

Abstract
Homeobox genes are known to be classic examples of the intimate relationship between embryogenesis and tumorigenesis, which are a family of transcriptional factors involved in determining cell identity during early development, and also dysregulated in many malignancies. Previously, HOXB7, HOXC6 and HOXC8 were found abnormally upregulated in esophageal squamous cell carcinoma (ESCC) tissues compared with normal mucosa and seen as poor prognostic predictors for ESCC patients, and were shown to promote cell proliferation and anti‐apoptosis in ESCC cells. These three HOX members have a high level of functional redundancy, making it difficult to target a single HOX gene. The aim of the present study was to explore whether ESCC cells are sensitive to HXR9 disrupting the interaction between multiple HOX proteins and their cofactor PBX, which is required for HOX functions. ESCC cell lines (KYSE70, KYSE150, KYSE450) were treated with HXR9 or CXR9, and coimmunoprecipitation and immunofluorescent colocalization were carried out to observe HOX/PBX dimer formation. To further investigate whether HXR9 disrupts the HOX pro‐oncogenic function, CCK‐8 assay and colony formation assay were carried out. Apoptosis was assessed by flow cytometry, and tumor growth in vivo was investigated in a xenograft model. RNA‐seq was used to study the transcriptome of HXR9‐treated cells. Results showed that HXR9 blocked HOX/PBX interaction, leading to subsequent transcription alteration of their potential target genes, which are involved in JAK‐signal transducer and activator of transcription (STAT) activation and apoptosis inducement. Meanwhile, HXR9 showed an antitumor phenotype, such as inhibiting cell proliferation, inducing cell apoptosis and significantly retarding tumor growth. Therefore, it is suggested that targeting HOX/PBX may be a novel effective treatment for ESCC.
Keywords: apoptosis, ESCC, HOX, HXR9, PBX
1. INTRODUCTION
Esophageal cancer is one of the most common cancers worldwide. According to the global cancer statistics of 2012, each year an estimated 456 000 people are newly diagnosed with esophageal cancer, resulting in 400 000 deaths.1 In China, the majority of cases are esophageal squamous cell carcinoma (ESCC), and most patients are diagnosed at a relatively advanced stage. Currently, the development of comprehensive perioperative therapies has greatly improved the efficacy of ESCC treatment, but the long‐term survival remains poor because of recurrence and resistance commonly occurring. Therefore, it is urgent to explore novel actionable targets and develop effective new therapy.
It is common knowledge that there is some similarity between embryogenesis and carcinogenesis.2, 3, 4 Moreover, some genes that regulate embryogenesis have also been shown to be abnormally expressed and implicated in carcinogenesis (eg, AFP in liver cancer, CEA in colorectal cancer), which include HOX genes. The HOX family comprises 39 HOX genes organized in four clusters that are localized at four different chromosomes and encode transcription regulatory proteins. Each cluster is divided into 13 regions according to their sequence similarity and relative position in the chromosome and arranged from the 3′ end to the 5′ end. Each gene is labeled with a number, such as HOXA1 to HOXA13. The genes positioned closer together show greater similarity of sequence and DNA binding specificity.5 During the last decade, dysregulated expression of HOX genes has been described in many solid tumors and derivative cell lines,6, 7 and overexpression of HOX genes was associated with poor prognosis.8, 9, 10, 11, 12 In our previous study, we found that 11 of 39 HOX genes were overexpressed in ESCC tissues compared with paired noncancerous mucosa,13 including HOXB7, HOXC6 and HOXC8. Moreover, we showed that these HOX genes promoted oncogenic properties in ESCC cells and presented negative survival significance in ESCC patients.14, 15 Specifically, knockdown of HOXB7, HOXC6 or HOXC8 resulted in antiproliferation and proapoptosis phenotype in ESCC cell lines, and induced cell cycle arrest in G1 phase, and inhibited tumor growth in a mice xenograft model.
HOX genes have distinct functions in a specific context during early development, and this functional complexity is also seen in tumorigenesis, with some HOX genes functioning as oncogenes and others as tumor suppressors.6 Specific reasons for these opposing functions are still unclear, However, it may be related to different regulation of target genes. DNA binding selectivity of HOX proteins is mediated by a homeodomain together with a defined set of cofactors including the PBX, MEIS and PREP families.16 Therefore, a high level of functional redundancy is seen among some HOX members, especially regarding the HOX genes localized in relative positions within the cluster. This is also true in ESCC, where a similar oncogenic function is common to HOXB7, HOXC6 and HOXC8. As a result of the functional redundancy, it is not only difficult to interpret the results of conventional knockdown results for single HOX genes, but it also makes targeting a single HOX gene very difficult. Therefore, exploring a way to target multiple HOX genes could potentially be a better strategy to explore the oncogenic role of HOX members by disrupting the interaction of HOX proteins with their cofactors. PBX is the defined as a cofactor binding to HOX members 1–917 which modifies DNA binding specificity and affinity and regulates the nuclear‐cytoplasm transport of HOX proteins.18, 19 The interaction is mediated by a highly conserved hexapeptide region in HOX proteins.18, 20 Previously, it was shown that a synthetic peptide known as HXR9 was capable of blocking the interaction between HOX and PBX proteins both in vitro and in vivo. HXR9 functioned as a competitive antagonist of the interaction by mimicking the conserved hexapeptide region.21 The present study aimed to investigate whether HXR9 could block the interaction between multiple HOX members (HOXB7, HOXC6, HOXC8) and PBX in ESCC cells and inhibit their oncogenic functions. Moreover, we attempted to search for the potential target genes in response to HXR9 treatment, which may be the clue to the mechanism underlying the effect of HOX/PBX inhibition.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture
Human ESCC cell lines KYSE70, KYSE150, KYSE450 were purchased from the Japanese Collection of Research Biosources cell bank (Osaka, Japan) and identified by standard STR analysis as well as matching with the ATCC (Manassas, VA, USA) and Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ; Braunschweig, Germany). All cell lines were cultured in RPMI‐1640 medium (Hyclone; GE Healthcare, Logan, UT, USA) supplemented with 10% heat‐inactivated FBS and 1% penicillin/streptomycin. Incubator was maintained at 37°C humidified atmosphere containing 5% CO2.
2.2. Synthesis of HXR9 and CXR9 peptides
The HOX/PBX interfering peptide HXR9 and control peptide (CXR9) were custom synthesized by Sangon Biotech Co. Ltd (Shanghai, China), with >98% purity. The powdered peptides were dissolved in ddH2O to a final concentration of 20 mmol/L, and stored at −20°C. HXR9 is an 18‐amino acid peptide consisting of the hexapeptide sequence that can bind with PBX and nine C‐terminal arginine residues (R9) that facilitate its entry into cells. CXR9 differs from HXR9 by a single amino acid so that it lacks a functional hexapeptide sequence but still includes the R9 sequence. Sequences of the peptides are as follows: HXR9: WYPWMKKHHRRRRRRRRR, CXR9: WYPAMKKHHRRRRRRRRR.
2.3. CCK‐8 assay
Five thousand cells/well were plated in 96‐well plates and treated with gradient dilutions of HXR9 or CXR9 (10, 20, 40, 60, 80, 160 μmol/L). After 24 hours of incubation, 10 μL CCK‐8 reagent (Dojindo Molecular Technologies Inc., Kumamoto, Japan) was added to each well for an additional 2 hours at 37°C. Finally, the absorbance was measured at 450 nm on a microplate reader (iMark; Bio‐Rad, Hercules, CA, USA).
2.4. Colony formation assay
Plated cells were treated with 60 μmol/L HXR9 or CXR9 for 8 hours prior to replating at 500 cells/well in six‐well plates. Cells were maintained at 37°C and the completed medium was replaced by fresh one every 4 days for the following 2 weeks. Subsequently, obtained colonies were visualized by methanol fixation and 0.1% crystal violet staining. Number of colonies was counted in control, CXR9‐treated and HXR9‐treated groups from at least three independent experiments.
2.5. Cell apoptosis analysis
Cells were plated at a density of 1 × 106 cells/well in six‐well plates and then treated with 60 μmol/L HXR9 or CXR9 for 2 hours. Cells were collected after being digested by EDTA‐free trypsin (Gibco, Waltham, MA, USA) and resuspended in binding buffer (Dojindo, Tokyo, Japan) to a density of 1 × 105 cells/100 μL. Annexin V‐FITC antibody (5 μL; Dojindo,) and PI (5 μL; Dojindo) were added and incubated for 15 minutes at room temperature in the dark. Finally, the samples were analyzed by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA) within 1 hour.
2.6. Western blotting
Cells were treated with 60 μmol/L HXR9 or CXR9 for 2 hours. Total proteins were extracted by using RIPA lysis buffer with protease inhibitor cocktail and separated by SDS‐PAGE, blotted onto PVDF, then immunoreacted with primary antibody overnight at 4°C. The primary antibodies used were anti‐PBX (sc‐28313 at 1:200; Santa Cruz Biotechnology, Dallas, TX, USA), anti‐HOXB7 (ab51237 at 1:50; Abcam, Cambridge, MA, USA), anti‐HOXC6 (ab151575 at 1:1000; Abcam), anti‐HOXC8 (ab86236 at 1:1000; Abcam), anti‐Caspase‐3 (#9662 at 1:1000; Cell Signaling Technology, Danvers, MA, USA), anti‐poly ADP ribose polymerase (anti‐PARP, #5625 at 1:1000; Cell Signaling Technology), anti‐c‐FOS (sc‐447 at 1:200; Santa Cruz Biotechnology), anti‐PI3K (#4249 at 1:1000; Cell Signaling Technology), anti‐AKT (#9272 at 1:1000; Cell Signaling Technology), anti‐p‐AKT (#5012 at 1:1000; Cell Signaling Technology), anti‐signal transducer and activator of transcription‐6 (anti‐STAT6, #5397 at 1:1000; Cell Signaling Technology), anti‐p‐STAT6 (#9364 at 1:1000; Cell Signaling Technology) and anti‐GAPDH (ZS‐25778 at 1:2000; ZSGB‐BIO, Beijing, China). Goat antirabbit IgG (ZB‐2301 at 1:5000; ZSGB‐BIO) and goat antimouse IgG (ZB‐2305 at 1:5000; ZSGB‐BIO) were used as the secondary antibodies.
2.7. Immunofluorescent colocalization
Cells were plated at a density of 1 × 105 cells/well in 12‐well plates with coverslips overnight and then treated with 60 μmol/L HXR9 or CXR9 for 2 hours. After being fixed in 3.7% paraformaldehyde for 15 minutes and permeabilized with 0.5% Triton X‐100 for 5 minutes, the coverslips were blocked in 5% normal serum and then incubated in primary antibody dilutions overnight at 4°C. The primary antibodies used were anti‐PBX (sc‐28313 at 1:50, mouse; Santa Cruz Biotechnology), anti‐HOXB7 (#40‐2000 at 1:50, rabbit; Thermo Fisher Scientific, Waltham, MA, USA), anti‐HOXC6 (#PA5‐65913 at 4 μg/mL, rabbit; Invitrogen, Carlsbad, CA, USA), and anti‐HOXC8 (ab86236 at 1:50, rabbit; Abcam). Then, coverslips were incubated in the appropriate fluorophore‐conjugated secondary antibody dilutions and counterstained with DAPI (D523 at 300 nmol/L; Invitrogen) dilution. The secondary antibodies used were FITC‐conjugated goat antimouse IgG (F2761 at 1:50; Invitrogen) and TRITC‐conjugated goat antirabbit IgG (T2769 at 1:50; Invitrogen). Immunofluorescence was visualized by Zeiss scanning microscope (Zeiss Germany, Oberkochen, Germany).
2.8. Coimmunoprecipitation
Cells were treated with 60 μmol/L HXR9 or CXR9 for 2 hours and proteins were extracted by using cold immunoprecipitation (IP) lysis buffer (Thermo Fisher Scientific) with protease inhibitor cocktail and pre‐cleared by Control Agarose Resin (26150; Thermo Fisher Scientific). The PBX antibody (sc‐28313 at 20 μg/mL; Santa Cruz Biotechnology) was immobilized using AminoLink Plus Coupling Resin (Thermo Fisher Scientific). Then the pre‐cleared lysate was incubated in antibody immobilized resin with gentle mixing overnight at 4°C. After incubation, the resin was eluted using elution buffer (21004; Thermo Fisher Scientific). Samples of elution buffer were prepared for western blotting analysis as described above.
2.9. Esophageal squamous cell carcinoma xenograft models
BALB/c nude mice were raised in the Laboratory Animal Unit of First Affiliated Hospital of PLA General Hospital, China. Each 8‐week‐old female BALB/c nude mouse was s.c. inoculated with 2.5 × 106 cells of KYSE70 and KYSE150 in 100 μL PBS into the right groin. Sizes of tumors and body weight were measured every 3 days. Tumor volume = [(length) × (width) × (width)]/2. When the average tumor volume reached approximately 100 mm3 for KYSE70 and 200 mm3 for KYSE150, the mice were randomly divided into two groups and received an initial dose of 100 mg/kg CXR9 or HXR9 (i.v. or intratumor), respectively, followed by twice weekly treatments at 10 mg/kg. Mice were monitored carefully; after 18 days, tumors were excised, and measured by a slide caliper for volume and weighed with an electronic analytical balance.
2.10. Immunohistochemistry
Nude mouse tumors were fixed in 4% formalin immediately after dissection. Subsequently, dehydration, transparency, soaking, and embedding were sequentially carried out, and tissues cut into 4‐μm sections. After routine deparaffinization and hydration, tissue sections were treated with 3% hydrogen peroxide for 10 minutes and then heated in citrate solution for antigen retrieval for 5 minutes. After antigen retrieval, the sections were incubated with 10% normal goat serum to block any nonspecific reaction and incubated in primary antibody dilutions overnight at 4°C. The primary antibodies used were anti‐Caspase‐3 (9662 at 1:1000; Cell Signaling, America), anti‐PARP (#5625 at 1:50; Cell Signaling Technology) and anti‐c‐FOS (TA806833 at 1:250; ZSGB‐BIO). Dako REAL EnVision Detection System, Peroxidase/DAB, Rabbit/Mouse (K5007; Dako, Glostrup, Denmark) was used as the secondary antibody for staining. Finally, the immunohistochemical signals were scored by two independent pathologists. Staining intensity was categorized by relative intensity as follows: 0, negative; 1, weak; 2, moderate; and 3, strong.
2.11. RNA sequencing
Total RNA from KYSE70 and KYSE150 treated with HXR9 or CXR9 (60 μM) with three replications was isolated using Trizol for the construction of a RNA‐seq library and sequencing. Details of RNA sequencing was described in our previous study.22
2.12. Statistical analysis
SPSS software (version 24.0; IBM SPSS, Armonk, NY, USA) was used for statistical analysis. Quantitative analysis was carried out with Image J analysis software (Version 1.30v; Wayne Rasband, NIH, Bethesda, MD, USA). Comparisons between groups for statistical significance were carried out with two‐tailed unpaired Student's t test. All values are expressed as mean ± SEM. Significance was set at P < .05 for all statistical analyses.
3. RESULTS
3.1. HOXB7, HOXC6 or HOXC8 show similar oncogenic functions in ESCC
Our previous studies reported that 11 of 39 HOX genes were overexpressed in esophageal cancer tissues, in which HOXB7, HOXC6 and HOXC8 are positioned closely together and might consequently show functional redundancy. It has been shown that ESCC patients with high expression of HOXB7/HOXC6/HOXC8 had poorer prognosis than those with low expression.14, 15 However, no evidence has established their oncogenic function in ESCC. Herein, we established cell strains with stable knockdown of single HOXB7/HOXC6/HOXC8 genes using RNAi to observe their oncogenic properties. Results of HOXB7 knockdown in ESCC cells have been reported in our previous study,15 which showed that cell proliferation rate dropped, cell growth rate decreased, colony‐formation ability reduced and tumorigenicity reduced remarkably. In the present study, we observed oncogenic function redundancy between HOXB7 and HOXC6/HOXC8. Specifically, compared to control cells, the viability of cells with stable knockdown of HOXC6 or HOXC8 was decreased by more than 60% (Figure S1A), and colony formation decreased by 58% and 67%, respectively (Figure S1B,C). Xenograft nude mouse model was established to evaluate whether HOXC6 and HOXC8 affect tumor growth in vivo. Weight of tumors from HOXC6 and HOXC8 knockdown cells was 218 ± 203 mg and 61 ± 165 mg, respectively, which were significantly lower than those derived from control cells (462 ± 358 mg; P < .01, Figure S1D,E). Moreover, knockdown of HOXC6 or HOXC8 induced cell cycle arrest in G1 phase. Specifically, shHOXC6‐ and shHOXC8‐transfected cells showed an increased proportion of cells in G1 phase compared to control cells (52.57% ± 2.29% and 69.02% ± 3.10% vs 38.50% ± 3.17%) (Figure S2), and the rate of cell apoptosis significantly increased to 32.80% ± 0.29% and 36.10% ± 0.35%, respectively, compared to 12.70% ± 0.31% for control cells (Figure S3).
3.2. Targeting HOX/PBX interaction in ESCC cell lines (KYSE70, KYSE150, KYSE450)
Given PBX binding to HOX modifies the selection of DNA binding sites and the identity of HOX target genes, a short peptide HXR9 was designed in previous work to disrupt HOX/PBX dimer formation. This peptide mimics the ‘hexapeptide’ sequence in HOX proteins and penetrates cells efficiently.23, 24, 25, 26 In the present study, we detected whether HXR9 could block HOX/PBX interaction in ESCC cells through coimmunoprecipitation and fluorescence colocalization assay. As a control, a second peptide CXR9 was used which lacks a functional hexapeptide portion.
As HOXB7, HOXC6, HOXC8 are known PBX‐binding partners, to test the specificity of HXR9 to block PBX/HOX dimer formation, we treated ESCC cell lines KYSE70, KYSE150 and KYSE450 with 60 μmol/L HXR9 or CXR9 for 2 hours and total protein was extracted. PBX was then immunoprecipitated using a monoclonal anti‐PBX antibody. Then the immunoprecipitation complex was collected and subjected to western blotting using anti‐HOXB7, anti‐HOXC6, and anti‐HOXC8. The results confirmed the existence of HOX/PBX heterodimers in untreated cells, and they were also present in CXR9‐treated cells but not in HXR9‐treated cells, indicating that HXR9 did, indeed, block the interaction between PBX and HOX (Figure 1A). However, HXR9 did not reduce the expression of HOX and PBX.
Figure 1.
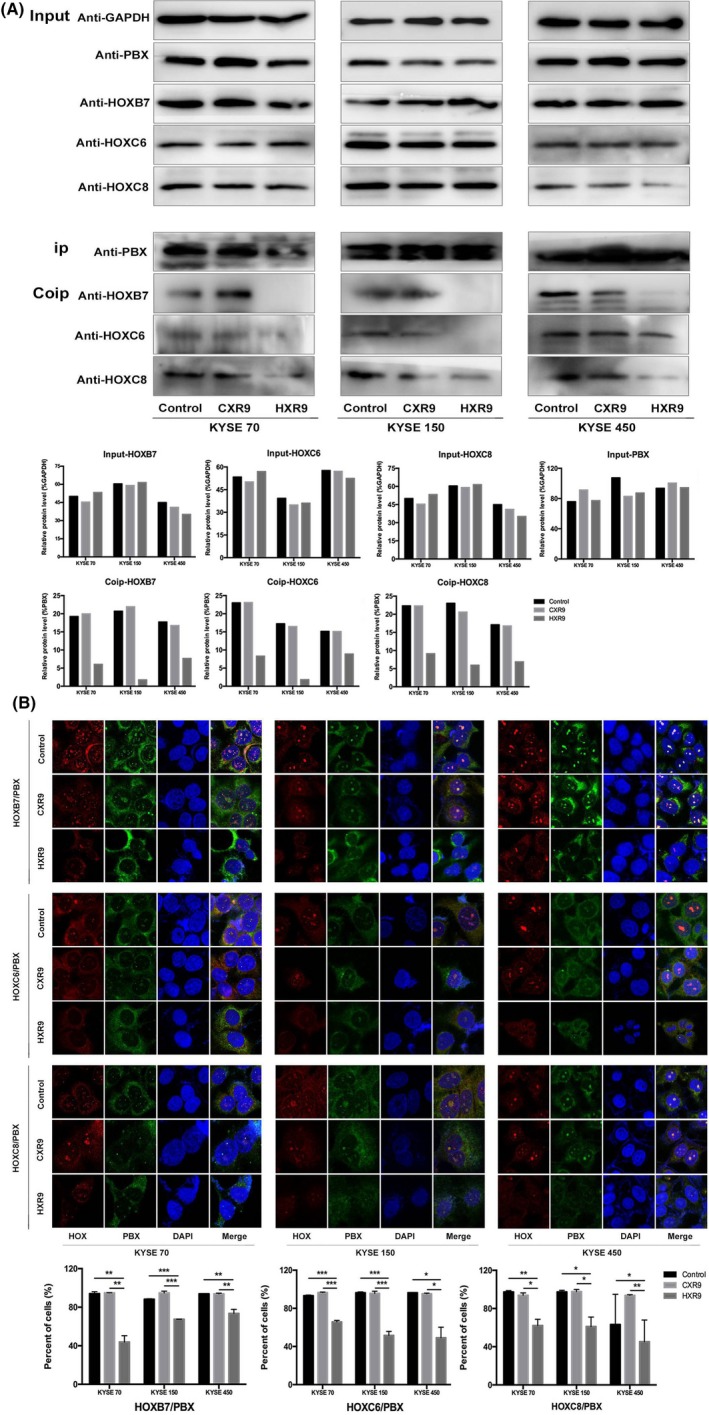
HXR9 disrupts HOX/PBX interaction in esophageal squamous cell carcinoma cells. KYSE70, KYSE150, KYSE450 were treated with 60 μmol/L HXR9 or CXR9 for 2 h. A, Protein was extracted and precipitated using anti‐PBX antibody. Precipitates were subjected to western blotting using anti‐HOXB7, anti‐HOXC6 and anti‐HOXC8. In HXR9‐treated cells, the binding of PBX to HOX was abrogated. Densitometry quantification of HOXB7, HOXC6 and HOXC8 expression in coimmunoprecipitation (Co‐IP) assay is presented. B, Cells were fixed and colabeled with anti‐PBX and anti‐HOXB7/anti‐HOXC6/anti‐HOXC8. PBX and HOX colocalization were analyzed by immunofluorescence microscopy. In HXR9‐treated cells, HOX/PBX dimer formation decreased remarkably compared to control or CXR9‐treated cells. Percentage of HOX/PBX colocalization foci is quantified. *P < 0.05, **P < 0.01 and ***P < 0.001
To further confirm that HOX/PBX dimer formation occurs in cells but not false bindings happening during the cell lysis, we treated ESCC cell lines KYSE70, KYSE150 and KYSE450 with 60 μmol/L HXR9 or CXR9 for 2 hours, and stained cells using anti‐PBX and anti‐HOX and then fluorescent‐labeled secondary antibody to observe their cellular localization. In the untreated cells, HOX and PBX protein were observed in the cytoplasm and nucleus, and their colocalization indicated the existence of HOX/PBX dimers, which were decreased remarkably in HXR9‐treated cells but not in CXR9‐treated cells (Figure 1B). In addition, we noticed that HOX mainly localized in the cytoplasm but not in the nucleus for HXR9‐treated cells, implying that HXR9 affects the nucleus‐cytoplasm translocation of HOX.
3.3. HXR9 blocks the pro‐proliferation function of HOXB7/HOXC6/HOXC8 in vitro and in vivo
We have mentioned that knock down of HOXB7/HOXC6/HOXC8 inhibited ESCC cell proliferation. In the present study, we wanted to test whether HXR9, a HOX/PBX antagonist, could have a similar effect through blocking HOX function. First, we treated ESCC cells with varying concentrations (10, 20, 40, 60, 80, 160 μmol/L) of HXR9 or CXR9 for 24 hours, then CCK‐8 assay was used to examine the proliferation rate and determine the IC50. The results indicated that the IC50 of HXR9 for KYSE70, KYSE150 and KYSE450 were 68.6, 66.76 and 78.19 μmol/L, whereas the IC50 of CXR9 were 8459, 3475, and 4220 μmol/L, respectively (Figure 2A). Thus, KYSE450 was relatively insensitive to HXR9 whereas KYSE70 and KYSE150 were significantly more sensitive, but no cell line was sensitive to CXR9. Moreover, non‐malignant human esophagus epithelial cell (HEEC) was significantly less sensitive to HXR9. The results signified that HXR9 was cytotoxic to ESCC cells. Then, we treated ESCC cell lines KYSE70, KYSE150 and KYSE450 with 60 μmol/L HXR9 or CXR9 and a colony formation assay was carried out. The colony formation rate of HXR9‐treated cells was significantly reduced, as compared with that of CXR9‐treated cells and control cells (Figure 2B). Level of colony formation in CXR9‐treated cells showed no significant difference compared with that of control cells. Meanwhile, we found that the PI3K‐AKT pathway was inhibited in HXR9‐treated cells, which was directly related to cell proliferation (Figure 2C).
Figure 2.
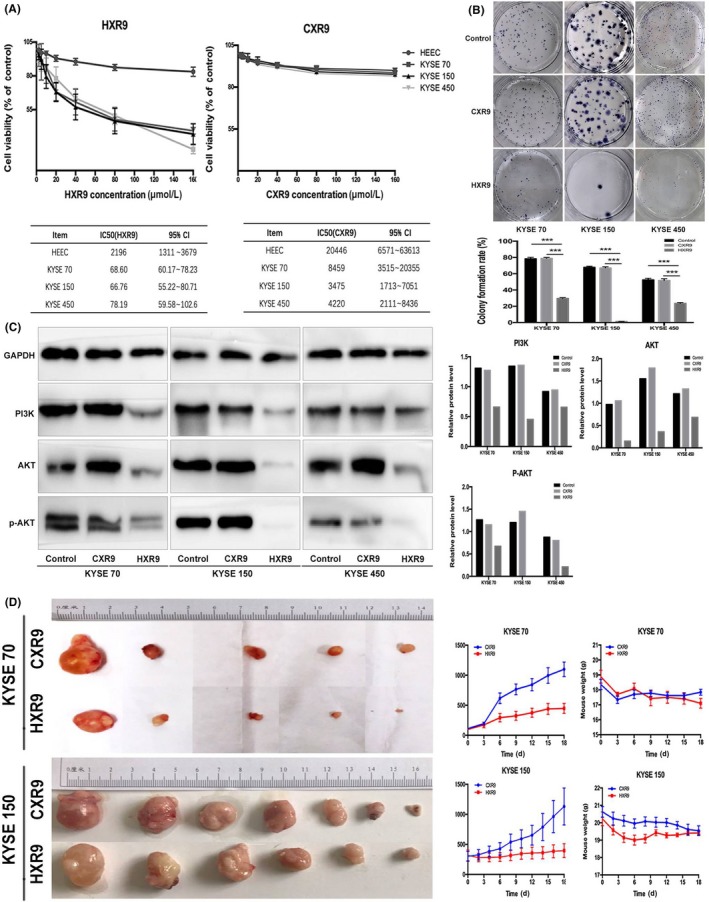
HXR9 inhibits esophageal squamous cell carcinoma cell proliferation and retards tumor growth in vivo. A, KYSE70, KYSE150, KYSE450 and HEEC (normal human esophagus epithelial cell) were seed in 96‐well plates, and then treated for 24 h with gradient concentrations of HXR9 or CXR9. Cell viability was assessed using CCK‐8 staining. Results are presented as percentage of viable cells (mean ± SEM), averaged from three independent experiments, each with four replicates. HXR9 inhibited proliferation of KYSE70, KYSE150 and KYSE450 compared to HEEC; all cell lines tested did not show apparent cytotoxicity for CXR9. B, KYSE70, KYSE150, KYSE450 were treated with HXR9 or CXR9 for 8 h prior to seeding in 6‐well plates. After 2 weeks, obtained colonies were visualized and counted. C, HXR9 treatment inhibited PI3K‐AKT pathway activation in all cell lines tested. Densitometry quantification of PI3K and AKT expression is presented. D, KYSE70 and KYSE150 cells were s.c. injected into the right groin of nude mice. When average tumor volume reached 100 mm3 for KYSE70 and 200 mm3 for KYSE150, mice were given an initial dose of HXR9 of 100 mg/kg (i.v. or injected directly into tumors), followed by twice weekly treatments at 10 mg/kg. Tumor volume was measured every 2–3 days. Tumor growth curves indicated that HXR9 slowed down tumor growth significantly compared to CXR9, and mice given HXR9 showed significant tumor volume reduction from day 6. However, the HXR9‐treated mice did not suffer greater weight loss. ***P < 0.05
In order to assess the efficacy of HXR9 in vivo, we established xenograft models of KYSE70 and KYSE150 cells in nude mice by s.c. injecting cells into the right groin. When the average tumor volume reached 100 mm3 for KYSE70 and 200 mm3 for KYSE150, mice were given an initial dose of HXR9 of 100 mg/kg (i.v. or injected directly into tumors), followed by twice weekly treatments at 10 mg/kg. Tumor volume was measured every 2–3 days. After 18 days, tumors were removed and weighed. As Figure 2D shows, growth of tumors treated with HXR9 was significantly slower than that of tumors treated with CXR9. On day 6, tumor growth inhibition (TGI) of xenograft tumor was 52.5% and 32.6% for KYSE70 and KYSE150, respectively. On day 18, TGI of xenograft tumor was 59.3% and 65.0% for KYSE70 and KYSE150, respectively. However, body weight loss in mice treated with HXR9 was not significantly greater than that in mice treated with CXR9, indicating that HXR9 had no overt toxicity to mice.
3.4. HXR9 induces apoptosis in ESCC cells in vitro and in vivo
In order to establish whether HXR9 induces apoptosis, HXR9‐ or CXR9‐treated cells were analyzed by FACS after staining with Annexin V‐FITC and propidium iodide (PI). It was shown that HXR9‐treated cells had a significant increment in cell apoptosis (Figure 3A). Also, HXR9‐treated cells underwent obvious morphological changes, which was observed under microscope, with cell shrinkage and pieces of cell debris (Figure 3B). However, there was no significant corresponding increase in Caspase‐3 and PARP activity over 2 hours with the same concentration of HXR9. Similar results were obtained for KYSE70, KYSE150 and KYSE450 cell lines (Figure 3C). Previous studies have suggested that upregulation of c‐fos could trigger apoptosis.21, 23, 24, 25, 26 c‐fos showed significant upregulation in HXR9‐treated KYSE70 and KYSE450. Meanwhile, we also found that p‐STAT6, a transcription activator of BCL2L1/BCL‐X(L) which is responsible for anti‐apoptosis, was downregulated in HXR9‐treated cells (Figure 3C). To investigate whether HXR9 triggers apoptosis in vivo, we detected the expression of Caspase‐3, PARP and c‐fos in tumors removed from xenograft models, and found that they were remarkably upregulated in HXR9‐treated tumors (Figure 3D).
Figure 3.
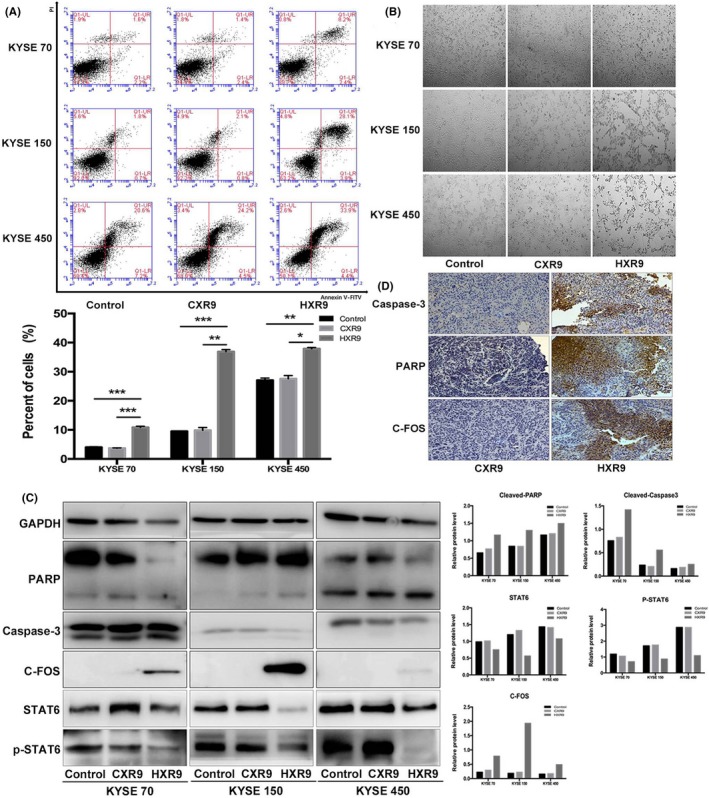
HXR9 induced esophageal squamous cell carcinoma cell apoptosis. A, Effect of HXR9 or CXR9 on cell apoptosis was assessed by flow cytometry using Annexin V/PE after treatment for 2 h. The apoptosis rate for HXR9‐treated cells was significantly higher than that for CXR9‐treated cells. B, Light micrographs of KYSE70, KYSE150 and KYSE450 cells treated with 60 μmol/L CXR9 or HXR9 for 2 h. A great deal of cell shrinkage and pieces of cell debris was observed in HXR9‐treated cells. C, Several apoptosis indicators were analyzed using western blotting. Caspase‐3 and poly ADP ribose polymerase (PARP) activity did not increase significantly over 2 h with HXR9 treatment. However, c‐fos showed significant upregulation in HXR9‐treated KYSE70 and KYSE450 cells. Meanwhile, p‐signal transducer and activator of transcription (p‐STAT)6 was downregulated in HXR9‐treated cells, which was an indicator of anti‐apoptosis. D, In tumors removed from animals, expression of Caspase‐3, PARP and c‐fos showed remarkable upregulation by HXR9 treatment. *P < 0.05, **P < 0.01 and ***P < 0.001
3.5. HXR9 causes transcription alteration
HXR9 is considered a specific, competitive inhibitor of HOX/PBX interaction by mimicking the “hexapeptide” sequence of HOX proteins and blocking HOX/PBX dimer formation, thus preventing HOX/PBX from binding to DNA. To identify possible target genes that are regulated by a HOX/PBX dimer, and are thus differentially expressed upon treatment with HXR9, we carried out RNA‐seq to study the transcriptome of HXR9‐treated cells (KYSE70 and KYSE150). Differential expression gene (DEG) analysis identified 234 genes significantly altered in HXR9‐treated KYSE70 cells, with 138 genes upregulated and 96 genes downregulated, while 177 genes were significantly altered in HXR9‐treated KYSE150 cells, with 99 genes upregulated and 78 genes downregulated. This result indicated that the HOX/PBX dimers functioned to activate or repress transcription in ESCC cells. Intriguingly, these genes included MPL proto‐oncogene, thrombopoietin receptor, interleukin (IL)‐15, IL‐23A and IL‐24, which encode cytokines involved in regulating the activation of the JAK‐STAT signaling pathway or induction of apoptosis. In order to confirm that the expression was altered in HXR9‐treated cells, real‐time PCR was used to measure the relative number of transcripts in RNA (Figure 4A). Specifically, IL‐15, IL‐23A, IL‐24 were downregulated and MPL was upregulated in response to HXR9 treatment. All of the altered genes were then classified based on their involved signaling pathways and biological functions using Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Figure 4B) and gene ontology (GO) analysis (Figure 4C). We found that HXR9 treatment caused a wide array of biological changes including the JAK‐STAT pathway. These results implied that the dysregulated signaling pathway component may be the transcriptional target of HOX/PBX and the underlying mechanism of HXR9 efficacy.
Figure 4.
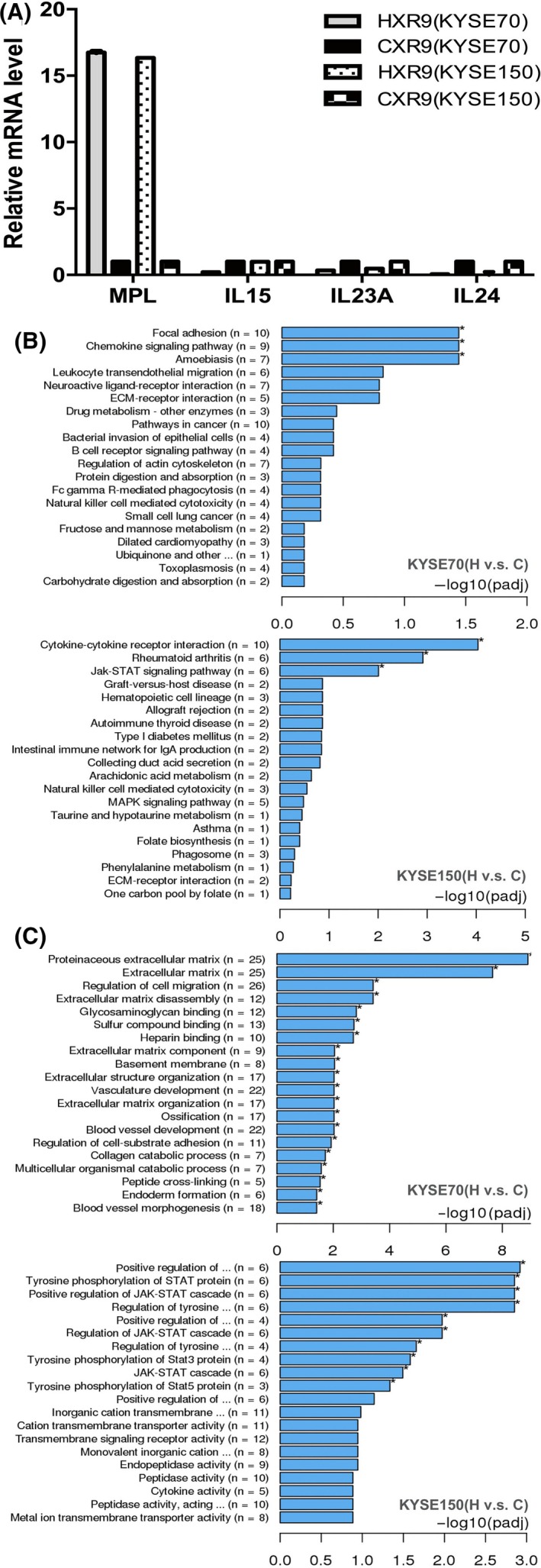
HXR9 causes transcription alteration. RNA was extracted from KYSE70 and KYSE150 cells treated with 60 μmol/L peptide for 2 h. A, Real‐time PCR of those genes identified altered by RNA‐seq in response to HXR9 treatment. Results are expressed as fold increase in transcript number in HXR9‐treated cells above the value for CXR9‐treated cells. B, Representation of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of significantly altered genes. C, Representation of gene ontology (GO) categories enrichment of altered genes
4. DISCUSSION
Previously, HOXB7, HOXC6 and HOXC8 were found to be dysregulated in ESCC tissues. To elucidate their significance in ESCC, it is necessary to clarify the association between their expression patterns and patient prognosis. It was shown that the survival time of patients with high HOXB7 or HOXC6 or HOXC8 expression was significantly shorter than that of patients with low expression, which is similar to the results of other groups.27, 28, 29 As reported, the consequences of dysregulated HOX genes in carcinogenesis can be interpreted as an extension of their normal function.30, 31, 32 Given that HOX genes can be viewed as global regulators of growth and differentiation, we investigated whether they could modulate the malignant phenotype in esophageal cancer. To this end, we established stable knockdown cell culture models by using RNAi, and explored their effect on cell proliferation, cell apoptosis, cell cycle progression and tumor growth in vivo. The results showed that cell viability and colony formation ability were significantly decreased in knockdown cells, as well as enhanced apoptosis, cell cycle arrest in G1 phase, and slowed tumor growth. This indicates that HOXB7/HOXC6/HOXC8 shows a high level of functional redundancy in causing ESCC, which commonly occurs during development. Consequently, it becomes difficult to interpret these data and determine the exact contribution of any of these individual genes to a malignant phenotype. In addition, the general difficulty in developing effective small molecule inhibitors against transcription factors have proven significant barriers to consider individual HOX genes as therapy targets.
However, the HOX/PBX dimer provides a potential solution to this problem. The functional redundancy of HOX is derived from the sequence similarity of the homeodomain in HOX protein and interaction with defined cofactors which stabilize HOX and modulate specificity of target DNA binding. HXR9, an inhibitor of the interaction between HOX and cofactor PBX, targets a large subset of HOX proteins (members of paralogue groups 1–9), which was previously shown to cause apoptosis and inhibit tumor survival in a variety of tumor types,23, 24, 25, 26 implying that the HOX/PBX interaction is a potential target for therapy. In the present study, we also showed that treating ESCC cells with HXR9 caused apoptosis and inhibited cell proliferation and tumor growth in all of the lines tested. It has been reported that a rapid increase in c‐fos expression as a response to HOX/PBX inhibition was activated to induce apoptosis,23, 24, 25, 33 which was also confirmed in our study. In addition, we found that PI3K‐AKT and JAK‐STAT pathway repression may be a response to antiproliferation and proapoptosis. Previously, it was reported that HOXB7 was possibly involved in regulating the downstream PI3K/AKT pathway in a study identifying potential chromatin binding sites of HOXB7.34 In addition, HOX/PBX inhibition also influenced transcriptional activity of a subset of genes, which varied between different cell lines. This means that HOX/PBX inhibition specifically modifies HOX function through diverse mechanisms. It is worth noting that several genes encoding cytokines such as MPL, IL‐15, and IL‐23A were altered, and these are involved in JAK‐STAT pathway activation. Upon binding of these cytokines, they are dimerized such that JAK kinase domains face each other in a productive conformation for transactivation and the cytokine receptors become phosphorylated in the cytoplasm tail.35 The phosphorylated receptors and JAK themselves become scaffolds for the members of the STAT family, which in turn are phosphorylated and homo/hetero‐dimerized before translocating to the nucleus. It was also demonstrated that HOX initially induced a cascade of signaling molecules including the JAK/STAT signaling pathway. Then, at a later stage, STAT activity feeds back directly to HOX, triggering transformation of the HOX cascade into a gene‐network during development.36 Thus, we hypothesize that HXR9 may disrupt HOX/PBX functions through modifying cytokine‐JAK‐STAT pathway activation.
Although growing evidence has suggested since 2007 that the HOX/PBX dimer is a potential therapeutic target in both solid and hematological malignancies,21 the mechanism by which HOX/PBX inhibition represses tumor survival remains to be fully elucidated.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This work was supported financially by National Key R&D Program of China (2017YFC0907500, 2017YFC0907504), and Beijing Municipal Administration of Hospitals Incubating Program (PX2018044) on laboratory reagents.
Shen L‐Y, Zhou T, Du Y‐B, Shi Q, Chen K‐N. Targeting HOX/PBX dimer formation as a potential therapeutic option in esophageal squamous cell carcinoma. Cancer Sci. 2019;110:1735–1745. 10.1111/cas.13993
Shen and Zhou contributed equally to this work.
REFERENCES
- 1. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics. CA Cancer J Clin. 2012;62(4):220‐241. [DOI] [PubMed] [Google Scholar]
- 2. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843‐850. [DOI] [PubMed] [Google Scholar]
- 3. Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349‐354. [DOI] [PubMed] [Google Scholar]
- 4. Teo WW, Merino VF, Cho S, et al. HOXA5 determines cell fate transition and impedes tumor initiation and progression in breast cancer through regulation of E‐cadherin and CD24. Oncogene. 2016;35(42):5539‐5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grier DG, Thompson A, Kwasniewska A, et al. The pathophysiology of HOX genes and their role in cancer. J Pathol. 2005;205:154‐171. [DOI] [PubMed] [Google Scholar]
- 6. Abate‐Shen C. Deregulated homeobox gene expression in cancer: cause or consequence? Nat Rev Cancer. 2002;2:777‐785. [DOI] [PubMed] [Google Scholar]
- 7. Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J Mol Med. 2014;92(8):811‐823. [DOI] [PubMed] [Google Scholar]
- 8. Zhai Y, Kuick R, Nan B, et al. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007;67:10163‐10172. [DOI] [PubMed] [Google Scholar]
- 9. Gu ZD, Shen LY, Wang H, et al. HOXA13 promotes cancer cell growth and predicts poor survival of patients with esophageal squamous cell carcinoma. Cancer Res. 2009;69(12):4969‐4973. [DOI] [PubMed] [Google Scholar]
- 10. Morgan R, Simpson G, Gray S, et al. HOX transcription factors are potential targets and markers in malignant mesothelioma. BMC Cancer. 2016;16:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kelly Z, Moller‐Levet C, McGrath S, et al. The prognostic significance of specific HOX gene expression patterns in ovarian cancer. Int J Cancer. 2016;139(7):1608‐1617. [DOI] [PubMed] [Google Scholar]
- 12. Se YB, Kim SH, Kim JY, et al. Underexpression of HOXA11 is associated with treatment resistance and poor prognosis in glioblastoma. Cancer Res Treat. 2017;49(2):387‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen KN, Gu ZD, Ke Y, et al. Expression of 11 HOX genes is deregulated in esophageal squamous cell carcinoma. Clin Cancer Res. 2005;11(3):1044‐1049. [PubMed] [Google Scholar]
- 14. Du YB, Dong B, Shen LY, et al. The survival predictive significance of HOXC6 and HOXC8 in esophageal squamous cell carcinoma. J Surg Res. 2014;188(2):442‐450. [DOI] [PubMed] [Google Scholar]
- 15. Li H, Shen LY, Yan WP, et al. Deregulated HOXB7 expression predicts poor prognosis of patients with esophageal squamous cell carcinoma and regulates cancer cell proliferation in vitro and in vivo. PLoS One. 2015;10(6):e0130551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Longobardi E, Penkov D, Mateos D, et al. Biochemistry of the tale transcription factors PREP, MEIS, and PBX in vertebrates. Dev Dyn. 2014;243:59‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mann RS, Affolter M. Hox proteins meet more partners. Curr Opin Genet Dev. 1998;8:423‐429. [DOI] [PubMed] [Google Scholar]
- 18. Piper DE, Batchelor AH, Chang CP, et al. Structure of a HoxB1–Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell. 1999;96(4):587‐597. [DOI] [PubMed] [Google Scholar]
- 19. Abu‐Shaar M, Ryoo HD, Mann RS. Control of the nuclear localization of Extradenticle by competing nuclear import and export signals. Genes Dev. 1999;13:935‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morgan R, In der Rieden P, Hooiveld MH, et al. Identifying HOX paralog groups by the PBX‐binding region. Trends Genet. 2000;16(2):66‐67. [DOI] [PubMed] [Google Scholar]
- 21. Morgan R, El‐Tanani M, Hunter KD, et al. Targeting HOX/PBX dimers in cancer. Oncotarget. 2017;19:32322‐32331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi Q, Shen LY, Dong B, et al. The identification of the ATR inhibitor VE‐822 as a therapeutic strategy for enhancing cisplatin chemosensitivity in esophageal squamous cell carcinoma. Cancer Lett. 2018;432:56‐68. [DOI] [PubMed] [Google Scholar]
- 23. Morgan R, Boxall A, Harrington KJ, et al. Targeting the HOX/PBX dimer in breast cancer. Breast Cancer Res Treat. 2012;136:389‐398. [DOI] [PubMed] [Google Scholar]
- 24. Morgan R, Pirard PM, Shears l, et al. Antagonism of HOX/PBX dimer formation blocks the in vivo proliferation of melanoma. Cancer Res. 2007;67:5806. [DOI] [PubMed] [Google Scholar]
- 25. Morgan R, Plowright L, Harrington KJ, et al. Targeting HOX and PBX transcription factors in ovarian cancer. BMC Cancer. 2010;10:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Plowright L, Harrington K, Pandha H, et al. HOX transcription factors are potential therapeutic targets in nonsmall‐cell lung cancer (targeting HOX genes in lung cancer). Br J Cancer. 2009;100:470‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Long QY, Zhou J, Zhang XL, et al. HOXB7 predicts poor clinical outcome in patients with advanced esophageal squamous cell cancer. Asian Pac J Cancer Prev. 2014;15(4):1563‐1566. [DOI] [PubMed] [Google Scholar]
- 28. Xie X, Zhang SS, Wen J, et al. Prognostic value of HOXB7 mRNA expression in human oesophageal squamous cell cancer. Biomarkers. 2013;18(4):297‐303. [DOI] [PubMed] [Google Scholar]
- 29. Malek R, Gajula RP, Williams RD, et al. TWIST1‐WDR5‐Hottip regulates Hoxa9 chromatin to facilitate prostate cancer metastasis. Cancer Res. 2017;77(12):3181‐3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang X, Liu G, Ding L, et al. HOXA3 promotes tumor growth of human colon cancer through activating EGFR/Ras/Raf/MEK/ERK signaling pathway. J Cell Biochem. 2018;119(3):2864‐2874. [DOI] [PubMed] [Google Scholar]
- 31. Guan Y, He Y, Lv S, et al. Overexpression of HOXC10 promotes glioblastoma cell progression to a poor prognosis via the PI3K/AKT signaling pathway. J Drug Target. 2018;16:1735‐19. [DOI] [PubMed] [Google Scholar]
- 32. Morgan R, Boxall A, Harrington KJ, et al. Targeting HOX transcription factors in prostate cancer. BMC Urol. 2014;14:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vainchenker W, Leroy E, Gilles L, et al. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res. 2018;7:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heinonen H, Lepikhova T, Sahu B, et al. Identification of several potential chromatin binding sites of HOXB7 and its downstream target genes in breast cancer. Int J Cancer. 2015;137(10):2374‐2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27(12):1984‐2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pinto PB, Espinosa‐Vázquez JM, Rivas ML, et al. JAK/STAT and Hox dynamic interactions in an organogenetic gene cascade. PLoS Genet. 2015;11(7):e1005412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials