

Abstract
DNA double‐strand breaks are a threat to genome integrity and cell viability. The nucleolytic processing of broken DNA ends plays a central role in dictating the repair processes that will mend these lesions. Usually, DNA end resection promotes repair by homologous recombination, whereas minimally processed ends are repaired by non‐homologous end joining. Important in this process is the chromatin‐binding protein 53BP1, which inhibits DNA end resection. How 53BP1 shields DNA ends from nucleases has been an enduring mystery. The recent discovery of shieldin, a four‐subunit protein complex with single‐stranded DNA‐binding activity, illuminated a strong candidate for the ultimate effector of 53BP1‐dependent end protection. Shieldin consists of REV7, a known 53BP1‐pathway component, and three hitherto uncharacterized proteins: C20orf196 (SHLD1), FAM35A (SHLD2), and CTC‐534A2.2 (SHLD3). Shieldin promotes many 53BP1‐associated activities, such as the protection of DNA ends, non‐homologous end joining, and immunoglobulin class switching. This review summarizes the identification of shieldin and the various models of shieldin action and highlights some outstanding questions requiring answers to gain a full molecular understanding of shieldin function.
Keywords: DNA repair, end resection, genome stability, homologous recombination, non‐homologous end joining
Subject Categories: DNA Replication, Repair & Recombination
Glossary
- 53BP1
p53‐binding protein 1
- AP
affinity purification
- APEX2
ascorbate peroxidase 2
- ATM
ataxia telangiectasia mutated
- ATMIN
ataxia telangiectasia‐mutated interactor
- BirA
bifunctional ligase/repressor A
- BLM
Bloom syndrome protein
- BRCA1
breast cancer type 1 susceptibility protein
- BrdU
bromodeoxyuridine
- C20orf196
chromosome 20 open reading frame 196
- CDC20
cell division cycle 20
- CRISPR/Cas9
clustered regularly interspaced short palindromic repeats/CRISPR‐associated 9
- CSR
class switch recombination
- CST
CTC1‐STN1‐TEN1
- CTC1
conserved telomere maintenance component 1
- CtIP
CtBP‐interacting protein
- DNA2
DNA replication helicase/nuclease 2
- DNA
deoxyribonucleic acid
- DSB
double‐strand break
- DT40
avian leukosis virus‐induced bursal lymphoma cell line derived from a Hyline SC chicken
- DYNLL1
dynein light chain LC8‐type 1
- eIF4E
eukaryotic translation initiation factor 4E
- EXO1
exonuclease 1
- FAM35A
family with sequence similarity 35, member A
- FHA
forkhead‐associated
- GFP
green fluorescent protein
- H2AK15
histone H2A lysine 15
- H4K20
histone H4 lysine 20
- HEK293T
human embryonic kidney 293 cells containing the SV40 T‐antigen.
- HORMA
HOP1, REV7, MAD2
- HR
homologous recombination
- ICL
interstrand crosslinking
- IR
ionizing radiation
- Ku70
Ku autoantigen, 70 kDa
- MAD2
mitotic arrest deficient 2
- MDC1
mediator of DNA damage checkpoint 1
- MMC
mitomycin C
- MRE11
meiotic recombination 11
- mRNA
messenger RNA
- MRN
MRE11‐RAD50‐NBS1
- MS
mass spectrometry
- NHEJ
non‐homologous end joining
- nM
nanomolar
- nt
nucleotides
- OB‐fold
oligosaccharide/oligonucleotide binding fold
- PARPi
Poly(ADP‐ribose) polymerase inhibitor
- Pol α‐primase
DNA polymerase alpha‐primase
- PTIP
PAX‐interacting protein 1
- RAD51
radiation sensitive 51
- RBM
REV7 binding motif
- REV7/MAD2L2
revertibility protein 7/mitotic arrest deficient 2‐like protein 2
- RIF1
RAP1‐interacting factor 1
- RINN
REV7‐interacting novel NHEJ regulator
- RNF168
RING finger protein 168
- RNF8
RING finger protein 8
- RPA1
replication protein A 70 kDa DNA‐binding subunit
- RPA
replication protein A
- SHLD
shieldin
- ssDNA
single‐stranded DNA
- TIRR
Tudor‐interacting repair regulator protein
- TLS
translesion DNA synthesis
- TPP1
TINT1/PTOP/PIP1
- TRAPPC13
trafficking protein particle complex subunit 13
- TRF2
telomeric repeat‐binding factor 2
- TRIP13
thyroid hormone receptor interactor 13
- V(D)J
variable, diversity, and joining gene segment
Introduction
53BP1 is a chromatin‐binding protein 1 that regulates DNA repair primarily by limiting long‐range 5′–3′ nucleolytic digestion of DNA ends, a process known as DNA end resection 2. The protection of DNA ends by the 53BP1‐dependent pathway promotes physiological or pathological DNA double‐strand break (DSB) repair by non‐homologous end joining (NHEJ) despite the fact that 53BP1 is not a core component of the NHEJ machinery 3. Indeed, 53BP1 is crucial for NHEJ‐driven biological processes such as immunoglobulin class switching 4, 5, the fusion of dysfunctional telomeres 6, and the chromosome aberrations caused by the exposure of BRCA1‐deficient cells to poly(ADP‐ribose) polymerase inhibitors (PARPi) 2 (Fig 1).
Figure 1. 53BP1 and shieldin act in various physiological contexts.
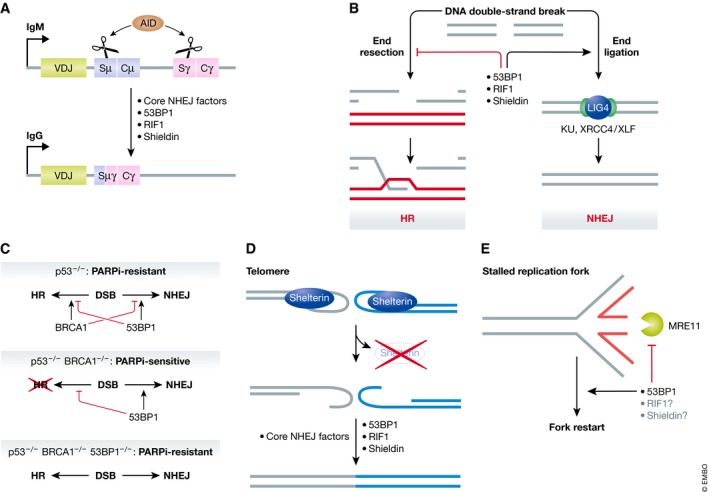
(A) 53BP1, RIF1, and shieldin mediate immunoglobulin class switch recombination. During B‐cell stimulation, the activation‐induced cytidine deaminase (AID) enzyme causes single‐stranded breaks at two switch regions within the immunoglobulin heavy chain locus. 53BP1, RIF1, and shieldin are essential for non‐homologous end joining (NHEJ)‐mediated fusion of the two distant switch regions, altering the antibody subtype expressed from the locus. (B) Genomic DNA double‐strand breaks can be repaired through two competing pathways: the resection‐dependent homologous recombination or resection‐independent direct ligation through NHEJ. The components of the 53BP1 pathway inhibit end resection and facilitate repair via NHEJ. (C) 53BP1 and BRCA1 antagonize each other. In a Δp53 background, BRCA1 promotes HR and inhibits NHEJ while 53BP1 promotes NHEJ and inhibits HR, resulting in both DSB repair pathways being active. HR‐proficient cells are resistant to PARP inhibition (PARPi). In the absence of BRCA1, 53BP1 inhibits HR, resulting in HR deficiency and PARPi sensitivity. Concurrent depletion of BRCA1 and 53BP1 results in the de‐repression of HR, resulting in PARPi resistance. BRCA1 depletion is lethal in p53‐proficient cells unless accompanied by a depletion of 53BP1. (D) Telomere dysfunction due to shelterin subunit depletion results in aberrant DNA end processing. TRF2 depletion results in 53BP1‐dependent fusion of telomeres. (E) 53BP1 prevents MRE11‐mediated degradation of stalled replication forks. During DNA replication, stalled replication forks can reverse into the “chicken‐foot” configuration depicted. The nascent DNA portion (red) is a substrate for MRE11‐mediated exonucleolytic degradation. 53BP1 prevents this degradation and promotes fork restart, while the involvement of RIF1 and shieldin in this context has not been characterized.
Intriguingly, 53BP1 is not necessary for all NHEJ‐dependent repair reactions. Indeed, 53BP1 is involved in only a subset of V(D)J recombination events 4, 5, 7 and analysis of isogenic DT40 cell knockouts indicated that 53BP1 loss causes milder radiosensitization than mutations in the core NHEJ factor Ku70 3. Conversely, the ability of 53BP1 to limit the formation of single‐stranded (ss) DNA at broken ends is not solely involved in regulating NHEJ. As an example, during the phases of the cell cycle where homologous recombination (HR) is active 8, 53BP1 influences the type of HR pathway used by modulating end resection 9. Recent work indicates that 53BP1 also shields nascent DNA from degradation at stalled replication forks (Fig 1) 10, 11. 53BP1 is also active at dysfunctional telomeres that have been depleted of shelterin complex subunits 6, 12. TRF2‐depleted telomeres undergo 53BP1‐ and NHEJ‐dependent fusion, accentuating the role of 53BP1 as an NHEJ factor 6. In contrast, 53BP1 prevents resection at TPP1‐depleted telomeres without promoting NHEJ‐driven fusion, suggesting a DNA end protection role independent of NHEJ 12. Therefore, an emerging view of 53BP1 points to a role as a resection antagonist rather than a dedicated NHEJ factor.
Remarkably, the loss of 53BP1 reverses the cell and organismal lethality associated with mutations in BRCA1 2, 13, 14, and loss‐of‐function mutations in 53BP1 lead to PARPi resistance in both cell and pre‐clinical mouse tumor models of BRCA1 deficiency 15, 16. Loss of 53BP1 in BRCA1‐deficient cells restores, to some degree, homologous recombination in a manner that depends on the activation of end resection 2. This extraordinary genetic interaction points to a unique antagonism between BRCA1 and 53BP1, a conclusion supported by cell biological studies where BRCA1 and 53BP1 appear to compete for accumulation at DNA damage sites 17, 18, 19, 20. These findings suggest that initiating end resection is a key decision point in DSB repair pathway choice, with a direct impact on the therapeutic efficacy of PARP inhibitors.
How 53BP1 impacts DNA repair has long been enigmatic, but it is certain that its action requires its recruitment to DSB sites 21, 22, 23. 53BP1 accumulates on the chromatin surrounding DSB sites by recognizing dually modified nucleosomes containing histone H4 methylated on its Lys20 residue and histone H2A ubiquitylated on Lys15 21, 24. Since H4K20 methylation is nearly ubiquitous, H2AK15 ubiquitylation by RNF168 provides the first DNA damage‐dependent signal leading to 53BP1 recruitment. 53BP1 must be at minimum a dimer to accrue on the chromatin flanking DSBs 21, 25, leading to a model where the 53BP1–nucleosome interaction enhances the ability of chromatin to inhibit DNA end resection 26.
However, the function of 53BP1 in DNA repair also requires interacting partners, indicating that its interaction with nucleosomes alone is not sufficient to block DNA end processing. 53BP1 is phosphorylated by ATM on over 25 sites concentrated in the N‐terminal half of the protein 27, 28. 53BP1 phosphorylation provides a second DNA damage‐induced signal leading to the activation of the DNA repair function of 53BP1 22, 23 and promotes its interaction with two proteins, PTIP 29 and RIF1 18, 19, 20, 30, 31. These two proteins are involved in limiting end resection at DSBs independently of each other 32. How RIF1 and PTIP collaborate to mediate 53BP1‐dependent DNA repair is not understood and is likely complex 33, but genetic studies suggest that it is RIF1, not PTIP, that promotes the function of 53BP1 in many NHEJ‐driven processes such as immunoglobulin class switching 32, 33.
In 2015, two reports identified the small HORMA domain‐containing protein REV7/MAD2L2 as a factor acting downstream of 53BP1 and RIF1 34, 35. Indeed, REV7 is critical for mediating the cytotoxic effects of PARPi in BRCA1‐deficient cells, immunoglobulin class switching, and fusion of dysfunctional telomeres 34, 35. The robust genetic data linking REV7 to 53BP1‐dependent DNA repair was as convincing as it was confusing: How can this small protein, better known for its function in translesion DNA synthesis (TLS) as part of DNA polymerase ζ 36, antagonize DNA end resection? These findings raised the distinct possibility that additional 53BP1 effectors remained to be identified. In retrospect, this possibility clearly resonated with many in the field since (unbeknownst to most) a race for the identification for new factors involved in 53BP1‐dependent DNA repair had just begun.
The hunt for the missing effectors of 53BP1
The search for the elusive 53BP1 effectors utilized various strategies that remarkably all converged on the same protein complex. One strategy involved CRISPR/Cas9‐based pooled genetic screens to identify factors whose mutation conferred resistance to PARPi in BRCA1‐mutated cells. These screens took advantage of the ability of 53BP1 mutations to suppress the sensitivity of BRCA1‐mutated cells to PARPi, reasoning that mutation of 53BP1 effectors should do the same 37, 38. Noordermeer et al additionally mined a screen aimed at finding genes that promote resistance to ionizing radiation (IR), which is mediated in large part by NHEJ‐dependent DNA repair 37. These screens identified the previously uncharacterized proteins C20orf196 and FAM35A as promoters of NHEJ and suppressors of HR. Additionally, Noordermeer et al also identified CTC‐534A2.2 as a factor acting alongside C20orf196 and FAM35A. CTC‐534A2.2 is a protein encoded by an alternative transcript emanating from the TRAPPC13 locus and was not annotated in many databases, which explains why it was identified only by a subset of the groups who were searching for 53BP1 effectors. These three hitherto uncharacterized proteins arose in vertebrates, with the complete set co‐occurring in species that perform immunoglobulin class switching 39.
As alternative approaches, all groups either based their searches on proximity labeling or affinity purification (AP) mass spectrometry (MS), or complemented their genetic screens with MS‐based approaches. In particular, fusions of 53BP1 or REV7 with either the APEX2 peroxidase 39 or a promiscuous form of the BirA biotin ligase 40, respectively, allowed for selective biotinylation of proteins in close proximity to these baits in cells. AP‐MS was also used to identify proteins interacting with REV7 37, 38, 40, 41, 42, or, after their initial identification, partners of FAM35A, C20orf196, and CTC‐534A2.2 37, 39, 42. In a more targeted approach, interaction partners of REV7 mutants specifically defective in CSR were also identified by AP‐MS 43.
C20orf196, FAM35A, CTC‐534A2.2, and REV7 form a stable complex even in the absence of exogenous DNA damage 37, 38, 39, 40, 43. This protein complex was named “shieldin”, a term originally coined a few years ago by Jiri Lukas to describe the idea that 53BP1 protects DNA ends in a manner analogous to the telomere end‐protecting complex shelterin 44. The previously uncharacterized components of shieldin were also given new names, using either the SHLD (shieldin) or the alternate RINN (REV7‐interacting novel NHEJ regulator) nomenclature: C20orf196 was renamed SHLD1/RINN3; FAM35A, SHLD2/RINN2; and CTC‐534A2.2, SHLD3/RINN1. For the sake of clarity, we will employ the SHLD1/2/3 nomenclature for the remainder of this review.
Shieldin promotes PARP inhibitor cytotoxicity in BRCA1‐mutated cells
Studies assessing depletion of the newly identified shieldin subunits revealed pronounced and highly consistent phenotypes. In particular, its role in antagonizing homologous recombination was validated exhaustively. Depleting any single subunit in various BRCA1‐deficient cell lines suppressed their sensitivity to PARPi to a degree comparable to that of 53BP1 or REV7 depletion 37, 38, 39, 43, 45. The potency of this effect was illustrated in vivo in mouse allograft experiments where Brca1‐null mammary tumor cells edited to mutate SHLD1 or SHLD2 prior to allografting were resistant to PARPi treatment 37. In addition, expression levels of SHLD1 and SHLD2 correlated with PARPi sensitivity in patient‐derived xenografts of BRCA1‐null tumors 38. These findings suggest that shieldin mutations modify PARPi responses in BRCA1‐mutated tumors.
CRISPR‐mediated knockout of Shld1 or Shld2 enabled p53‐proficient mouse embryonic stem cells to survive Brca1 loss 37, recapitulating the profound genetic interaction observed between 53BP1 and BRCA1 2, 13, 14. Whether or not shieldin gene mutations will suppress embryonic lethality caused by BRCA1 loss remains to be determined. Nevertheless, the concomitant loss of BRCA1 with any of the newly identified shieldin subunits restores HR as observed by gene conversion assays and RAD51 ionizing radiation‐induced focus formation 37, 38, 39, 40, 42. These phenotypes closely mirror those of 53BP1 or REV7 loss in the context of HR suppression 2, 34, further suggesting that shieldin acts in the same pathway as 53BP1.
Shieldin promotes 53BP1‐dependent NHEJ
The role of 53BP1 in promoting NHEJ in a variety of physiological and pathological contexts is also shared by shieldin. Shieldin loss confers sensitivity to IR, the DNA topoisomerase II inhibitor etoposide, and the radiomimetic drug bleomycin 37, 38, 39, 40, 42, as was seen previously with the loss of REV7 35. More direct NHEJ measurements via random plasmid integration and the EJ5‐GFP reporter assay revealed that every shieldin subunit, including REV7, contributes to NHEJ 35, 37, 38, 39, 40, 41, 42. Although 53BP1 is not a core NHEJ component, it is essential for the long‐range fusion of deprotected telomeres in cells deficient for the shelterin complex subunit TRF2 6. This feature is shared with shieldin, where loss of any subunit reduces the fusion of deprotected telomeres 35, 38, 39.
Shieldin also participates in 53BP1‐dependent immunoglobulin class switching (also known as class switch recombination or CSR). CSR involves the long‐range end joining of two DSBs within the immunoglobulin heavy chain‐coding gene, which generates a large deletion that alters the antibody subtype (Fig 1A) 46. 53BP1 is essential for CSR 4, 5 and loss of any shieldin component, including REV7, similarly impairs this process 34, 35, 37, 38, 39, 40, 43. Of further importance to immune system development, 53BP1‐deficient mice also have decreased numbers of B lymphocytes due to partially defective V(D)J recombination 7. However, in a departure from perfectly phenocopying of 53BP1 loss, genetic ablation of REV7 did not affect B‐cell numbers, suggesting that shieldin does not participate in this pathway 43.
Epistasis between the 53BP1‐RIF1 axis and shieldin
The genetic evidence presented in these multiple contemporaneous studies paints a compelling picture of shieldin sharing the same functions as 53BP1, and multiple lines of evidence indicate that shieldin acts genetically as part of a 53BP1‐RIF1‐shieldin pathway. Shieldin genes are epistatic with 53BP1 or RIF1 with respect to CSR 37, sensitivity to DSB‐inducing drugs 42, and rescue of HR in BRCA1‐mutated cells 37, 38. Shieldin components are also epistatic to each other, consistent with them being subunits of the same complex 37, 38, 42, 43.
Several intriguing results deviate from perfect epistasis within this pathway. In one study, BRCA1/SHLD1 and BRCA1/SHLD2 double‐knockout cells were observed to be much more sensitive to ionizing radiation than BRCA1/53BP1 knockout cells 38. Different subunits of shieldin may also have slightly different roles. Knocking out SHLD1 in DT40 cells has a modest but reproducible sensitivity to the topoisomerase I poison camptothecin, while a SHLD2 knockout has no effect 42. Additionally, one study found that SHLD1 and SHLD2 knockout cells are sensitive to the DNA interstrand crosslinking (ICL) agent cisplatin, a phenotype often observed in cells with defects in the TLS, Fanconi anemia, or HR pathways 38, 47. However, other reports show that SHLD3 knockout does not affect response to ICL agents 43 and that REV7 involvement in DSB repair pathway choice is distinct from its role in TLS 34, 35, 43. Whether these observations represent true mechanistic differences or clonal/experimental variation remains to be determined, but they raise the possibility that some of the newly characterized shieldin components might have functions outside the 53BP1‐RIF1‐shieldin pathway, just as REV7 does.
REV7 in the context of shieldin
Aside from its role as a subunit of shieldin, REV7 is best known as an integral member of the TLS polymerase Pol ζ complex, where it binds to REV1 and REV3L 48. This complex responds to DNA damage caused by a variety of lesions but is particularly important for the tolerance of DNA interstrand crosslinks such as those caused by the drugs cisplatin or mitomycin C (MMC) 49, 50. Indeed, biallelic mutations in REV7 were found in a patient displaying a Fanconi anemia‐like syndrome, a disease characterized by interstrand crosslink sensitivity, highlighting the importance of Pol ζ for the tolerance of such lesions 51.
REV7 is a 211‐amino acid residue protein consisting entirely of a HORMA domain (Fig 2A). Within Pol ζ, REV3L has two conserved REV7‐binding motifs (RBMs) defined by a P‐x‐x‐x‐p‐P motif (x represents any amino acid, uppercase P represents a proline residue essential for the interaction, while lowercase p represents a less important proline residue) 52, 53, 54. Analysis of REV7 mutants by Ghezraoui et al revealed that the Y63A and W171A mutants are unable to bind REV3L and are inactive in TLS, as shown by their inability to rescue the sensitivity of REV7‐null cells to MMC 43. However, REV7‐W171A, but not the Y63A variant, was able to rescue the CSR defect of a REV7 deletion, consistent with the finding that REV7 has distinct contributions to TLS and DSB repair 34, 35, 43. Importantly, the W171A mutant, but not the Y63A mutant, can bind SHLD3, providing a compelling rationale for the observed separation of function 43. Two RBMs are found in the SHLD3 N‐terminus; REV7 interacts with a fragment of SHLD3 (amino acids 28–83) that harbors an RBM necessary for REV7 binding 39, 43. SHLD3 interacts with RIF1 in co‐immunoprecipitation experiments, while REV7 bridges SHLD3 to the rest of the complex by directly binding SHLD2 37, 39, 43. These findings suggest that SHLD3 and REV7 are the most 53BP1‐RIF1‐proximal elements within shieldin and form a localization module within the complex.
Figure 2. Schematic of shieldin subunits and the architecture of the complex.
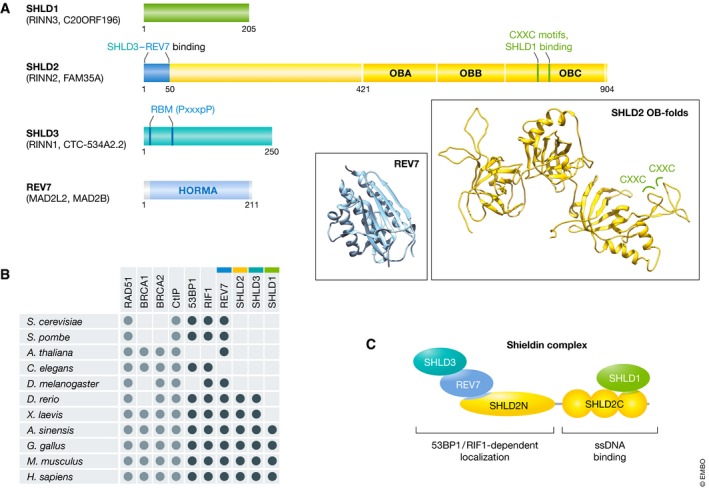
(A) Amino acid residues of interest are numbered over each shieldin component. The three predicted tandem oligonucleotide/oligosaccharide‐binding (OB) folds in SHLD2 and the HORMA domain of REV7 are depicted. SHLD2 contains a CXXC zinc‐finger motif while SHLD3 has a REV7‐binding motif (RBM). Within the RBM (PxxxpP), P is an essential proline, p denotes an optional proline, and x represents any amino acid. Ribbon structures: The predicted structure of the SHLD2 OB‐folds (generated by homology modeling using the RPA1 structure; PDB:4GNX) with two CXXC zinc‐finger motifs highlighted is shown in yellow. The structure of REV7 (PDB:3ABE) is shown in cyan. (B) Evolutionary conservation of shieldin and other proteins involved in DNA double‐strand break repair pathway choice, based on known orthologues and BLAST homology search. (C) Functional architecture of the shieldin complex. SHLD3 and REV7 associate with the SHLD2 N‐terminus, forming the 53BP1‐ and RIF1‐dependent localization module. Meanwhile, SHLD1 associates with the SHLD2 C‐terminus, forming the ssDNA‐binding module.
Like other HORMA‐domain proteins, REV7 contains a stereotypical “safety belt” that encircles the domain's binding partners in its C‐terminus 36. This safety belt region of REV7 facilitates its interaction with SHLD3 43. In the case of the REV7 paralog MAD2, the safety belt is remodeled from a closed to an open conformation by the AAA+ ATPase TRIP13 55, 56. This remodeling into the open conformation ablates binding of MAD2 to its binding partner CDC20, modulating its function through conformational modification 55, 56, 57. The idea that the REV7 safety belt may also be remodeled to modulate the 53BP1‐RIF1‐shieldin pathway is a tantalizing possibility that should be thoroughly investigated, especially since TRIP13 can be co‐immunoprecipitated with REV7 37.
Shieldin is a triple OB‐fold‐containing complex
Aside from REV7, none of the other shieldin components had been previously characterized. SHLD1 and SHLD3 are, like REV7, relatively small proteins of 205‐ and 250‐amino acid residues in size, respectively, whereas SHLD2 is the largest subunit at 904 residues (Fig 2A). Shieldin homologues arose relatively late in evolution compared to other DSB repair pathway choice proteins, with conserved sequences found primarily in vertebrates (Fig 2B). Structure‐based homology searches and predictions suggest that the C‐terminal half of SHLD2 forms three tandem oligosaccharide/oligonucleotide binding (OB) folds 37, 38, 40, 42, while a region of SHLD3 has limited homology to the mRNA cap‐binding domain of the translation elongation initiation factor eIF4E 43.
The N‐terminal half of SHLD2 is predicted to be disordered, with a conserved region in the first 100 residues. In particular, the first 50 residues of SHLD2 are sufficient and necessary for interaction with SHLD3 and REV7 37, 39. Within this region of SHLD2, two conserved prolines (P14 and P17) are essential for REV7 interaction 42. The C‐terminal half of SHLD2 is predicted at high confidence to form three tandem OB‐folds similar to those found in RPA1 and CTC1 37, 38, 40, 42, the two largest subunits of the RPA (replication protein A) and CST (CTC1‐STN1‐TEN1) ssDNA‐binding complexes, respectively. OB‐folds are ssDNA‐binding domains that are found in multiple proteins involved in genome stability 58. One of the loops in the third OB‐fold domain is predicted to form two CXXC‐type zinc‐finger motifs 38. This putative zinc‐finger‐containing region is important for the association of SHLD2 with SHLD1 38. One report predicted that SHLD1 contains a winged helix domain in its C‐terminus that is similar to the one present in the STN1 subunit of the CST complex 38. The relatively large size of SHLD2 compared to that of the other shieldin subunits suggests that it serves as the core scaffold of the complex, and the presence of tandem OB‐folds in its C‐terminus may point toward a direct mechanism of action for shieldin through ssDNA binding.
Shieldin is recruited to DSB sites and represses resection
As expected of an effector of 53BP1, each subunit of shieldin, including REV7, accumulates at DSB sites 37, 38, 39, 40, 42 downstream of 53BP1 and RIF1 37, 38. In agreement with the protein–protein interaction studies, assays of recruitment to DNA damage sites showed that SHLD3 is the subunit most proximal to RIF1, followed by REV7, SHLD2, and SHLD1 37, 38. In fact, shieldin can be roughly divided into two modules: one module composed of SHLD3‐REV7 and the N‐terminal 50 residues of SHLD2 forming a “localization module”, whereas the complex formed by SHLD1 and C‐terminal OB‐folds of SHLD2 form a ssDNA‐binding module, as will be discussed in detail below (Fig 2B).
The decision point of DSB pathway choice revolves around end resection 8, 59, 60. HR requires extensive degradation of the 5′ strand relative to the DSB, generating long tracts of ssDNA used for RAD51‐mediated homology searching 61. Initiation of resection in mammals occurs in a two‐step process 62, 63: First, the MRE11‐RAD50‐NBS1 (MRN) resection complex induces endonuclease‐generated nicks on the 5′‐terminated strands on either side of the break with the aid of CtIP 64, 65, 66. The resulting nick is then expanded through the 3′–5′ exonuclease activity of MRN and the 5′–3′ exonuclease activity of EXO1 or DNA2‐BLM 67, 68. The resulting large tracts of ssDNA are bound by RPA, which is then replaced by RAD51 to initiate homology searching, strand invasion, and copying of homologous sequences.
Multiple lines of evidence indicate that shieldin antagonizes DNA end resection. First, depletion of shieldin subunits increases the levels of phosphorylated RPA after induction of DSBs 35, 37, 38, 40, which is a surrogate readout for ssDNA formation 69. Similarly, induction of DSBs in shieldin‐depleted cells results in increased numbers of RPA foci measured by immunofluorescence 34, 38, 39. Secondly, the levels of RPA bound at the immunoglobulin gene switch regions are increased following induction of class switching as determined by chromatin immunoprecipitation 34, 43. Thirdly, cells with shieldin gene knockouts display an increased amount of ssDNA after camptothecin treatment as measured by native BrdU labeling, also reflective of extensive DNA end resection 38, 39. Finally, measurement of resection by native Southern blotting at deprotected telomeres provides a direct assessment of end resection, and analysis of shieldin depletion using the Cre‐mediated removal of Tpp1 or Trf2 showed that loss of shieldin increases the formation of ssDNA at deprotected telomeres 45. Therefore, like 53BP1, shieldin opposes resection, but the key question remains whether it does so directly through an inherent activity of the complex or indirectly through the recruitment of other factors.
SHLD2 ssDNA‐binding activity is important for shieldin function
The predicted presence of the OB‐fold domains in the SHLD2 C‐terminus provided the first clue into the biochemical activity of shieldin. Expression of a SHLD2 variant lacking its OB‐folds fails to complement the IR sensitivity of SHLD2‐knockout cells or restore PARPi sensitivity in the SHLD2/BRCA1 double‐knockout cells 38. These findings suggest that the SHLD2 OB‐folds are critical for the function of the complex.
If the OB‐folds of SHLD2 are critical effectors of the 53BP1‐RIF1 pathway, it then follows that a major role for 53BP1 in suppressing HR is the recruitment of SHLD2 to sites of DNA damage. To test this possibility, Noordermeer et al 37 artificially recruited SHLD2 to DSB sites via its fusion to the FHA domain of RNF8. The resulting FHA‐SHLD2 fusion impaired RAD51 IR‐induced focus formation in BRCA1/53BP1‐double‐knockout cells, consistent with an inhibition of HR 37. Importantly, mutations designed to remove key ssDNA‐interacting aromatic residues in the SHLD2 OB‐folds resulted in a fusion protein that was unable to suppress HR. Furthermore, fusion of a truncated variant of SHLD2, consisting solely of the OB‐folds, to the RNF8 FHA domain, was sufficient to fully recapitulate the inhibition of HR in BRCA1/53BP1‐double‐knockout cells 37. A similar set of mutants assessed by Dev et al 38 were unable to complement the IR sensitivity of SHLD2 knockout cells. Collectively, these results identify the predicted OB‐fold domains of SHLD2 as critical elements of the 53BP1‐RIF1‐shieldin pathway.
Although structural information on the SHLD2 OB‐folds is currently unavailable, the purified SHLD2 C‐terminus binds to DNA, with a strong preference for ssDNA that is consistent with the binding properties of other OB‐fold proteins 37, 38, 40, 42. ssDNA binding in vitro is abolished by the same aromatic residue mutations that disable the ability of SHLD2 to suppress HR, underlining ssDNA binding as a key function of SHLD2 37, 38. In all cases, ssDNA binding was determined using ssDNA templates > 50 nt 37, 38, 40, 42 and one report observed that the SHLD2 C‐terminus cannot bind 30‐nt substrates 42. The requirement for long ssDNA substrates is surprising, as tandem OB‐fold‐containing proteins and complexes often reach their peak binding affinity at a substrate length of 35 nt or less 70, 71, 72.
Intriguingly, the affinity of SHLD2 for ssDNA appears highly variable dependent on context. The SHLD2 C‐terminus purified in complex with SHLD1 from HEK293T cells binds ssDNA with a dissociation constant of approximately 10 nM 37, an intermediate affinity between RPA (< 1 nM) and RAD51 (> 100 nM) 71, 73. However, the SHLD2 C‐terminus expressed in Escherichia coli has 1–2 orders of magnitude lower affinity for ssDNA than the protein complex purified from human cells 38, 42. Co‐expression with SHLD1 increases the stability of the SHLD2 C‐terminus in mammalian cells 37, but whether SHLD1 stimulates the affinity of SHLD2 for ssDNA remains an open question. Alternatively, there may either be mammalian‐specific post‐translational modifications that increase the affinity of SHLD2 for ssDNA or additional components of shieldin that remain unidentified.
The shieldin paradox
The extensive genetic and biochemical characterization of shieldin presented above converges into one central paradox: Why would a complex that prevents end‐resection function by binding to ssDNA? NHEJ has mechanisms for processing short overhangs 74, but ssDNA longer than 20–30 nt is characteristic of resection. If shieldin binds to ssDNA after the initiation of resection, how would the complex interrupt a processive nuclease acting upon its substrate? Resolving this paradox is key to understanding shieldin function and may reveal a key step in DSB repair pathway choice.
One possibility to solve this paradox is that shieldin promotes fill‐in synthesis at resected ends rather than blocking end‐resection nucleases per se. Indeed, shieldin interacts with the CST complex, which also antagonizes end resection 45. During telomere replication, CST interacts with the polymerase alpha‐primase complex (Pol α‐primase) to synthesize DNA, filling in the excessively long overhangs of nascent telomeres 75. CST‐Polα is recruited to telomeres through the OB‐fold‐containing shelterin complex 75. Analogously, Mirman et al found that CST is recruited to DSBs in a 53BP1‐ and shieldin‐dependent manner, with knockdown of CST components promoting resection and suppressing the sensitivity of BRCA1‐null cells to PARPi 45, 76. Furthermore, the suppression of PARPi sensitivity through CST knockdown is epistatic to either 53BP1 or REV7 knockout 45. Radial chromosome formation in PARPi‐treated BRCA1‐null cells, a characteristic sign of PARPi toxicity, is suppressed by inhibition of Polα 45. These findings provide a strong argument toward the involvement of CST‐Polα in HR suppression through the 53BP1‐RIF1‐shieldin pathway.
Despite being a compelling mechanism of action to antagonize resection, the role of fill‐in synthesis by CST‐Polα also raises multiple questions with respect to the 53BP1‐shieldin pathway. First, it is unclear how CST‐Polα‐mediated DNA synthesis and SHLD2‐mediated ssDNA binding are integrated to modulate end resection. Second, as EXO1 and DNA2‐BLM can generate long ssDNA tracts, the poor processivity of Pol α‐primase 77 and its lack of proofreading activity 78 seems problematic unless more processive polymerases take over after synthesis is initiated. Regardless of these possibilities, we view the identification of shieldin mutants that are unable to interact with CST as an important first step in helping untangle the respective contributions of CST‐Polα and SHLD2 ssDNA binding in this process.
Another means by which shieldin may antagonize resection is by inhibiting the nucleases involved in long‐range resection. Resection at blocked DNA ends is initiated by the combined action of MRN and CtIP, which generates an endonuclease cleavage between 20 and 40 nucleotides away from the blockage, which can include nucleosomes or the DNA end‐binding factor Ku (Fig 3A) 66, 79. This endonuclease cut is then extended toward the break using by the 3′–5′ exonuclease activity of MRE11 followed by the long‐range 5′–3′ resection away from the break by EXO1 and DNA2/BLM (Fig 3A). The initial MRE11 processing generates ssDNA of similar length to the preferred shieldin substrate, and may provide an opportunity to interrupt the switch between short‐ to long‐range resection, possibly through competition with RPA or steric occlusion of EXO1 and DNA2‐BLM (Fig 3B). The MRE11‐resected product is also short enough to be conceivably repaired by Pol α‐primase (Fig 3C). After Polα fill‐in of the ssDNA generated by MRN resection, the break could then be repaired through NHEJ (Fig 3D). Furthermore, the NHEJ‐promoting Ku complex also protects DNA ends, but is removed by the MRE11‐dependent resection initiation step 80. Consistent with this model of Ku and shieldin acting on different steps of resection, their mutations are not epistatic to each other 42. Since end resection can be recapitulated in vitro 81, it should be feasible to similarly reconstitute shieldin‐dependent end protection.
Figure 3. Proposed 53BP1‐RIF1‐shieldin mechanism of action in DNA double‐strand break repair.
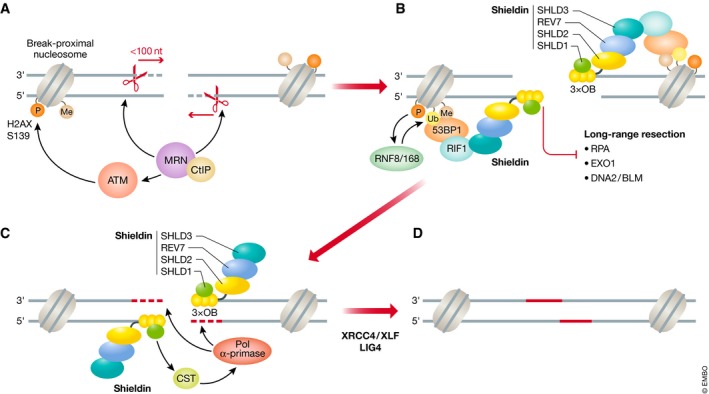
(A) The MRE11‐RAD50‐NBS1 (MRN) complex and its accessory factor CtIP is recruited to DNA double‐strand break sites and introduces an endonuclease nick on the 5′‐terminated strand, which is expanded toward the break via the 3′–5′ exonuclease activity of MRE11. The nucleolytic activity of MRN results in short (< 100 nucleotides) tracts of single‐stranded (ss) DNA around the break. The NBS1 subunit of MRN also recruits and activates the ataxia telangiectasia‐mutated (ATM) kinase that phosphorylates histone H2AX at serine 139 (γH2A.X) on nucleosomes surrounding the DSB. (B) γH2AX recruits the RNF8 ubiquitin ligase through MDC1 binding (not shown). RNF8 catalyzes the K63‐linked polyubiquitination of histone H1, which in turn recruits RNF168. RNF168 ubiquitinates histone H2A lysine 15 which, in concert with constitutive H4 lysine 20 methylation, recruits 53BP1. 53BP1 recruits RIF1 in an ATM‐dependent manner, which localizes the shieldin complex to the DSB. The three tandem OB‐folds of SHLD2 binds the short ssDNA tract and inhibits long‐range 5′–3′ resection mediated by EXO1 and DNA2/BLM nucleases. (C) Shieldin then recruits the CTC1‐STN1‐TEN1 (CST) complex and its binding partner, the polymerase α‐primase (Pol α‐primase) complex to the DSB site. Pol α‐primase fills in the short ssDNA tract by synthesizing new DNA (red). (D) The DSB is subsequently repaired by non‐homologous end joining via DNA ligase IV (LIG4) and its accessory factors XRCC4/XLF. P, Ub, and Me represent phosphorylated, ubiquitinated, and methylated histones, respectively. S1 represents SHLD1. Black dashed lines indicate short range resection, while red dashed lines indicate fill‐in synthesis.
Future perspectives
The discovery of shieldin is a new and exciting chapter in our understanding of the regulation of DSB repair. Until the identification of shieldin, there were very few hints as to the molecular mechanism of end protection by the 53BP1 pathway. In our opinion, shieldin is likely to represent the ultimate effector of 53BP1, but several key questions must be answered. First and foremost, how does shieldin oppose resection at the molecular level? How does it cooperate with CST? How is it regulated by the cell cycle? Furthermore, orthologues of 53BP1 have been described in many species lacking shieldin (Fig 2B), with the yeast orthologues having been shown to oppose DNA end resection 82, 83. Comparison of the mechanisms behind 53BP1 antagonism of resection in the presence or absence of shieldin will provide valuable insights into the circumstances leading to shieldin evolution.
Other questions of importance relate to potential functions of shieldin outside DSB repair: Is shieldin involved in 53BP1‐independent processes such as the regulation of DNA replication timing by RIF1 84, 85 or is it involved in other processes regulated by 53BP1 such as the protection of DNA replication forks 10, 11, 86? Finally, the finding that shieldin is critical to mediate the cytotoxicity of PARPi in BRCA1‐deficient cells has obvious translational potential. The high incidence of PARPi resistance arising in the clinic, many of which cannot be explained by mutations restoring BRCA1 function, suggests that a variety of factors mediate PARPi lethality in BRCA1‐deficient cells 87. Disruption of any one these factors may subsequently lead to a diminished response to PARPi‐based therapy. The studies discussed in this review provide compelling in vitro evidence of shieldin being one such factor, but it remains unclear whether loss of shieldin subunits will represent a significant mode of acquired resistance to PARPi in the clinic. Remaining on the topic of PARPi resistance, the ATMIN‐DYNLL1 pathway has recently been identified as another mechanism enforcing cytotoxicity of PARPi in BRCA1‐deficient cells 88, 89. ATMIN is a transcription factor that controls the expression of DYNLL1, a 53BP1‐interacting protein, which supports many of its known activities 88, 89). One study proposed that DYNLL1 partially mediates the oligomerization of 53BP1, which is essential for its recruitment to DSBs 89, while another report suggests that DYNLL1 directly interacts with MRN to inhibit its nuclease activity 88. These findings raise the question of how the mechanisms of shieldin‐ and DYNLL1‐mediated regulation of end resection functionally interact. Does DYNLL1 act upstream or parallel to shieldin, as posited by the oligomerization and MRN inhibitor models, respectively? The answers to these questions will certainly bring us much closer to a long‐awaited mechanistic understanding of the regulation of DSB repair in mammalian cells.
Box 1: In need of answers.
What is the mechanism of action behind shieldin's inhibition of DNA end resection?
Is CST necessary for shieldin's function or do the two complexes have distinct contributions to the antagonism of end resection?
Are there cell‐cycle‐specific mechanisms that regulate shieldin?
Is shieldin regulated by TRIP13 through structural remodeling of REV7?
How do 53BP1 orthologues regulate end resection in organisms lacking shieldin?
Does shieldin have a role in stalled replication fork protection?
How does shieldin functionally interact with other 53BP1‐associated factors such as PTIP, DYNLL1, or TIRR?
Do shieldin and BRCA1 antagonize each other and if so, what is the mechanism of regulation?
Conflict of interest
DS has no conflict of interest to declare; DD declares that he is a founder and shareholder of Repare Therapeutics.
Acknowledgements
We thank R. Szilard, T. Goullet de Rugy, and J.R. Chapman for advice and critical reading of the manuscript. D.S. is funded by a research fellowship from the Canadian Institutes of Health Research. D.D. is the Thomas Kierans Chair in Mechanisms of Cancer Development and a Canada Research Chair (Tier 1) in the Molecular Mechanisms of Genome Integrity. Work pertaining to shieldin is supported by grants from CIHR (FDN143343), Canadian Cancer Society (CCS grant #705644 and #70389), and OICR (OICR‐OC‐TRI).
EMBO Reports (2019) 20: e47560
See the Glossary for abbreviations used in this article.
References
- 1. Panier S, Boulton SJ (2014) Double‐strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15: 7–18 [DOI] [PubMed] [Google Scholar]
- 2. Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez‐Capetillo O, Cao L et al (2010) 53BP1 inhibits homologous recombination in Brca1‐deficient cells by blocking resection of DNA breaks. Cell 141: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, Taniguchi Y (2006) Genetic dissection of vertebrate 53BP1: a major role in non‐homologous end joining of DNA double strand breaks. DNA Repair 5: 741–749 [DOI] [PubMed] [Google Scholar]
- 4. Ward IM, Reina‐San‐Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F et al (2004) 53BP1 is required for class switch recombination. J Cell Biol 165: 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB (2004) 53BP1 links DNA damage‐response pathways to immunoglobulin heavy chain class‐switch recombination. Nat Immunol 5: 481–487 [DOI] [PubMed] [Google Scholar]
- 6. Dimitrova N, Chen YC, Spector DL, de Lange T (2008) 53BP1 promotes non‐homologous end joining of telomeres by increasing chromatin mobility. Nature 456: 524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC et al (2008) 53BP1 facilitates long‐range DNA end‐joining during V(D)J recombination. Nature 456: 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hustedt N, Durocher D (2016) The control of DNA repair by the cell cycle. Nat Cell Biol 19: 1–9 [DOI] [PubMed] [Google Scholar]
- 9. Ochs F, Somyajit K, Altmeyer M, Rask MB, Lukas J, Lukas C (2016) 53BP1 fosters fidelity of homology‐directed DNA repair. Nat Struct Mol Biol 23: 714–721 [DOI] [PubMed] [Google Scholar]
- 10. Schmid JA, Berti M, Walser F, Raso MC, Schmid F, Krietsch J, Stoy H, Zwicky K, Ursich S, Freire R et al (2018) Histone ubiquitination by the DNA damage response is required for efficient DNA replication in unperturbed S phase. Mol Cell 71: 897–910 e898 [DOI] [PubMed] [Google Scholar]
- 11. Her J, Ray C, Altshuler J, Zheng H, Bunting SF (2018) 53BP1 mediates ATR‐Chk1 signaling and protects replication forks under conditions of replication stress. Mol Cell Biol 38: e00472–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kibe T, Zimmermann M, de Lange T (2016) TPP1 blocks an ATR‐mediated resection mechanism at telomeres. Mol Cell 61: 236–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A et al (2009) A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 35: 534–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q et al (2010) 53BP1 loss rescues BRCA1 deficiency and is associated with triple‐negative and BRCA‐mutated breast cancers. Nat Struct Mol Biol 17: 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A et al (2013) Loss of 53BP1 causes PARP inhibitor resistance in Brca1‐mutated mouse mammary tumors. Cancer Discov 3: 68–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez‐Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A et al (2012) BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell 46: 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chapman JR, Sossick AJ, Boulton SJ, Jackson SP (2012) BRCA1‐associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 125(Pt 15): 3529–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T (2013) 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339: 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Escribano‐Diaz C, Orthwein A, Fradet‐Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD et al (2013) A cell cycle‐dependent regulatory circuit composed of 53BP1‐RIF1 and BRCA1‐CtIP controls DNA repair pathway choice. Mol Cell 49: 872–883 [DOI] [PubMed] [Google Scholar]
- 20. Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas‐Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ (2013) RIF1 is essential for 53BP1‐dependent nonhomologous end joining and suppression of DNA double‐strand break resection. Mol Cell 49: 858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fradet‐Turcotte A, Canny MD, Escribano‐Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski‐Leblanc J, Noordermeer SM, Sicheri F et al (2013) 53BP1 is a reader of the DNA‐damage‐induced H2A Lys 15 ubiquitin mark. Nature 499: 50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, de Lange T (2013) Role of 53BP1 oligomerization in regulating double‐strand break repair. Proc Natl Acad Sci USA 110: 2146–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E et al (2011) Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell 42: 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G (2006) Structural basis for the methylation state‐specific recognition of histone H4‐K20 by 53BP1 and Crb2 in DNA repair. Cell 127: 1361–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zgheib O, Pataky K, Brugger J, Halazonetis TD (2009) An oligomerized 53BP1 tudor domain suffices for recognition of DNA double‐strand breaks. Mol Cell Biol 29: 1050–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adkins NL, Niu H, Sung P, Peterson CL (2013) Nucleosome dynamics regulates DNA processing. Nat Struct Mol Biol 20: 836–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anderson L, Henderson C, Adachi Y (2001) Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 21: 1719–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jowsey P, Morrice NA, Hastie CJ, McLauchlan H, Toth R, Rouse J (2007) Characterisation of the sites of DNA damage‐induced 53BP1 phosphorylation catalysed by ATM and ATR. DNA Repair 6: 1536–1544 [DOI] [PubMed] [Google Scholar]
- 29. Munoz IM, Jowsey PA, Toth R, Rouse J (2007) Phospho‐epitope binding by the BRCT domains of hPTIP controls multiple aspects of the cellular response to DNA damage. Nucleic Acids Res 35: 5312–5322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feng L, Fong KW, Wang J, Wang W, Chen J (2013) RIF1 counteracts BRCA1‐mediated end resection during DNA repair. J Biol Chem 288: 11135–11143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT et al (2013) Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339: 711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Callen E, Di Virgilio M, Kruhlak MJ, Nieto‐Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM et al (2013) 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153: 1266–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Escribano‐Diaz C, Durocher D (2013) DNA repair pathway choice‐a PTIP of the hat to 53BP1. EMBO Rep 14: 665–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M et al (2015) REV7 counteracts DNA double‐strand break resection and affects PARP inhibition. Nature 521: 541–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boersma V, Moatti N, Segura‐Bayona S, Peuscher MH, van der Torre J, Wevers BA, Orthwein A, Durocher D, Jacobs JJ (2015) MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 521: 537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosenberg SC, Corbett KD (2015) The multifaceted roles of the HORMA domain in cellular signaling. J Cell Biol 211: 745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez‐Quilon A, Moatti N, Zimmermann M et al (2018) The shieldin complex mediates 53BP1‐dependent DNA repair. Nature 560: 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka‐Clift M, Wei W, Ostermaier M et al (2018) Shieldin complex promotes DNA end‐joining and counters homologous recombination in BRCA1‐null cells. Nat Cell Biol 20: 954–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A et al (2018) DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173: 972–988 e923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Findlay S, Heath J, Luo VM, Malina A, Morin T, Coulombe Y, Djerir B, Li Z, Samiei A, Simo‐Cheyou E et al (2018) SHLD2/FAM35A co‐operates with REV7 to coordinate DNA double‐strand break repair pathway choice. EMBO J 37: e100158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tomida J, Takata KI, Bhetawal S, Person MD, Chao HP, Tang DG, Wood RD (2018) FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1‐defective cells. EMBO J 37: e99543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao S, Feng S, Ning S, Liu J, Zhao H, Xu Y, Shang J, Li K, Li Q, Guo R et al (2018) An OB‐fold complex controls the repair pathways for DNA double‐strand breaks. Nat Commun 9: 3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar‐Lele M, Sanchiz‐Calvo M, Fueyo‐Marcos E et al (2018) 53BP1 cooperation with the REV7‐shieldin complex underpins DNA structure‐specific NHEJ. Nature 560: 122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Lange T (2018) Shelterin‐mediated telomere protection. Annu Rev Genet 52: 223–247 [DOI] [PubMed] [Google Scholar]
- 45. Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, de Lange T (2018) 53BP1‐RIF1‐shieldin counteracts DSB resection through CST‐ and Polalpha‐dependent fill‐in. Nature 560: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Methot SP, Di Noia JM (2017) Molecular mechanisms of somatic hypermutation and class switch recombination. Adv Immunol 133: 37–87 [DOI] [PubMed] [Google Scholar]
- 47. Kottemann MC, Smogorzewska A (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493: 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murakumo Y, Ogura Y, Ishii H, Numata S, Ichihara M, Croce CM, Fishel R, Takahashi M (2001) Interactions in the error‐prone postreplication repair proteins hREV1, hREV3, and hREV7. J Biol Chem 276: 35644–35651 [DOI] [PubMed] [Google Scholar]
- 49. Wittschieben JP, Reshmi SC, Gollin SM, Wood RD (2006) Loss of DNA polymerase zeta causes chromosomal instability in mammalian cells. Cancer Res 66: 134–142 [DOI] [PubMed] [Google Scholar]
- 50. Makarova AV, Burgers PM (2015) Eukaryotic DNA polymerase zeta. DNA Repair 29: 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bluteau D, Masliah‐Planchon J, Clairmont C, Rousseau A, Ceccaldi R, Dubois d'Enghien C, Bluteau O, Cuccuini W, Gachet S, Peffault de Latour R et al (2016) Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest 126: 3580–3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hara K, Hashimoto H, Murakumo Y, Kobayashi S, Kogame T, Unzai S, Akashi S, Takeda S, Shimizu T, Sato M (2010) Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase zeta and REV1. J Biol Chem 285: 12299–12307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hanafusa T, Habu T, Tomida J, Ohashi E, Murakumo Y, Ohmori H (2010) Overlapping in short motif sequences for binding to human REV7 and MAD2 proteins. Genes Cells 15: 281–296 [DOI] [PubMed] [Google Scholar]
- 54. Tomida J, Takata K, Lange SS, Schibler AC, Yousefzadeh MJ, Bhetawal S, Dent SY, Wood RD (2015) REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase zeta. Nucleic Acids Res 43: 1000–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ye Q, Rosenberg SC, Moeller A, Speir JA, Su TY, Corbett KD (2015) TRIP13 is a protein‐remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife 4: e07367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eytan E, Wang K, Miniowitz‐Shemtov S, Sitry‐Shevah D, Kaisari S, Yen TJ, Liu ST, Hershko A (2014) Disassembly of mitotic checkpoint complexes by the joint action of the AAA‐ATPase TRIP13 and p31(comet). Proc Natl Acad Sci USA 111: 12019–12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alfieri C, Chang L, Barford D (2018) Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature 559: 274–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bochkarev A, Bochkareva E (2004) From RPA to BRCA2: lessons from single‐stranded DNA binding by the OB‐fold. Curr Opin Struct Biol 14: 36–42 [DOI] [PubMed] [Google Scholar]
- 59. Chapman JR, Taylor MR, Boulton SJ (2012) Playing the end game: DNA double‐strand break repair pathway choice. Mol Cell 47: 497–510 [DOI] [PubMed] [Google Scholar]
- 60. Symington LS, Gautier J (2011) Double‐strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271 [DOI] [PubMed] [Google Scholar]
- 61. Kowalczykowski SC (2015) An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb Perspect Biol 7: a016410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mimitou EP, Symington LS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double‐strand break processing. Nature 455: 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhu Z, Chung WH, Shim EY, Lee SE, Ira G (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double‐strand break ends. Cell 134: 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450: 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cannavo E, Cejka P (2014) Sae2 promotes dsDNA endonuclease activity within Mre11‐Rad50‐Xrs2 to resect DNA breaks. Nature 514: 122–125 [DOI] [PubMed] [Google Scholar]
- 66. Anand R, Ranjha L, Cannavo E, Cejka P (2016) Phosphorylated CtIP functions as a co‐factor of the MRE11‐RAD50‐NBS1 endonuclease in DNA end resection. Mol Cell 64: 940–950 [DOI] [PubMed] [Google Scholar]
- 67. Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC (2011) BLM‐DNA2‐RPA‐MRN and EXO1‐BLM‐RPA‐MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25: 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gravel S, Chapman JR, Magill C, Jackson SP (2008) DNA helicases Sgs1 and BLM promote DNA double‐strand break resection. Genes Dev 22: 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Marechal A, Zou L (2014) RPA‐coated single‐stranded DNA as a platform for post‐translational modifications in the DNA damage response. Cell Res 514: 122–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP (2002) BRCA2 function in DNA binding and recombination from a BRCA2‐DSS1‐ssDNA structure. Science 297: 1837–1848 [DOI] [PubMed] [Google Scholar]
- 71. Kim C, Snyder RO, Wold MS (1992) Binding properties of replication protein A from human and yeast cells. Mol Cell Biol 12: 3050–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lei M, Podell ER, Cech TR (2004) Structure of human POT1 bound to telomeric single‐stranded DNA provides a model for chromosome end‐protection. Nat Struct Mol Biol 11: 1223–1229 [DOI] [PubMed] [Google Scholar]
- 73. Zaitseva EM, Zaitsev EN, Kowalczykowski SC (1999) The DNA binding properties of Saccharomyces cerevisiae Rad51 protein. J Biol Chem 274: 2907–2915 [DOI] [PubMed] [Google Scholar]
- 74. Pannunzio NR, Watanabe G, Lieber MR (2018) Nonhomologous DNA end‐joining for repair of DNA double‐strand breaks. J Biol Chem 293: 10512–10523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu P, Takai H, de Lange T (2012) Telomeric 3′ overhangs derive from resection by Exo1 and Apollo and fill‐in by POT1b‐associated CST. Cell 150: 39–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Barazas M, Annunziato S, Pettitt SJ, de Krijger I, Ghezraoui H, Roobol SJ, Lutz C, Frankum J, Song FF, Brough R et al (2018) The CST complex mediates end protection at double‐strand breaks and promotes PARP inhibitor sensitivity in BRCA1‐deficient cells. Cell Rep 23: 2107–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hohn KT, Grosse F (1987) Processivity of the DNA polymerase alpha‐primase complex from calf thymus. Biochemistry 26: 2870–2878 [DOI] [PubMed] [Google Scholar]
- 78. Perrino FW, Loeb LA (1990) Hydrolysis of 3′‐terminal mispairs in vitro by the 3′—5′ exonuclease of DNA polymerase delta permits subsequent extension by DNA polymerase alpha. Biochemistry 29: 5226–5231 [DOI] [PubMed] [Google Scholar]
- 79. Daley JM, Jimenez‐Sainz J, Wang W, Miller AS, Xue X, Nguyen KA, Jensen RB, Sung P (2017) Enhancement of BLM‐DNA2‐mediated long‐range DNA end resection by CtIP. Cell Rep 21: 324–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Langerak P, Mejia‐Ramirez E, Limbo O, Russell P (2011) Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double‐strand breaks. PLoS Genet 7: e1002271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pinto C, Anand R, Cejka P (2018) Methods to study DNA end resection II: biochemical reconstitution assays. Methods Enzymol 600: 67–106 [DOI] [PubMed] [Google Scholar]
- 82. Lazzaro F, Sapountzi V, Granata M, Pellicioli A, Vaze M, Haber JE, Plevani P, Lydall D, Muzi‐Falconi M (2008) Histone methyltransferase Dot1 and Rad9 inhibit single‐stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J 27: 1502–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Leland BA, Chen AC, Zhao AY, Wharton RC, King MC (2018) Rev7 and 53BP1/Crb2 prevent RecQ helicase‐dependent hyper‐resection of DNA double‐strand breaks. Elife 7: e33402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Buonomo SB, Wu Y, Ferguson D, de Lange T (2009) Mammalian Rif1 contributes to replication stress survival and homology‐directed repair. J Cell Biol 187: 385–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella‐Mellwig R, Anthony C, Almouzni G, Gilbert DM, Buonomo SB (2012) Mouse Rif1 is a key regulator of the replication‐timing programme in mammalian cells. EMBO J 31: 3678–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Xu Y, Ning S, Wei Z, Xu R, Xu X, Xing M, Guo R, Xu D (2017) 53BP1 and BRCA1 control pathway choice for stalled replication restart. Elife 6: e30523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ang JE, Gourley C, Powell CB, High H, Shapira‐Frommer R, Castonguay V, De Greve J, Atkinson T, Yap TA, Sandhu S et al (2013) Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi‐institutional study. Clin Cancer Res 19: 5485–5493 [DOI] [PubMed] [Google Scholar]
- 88. He YJ, Meghani K, Caron MC, Yang C, Ronato DA, Bian J, Sharma A, Moore J, Niraj J, Detappe A et al (2018) DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1‐deficient cells. Nature 563: 522–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Becker JR, Cuella‐Martin R, Barazas M, Liu R, Oliveira C, Oliver AW, Bilham K, Holt AB, Blackford AN, Heierhorst J et al (2018) The ASCIZ‐DYNLL1 axis promotes 53BP1‐dependent non‐homologous end joining and PARP inhibitor sensitivity. Nat Commun 9: 5406 [DOI] [PMC free article] [PubMed] [Google Scholar]