug conjugate eradicates high‐grade pulmonary neuroendocrine tumor‐initiating cells in vivo. Sci Transl Med 2015;7:302ra136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henke RM, Meredith DM, Borromeo MD, Savage TK, Johnson JE. Ascl1 and Neurog2 form novel complexes and regulate Delta‐like3 (Dll3) expression in the neural tube. Dev Biol. 2009;328:529‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Augustyn A, Borromeo M, Wang T, et al. ASCL1 is a lineage oncogene providing therapeutic targets for high‐grade neuroendocrine lung cancers. Proc Nat Acad Sci USA. 2014;111:14788‐14793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Osada H, Tatematsu Y, Yatabe Y, Horio Y, Takahashi T. ASH1 gene is a specific therapeutic target for lung cancers with neuroendocrine features. Cancer Res. 2005;65:10680‐10685. [DOI] [PubMed] [Google Scholar]
- 17. Jiang T, Collins BJ, Jin N, et al. Achaete‐scute complex homologue 1 regulates tumor‐initiating capacity in human small cell lung cancer. Cancer Res. 2009;69:845‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cao C, Mu Y, Hallahan D, Lu B. XIAP and survivin as therapeutic targets for radiation sensitization in preclinical models of lung cancer. Oncogene. 2004;23:7047‐7052. [DOI] [PubMed] [Google Scholar]
- 19. Sriuranpong V, Borges MW, Strock CL, et al. Notch signaling induces rapid degradation of achaete‐scute homolog 1. Mol Cell Biol. 2002;22:3129‐3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ball DW. Achaete‐scute homolog‐1 and Notch in lung neuroendocrine development and cancer. Cancer Lett. 2004;204:159‐169. [DOI] [PubMed] [Google Scholar]
- 21. Wang Z, Li Y, Kong D, Sarkar FH. The role of Notch signaling pathway in epithelial‐mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets. 2010;11:745‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan X, Wu H, Han N, et al. Notch signaling and EMT in non‐small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol. 2014;7:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu J, Lamouille S, Derynck R. TGF‐beta‐induced epithelial to mesenchymal transition. Cell Res. 2009;19:156‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y, Shi J, Chai K, Ying X, Zhou BP. The role of Snail in EMT and tumorigenesis. Curr Cancer Drug Targets. 2013;13:963‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu YD, Zhou BP. Snail: more than EMT. Cell Adh Migr. 2010;4:199‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rudin CM, Pietanza MC, Bauer TM, et al. Rovalpituzumab tesirine, a DLL3‐targeted antibody‐drug conjugate, in recurrent small‐cell lung cancer: a first‐in‐human, first‐in‐class, open‐label, phase 1 study. Lancet Oncol. 2017;18:42‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanaka K, Isse K, Fujihira T, et al. Prevalence of Delta‐like protein 3 expression in patients with small cell lung cancer. Lung Cancer. 2018;115:116‐120. [DOI] [PubMed] [Google Scholar]
- 28. Geffers I, Serth K, Chapman G, et al. Divergent functions and distinct localization of the Notch ligands DLL1 and DLL3 in vivo. J Cell Biol. 2007;178:465‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chapman G, Sparrow DB, Kremmer E, Dunwoodie SL. Notch inhibition by the ligand Delta‐like 3 defines the mechanism of abnormal vertebral segmentation in spondylocostal dysostosis. Hum Mol Genet. 2011;20:905‐916. [DOI] [PubMed] [Google Scholar]
- 30. Maemura K, Yoshikawa H, Yokoyama K, et al. Delta‐like 3 is silenced by methylation and induces apoptosis in human hepatocellular carcinoma. Int J Oncol. 2013;42:817‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dunwoodie SL, Clements M, Sparrow DB, Sa X, Conlon RA, Beddington RSP. Axial skeletal defects caused by mutation in the spondylocostal dysplasia/pudgy gene Dll3 are associated with disruption of the segmentation clock within the presomitic mesoderm. Development. 2002;129:1795‐1806. [DOI] [PubMed] [Google Scholar]
- 32. Yang MH, Chang SY, Chiou SH, et al. Overexpression of NBS1 induces epithelial‐mesenchymal transition and co‐expression of NBS1 and Snail predicts metastasis of head and neck cancer. Oncogene. 2007;26:1459‐1467. [DOI] [PubMed] [Google Scholar]
- 33. Merikallio H, Turpeenniemi‐Hujanen T, Paakko P, et al. Snail promotes an invasive phenotype in lung carcinoma. Respir Res. 2012;13:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ocana OH, Corcoles R, Fabra A, et al. Metastatic colonization requires the repression of the epithelial‐mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22:709‐724. [DOI] [PubMed] [Google Scholar]
- 35. Nieto MA, Huang RYJ, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21‐45. [DOI] [PubMed] [Google Scholar]
- 36. Barrallo‐Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151‐3161. [DOI] [PubMed] [Google Scholar]
- 37. Gao J, Zhu Y, Nilsson M, Sundfeldt K. TGF‐beta isoforms induce EMT independent migration of ovarian cancer cells. Cancer Cell Int. 2014;14:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lundgren K, Nordenskjold B, Landberg G. Hypoxia, Snail and incomplete epithelial‐mesenchymal transition in breast cancer. Br J Cancer. 2009;101:1769‐1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer Cell. 2014;25:318‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials