

Abstract
Background
Previous research has revealed that Krüppel‐like factor 5 (KLF5) may affect DNA damage repair pathways; however, the associated molecular mechanisms are unclear.
Methods
The expression of KLF5 was studied by immunohistochemical staining in paired tumour and normal tissues from 90 patients with ESCC. We studied the effects of KLF5 knockdown on cell proliferation and apoptosis with or without cisplatin treatment in A549 and H1299 cell lines. Moreover, we examined the effect of KLF5 on the DNA damage response.
Results
KLF5 was significantly overexpressed in non‐small cell lung cancer (NSCLC) tissues, and high KLF5 expression predicted poor prognosis for NSCLC patients. The inhibition of KLF5 markedly augmented cisplatin‐induced cell apoptosis. In addition, we observed that KLF5 knockdown could decrease DNA repair potential by inhibiting H2AX S139 phosphorylation in response to cisplatin. Moreover, silencing of KLF5 in NSCLC cell lines inhibited the phosphorylation of checkpoint kinases Chk1 S345 and Chk2 T68. KLF5 knockdown permits cells with broken or damaged DNA strands to enter mitosis by inhibiting the activation of H2AX, Chk1 and Chk2, resulting in mitotic catastrophe.
Conclusion
KLF5 plays a significant role in the DNA damage response by regulating DNA damage checkpoint proteins. Inhibition of KLF5 may be a potential therapeutic target for NSCLC patients with cisplatin resistance.
Keywords: DNA damage response, DNA repair, Krüppel‐like factor 5, NSCLC
Introduction
Non‐small cell lung cancer (NSCLC) is one of the leading causes of cancer‐related death worldwide.1 Despite recent therapeutic advances, platinum‐based chemotherapy is still a widely used adjuvant therapeutic strategy for patients with advanced NSCLC.2 Cisplatin is one of the most commonly used chemotherapy drugs, but its use is limited because of acquired drug resistance. Multiple cellular self‐defense adaptations, such as reduced uptake and increased drug efflux, protein inactivation, and increased damage repair, are responsible for cisplatin resistance.3, 4 However, how to overcome cisplatin resistance remains a conundrum.
DNA is considered a major target for cisplatin. Cisplatin interacts with DNA mainly in the form of Pt‐d(GpG) di‐adducts, which inhibit cell proliferation and activate the DNA damage response (DDR).5, 6 The DNA damage checkpoint is a complex signal transduction pathway that includes ATM, ATR, Chk1, Chk2, the MRN complex, and other checkpoint proteins.7, 8 Once the DDR is activated, cell cycle arrest is initiated to repair DNA or induce apoptosis. Disruption of DDR has been identified in various cancers, and strategies targeting DDR have been exploited for the treatment of cancer with radiotherapies or chemotherapies.9
KLF5 is a critical member of the KLF family, which contains a triple zinc finger DNA‐binding domain. KLF5 regulates numerous key cellular processes, including proliferation, angiogenesis, pluripotency, inflammation, and migration.10 In cancer, KLF5 is typically pro‐proliferative by regulating cell cycle progression.11, 12 Recently, studies have demonstrated that knockdown of KLF5 suppresses hypoxia‐induced cisplatin resistance in NSCLC cells.13 In the intestine, KLF5 may affect DNA damage repair pathways.14 Given this background, we hypothesized that KLF5 may regulate cisplatin resistance through the DDR in NSCLC.
In this study, we compared KLF5 expression in NSCLC and adjacent normal lung tissues. KLF5 was knocked down to assess its influence on cell growth with or without cisplatin treatment. Furthermore, we sought to investigate the potential role of KLF5 in DDR. To our knowledge, this is the first report to confirm that KLF5 exerts a regulatory effect on key enzymes in the DDR.
Methods
Cell lines and cell culture
NSCLC cell lines H1299 and A549 were cultivated in RPMI 1640 (Corning, Logan, UT, USA) supplemented with 10% fetal bovine serum (Corning, Mediatech Inc., Manassas, VA, USA). The cell lines were cultured at 37°C with 5% CO2. All of the cell lines used in the study were authenticated by short tandem repeat detection and tested for the presence of mycoplasma.
Patient samples
The follow‐up data of 90 patients diagnosed with lung adenocarcinoma between January 2010 and December 2016 were collected from the same hospital. Information including age, gender, and tumor node metastasis (TNM) stage was collected from medical records. Informed consent was obtained from all patients, and the Research Ethics Committee of the Cancer Hospital, Chinese Academy of Medical Sciences approved the research application.
Antibodies and materials
Antibodies (poly [ADP‐ribose] polymerase [PARP], cleaved PARP, cleaved caspase 3, pH2AX, Chk1, p‐Chk1, Chk2, p‐Chk2, ATM, p‐ATM, ATR, p‐ATR, MRE11, NBS1, RAD50, and glyceraldehyde 3‐phosphate dehydrogenase [GAPDH]) used for Western blot analysis were purchased from Cell Signaling Technology (Danvers, MA, USA). KLF5 antibody was purchased from Abcam (ab137676; Cambridge, MA, USA). Cisplatin was purchased from Sigma‐Aldrich (St. Louis, MO, USA) and dissolved in 0.9% NaCl solution.
Immunohistochemical staining
NSCLC and adjacent normal lung tissues were surgically resected and collected from the Department of Thoracic Surgery (Cancer Hospital, Chinese Academy of Medical Sciences). Briefly, formalin‐fixed and paraffin‐embedded tissue sections were incubated with an antibody against KLF5 overnight at 4°C and then incubated with an appropriate secondary antibody. We quantitatively scored the tissues according to the percentage of positive staining cells and the staining intensity.15 A pathologist and two experienced researchers independently scored the slides.
Immunofluorescence
Cells (1 × 105) were seeded in chamber slides and cultured overnight. After cisplatin or vehicle treatment, cells were fixed and stained with phosphorylated histone 2AX (pH2AX) antibody overnight at 4°C. The next day, the cells were washed with phosphate buffered saline and then incubated with an appropriate secondary antibody. DAPI (4′,6‐diamidino‐2‐phenylindole, Sigma) was used to counterstain the nuclei. The images were captured via immunofluorescence microscopy using the same exposure time.
Lentivirus vector and reagents
Two short‐hairpin RNAs (shRNAs), 5′‐cctataattccagagcataaa‐3′ and 5′‐ccctgagttcaccagtatatt‐3′, were synthesized for KLF5 suppression. They were respectively inserted into the pLKO.1‐puro lentiviral shRNA vector (Generay Biotech Co., Shanghai, China) to produce lentivirus in 293T cells. A549 and H1299 cells were infected with concentrated virus and cultured with complete culture media after 24 hours, followed by selection with puromycin for seven days.
Western blot analysis
A549 and H1299 cells were collected following cisplatin or vehicle treatment. Cells lysates were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and electrotransferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The membranes were incubated with primary antibodies overnight at 4°C and were then incubated with appropriate secondary antibodies.
Cell viability assay
Cell viability was assessed by cell counting kit‐8 (CCK‐8) assay (Dojindo, Kumamoto, Japan) according to the manufacturer's instructions. Briefly, A549 or H1299 cells were seeded in 96‐well plates and treated with different doses of cisplatin or the vehicle. Cells were allowed to grow for two days. The optical density was measured at 450 nm.
For colony formation assays, cells were seeded in six‐well plates (600 cells/well) and were allowed to grow for seven days. Cells were then treated with cisplatin (20μM) or the vehicle for 36 hours. Thereafter, colonies were fixed and stained for visualization and counting.
Cell apoptosis analysis
Cell apoptosis was assessed using an Annexin V‐FITC Apoptosis Detection Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. Briefly, A539 and H1299 cells were treated with or without cisplatin (20μM) for 48 hours. Cells were then collected, washed with binding buffer, and stained with Annexin V‐FITC and propidium iodide, followed by quantification via flow cytometry.
Statistical analysis
All data were analyzed using GraphPad Prism 6.0 software. Data from specific experiments were compared by Student's t or X 2 tests, or Cox regression analysis. Survival curves were plotted using the Kaplan–Meier method and log‐rank test. Data were presented as mean ± standard deviation and *P < 0.05 was considered statistically significant.
Results
KLF5 is overexpressed in non‐small cell lung cancer (NSCLC) and is correlated with poor prognosis
We used immunohistochemical (IHC) staining to detect KLF5 expression in 90 pairs of lung cancer and adjacent normal tissues. KLF5 expression was significantly higher in cancer tissues than in adjacent normal tissues (Fig 1a, Fig S1). IHC staining also suggested that KLF5 was mainly localized in the nucleus in NSCLC tissues (Fig 1b). We then conducted Western blot analysis of five pairs of randomly selected samples to detect KLF5 protein expression, and the results revealed that most tumor tissues showed higher KLF5 expression than paired normal tissues (Fig 1c).
Figure 1.

KLF5 is overexpressed in non‐small cell lung cancer (NSCLC) and is correlated with poor prognosis. (a) Relative expression levels of KLF5 in NSCLC and adjacent normal tissues (n = 90) were analyzed via immunohistochemical staining (***P < 0.001). (b) Representative images of immunohistological staining of KLF5 in NSCLC tissues. Scale bar = 20 μm. (c) Western blot analysis of KLF5 expression in NSCLC and adjacent normal tissues. (d) Kaplan–Meier analysis suggested that high KLF5 levels predict a poor overall survival rate (Log‐rank, *P = 0.019).  Low KLF5 (n = 55),
Low KLF5 (n = 55),  high KLF5 (n = 35). GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
high KLF5 (n = 35). GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
Furthermore, we examined the relationship between KLF5 expression levels and patient survival. KLF5 expression levels were significantly correlated with patient survival (Fig 1d), and high KLF5 expression levels showed excess hazard ratios for death compared to low expression levels (Table 1). A multivariate Cox regression model revealed a statistically significant reduction in survival with increased KLF5 expression levels, independent of other important variables in NSCLC (Table 1).
Table 1.
Univariate and multivariate analysis of KLF5 expression and other important variables in NSCLC
| n | Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|---|
| KLF5 | |||||
| Characteristics | HR (95% CI) | P | HR (95% CI) | P | |
| Age (years) | |||||
| < 60 | 45 | (Referent) | (Referent) | (Referent) | (Referent) |
| ≥ 60 | 45 | 1.787 (1.066 to 2.999) | 0.026 | 1.556 (0.921 to 2.626) | 0.098 |
| Gender | |||||
| Male | 58 | (Referent) | (Referent) | (Referent) | (Referent) |
| Female | 32 | 0.829 (0.483 to 1.421) | 0.495 | 0.700 (0.401 to 1.220) | 0.208 |
| TNM stage | |||||
| I and II | 47 | (Referent) | (Referent) | (Referent) | (Referent) |
| III and IV | 43 | 1.866 (1.112 to 3.132) | 0.018 | 1.841 (1.096 to 3.091) | 0.021 |
| KLF5 | |||||
| Low | 55 | (Referent) | (Referent) | (Referent) | (Referent) |
| High | 35 | 1.837 (1.097 to 3.078) | 0.019 | 1.810 (1.080 to 3.034) | 0.024 |
P values were determined from the Cox proportional hazards model using two‐sided Wald tests. Multivariable model: age, gender, tumor node metastasis (TNM) stage, and KLF5 expression level. CI, confidence interval; HR, hazard ratio; NSCLC, non‐small cell lung cancer.
KLF5 knockdown increases cisplatin‐induced cell apoptosis in NSCLC cell lines
To determine the effect of KLF5 on the proliferation of NSCLC cells, we knocked down KLF5 in A549 and H1299 cell lines via lentiviral infection (Fig 2a). Cell viability was evaluated by CCK‐8 assay. Our results showed that the proliferation rate of cells subjected to KLF5 knockdown was not significantly different from the control under vehicle treatment (Fig 2b). However, under different doses of cisplatin treatment, KLF5 knockdown significantly suppressed the proliferative ability of A549 and H1299 cells compared to the control (Fig 2c). We then reevaluated cell proliferation using colony formation assay and found that the number and size of colonies in the KLF5‐knockdown groups were markedly lower and smaller than in the control group under cisplatin treatment, respectively (Fig 2d). These results show that KLF5 knockdown led to reduced cell proliferative ability in response to cisplatin.
Figure 2.

KLF5 knockdown reduces cell survival rates in response to cisplatin in non‐small cell lung cancer (NSCLC) cell lines. (a) Knockdown of KLF5 with two short‐hairpin RNAs in A549 and H1299 cell lines. (b) Relative viability of cells subjected to KLF5 knockdown, measured via cell counting kit‐8 (CCK‐8) assay. (c) Relative viability of cells treated with different doses of cisplatin for 48 hours, measured via the CCK‐8 assay in indicated cell lines. (d) The effect of KLF5 on proliferation under cisplatin or vehicle treatment was measured by colony formation assay.  shvec,
shvec,  shKLF5#1,
shKLF5#1,  shKLF5#2. Data are shown as mean ± standard deviation. P values were determined by two‐tailed Student's t test (ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001). GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
shKLF5#2. Data are shown as mean ± standard deviation. P values were determined by two‐tailed Student's t test (ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001). GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
The effect of KLF5 on NSCLC cell apoptosis was evaluated by flow cytometry analysis. KLF5 knockdown had no significant effect on the cell apoptotic rate (Fig 3a) except after 48 hours of treatment with cisplatin, at which time the rate significantly increased (Fig 3b). To further investigate the effect of KLF5 knockdown on cell apoptosis, apoptosis‐related proteins were detected by Western blot analysis. We found that KLF5 knockdown significantly increased the levels of the intrinsic apoptosis‐inducing proteins, cleaved PARP and cleaved caspase 3, after treatment with cisplatin (Fig 3c). These results demonstrate that KLF5 knockdown increases cell apoptosis induced by cisplatin.
Figure 3.

KLF5 knockdown promotes cisplatin‐induced cell apoptosis in non‐small cell lung cancer (NSCLC) cell lines. (a,b) Cells subjected to KLF5 knockdown were treated with cisplatin (20μM) or the vehicle for 48 hours and analyzed via flow cytometry. Apoptosis was measured with FACS‐based annexin‐V/propidium iodide (PI) double staining. The proportion of apoptotic cells within the total number of cells was used to evaluate cell apoptosis. (c) The expression of apoptosis‐inducing proteins was analyzed by Western blotting following cisplatin (20μM) treatment. Data are shown as mean ± SD. P values were determined by two‐tailed Student's t test (ns, not significant; ***P < 0.001). GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; PARP, poly (ADP‐ribose) polymerase; PI, propidium iodide.
KLF5 knockdown decreases DNA repair potential in NSCLC cell lines
The activation of checkpoint pathways in response to DNA damage leads to cell cycle arrest to repair damaged DNA. pH2AX, a widely used indicator of the presence of damaged DNA, is required for checkpoint‐mediated cell cycle arrest and DNA repair following double‐stranded DNA breaks.16 We assessed the expression of nuclear pH2AX (S139) through immunofluorescence analysis. Under cisplatin treatment, the pH2AX staining intensity in A549 and H1299 cells increased significantly compared to the control (Fig 4a). However, when cells were targeted with KLF5 shRNA, the pH2AX nuclear expression induced by cisplatin was attenuated (Fig 4a,b). We confirmed this result through Western blot analysis (Fig 5a). These results reveal that KLF5 knockdown decreases DNA repair potential by downregulating pH2AX S139 phosphorylation levels.
Figure 4.

KLF5 knockdown decreases the DNA repair potential by regulating H2AX S139 phosphorylation. (a) Immunofluorescence analysis showed increased phosphorylation of H2AX after cisplatin (10μM) treatment for 24 hours. As shown in the figure, KLF5 knockdown abrogates cisplatin‐induced H2AX S139 phosphorylation. All images were captured using the same exposure time. Scale bar = 20μm. (b) Cells with pH2AX nuclear foci staining were quantified and statistically analyzed. Data are shown as mean ± standard deviation. P values were determined by two‐tailed Student's t test (ns, not significant; *P < 0.05; **P < 0.01). DAPI, 4′,6‐diamidino‐2‐phenylindole.
Figure 5.
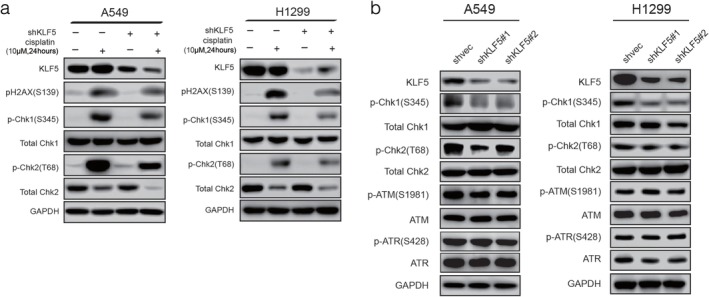
KLF5 knockdown regulates checkpoint proteins in the DNA damage response (DDR). (a) Western blotting showed that KLF5 knockdown attenuated the activating phosphorylation of H2AX (S139), Chk1 (S345), and Chk2 (T68) after cisplatin (10 μM) treatment for 24 hours. (b) Knockdown of KLF5 affects phosphorylation of Chk1 and Chk2, but not ATM or ATR. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
KLF5 knockdown regulates DNA damage checkpoint kinases in NSCLC cell lines
To investigate the role of KLF5 in checkpoint activation, we examined the effect of KLF5 knockdown on the activating phosphorylation of several key checkpoint proteins, including ATM, ATR, and downstream checkpoint proteins (Chk1, Chk2, and MRN complex) in A549 and H1299 cells. KLF5 silencing attenuated the activating phosphorylation of Chk1 (S345) and Chk2 (T68) but not ATM (S1981) in response to DNA damage induced by cisplatin (Fig 5a). KLF5 knockdown did not alter the total protein levels of these checkpoint proteins in A549 and H1299 cell lines (Fig 5b,Fig S2); however, the total Chk2 protein levels significantly decreased under cisplatin treatment (Fig 5a). Consistent with the results of a previous study, DNA damage‐induced autophosphorylation on S379 led to the ubiquitination of Chk2 by a Cullin1‐containing E3 ligase, which could cause degradation of Chk2.17 These results demonstrate that KLF5 knockdown indeed attenuates DNA damage checkpoint activation in NSCLC cells in response to cisplatin.
Discussion
Lung cancer development and progression are multistep processes characterized by aberrant genetic changes and protein expression. Recent studies have revealed that KLF5 is one of a group of new significantly mutated genes in NSCLC.18 Cancer type‐specific hotspot mutations within a zinc‐finger DNA binding domain of KLF5 change its DNA binding specificity and affect cellular transcription.19 Multiple studies have shown that KLF5 plays a role in cisplatin resistance in various tumors. In colorectal cancer, KLF5 is predictive for cell line and patient response to platinum agents.20 In ovarian cancer cell lines, silencing KLF5 by small interfering RNA sensitizes cells to apoptosis induced by cisplatin.21 A previous study reported that KLF5 knockdown could suppress hypoxia‐induced cisplatin resistance in NSCLC, probably via inactivation of the PI3K/Akt/mTOR pathway;13 however, the study failed to mention the prognostic value of KLF5 and its underlying roles in the DDR in NSCLC.
In this study, we showed that KLF5 is upregulated in NSCLC via IHC staining in 90 pairs of NSCLC and adjacent normal tissues. KLF5 expression levels were significantly correlated with survival duration. These results indicate that KLF5 overexpression is a common feature in NSCLC and may serve as a valuable prognostic biomarker. To further investigate the function of KLF5, we stably knocked down KLF5 in NSCLC cell lines. Our results showed that KLF5 knockdown significantly reduced the survival rate in response to cisplatin by upregulating apoptosis‐inducing proteins, cleaved PARP and cleaved caspase 3, in A549 and H1299 cell lines. Thus, KLF5 may serve as a biomarker to predict cisplatin treatment response.
Many anticancer agents cause DNA damage, resulting in the activation of DNA damage checkpoints and proliferation arrest. As a member of the histone H2A family, H2AX is required for DNA damage signal amplification and the subsequent recruitment of repair factors to nuclear foci after DNA damage.22 The suppression of pH2AX decreases DNA repair potential. In our study, KLF5 knockdown attenuated cisplatin‐induced H2AX S139 phosphorylation in NSCLC cells, suggesting that KLF5 knockdown can decrease the DNA repair potential in NSCLC cells.
In response to DNA injury, critical kinases Chk1 and Chk2 were activated. ATR phosphorylates Chk1 at S317 and S345, resulting in a marked increase in Chk1 activity.23 ATM phosphorylates Chk2 at T68, which is thought to activate Chk2.24 Both Chk1 and Chk2 activation converge to inactivate members of the Cdc25 family and then arrest cells in the G2/M phase.25 In this study, phosphorylation of Chk1 (S345) and Chk2 (T68) was significantly increased under cisplatin treatment. KLF5 knockdown attenuated the phosphorylation of Chk1 and Chk2 induced by cisplatin and reversed their inhibitory effect on the cell cycle. In combination with pH2AX (S139) suppression, KLF5 knockdown permits cells with broken or damaged DNA strands to enter mitosis, resulting in mitotic catastrophe, leading to cell death.26 We investigated two major kinases, ATM and ATR, which phosphorylate Chk2 and Chk1, and found that the phosphorylation level of ATM (S1891) or ATR (S428) was not affected by KLF5 suppression. We propose that other regulatory mechanisms might be involved. Manila et al. demonstrated that IGF‐1 decreases radiation‐induced phosphorylation of Chk1 at S345 or S296 via the PI3K/AKT pathway.27 Notably, various studies have confirmed that KLF5 plays critical roles in the PI3K/Akt/mTOR pathway.11, 13 Moreover, phosphorylation of Chk2 at T68 in response to DNA damage occurs in ATM‐deficient cells, suggesting that kinases other than ATM may be involved.28 Thus, this study provides proof for further investigation of the role of KLF5 in regulating the phosphorylation of Chk1 and Chk2.
In conclusion, our results show that KLF5 knockdown can inhibit DNA repair in NSCLC cell lines by suppressing phosphorylation of H2AX, Chk1, and Chk2. Moreover, KLF5 is commonly overexpressed in NSCLC tissues, and high KLF5 predicts poor overall survival. Our study indicated that KLF5 may be a potential therapeutic target in patients with cisplatin resistance.
Disclosure
No authors report any conflict of interest.
Supporting information
Supplementary Fig. 1 Hematoxylin and eosin (H&E) and immunohistological staining of KLF5 in non‐small cell lung cancer (NSCLC) and adjacent normal tissues from four different cases. Scale bar = 20μm.
Supplementary Fig. 2 Western blot analysis of the MRN complex in A549 and H1299 cell lines subjected to KLF5 knockdown.
Acknowledgments
This study was supported by the National Key R&D Programme of China (2017YFC1308700, 2017YFC1311000, and 2018YFC1312100).
We thank Wei Wang and Xueying Yang for their technical support.
Contributor Information
Yibo Gao, Email: gaoyibo@cicams.ac.cn.
Jie He, Email: prof.jiehe@gmail.com.
References
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87–108. [DOI] [PubMed] [Google Scholar]
- 2. Wu Z, Fournel L, Stadler N et al Modulation of lung cancer cell plasticity and heterogeneity with the restoration of cisplatin sensitivity by neurotensin antibody. Cancer Lett 2018; 444: 147–61. [DOI] [PubMed] [Google Scholar]
- 3. Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 2005; 4: 307–20. [DOI] [PubMed] [Google Scholar]
- 4. Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res 2016; 106: 27–36. [DOI] [PubMed] [Google Scholar]
- 5. Yang Y, Adebali O, Wu G et al Cisplatin‐DNA adduct repair of transcribed genes is controlled by two circadian programs in mouse tissues. Proc Natl Acad Sci U S A 2018; 115: E4777–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yimit A, Adebali O, Sancar A, Jiang Y. Differential damage and repair of DNA‐adducts induced by anti‐cancer drug cisplatin across mouse organs. Nat Commun 2019; 10: 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bartek J, Lukas J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr Opin Cell Biol 2007; 19: 238–45. [DOI] [PubMed] [Google Scholar]
- 8. Williams RS, Williams JS, Tainer JA. Mre11‐Rad50‐Nbs1 is a keystone complex connecting DNA repair machinery, double‐strand break signaling, and the chromatin template. Biochem Cell Biol 2007; 85: 509–20. [DOI] [PubMed] [Google Scholar]
- 9. Pearl LH, Schierz AC, Ward SE, al‐Lazikani B, Pearl FMG. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 2015; 15: 166–80. [DOI] [PubMed] [Google Scholar]
- 10. Tetreault MP, Yang Y, Katz JP. Kruppel‐like factors in cancer. Nat Rev Cancer 2013; 13: 701–13. [DOI] [PubMed] [Google Scholar]
- 11. An T, Dong T, Zhou H et al The transcription factor Kruppel‐like factor 5 promotes cell growth and metastasis via activating PI3K/AKT/snail signaling in hepatocellular carcinoma. Biochem Biophys Res Commun 2019; 508: 159–68. [DOI] [PubMed] [Google Scholar]
- 12. He P, Yang JW, Yang VW, Bialkowska AB. Kruppel‐like factor 5, increased in pancreatic ductal adenocarcinoma, promotes proliferation, acinar‐to‐ductal metaplasia, pancreatic intraepithelial neoplasia, and tumor growth in mice. Gastroenterology 2018; 154: 1494–1508 e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gong T, Cui L, Wang H, Wang H, Han N. Knockdown of KLF5 suppresses hypoxia‐induced resistance to cisplatin in NSCLC cells by regulating HIF‐1alpha‐dependent glycolysis through inactivation of the PI3K/Akt/mTOR pathway. J Transl Med 2018; 16: 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li M, Gu Y, Ma YC et al Kruppel‐like factor 5 promotes epithelial proliferation and DNA damage repair in the intestine of irradiated mice. Int J Biol Sci 2015; 11: 1458–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang W, Zheng Y, Xia Y et al ERK1/2‐dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol 2012; 14: 1295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan J, Adamski R, Chen J. Focus on histone variant H2AX: To be or not to be. FEBS Lett 2010; 584: 3717–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lovly CM, Yan L, Ryan CE, Takada S, Piwnica‐Worms H. Regulation of Chk2 ubiquitination and signaling through autophosphorylation of serine 379. Mol Cell Biol 2008; 28: 5874–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Campbell JD, Alexandrov A, Kim J et al Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 2016; 48: 607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang X, Choi PS, Francis JM et al Somatic superenhancer duplications and hotspot mutations lead to oncogenic activation of the KLF5 transcription factor. Cancer Discov 2018; 8: 108–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Atkinson C.J., Kawamata F., Liu C., et al. EGFR and prion protein promote signaling via FOXO3a‐KLF5 resulting in clinical resistance to platinum agents in colorectal cancer, Mol Oncol 2018; doi: 10.1002/1878‐0261.12411 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dong Z, Yang L, Lai D. KLF5 strengthens drug resistance of ovarian cancer stem‐like cells by regulating survivin expression. Cell Prolif 2013; 46: 425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu C, Zhu F, Cho YY et al Cell apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase‐3. Mol Cell 2006; 23: 121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao H, Piwnica‐Worms H. ATR‐mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21: 4129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ahn JY, Schwarz JK, Piwnica‐Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res 2000; 60: 5934–6. [PubMed] [Google Scholar]
- 25. Wang JN, Che Y, Yuan ZY et al Acetyl‐macrocalin B suppresses tumor growth in esophageal squamous cell carcinoma and exhibits synergistic anti‐cancer effects with the Chk1/2 inhibitor AZD7762. Toxicol Appl Pharmacol 2019; 365: 71–83. [DOI] [PubMed] [Google Scholar]
- 26. Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53‐deficient cells rely on ATM‐ and ATR‐mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007; 11: 175–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Manila NG, Kaida A, Nakahama KI, Miura M. Insulin‐like growth factor I receptor regulates the radiation‐induced G2/M checkpoint in HeLa cells. Biochem Biophys Res Commun 2018; 503: 2977–83. [DOI] [PubMed] [Google Scholar]
- 28. Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 2009; 21: 245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 Hematoxylin and eosin (H&E) and immunohistological staining of KLF5 in non‐small cell lung cancer (NSCLC) and adjacent normal tissues from four different cases. Scale bar = 20μm.
Supplementary Fig. 2 Western blot analysis of the MRN complex in A549 and H1299 cell lines subjected to KLF5 knockdown.