

Abstract
Blockade of programmed cell death ligand‐1 with durvalumab has shown efficacy and safety in large, international studies of patients with advanced solid tumors. A phase 1, non‐randomized, open‐label multicenter study was initiated to evaluate durvalumab in a Japanese population. The first part of this study used a standard 3 + 3 dose‐escalation design to determine the optimal dosing schedule of durvalumab. Primary objective was evaluation of safety and tolerability of durvalumab monotherapy. Secondary objectives were to evaluate maximum tolerated dose (MTD), immunogenicity, pharmacokinetics, and efficacy. Twenty‐two patients (median age, 61.5 years; range, 41‐76; 64% male) received durvalumab at doses of 1, 3, or 10 mg/kg every 2 weeks (q2w), 15 mg/kg q3w, or 20 mg/kg q4w. Twenty patients discontinued before completing 12 months of treatment as a result of progressive disease and two due to adverse events (AE). The most common treatment‐related AE (trAE) were rash (18%) and pruritus (14%); two patients had grade ≥3 trAE including one patient each with hyponatremia and hypothyroidism. No patient experienced a dose‐limiting toxicity (DLT) during the DLT evaluation period and the MTD was not identified. There were no AE leading to a fatal outcome during study treatment. Durvalumab showed dose‐proportional pharmacokinetics across the 1‐20 mg/kg dose range; incidence of positive titers for antidrug antibodies was 9%. One patient with lung cancer had a partial response and disease control rate at 12 weeks was 36%. In conclusion, durvalumab at the doses and regimens evaluated was safe and well tolerated in Japanese patients with advanced solid tumors.
Keywords: advanced solid tumor, dose escalation, durvalumab, immune checkpoint blockade, PD‐L1
Abbreviations
- ADA
antidrug antibody
- AESI
adverse event of special interest
- AUC0‐t
area under the concentration‐time curve from time zero to t
- Cmax
maximum concentration
- CR
complete response
- CTLA‐4
cytotoxic T‐lymphocyte–associated protein‐4
- DCR
disease control rate
- DLT
dose‐limiting toxicity
- imAE
immune‐mediated adverse event
- MTD
maximum tolerated dose
- NSCLC
non–small‐cell lung cancer
- ORR
objective response rate
- OS
overall survival
- PD‐1
programmed cell death‐1 receptor
- PD
progressive disease
- PD‐L1
programmed cell death ligand‐1
- PFS
progression‐free survival
- PK
pharmacokinetics
- PR
partial response
- q2w
every 2 weeks
- q3w
every 3 weeks
- q4w
every 4 weeks
- SAE
serious adverse event
- SD
stable disease
- sPD‐L1
soluble programmed cell death ligand‐1
- TC
tumor cell
- Tmax
time to C max
- trAE
treatment‐related adverse event
- tx
treatment
- UC
urothelial bladder carcinoma
1. INTRODUCTION
The programmed cell death‐1 receptor and ligand (PD‐L1) pathway has recently emerged as a prominent therapeutic target for preventing tumor escape from immune surveillance in cancer.1, 2 Physiologically, PD‐1/PD‐L1 expression constitutes an important checkpoint for immune tolerance. However, in the tumor microenvironment, this pathway allows TC to circumvent host immunity, leading to tumor progression and survival through effects on T cells that include dysfunction, exhaustion, neutralization, and immunosuppression.3
The clinical relevance of the PD‐1/PD‐L1 pathway has been demonstrated in a range of solid tumors following the therapeutic success of PD‐1 and PD‐L1 checkpoint blockade, with beneficial outcomes associated with mutational burden of TC, presence of tumor‐infiltrating immune cells, and PD‐L1 expression.4 Overexpression of PD‐L1, in particular, has been associated with poor prognosis in different tumor types and thus represents a rational target for cancer immunotherapy.5, 6, 7
In addition to its membrane expression, PD‐L1 has a soluble form (sPD‐L1) that has been shown to have PD‐1–binding capacity, with its concentration in plasma correlating with tumor aggressiveness and outcome in different tumor types.8, 9, 10, 11 A recent meta‐analysis of patients with solid tumors confirmed that high circulating concentrations of sPD‐L1 predicted shorter OS, indicating that a high sPD‐L1 level may serve as a prognostic biomarker.12
Anti–PD‐L1 blockade with durvalumab, a human IgG1 mAb that blocks PD‐L1 binding to PD‐1 and CD‐80, has demonstrated efficacy and safety in large, international studies of patients with advanced/metastatic NSCLC and UC.13, 14, 15, 16 Patients with locally advanced, unresectable NSCLC (stage 3) who were treated with durvalumab after platinum‐based chemoradiotherapy in a phase 3 trial experienced significantly longer OS, PFS, and time to distant metastasis compared with placebo.13, 16 Among 191 patients with locally advanced or metastatic UC, ORR with single‐agent durvalumab (10 mg/kg q2w) was 17.8% and median PFS and OS were 1.5 and 18.2 months, respectively. Grade 3/4 trAE occurred in 13 patients (6.8%); grade 3/4 imAE occurred in four patients (2.1%); and trAE led to discontinuation of three patients (1.6%), two of whom had imAE that led to death (autoimmune hepatitis and pneumonitis).14, 15 This encouraging antitumor activity and manageable safety profile resulted in the approval of durvalumab in the USA for treatment of patients with locally advanced or metastatic UC who have disease progression during or following platinum‐containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum‐containing chemotherapy as well as for patients with unresectable stage 3 NSCLC whose disease has not progressed following concurrent platinum‐based chemotherapy and radiation therapy;13, 14, 15, 16 durvalumab has also been approved in several other countries worldwide, including Japan.
The first portion of the phase 1 Japan 02 Study is a dose‐escalation phase designed to assess the safety, tolerability, and PK of escalating doses and different dosing schedules of durvalumab as monotherapy in Japanese patients with advanced solid tumors. Upon completion of the dose‐escalation phase, a dose‐expansion phase will further evaluate the safety and efficacy of durvalumab at selected doses in Japanese patients with biliary tract carcinoma, esophageal carcinoma, or squamous cell carcinoma of the head and neck (HNSCC), including patients from other Asian countries. Herein, we report the findings of the dose‐escalation phase for durvalumab as monotherapy in Japanese patients.
2. PATIENTS AND METHODS
2.1. Study design and objectives
This study was a phase 1, non‐randomized, open‐label, multicenter study (NCT01938612) in which durvalumab was given i.v. to patients with advanced solid tumors according to a standard 3 + 3 dose‐escalation design. Patients continued durvalumab treatment for up to 12 months or until disease progression, whichever occurred first. Findings of the dose‐expansion phase of the study will be reported when complete. Up to 24 Japanese patients were planned for enrollment incrementally by dose group, with at least three and up to six evaluable patients planned for each dose group. Primary objective was to evaluate the safety and tolerability of durvalumab monotherapy. Secondary objectives were to identify MTD, determine immunogenicity, and evaluate the PK and antitumor activity of durvalumab as monotherapy. The protocol for this study was reviewed and approved by the appropriate review committees for each institution within which this work was undertaken. This study conforms to the provisions of the Declaration of Helsinki (as revised in Fortaleza, Brazil, October 2013).
2.2. Patients
Males or females aged 20 years or older were eligible for enrollment. In the dose‐escalation phase, patients with advanced solid tumors refractory to standard treatment, intolerant of standard treatment, or for whom no standard therapy exists, were included. In addition, eligible patients had to have at least one measurable lesion by RECIST v1.1 criteria, an ECOG status of 0 or 1, a minimum life expectancy of 16 weeks, adequate organ and bone marrow function, and available archived tumor tissue sample or fresh biopsy of a lesion that may not be used for archival tumor assessment. Patients were to be excluded if they fulfilled any of the following criteria: treatment with any immunotherapy or investigational anticancer therapy within 4 weeks prior to the first dose of study drug or, in the case of mAb therapy, within 6 weeks prior to the first dose of study drug; treatment with concurrent chemotherapy, immunotherapy, biological or hormonal therapy for cancer, except concurrent use of hormones for non–cancer‐related conditions; current or prior use of immunosuppressive medications within 28 days of the first dose of study drug, except intranasal or inhaled corticosteroids or systemic corticosteroids at physiological doses not to exceed 10 mg/day prednisolone or equivalent; use of live attenuated vaccination within 30 days prior to study enrollment or within 30 days of receiving study drug; prior exposure to any anti–PD‐1, anti–PD‐L1, or anti–CTLA‐4 antibody; major surgical procedure within 30 days prior to first dose of study drug or recovering from prior surgery; toxicity from prior anticancer therapy not resolved to NCI Common Terminology Criteria for Adverse Events (CTCAE) v4.03 grade 0 or 1, or any prior grade ≥3 imAE while receiving immunotherapy; any symptomatic or untreated central nervous system metastases requiring concurrent treatment, other invasive malignancy within 5 years prior to study enrollment, uncontrolled concomitant illness, active or prior documented autoimmune disease within the past 2 years, history of primary immunodeficiency, or any condition that in the investigator's opinion would interfere with evaluation of the study drug. All patients were required to provide their written informed consent prior to any study procedures.
2.3. Treatment schedule
Dose‐limiting toxicity was assessed during the DLT evaluation period of 4 weeks in groups 1, 2, 3, and 5, and 3 weeks in group 4 after first dosing with durvalumab. Group 1 received durvalumab 1.0 mg/kg by i.v. infusion q2w. If no DLT was observed in the first three evaluable patients, then dose escalation was planned to occur at the next dose level; if one patient experienced a DLT in a group of three evaluable patients, the group was expanded to include three additional evaluable patients. If only one DLT occurred in a group of six evaluable patients for group 1, then dose escalation was planned with enrollment of patients to group 2, who received 3.0 mg/kg q2w and subsequently to group 3, who received 10 mg/kg q2w (Figure S1). If two or more patients experienced a DLT in a group of up to six patients, dose escalation was stopped irrespective of the number of patients enrolled, with a lower intermediary dose considered in order to define the MTD. After completion of the durvalumab q2w dose escalation, separate q3w (group 4) and q4w dose (group 5) escalations were started at the equivalent dosing rate (average mg/kg per wk) to the optimal biological dose, or highest dose tested if an optimal biological dose was not identified. If an MTD was reached prior to completing the q2w dose escalation, the q3w and q4w starting doses were to be equivalent to 1 dose level below the q2w MTD.
2.4. Study procedures and assessments
Patients underwent initial screening within 28 days prior to the first dose of study drug. A 3‐hour post‐infusion observation was carried out on the day of first dosing with durvalumab. For subsequent doses, the 3‐hour post‐infusion period was required only for patients who experienced an infusion‐related reaction. Patients additionally underwent safety, laboratory, and efficacy evaluations during treatment, at the end of treatment, and during the follow‐up period.
Adverse events were based upon investigator assessment; AE, SAE, and concomitant treatment assessments were conducted post‐dosing. AESI were determined using clinical concepts and selected individual MedDRA preferred terms. To fully characterize the AESI for durvalumab, AESI were reviewed and confirmed and imAE were reported. A confirmed imAE was defined as a suspected imAE that, after medical review by the sponsor, was consistent with an immune‐mediated mechanism of action, and where there was no clear alternative etiology. Serological, immunological, and histological (biopsy) data, as appropriate, were used to support characterization of an imAE.
Dose‐limiting toxicities were those toxicities that occurred from the time of first dose of durvalumab to prior to giving the third dose (or prior to the second dose for the q4w dose group) and were defined as any grade ≥3 treatment‐related toxicity that could include grade ≥3 colitis or a grade ≥3 imAE, such as rash, pruritus, or diarrhea, that were not improved to grade ≤2 within 3 days of onset despite maximal supportive care. MTD was defined as the previous dose level below any dose level at which ≥2 of up to six evaluable patients experienced a DLT.
Tumor assessments for efficacy evaluations were conducted at screening, at week 7 (day 43) and at weeks 13, 17, and 25 and then every 8 weeks (or 9 weeks for q3w dosing). DCR was assessed at 6, 12, or 24 weeks based on the percentage of patients with CR or PR or SD for a minimum of 6, 12, or 24 weeks, following treatment initiation. Patients who, up to the end of the 12‐month treatment period, achieved and maintained disease control defined as CR, PR, or SD according to RECIST v1.1 were eligible for follow up. Patients who discontinued durvalumab during initial treatment or retreatment entered a 3‐month follow up for evaluation of safety and survival.
Assessment of sPD‐L1 concentration was made from blood samples taken at screening, dose 1, dose 2, and on other dose days up to week 7. The number of observations above the lower limit of quantification (LLOQ) was recorded together with the median (minimum, maximum) titers.
Programmed cell death ligand‐1 status was determined from archival or fresh tumor samples taken at screening using the VENTANA PD‐L1 (SP263) Assay (Ventana Medical Systems, Inc., Tucson, AZ, USA), with PD‐L1–low/negative expression (TC <25%) defined as less than 25% TC expressing PD‐L1 at any staining intensity above background. Serum PK parameters were assessed for durvalumab and summarized for each dose group. Parameters of interest were drug exposure determined as AUC0‐t, and C max, C max/dose and T max. ADA titers were determined for durvalumab, with maximum titer summarized by median (minimum, maximum) for each dose group.
2.5. Statistical analyses
Analyses were carried out by dose level using descriptive statistics prepared for patient demographics and baseline characteristics, and safety data. Categorical data were summarized by number and percentage of patients, and continuous variables were summarized by number of observations, arithmetic mean, standard deviation, median, minimum, and maximum values. Baseline was defined as the last non‐missing observation collected on or prior to the date of the first dose of study drug. Missing data were not imputed and patients with missing data were excluded from the summary of any given variable. The safety analysis set (SAS) included all patients who received at least 1 dose of durvalumab. The response evaluable set (RES) included all patients who received any dose of durvalumab prior to data cutoff and who had a baseline disease assessment with measurable disease per RECIST 1.1 as assessed by the study investigators. The DLT evaluable set included all patients enrolled in the dose‐escalation phase who received ≥2 doses of durvalumab for q2w and q3w dose groups and ≥1 dose of durvalumab for the q4w group and completed the safety follow up through the DLT evaluable period or experienced any DLT. The ADA analysis set included all patients who received ≥1 dose of durvalumab and who had ADA data available. PK data were analyzed based on the PK analysis set, with data summarized using descriptive statistics. Data underlying the findings described in this article may be obtained in accordance with AstraZeneca's data sharing policy described at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
3. RESULTS
3.1. Patients
Twenty‐five patients were enrolled into the study of which three patients failed to meet eligibility criteria during the screening period after signing informed consent and were not assigned to treatment. Of the 22 patients assigned to treatment, four patients received 1.0 mg/kg q2w durvalumab, four patients received 3.0 mg/kg q2w durvalumab, and four patients received 10 mg/kg q2w durvalumab. Six patients were treated with durvalumab 15 mg/kg q3w, and four patients were treated with durvalumab 20 mg/kg q4w. None of the 22 patients completed 12 months of treatment, with 20 (91%) patients discontinuing before the maximum of 12 months of treatment as a result of PD and two (9%) as a result of AE. At the data cutoff of June 3, 2017, four (18%) patients remained on follow up.
All 22 patients were included in the SAS, RES, PK, and ADA analysis sets. Two patients were excluded from the DLT evaluable set as they did not meet the protocol‐defined AE follow‐up duration; one patient receiving 1.0 mg/kg q2w discontinued before the second durvalumab dose as a result of PD and one patient receiving 3.0 mg/kg q2w did not receive a second durvalumab dose.
Baseline demographics are summarized in Table 1. Patients were predominantly male (64%), with a median age of 61.5 years (range, 41‐76 years). Thirteen (59%) patients and nine (41%) patients had ECOG performance status of 0 and 1, respectively. No data were available on patients' smoking status. All patients had stage 4 disease at study entry, with tumor types summarized in Table 1. All patients had tumors with TC <25% PD‐L1 expression. Median number of prior therapies ranged from 2 to 3.5 among the treatment cohorts. The best response to previous therapy prior to study entry was PD for 11 (50%) patients and SD for seven (32%); four (18%) patients were either treatment‐naïve or their best response was not reported.
Table 1.
Patient demographics and disease characteristics
| 1.0 mg/kg q2w (Group 1, n = 4) | 3.0 mg/kg q2w (Group 2, n = 4) | 10 mg/kg q2w (Group 3, n = 4) | 15 mg/kg q3w (Group 4, n = 6) | 20 mg/kg q4w (Group 5, n = 4) | Total (N = 22) | |
|---|---|---|---|---|---|---|
| Median age, y (range) | 53.5 (42‐68) | 57.0 (45‐76) | 68.5 (66‐75) | 62.0 (48‐72) | 61.5 (41‐66) | 61.5 (41‐76) |
| Gender, n (%) | ||||||
| Male | 1 (25) | 4 (100) | 3 (75) | 2 (33) | 4 (100) | 14 (64) |
| Female | 3 (75) | 0 | 1 (25) | 4 (67) | 0 | 8 (36) |
| ECOG performance status, n (%) | ||||||
| 0 | 2 (50) | 2 (50) | 3 (75) | 4 (67) | 2 (50) | 13 (59) |
| 1 | 2 (50) | 2 (50) | 1 (25) | 2 (33) | 2 (50) | 9 (41) |
| Median no. of prior chemotherapies, n (range) | 3.0 (1‐5) | 3.0 (1‐5) | 2.0 (1‐5) | 3.5 (2‐5) | 3.5 (0‐6) | 3.0 (0‐6) |
| Tumor type, n (%) | ||||||
| Gastric/gastroesophageal | 0 | 1 (25) | 1 (25) | 1 (17) | 1 (25) | 4 (18) |
| NSCLC | 1 (25) | 1 (25) | 2 (50) | 0 | 0 | 3 (14) |
| Cervical cancer | 2 (50) | 0 | 0 | 0 | 0 | 2 (9) |
| Ovarian cancer | 1 (25) | 0 | 0 | 1 (17) | 0 | 2 (9) |
| Malignant melanoma | 1 (25) | 0 | 1 (25) | 0 | 0 | 2 (9) |
| Breast cancer | 0 | 0 | 0 | 1 (17) | 0 | 1 (5) |
| Thyroid cancer | 0 | 1 (25) | 0 | 0 | 0 | 1 (5) |
| Othera | 0 | 1 (25) | 0 | 3 (50) | 3 (75) | 7 (32) |
CR, complete response; NSCLC, non–small‐cell lung cancer; PD, progressive disease; PR, partial response; q2w, every 2 weeks; q3w, every 3 weeks; q4w, every 4 weeks; SD, stable disease.
Urachal carcinoma, lung adenocarcinoma, intrahepatic bile duct carcinoma, pancreatic cancer, hypopharyngeal cancer, thymic cancer, and cancer of unknown primary.
Median duration of durvalumab treatment overall was 17.1 weeks (range, 1.9‐50.1 weeks), with a median treatment duration of 15.6 weeks for patients receiving 1.0 mg/kg q2w, 15.0 weeks for patients receiving 3.0 mg/kg q2w, 19.1 weeks for patients receiving 10.0 mg/kg q2w, 22.4 weeks for patients receiving 15 mg/kg q3w, and 14.3 weeks for patients receiving 20 mg/kg q4w. Among all patients, median number of durvalumab doses was 6.0 (range, 1‐25). At the time of data cutoff, no patient had any dose delays or dose interruptions.
3.2. Safety
Treatment‐related adverse events that were most frequently reported in the entire dose‐escalation population were rash (4 [18%] patients), pruritus, constipation, nausea, stomatitis, and pyrexia (3 [14%] patients each) (Table 2). Two (9%) patients had a trAE of grade ≥3, including one patient receiving 3.0 mg/kg q2w with hyponatremia and one patient receiving 10.0 mg/kg q2w with hypothyroidism (grade 3). This event was considered an SAE. Another trAE of grade 2 pneumonitis was considered an SAE and occurred in a patient who received durvalumab 3.0 mg/kg q2w. Two (9%) patients discontinued therapy as a result of a trAE, including one patient receiving 10 mg/kg q2w with non‐serious grade 1 pneumonitis and one patient receiving 20 mg/kg q4w with colitis. There were no deaths related to study treatment. The most frequently occurring AESI were rash (4 [18%] patients) and pruritus (3 [14%] patients). No DLT were observed for durvalumab at the doses given.
Table 2.
Treatment‐related adverse eventsa
| 1.0 mg/kg q2w (Group 1, n = 4) | 3.0 mg/kg q2w (Group 2, n = 4) | 10 mg/kg q2w (Group 3, n = 4) | 15 mg/kg q3w (Group 4, n = 6) | 20 mg/kg q4w (Group 5, n = 4) | Total (N = 22) | |
|---|---|---|---|---|---|---|
| Any trAE, n (%) | 3 (75) | 3 (75) | 3 (75) | 5 (83) | 2 (50) | 16 (73) |
| Rash | 1 (25) | 0 | 1 (25) | 1 (17) | 1 (25) | 4 (18) |
| Pruritus | 0 | 2 (50) | 0 | 1 (17) | 0 | 3 (14) |
| Constipation | 0 | 2 (50) | 0 | 1 (17) | 0 | 3 (14) |
| Nausea | 0 | 1 (25) | 0 | 0 | 2 (50) | 3 (14) |
| Stomatitis | 0 | 1 (25) | 0 | 1 (17) | 1 (25) | 3 (14) |
| Pyrexia | 1 (25) | 0 | 1 (25) | 0 | 1 (25) | 3 (14) |
q2w, every 2 weeks; q3w, every 3 weeks; q4w, every 4 weeks; trAE, treatment‐related adverse event.
Safety analysis set.
3.3. Pharmacokinetics and immunogenicity
Durvalumab showed dose‐proportional PK across the 1.0‐20 mg/kg dose range, with a mean (standard deviation) C max of 21.2 (4.78) μg/mL for the 1.0 mg/kg q2w dose group, 56.4 (14.1) μg/mL for the 3.0 mg/kg q2w dose group, 157 (74.5) μg/mL for the 10 mg/kg q2w dose group, 258 (50.2) μg/mL for the 15 mg/kg q3w dose group, and 319 (84.0) μg/mL for the 20 mg/kg q4w dose group. A dose‐proportional increase in AUC0‐t was observed within 1‐10 mg/kg of the q2w dose (Table 3). T max was similar across the dose groups (Table 3).
Table 3.
Summary of pharmacokinetic parameters of durvalumaba
| 1.0 mg/kg q2w | 3.0 mg/kg q2w | 10 mg/kg q2w | 15 mg/kg q3w | 20 mg/kg q4w | |
|---|---|---|---|---|---|
| C max, μg/mL | |||||
| n | 4 | 4 | 3 | 3 | 4 |
| Geometric mean (CV, %) | 20.8 (24.1) | 54.9 (27.9) | 145 (51.2) | 254 (20.8) | 311 (26.6) |
| Mean (SD) | 21.2 (4.78) | 56.4 (14.1) | 157 (74.5) | 258 (50.2) | 319 (84.0) |
| C max/dose, μ/mL·mg | |||||
| n | 4 | 4 | 3 | 3 | 4 |
| Geometric mean (CV, %) | 0.370 (19.4) | 0.356 (15.5) | 0.268 (32.3) | 0.375 (7.4) | 0.254 (23.2) |
| Mean (SD) | 0.375 (0.0694) | 0.359 (0.0535) | 0.277 (0.0916) | 0.376 (0.0281) | 0.259 (0.0640) |
| AUC0‐t, d·μg/mLb | |||||
| n | 4 | 4 | 3 | 3 | 4 |
| Geometric mean (CV, %) | 150 (30.4) | 405 (21.8) | 826 (51.4) | 2380 (16.9) | 2440 (31.8) |
| Mean (SD) | 155 (48.4) | 412 (83.7) | 885 (358) | 2400 (382) | 2540 (848) |
| AUC0‐t/dose, d·μg/mL·mg | |||||
| n | 4 | 4 | 3 | 3 | 4 |
| Geometric mean (CV, %) | 2.67 (32.8) | 2.63 (8.2) | 1.52 (30.3) | 3.51 (11.6) | 1.99 (35.7) |
| Mean (SD) | 2.78 (0.868) | 2.63 (0.209) | 1.56 (0.424) | 3.52 (0.413) | 2.09 (0.732) |
| T max, d | |||||
| n | 4 | 4 | 3 | 3 | 4 |
| Median (min, max) | 0.046 (0.045, 0.048) | 0.044 (0.043, 0.045) | 0.047 (0.044, 0.073) | 0.044 (0.044, 0.045) | 0.046 (0.043, 0.12) |
AUC0‐t, area under the concentration‐time curve from time zero to t; C max, maximum plasma concentration; CV, coefficient of variation; max, maximum; min, minimum; q2w, every 2 weeks; q3w, every 3 weeks; q4w, every 4 weeks; T max, time to C max.
Pharmacokinetics analysis set.
AUC0‐14 for 1, 3 and 10 mg/kg, AUC0‐21 for 15 mg/kg, and AUC0‐28 for 20 mg/kg.
Antidrug antibody response to durvalumab including immunogenicity titers of ADA‐positive samples was determined for each dose group (Table S1). At baseline, two of 22 (9%) patients had a positive ADA titer to durvalumab, including one patient each in the 3.0 mg/kg q2w and 20 mg/kg q4w dose groups who had median titers at baseline of 1.0 and 4.0, respectively; for both patients, post‐baseline samples were ADA‐negative. Post‐baseline data showed a low incidence of ADA‐positive samples, with two (9%) patients who were ADA‐negative at baseline developing positive titers, including one patient each in the durvalumab 1.0 and 10.0 mg/kg q2w dose groups. ADA titers were negative at baseline and persistently positive post‐durvalumab treatment for both patients, with the patient in the 1.0 mg/kg q2w dose group additionally positive for neutralizing antibodies post‐baseline. No patients showed transient ADA‐positive titers.
3.4. Efficacy
Objective response rate was 4.5%. One patient in the 10 mg/kg q2w dose group had a PR, with a duration of response of 4.2 months; this patient had NSCLC.
Disease control rate at 6, 12, and 24 weeks for all patients in the study was 64%, 36%, and 23%, respectively, and is summarized for each dose group in Table S2. This included two of four patients each in the 1.0 mg/kg q2w and 10.0 mg/kg q2w dose groups, one of four patients each in the 3.0 mg/kg q2w and 20 mg/kg q4w dose groups, and two of six patients in the 15 mg/kg q3w dose group. Reduction in tumor size ≥30% was observed in two patients in the 10 mg/kg q2w dose group (including the patient with a PR). Change in target lesion size based on investigator assessments is summarized by dose group in Figure 1.
Figure 1.
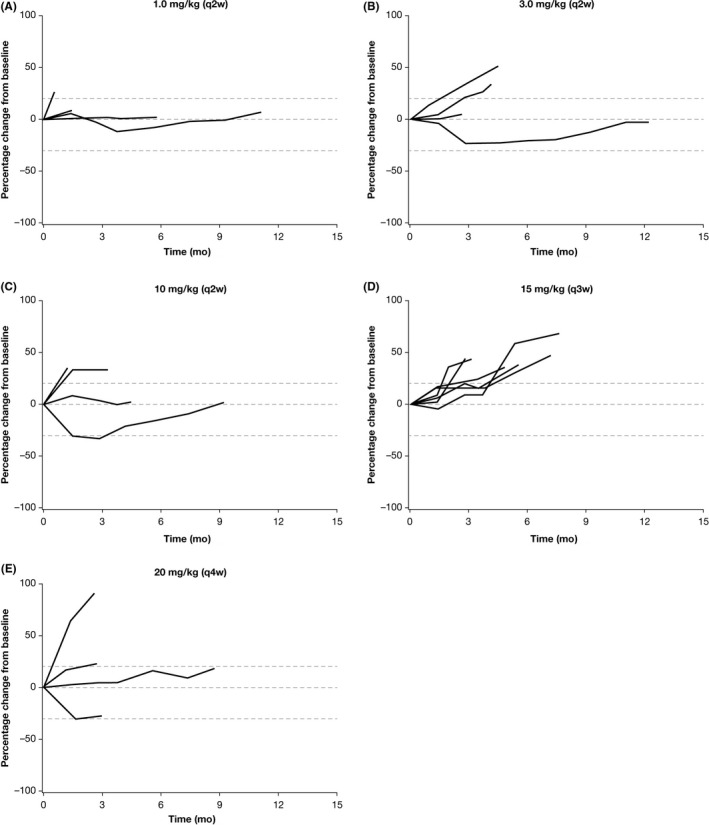
Change in target lesion size based on investigator assessments. A, 1.0 mg/kg q2w. B, 3.0 mg/kg q2w. C, 10.0 mg/kg q2w. D, 15.0 mg/kg q3w. E, 20.0 mg/kg q4w. q2w, every 2 weeks; q3w, every 3 weeks
Soluble programmed cell death ligand‐1 concentrations are summarized for each dose group based on assessments at screening, dose 1/day 1 pre‐infusion and at end of infusion, end of treatment, and 3‐months post‐end of treatment (Table S3). Median titers of sPD‐L1 ranging from 103 to 158 at screening and 90.7‐159 pre‐infusion (dose 1, day 1) were detected across the dose groups. Median sPD‐L1 titer was below the LLOQ at the end of infusion on day 1 of dose 1 and again at the end of treatment for all dose groups. At 3 months after the end of treatment, one of one evaluable patient in the 1.0 mg/kg q2w dose group and one of two evaluable patients in the 20 mg/kg q4w dose group had a detectable sPD‐L1 titer, with titers of 82.3 and 142, respectively.
4. DISCUSSION
Overall, durvalumab was generally well tolerated without DLT in these patients with a range of solid tumor types, including gastric/gastroesophageal cancer, NSCLC, and other advanced solid tumors. Number of patients with trAE was similar across the dose groups, and consistent with previous reports of PD‐1/PD‐L1 blockade in general17, 18, 19, 20, 21 and durvalumab specifically;13, 15, 22, 23 the most frequently occurring trAE in accordance with their immune‐mediated mechanism were rash and pruritus. Few treatment‐related grade ≥3 events occurred in any of the dosing groups. MTD of durvalumab monotherapy was not achieved because of the absence of DLT across a q2w to q4w dose schedule and dose range of 1.0‐20 mg/kg. This suggests that the MTD for durvalumab in Japanese patients could exceed 10 mg/kg in a q2w regimen and 20 mg/kg in a q4w regimen. As there was no dose‐response between PD‐L1/PD‐1 axis blockade and antitumor effects, and as 10 and 20 mg/kg were the highest doses evaluated for the q2w and q4w regimens, respectively, these doses were selected for study expansion. Furthermore, as toxicity of durvalumab was not a function of dose or exposure, higher doses were not assessed.
A low incidence of positive ADA to durvalumab was observed. ADA did not appear to impact the PK of durvalumab and efficacy. This has been seen in other studies with durvalumab, given in combination with tremelimumab; low levels of ADA were observed following treatment.22 Also, in these studies, no association between ADA and tolerability or antitumor activity was determined. Although all patients enrolled in this portion of the study did not show high expression levels for PD‐L1 (all had 25% or fewer TC that expressed PD‐L1), preliminary antitumor activity was observed, with one PR in a patient with NSCLC, and 36% and 23% of patients overall achieving disease control at 12 and 24 weeks, respectively. Pharmacokinetic evaluation showed a dose‐proportional relationship for C max over the 1.0‐20 mg/kg dose range and for AUC0‐t over the 1‐10 mg/kg q2w dose range. As in a previous study,24 no relationship was observed between drug exposure and safety, with higher drug exposure not associated with an increased risk of AE. Absence of DLT and a MTD of durvalumab is also consistent with other reports.25 In a population PK analysis, the PK characteristics of durvalumab were best described using a two‐compartment model with nonlinear elimination kinetics at doses <3 mg/kg and linear kinetics at higher doses.26
The pharmacodynamic effects of durvalumab were also evaluated using sPD‐L1 plasma concentration as a potential predictive biomarker. Although the small sample size and limited treatment response prevented any correlations between baseline sPD‐L1 concentration, dose, and outcomes, the present findings did show evidence of an immediate reduction in sPD‐L1 concentration with durvalumab treatment that was sustained in most dose groups throughout follow up and could therefore be of potential use in evaluating durvalumab dosing in individual patients.
With the increasing role of immunotherapies (such as immune checkpoint blockade with anti–PD‐L1 agents) in the treatment of a variety of advanced solid tumors, it is important to confirm the generalizability of findings in ethnically diverse patient groups. For example, in patients with gastric or gastroesophageal junction cancer, the anti–PD‐1 agent nivolumab increased OS compared with placebo in Asian patients confirming previous findings of nivolumab and leading to its regulatory approval in Japan.27
In conclusion, durvalumab at the doses and regimens evaluated was safe and well tolerated in Japanese patients with advanced solid tumors. Durvalumab is being further evaluated both as monotherapy and in combination with the anti–CTLA‐4 mAb, tremelimumab, in a dose‐expansion phase of study 2, which includes additional patients from Japan and other Asian countries and focuses on patients with squamous cell carcinoma of the head and neck, biliary tract carcinoma, and esophageal carcinoma. The dose and schedule selected for this second phase of the study was durvalumab 10 mg/kg q2w by i.v. infusion as monotherapy, and durvalumab 20 mg/kg q4w in combination with tremelimumab 1.0 mg/kg q4w for patients with biliary tract carcinoma, and esophageal carcinoma.
CONFLICTS OF INTEREST
Yutaka Fujiwara from AbbVie, AstraZeneca, Bristol‐Myers Squibb, Chugai Pharma, Daiichi Sankyo, Eisai, Incyte, Lilly, Merck Serono, MSD, Novartis (research funding), AstraZeneca, Bristol‐Myers Squibb, MSD, Ono Pharmaceutical (honoraria). Haruo Iguchi from AstraZeneca (research funding), Lilly, Nihon Medi‐Physics, Taiho Pharmaceutical, Yakult (honoraria). Noboru Yamamoto from AstraZeneca (research funding). Manabu Hayama, Shinya Ueda, Masahiro Nii, Keiko Komuro, Mariko Sugimoto and Gordana Vlahovic from AstraZeneca (employees). Gordana Vlahovic from Genentech/Roche, Pfizer (honoraria), Bristol‐Myers Squibb, Genentech/Roche, Pfizer (Consulting or Advisory Role), Genentech/Roche Pfizer, (Speakers' Bureau), Bristol‐Myers Squibb (Research Funding), Bristol‐Myers Squibb, Genentech/Roche, Pfizer (travel, accommodations, expenses). Toshiyuki Kozuki from AstraZeneca, Chugai Pharma, Kyowa Hakko Kirin, Lilly, Roche Pharma AG, Taiho Pharmaceutical (honoraria), AstraZeneca (research funding).
Supporting information
{kind=link}
ACKNOWLEDGMENTS
This study was funded by AstraZeneca. The authors would like to thank the patients, their families and caregivers, and all investigators involved in this study. Medical writing support, which was in accordance with Good Publication Practice (GPP3) guidelines, was provided by Jubilee Stewart, PhD, and was funded by AstraZeneca.
Fujiwara Y, Iguchi H, Yamamoto N, et al. Tolerability and efficacy of durvalumab in Japanese patients with advanced solid tumors. Cancer Sci. 2019;110:1715–1723. 10.1111/cas.14003
Trial Registration: clinicaltrials.gov identifier: NCT01938612.
Previously National Hospital Organization Shikoku Cancer Center during the conduct of this study.
REFERENCES
- 1. Frydenlund N, Mahalingam M. PD‐L1 and immune escape: insights from melanoma and other lineage‐unrelated malignancies. Hum Pathol. 2017;66:13‐33. [DOI] [PubMed] [Google Scholar]
- 2. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974‐1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alsaab HO, Sau S, Alzhrani R, et al. PD‐1 and PD‐L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kleinovink JW, Marijt KA, Schoonderwoerd MJA, van Hall T, Ossendorp F, Fransen MF. PD‐L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology. 2017;6:e1294299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jung HI, Jeong D, Ji S, et al. Overexpression of PD‐L1 and PD‐L2 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res Treat. 2017;49:246‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakanishi J, Wada Y, Matsumoto K, Azuma M, Kikuchi K, Ueda S. Overexpression of B7‐H1 (PD‐L1) significantly associates with tumor grade and postoperative prognosis in human urothelial cancers. Cancer Immunol Immunother. 2007;56:1173‐1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Okita R, Maeda A, Shimizu K, Nojima Y, Saisho S, Nakata M. PD‐L1 overexpression is partially regulated by EGFR/HER2 signaling and associated with poor prognosis in patients with non‐small‐cell lung cancer. Cancer Immunol Immunother. 2017;66:865‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim HJ, Park S, Kim KJ, Seong J. Clinical significance of soluble programmed cell death ligand‐1 (sPD‐L1) in hepatocellular carcinoma patients treated with radiotherapy. Radiother Oncol. 2018;129:130‐135. [DOI] [PubMed] [Google Scholar]
- 9. Okuma Y, Hosomi Y, Nakahara Y, Watanabe K, Sagawa Y, Homma S. High plasma levels of soluble programmed cell death ligand 1 are prognostic for reduced survival in advanced lung cancer. Lung Cancer. 2017;104:1715‐6. [DOI] [PubMed] [Google Scholar]
- 10. Zhou J, Mahoney KM, Giobbie‐Hurder A, et al. Soluble PD‐L1 as a biomarker in malignant melanoma treated with checkpoint blockade. Cancer Immunol Res. 2017;5:480‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takeuchi M, Doi T, Obayashi K, et al. Soluble PD‐L1 with PD‐1‐binding capacity exists in the plasma of patients with non‐small cell lung cancer. Immunol Lett. 2018;196:155‐160. [DOI] [PubMed] [Google Scholar]
- 12. Wei W, Xu B, Wang Y, Wu C, Jiang J, Wu C. Prognostic significance of circulating soluble programmed death ligand‐1 in patients with solid tumors: a meta‐analysis. Medicine (Baltimore). 2018;97:e9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non‐small‐cell lung cancer. N Engl J Med. 2017;377:1919‐1929. [DOI] [PubMed] [Google Scholar]
- 14. Massard C, Gordon MS, Sharma S, et al. Safety and efficacy of durvalumab (MEDI4736), an anti‐programmed cell death ligand‐1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J Clin Oncol. 2016;34:3119‐3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Powles T, O'Donnell PH, Massard C, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open‐label study. JAMA Oncol. 2017;3:e172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Antonia SJ, Villegas A, Daniel D, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379:2342‐2350. [DOI] [PubMed] [Google Scholar]
- 17. Hahn AW, Gill DM, Agarwal N, Maughan BL. PD‐1 checkpoint inhibition: toxicities and management. Urol Oncol. 2017;35:701‐707. [DOI] [PubMed] [Google Scholar]
- 18. Hu YB, Zhang Q, Li HJ, et al. Evaluation of rare but severe immune related adverse effects in PD‐1 and PD‐L1 inhibitors in non‐small cell lung cancer: a meta‐analysis. Transl Lung Cancer Res. 2017;6:S8‐S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khunger M, Rakshit S, Pasupuleti V, et al. Incidence of pneumonitis with use of programmed death 1 and programmed death‐ligand 1 inhibitors in non‐small cell lung cancer: a systematic review and meta‐analysis of trials. Chest. 2017;152:271‐281. [DOI] [PubMed] [Google Scholar]
- 20. Pillai RN, Behera M, Owonikoko TK, et al. Comparison of the toxicity profile of PD‐1 versus PD‐L1 inhibitors in non‐small cell lung cancer: a systematic analysis of the literature. Cancer. 2018;124:271‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang PF, Chen Y, Song SY, et al. Immune‐related adverse events associated with Anti‐PD‐1/PD‐L1 treatment for malignancies: a meta‐analysis. Front Pharmacol. 2017;8:730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Antonia S, Goldberg SB, Balmanoukian A, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non‐small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 2016;17:299‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Garassino MC, Cho BC, Kim JH, et al. Durvalumab as third‐line or later treatment for advanced non‐small‐cell lung cancer (ATLANTIC): an open‐label, single‐arm, phase 2 study. Lancet Oncol. 2018;19:521‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin C, Zheng Y, Jin X, et al. Exposure‐efficacy and safety analysis of durvalumab in patients with urothelial carcinoma (UC) and other solid tumors. J Clin Oncol. 2017;35(15 Suppl):2568.28514183 [Google Scholar]
- 25. Sheng J, Srivastava S, Sanghavi K, et al. Clinical pharmacology considerations for the development of immune checkpoint inhibitors. J Clin Pharmacol. 2017;57(Suppl 10):S26‐S42. [DOI] [PubMed] [Google Scholar]
- 26. Baverel PG, Dubois VFS, Jin CY, et al. Population pharmacokinetics of durvalumab in cancer patients and association with longitudinal biomarkers of disease status. Clin Pharmacol Ther. 2018;103:631‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taieb J, Moehler M, Boku N, et al. Evolution of checkpoint inhibitors for the treatment of metastatic gastric cancers: current status and future perspectives. Cancer Treat Rev. 2018;66:104‐113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials