

Abstract
Females develop kidney stones less frequently than males do. However, it is unclear if this gender difference is related to altered estrogen/estrogen receptor (ER) signaling. Here, we found that ER beta (ERβ) signals could suppress hepatic oxalate biosynthesis via transcriptional upregulation of the glyoxylate aminotransferase (AGT1) expression. Results from multiple in vitro renal cell lines also found that ERβ could function via suppressing the oxalate-induced injury through increasing the reactive oxygen species (ROS) production that led to a decrease of the renal calcium oxalate (CaOx) crystal deposition. Mechanism study results showed that ERβ suppressed oxalate-induced oxidative stress via transcriptional suppression of the NADPH oxidase subunit 2 (NOX2) through direct binding to the estrogen response elements (EREs) on the NOX2 5′ promoter. We further applied two in vivo mouse models with glyoxylate-induced renal CaOx crystal deposition and one rat model with 5% hydroxyl-L-proline-induced renal CaOx crystal deposition. Our data demonstrated that mice lacking ERβ (ERβKO) as well as mice or rats treated with ERβ antagonist PHTPP had increased renal CaOx crystal deposition with increased urinary oxalate excretion and renal ROS production. Importantly, targeting ERβ-regulated NOX2 with the NADPH oxidase inhibitor, apocynin, can suppress the renal CaOx crystal deposition in the in vivo mouse model. Together, results from multiple in vitro cell lines and in vivo mouse/rat models all demonstrate that ERβ may protect against renal CaOx crystal deposition via inhibiting the hepatic oxalate biosynthesis and oxidative stress-induced renal injury.
1. Introduction
Kidney stone is a common disorder that poses a significant health care burden in a working-age population [1]. The incidence of kidney stone disease is lower in premenopausal women than age-matched men, as well as in female animal models with renal calcium oxalate (CaOx) crystal deposition [2–6]. However, this disparity becomes less prominent in the sixth decade of life in parallel with the onset of menopause in women [7, 8]. These studies may indicate a protective function of 17β-estradiol (E2) in the kidney stone formation. However, results from the Women's Health Initiative (NHI) study and Nurse's Health Study all failed to find a positive linkage of hormone replacement therapy (HRT) with the kidney stone prevention [7–9]. It is still not clear why the beneficial effects of HRT were lost in these postmenopausal females. Turner and Kerber [9] proposed that long periods of hypoestrogenicity due to menopause may exacerbate the deterioration of the normal physiological ER function in the kidney, which may make “re-estrogenization”, by HRT, less beneficial. The possible reasons could be the reduced protein expressions of estrogen receptors (ERs) or their accessory factors in these women.
Estrogen actions are mediated mainly by two classical ER subtypes, ERα and ERβ. Both ERα and ERβ belong to the nuclear receptor superfamily and are genetically and functionally distinct [10]. Distribution of ERα and ERβ also varies in different tissues and may play distinct roles in various disorders including hypertension [11] and brain injury [12]. At present, there is limited knowledge whether E2 may alter the development of kidney stones, and if it does, it remains unclear whether ERα, ERβ, or both may be involved. In addition to inducing the nuclear genomic function, estrogens may induce rapid nongenomic responses from membrane-associated receptors, such as growth factor receptors and G protein-coupled estrogen receptor [13]. It remains to be investigated how E2 and its receptors affect the kidney stone formation.
The results from our previous nationwide cross-sectional survey on the prevalence of kidney stone formers in China of 3792 men and 5518 women revealed that the prevalence of kidney stone formers was 6.6% for men and 5.2% for women, suggesting that the gender difference existed among the kidney stone formers [14], with a lower incidence of kidney stone formers in the females. Together, all animal study data and clinical data analyses suggest that estrogens and ERs may play critical roles in renal CaOx crystal formation; however, the underlying mechanisms remain largely unclear.
CaOx kidney stones are the most prevalent solid-phase stones with a recurrence rate of approximately 40% at 5 years after the first treatment [15]. Hyperoxaluria plays key roles in CaOx kidney stone formation. The liver is the main organ of endogenous oxalate synthesis, in which glyoxylate aminotransferase (AGT1), glyoxylate aminotransferase 2 (AGT2), and glycolate oxidase (GO) are involved [16]. In addition, an increase of reactive oxygen species (ROS) and NADPH oxidase in the kidney gives rise to inflammation and injury of renal tubular cells, which may promote CaOx crystal formation [17].
Using multiple in vitro cell studies and in vivo mouse/rat models, here we found that ERβ could play a protective role in suppressing renal CaOx crystal deposition via inhibiting the hepatic oxalate biosynthesis and oxidative stress-induced renal cell injury.
2. Results
2.1. ERβ Decreases Hepatic Oxalate Biosynthesis via Increasing AGT1 Expression in Hepatocytes
To determine if the ER functions were involved in renal CaOx crystal deposition, we first examined the ER effects on the liver oxalate biosynthesis in the liver cells, and it was important to clarify the nonperoxisomal metabolism associated with oxalate synthesis in human hepatocytes [18]. Using the shRNA knockdown strategy, our results revealed that knockdown of the ERβ, but not the ERα, led to an increased oxalate excretion (Figure 1(a)). Using the lentiviral ER-cDNA overexpression (oe) method we found that oeERβ, but not oeERα, led to decreased oxalate excretion in the culture media of HepG2 cells. The above data suggests that hepatic ERβ, and not ERα, might downregulate oxalate biosynthesis, which consequently led to decreased oxalate excretion.
Figure 1.

ERβ decreases oxalate biosynthesis via increasing AGT1 expression in hepatocytes. (a) Oxalate level measurement in culture media of two sets of HepG2 cells: (i) shLuc, shERα, and shERβ and (ii) vector, oeERα, and oeERβ. sh = small hairpin RNA; oe = overexpression. (b) The mRNA expressions of the enzymes AGT1, AGT2, and GO upon manipulation of the ERβ level in HepG2 cells. (c) Protein expressions of the enzymes AGT1 and AGT2 upon manipulation of the ERβ level in HepG2 cells. (d) ChIP assay results in HepG2 cells showed that overexpression of ERβ (b) increased the physical binding between ERβ and both ERE I and ERE II. (e). Wild-type or two ERE-mutant AGT1 promoter reporter constructs were cotransfected with pRL-TK at a ratio of 1000 : 1 into HepG2-vector and HepG2-oeERβ cells (left panel). Luciferase reporter assay was performed after 48 hr incubation (right panel). Data in (a), (b), and (e) are presented as mean ± SD, n.s: no significance. ∗ P < 0.05, ∗∗ P < 0.01, and ∗∗∗ P < 0.001.
We then examined the expression of enzymes involved in the biosynthesis of oxalate in liver and found that targeting ERβ with ERβ-shRNA led to little effect on the expression of GO. However, altering the ERβ via ERβ-shRNA or ERβ-cDNA led to a significant alteration of both mRNA and protein expressions of AGT1, another key molecule [19] in controlling the removal of glyoxylate and prevention of its conversion to oxalate in the liver (Figures 1(b) and 1(c)).
Together, results from Figures 1(a)–1(c) suggest that ERβ could downregulate the liver oxalate biosynthesis via increasing AGT1 expression.
2.2. Mechanism Dissection of Why ERβ Can Increase AGT1 Expression in Hepatocytes: via Transcriptional Regulation
As we found ERβ altered the AGT1 expression in both mRNA and protein levels we then assayed ERβ's role in regulating AGT1 at the transcriptional level. We first searched for potential estrogen response elements (EREs) on the ~3.9 kb of the AGT1 promoter region using JASPAR database and found two putative EREs located within the AGT1 promoter region (Figure 1(d), upper panel). We then applied chromatin immunoprecipitation (ChIP) in vivo binding assay to verify their capacity of ERβ binding to EREs, and results revealed that ERβ could bind to the ERE located on sites -3774 to -3760 and -144 to -130 bp away from the transcription start site of AGT1 in ERβ-overexpressed HepG2 cells (Figure 1(d), lower panels). Importantly, ERβ could increase the luciferase activity of the pGL3 construct of ~3.9 kb of the AGT1 promoter with wild-type ERE, but not with mutant ERE (Figure 1(e)). Together, results from Figures 1(a)–1(e) suggest that ERβ can decrease liver oxalate biosynthesis via increasing the AGT1 expression at the transcriptional regulation of the AGT1 promoter.
2.3. ERβ Decreases the Oxalate-Induced Oxidative Stress in the Human Renal Tubular Cells
In addition to suppressing oxalate biosynthesis in the liver, we were interested in studying ERβ's effects on altering the oxidative stress-induced renal cell injury as one of the potential factors impacting the renal CaOx crystal deposition.
Early studies indicated that increased renal oxalate might increase the renal injury, which might then further increase the renal CaOx crystal deposition [20, 21]. Other studies also suggested that urinary oxalate excretion in normal people without kidney stone diseases ranged from 0.31 to 0.38 mM [22], which might then increase to 0.46 to 0.53 mM in idiopathic CaOx stone patients and further increase to 0.79 to 0.90 mM in enteric hyperoxaluria patients and 0.92 to 1.43 mM in bariatric surgery patients [23]. We decided to follow previous studies [24, 25] using 0.75 mM oxalate for exploring hyperoxaluria-induced renal CaOx crystal deposition in in vitro renal cell studies.
We first found that ERβ knockdown (using shERβ), but not ERα knockdown, could alter oxidative stress via increasing intracellular superoxide levels (Figures 2(a)–2(c)) with elevated H2O2 release (Figure 2(d)) in renal HK-2 and HKC-8 cells after exposure to 0.75 mM oxalate for 6 hr, suggesting that targeting ERβ can alter the oxidative stress in renal cells with higher oxalate concentration.
Figure 2.

Depletion of renal ERβ made renal epithelial cells more vulnerable to oxalate-induced ROS production and cell injury. (a) HK-2 and HKC-8 cells were transduced with lentiviral shLuc (control) or shERα or shERβ and treated with normal DMEM media or DMEM media containing 0.75 mM oxalate for 6 hr, and the ROS production in the cells was detected by dihydroethidium (DHE) staining under a fluorescence microscope. The images are representative of typical staining. (b) Digital scans of DHE-stained cells were quantified using ImageJ software by comparing cells with shERα or shERβ to shLuc. One-way ANOVA was applied for statistic analysis. (c) Western blots show ERα or ERβ knockdown efficiency in HK-2 and HKC-8 cells. (d) Detection of H2O2 levels in culture media of the shERα or shERβ or control (shLuc) renal tubular epithelial cells after challenge with 0.75 mM oxalate for 6 hr; Student's t tests, compared to the shLuc group. (e) LDH release measurement in the ERα or ERβ knocked-down renal tubular epithelial cells treated with 0.75 mM oxalate for 6 hr. (f) The qRT-PCR analysis of inflammation-related gene expression in HK-2 and HKC-8 cells with/without sh ERβ after 0.75 mM oxalate treatment for 6 hr. (g–j) Effect of ER antagonists on oxalate induced oxidative stress and cell injury. HK-2 cells were exposed to DMSO (Ctrl), 0.75 mM oxalate, oxalate + 10 nM E2, oxalate + 10 nM E2 + 10 μM ICI, oxalate + 10 nM E2 + 10 μM MPP, or oxalate + 10 nM E2 + 10 μM PHTPP for 6 hr. (g) The ROS production in the cells was detected by DHE staining under a fluorescence microscope. The images are representative of typical staining. (h) Digital scans of DHE-stained cells were quantified using ImageJ software; one-way ANOVA, compared to the control group. (i) Detection of H2O2 levels in culture media of each group. (j) LDH release measurement in cells with different treatments. For (b)–(f) and (h)–(j), data are presented as mean ± SD. ns = not significant. ∗ P < 0.05, ∗∗ P < 0.01, ∗∗∗ P < 0.001, and ∗∗∗∗ P < 0.0001.
Next, we found that knocking down ERβ, but not ERα, in HK-2 cells also increased the oxidative stress-induced renal cell injury (cytotoxicity) via measuring LDH activity after exposure to 0.75 mM oxalate for 6 hr (Figure 2(e)). Similar results were also obtained when we used the 2nd ERβ-shRNA (Supplementary Figure 1).
Importantly, we also that found knockdown of ERβ in HK-2 and HKC-8 cells led to an increased expression of oxidative stress/inflammation-related genes, including osteopontin (OPN), interleukin-6 (IL-6), and monocyte chemotactic protein 1 (MCP-1), at mRNA levels after exposure to 0.75 mM oxalate for 6 hr (Figure 2(f)).
Furthermore, we tested 3 different ER antagonists: (i) ICI 182,780 is an antagonist for both ERα and ERβ, (ii) 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo(1,5-a)pyrimidin-3-yl]phenol (PHTPP) is a ERβ-selective antagonist, and (iii) methylpiperidino pyrazole (MPP) is a selective ERα antagonist. HK-2 cells were cultured under media with CS-FBS for 48 hr; as expected, 10 nM E2 treatment could downregulate oxalate-mediated oxidative stress and cell injury via reducing the superoxide production. When HK-2 cells were further treated with 10 μM antagonists, we found that ICI 182,780 or ERβ special antagonist PHTPP, but not the ERα antagonist MPP, could significantly increase oxalate-mediated oxidative stress and cell injury via inducing the superoxide production (Figures 2(g)–2(j)).
Together, results from Figures 2(a)–2(j) demonstrate that targeting E2/ERβ signaling via (i) antiestrogens to suppress E2-activated ERβ or (ii) knocking down ERβ by shRNA to suppress the ERβ all led to increasing oxidative stress-induced cell injury in human renal epithelial cells (with 0.75 mM oxalate treatment), suggesting that activation of ERβ functions may protect renal tubular cells against oxidative stress-induced renal injury in patients with hyperoxaluria.
2.4. Mechanism Dissection of Why ERβ Suppresses the Oxidative Stress-Induced Renal Cell Injury: via Suppressing the NADPH Oxidase Subunit NOX2 Expression
To dissect the mechanism(s) of how ERβ can decrease the oxalate-mediated oxidative stress-induced renal cell injury, we first focused on NADPH oxidases, the key components related to ROS in renal tubular epithelial cells [26], including NOX1-5, p22-phox, p47-phox, p67-phox, and Rac1 [27]. Among those genes, NOX2 expression is upregulated in both cell lines, yet NOX4 expression is increased in only the HKC-8 cells; therefore, we choose NOX2 for further functional studies. Using qRT-PCR and western blot assays, we found that knocking down ERβ in HK-2 or HKC-8 cells led to an increase in the expression of NOX2 at mRNA and protein levels after exposure to 0.75 mM oxalate for 6 hr (Figures 3(a) and 3(b), respectively). Consistent results were also obtained when we replaced ERβ-shRNA with ER antagonist ICI or ERβ special antagonist PHTPP (see Figure 4(a)).
Figure 3.

ERβ inhibits oxalate-induced oxidative stress via modulating NOX2 expression. (a) The qRT-PCR analysis of NADPH oxidase subunits after knocking down ERβ in HK-2 and HKC-8 cells. Cells were transduced with shERβ or with shLuc as control after 72 hr incubation, and then treated with 0.75 mM oxalate for 6 hr. (b) Western blot analysis of NOX2 expression in renal cells with/without shERβ after 6 hr exposure to 0.75 mM oxalate. (c) Rescue assay using HK-2 and HKC-8 cells with/without shERβ. shNOX2 showed partially reversed knockdown of ERβ- (shERβ-) induced ROS production. All cells were exposed to 0.75 mM oxalate for 6 hr prior to collection for data analysis. Upper panels show representative images of DHE staining, and quantification is shown in the lower left panel. Western blot in the lower right panel shows NOX2 knockdown efficiency. (d) After treating with 0.75 mM oxalate for 6 hr, shNOX2 can partly reverse the silenced ERβ-induced H2O2 production in conditioned media. (e) Rescue assay using HK-2 and HKC-8 cells with/wthout shNOX2 showed partially reversed shERβ-induced LDH release. All the cells in Figures 3(a)–3(e) were exposed to 0.75 mM oxalate for 6 hr prior to collection for data analysis. For (a), (c), (d), and (e), data are presented as mean ± SD. ∗ P < 0.05, ∗∗ P < 0.01, and ∗∗∗ P < 0.001.
Figure 4.

Mechanism dissection on how ERβ regulates NOX2 expression. (a) NOX2 mRNA levels were regulated by E2 or ERβ antagonists in HK-2 and HKC-8 cells. Cells were treated for 6 hr with vehicle (control), 0.75 mM oxalate, 0.75 mM oxalate + 10 nM E2, 0.75 mM oxalate + 10 nM E2 + 10 μM ICI, or 0.75 mM oxalate + 10 nM E2 + 10 μM PHTPP for q-PCR of NOX2 mRNA levels. Bonferroni's test was applied for statistical analysis of results. (b) Predicted 3 potential EREs located in the 3 kb-NOX2 promoter region. (c) ChIP was performed in HK-2 cells, and only ERE I of the NOX2 promoter could be bound by ERβ. (d) ERE I wild-type or mutant of NOX2 promoter reporter constructs were cotransfected with pRL-TK at a ratio of 1000 : 1 into HK-2-shLuc and HK-2-shERβ cells. Luciferase reporter assay was performed after 48 hr incubation. For (a) and (d), data shown are mean ± SD. ∗ P < 0.05 and ∗∗ P < 0.01. n.s. = not significant.
We then applied an interruption approach using NOX2-shRNA to prove that ERβ could function via decreasing NOX2 expression to inhibit oxalate-mediated, oxidative stress-induced, renal injury. As expected, the results from 3 different assays including dihydroethidium (DHE) staining (Figure 3(c)), H2O2 release assay (Figure 3(d)), and LDH assay (Figure 3(e)) all revealed that knocking down NOX2 with NOX2-shRNA led to partial reversal of the ERβ-shRNA-increased, oxalate-mediated, oxidative stress-induced cell injury in the renal HK-2 and HKC-8 cells.
Together, results from Figures 3(a)–3(e) suggest that ERβ suppresses the oxalate-mediated oxidative stress-induced renal cell injury via suppressing the NOX2 expression.
2.5. Mechanism Dissection of How ERβ Suppresses NOX2 Expression: via Transcriptional Regulation
To further dissect the molecular mechanism of how ERβ regulates NOX2 expression in renal tubular epithelial cells, we first noticed that the NOX2 mRNA expressions were increased in both HK-2 and HKC-8 cells after 6 hr of 0.75 mM oxalate exposure (Figure 4(a)). As expected, treating with 10 nM E2 prevented oxalate-induced NOX2, and this reduction was reversed after treating with nonselective ER antagonist ICI or selective ERβ antagonist PHTPP, suggesting that a transcriptional mechanism may contribute to NOX2 downregulation by ERβ (Figure 4(a)).
We then searched for potential EREs on the 3 kb of the NOX2 promoter region using the JASPAR database, and results revealed three putative EREs located within the NOX2 promoter region (Figure 4(b)). We then performed the ChIP in vivo binding assay, and results revealed that ERβ could bind to ERE-I located -842 to -825 bp upstream of the transcriptional start site of NOX2 in HK-2 cells (Figure 4(c)). Furthermore, using the luciferase assay to examine 1.3 kb of the NOX2 promoter region that was linked to the pGL3-luciferase reporter plasmid, we found that knocking down ERβ with shRNA (shERβ) in the HK-2 cells could increase the luciferase expression of wild-type NOX2 promoter construct, but not the NOX2 promoter construct with mutated ERE-1 (Figure 4(d)).
Together, results from Figures 4(a)–4(d) suggest that ERβ can suppress NOX2 expression at the transcriptional level via binding to the ERE located in its 5′ promoter region.
2.6. Using In Vivo Mouse Models with PHTPP Injection to Increase the Renal CaOx Crystal Deposition
To confirm all above in vitro cell line results with the in vivo mouse model, we first investigated whether PHTPP, a ERβ selective antagonist, could promote renal CaOx crystal deposition in a mouse model. C57BL/6J female mice were treated with DMSO or PHTPP (10 μl of 1 × 10−2 M PHTPP per mouse) via intraperitoneal injection on days 1, 3, 5, 7, and 9. In addition, all mice were given daily intraperitoneal injections of glyoxalate (80 mg/kg/day) from days 3 to 10 to induce renal CaOx crystal deposition (Figure 5(a), upper panel) [28]. The results revealed that mice with ERβ selective antagonist PHTPP treatment had more renal CaOx crystal deposition compared to controls (Figure 5(a), lower panel).
Figure 5.

In vivo mouse model data confirm that ERβ deficiency promotes renal CaOx crystal deposition via increasing the hepatic oxalate biosynthesis and renal oxidative stress. (a) Diagram describing the injection schedule for mock control, a selective ERβ antagonist PHTPP, and glyoxylate (upper panel). Crystal staining (lower panels) showing the deposition of CaOx crystals in kidney tissues of the PHTPP-treated and mock-control mice. CaOx crystal formation was detected by Pizzolato staining. Crystallization in each kidney section was quantified by calculating the ratio (percent) of the area containing crystals to the entire kidney section using ImageJ software. (b) Staining (left panels) and quantification (right panels) results for CaOx crystal deposition as detected by Pizzolato staining in kidney tissues of the ERβKO and their WT female littermate mice. (c) Detection of 24 hr oxalate excretion in urine samples collected from the ERβKO and WT female mice prior to sacrifice. (d) Western blot of 60 μg liver protein from ERβKO and WT mice probed with affinity-purified rabbit antibody raised against recombinant mouse AGT1 shows lower expression of AGT1 in the ERβKO mice. (e) Fresh-frozen renal tissues were stained for 30 min with DHE. Representative sections from the ERβKO and their WT female littermate mice are shown (left panels). Right bar graphs show quantification of superoxide generation. (f) H2O2 concentration in urine sample from ERβKO and WT female mice. (g) Representative micrographs of NOX2 immunostaining in renal tissue from the ERβKO and their WT littermate mice. The NOX2 protein was mainly located in renal tubular cells. Quantification of NOX2 is shown in the right panel. Scores were classified as 0 to 3, based on the intensity of staining and the percentage of positive cells. (h) Inhibitory effect of apocynin treatment in mice. CaOx crystal formation in ERβKO mice was significantly decreased after in vivo apocynin treatment. (i) Apocynin treatment reduced H2O2 concentration in urine samples from ERβKO mice. For (a)–(c) and (e)–(i), data are presented as mean ± SD. ∗ P < 0.05 and ∗∗ P < 0.01.
2.7. Using In Vivo ERKO Mouse Models Showing Deletion of ERβ Gene Led to Increase in Renal CaOx Crystal Deposition
We then applied the 2nd mouse model using ERβ knockout (ERβKO) mice via breeding heterozygous ERβKO (ERβ +/-) male and female mice to generate the homozygote ERβKO (ERβ −/−) mice that lack both copies of the ERβ genes (see Supplementary Figure 2 for breeding and genotyping of the homozygote ERβKO mice). These ERβKO mice, plus their control wild-type (WT) mice, were given daily intraperitoneal injections of glyoxalate (80 mg/kg/day) for 7 days to induce intrarenal CaOx crystal deposition in the parenchyma. The results from Pizzolato staining revealed that ERβKO mice had more renal CaOx crystal deposition than the WT littermate mice (Figure 5(b)).
Metabolism cages were used for control of diet and excretion of each mouse. One day before sacrifice, we collected urine samples and examined the 24 hr urinary oxalate excretion in ERβKO and WT mice and found that ERβKO mice had higher urinary oxalate than the WT littermate controls (Figure 5(c)). Consistently, we found that AGT1 expression was lower in the livers collected from ERβKO mice than in WT mice after sacrifice (Figure 5(d)).
We then examined oxidative stress via measuring the ROS production in these mouse kidneys using the reporter molecule DHE, and results revealed that the kidney sections of ERβKO mice had significantly stronger nuclear fluorescent signals as compared with those of WT mice (Figure 5(e)). As expected, prior to sacrifice, we found a much higher H2O2 concentration in the urine of ERβKO mice than found in WT mice (Figure 5(f)), and results from the NOX2 IHC data were scored and results showed a higher NOX2 expression in renal tubular cells of ERβKO mice compared to WT mice (Figure 5(g)).
Taken together, the results from two different mouse models confirm the in vitro cell line data showing that targeting E2/ERβ signaling with either antiestrogen (PHTPP) or ERβ (knockout gene) all lead to increased renal CaOx crystal deposition with increased urinary oxalate via altering the liver oxalate biosynthesis and renal oxidative stress-induced renal cell injury.
2.8. Using In Vivo ERKO Mouse Models to Prove the Treatment with NADPH Oxidase Inhibitor Apocynin Can Suppress the CaOx Crystal Deposition on the Parenchyma
Then, to connect all above in vitro/in vivo studies to potential future clinical applications, we applied the ERβKO mouse model and treated the mice with the small molecule NADPH oxidase inhibitor, apocynin [29–31]. Results revealed that injecting apocynin could partly reverse the ERβKO-increased renal CaOx crystal deposition (Figure 5(h)) and oxidative stress (via measuring the level of H2O2 in urine) (Figure 5(i)), suggesting that targeting the ERβ-regulated NOX2 expression with apocynin may present a potential novel therapy to suppress renal CaOx crystal deposition.
2.9. Using In Vivo Rat Models to Confirm ERβ Can Decrease the Intrarenal CaOx Crystal Deposition on the Parenchyma
To overcome the potential glyoxalate side effects to damage the kidney in the mouse [32], we also applied the 4th animal model using rats fed with hydroxy-L-proline (HLP) for 8 weeks to induce CaOx crystal deposition, since early studies indicated that this rat model has little acute renal injury and more closely mimicks the renal CaOx crystal deposition occurring in human kidney stone formers [33, 34].
Eight-week-old female rats were fed with chow mixed with 5% HLP (weight/weight HLP/chow) to induce the hyperoxaluria and CaOx crystal deposition (Figure 6(a)) and then treated with PHTPP (850 μg/kg per rat) or DMSO mock control via intraperitoneal injection every other day for 8 weeks. Kidneys were collected for examining the renal CaOx crystal deposition and results revealed that little renal CaOx crystal deposition was found in the control DMSO-treated group. In contrast, renal CaOx crystal deposition was prevalent in rats treated with PHTPP (Figure 6(b)).
Figure 6.

Suppressing ERβ activity could increase renal CaOx crystal formation in HLP-induced rat model. (a) A diagram describing the injection schedule for PHTPP/DMSO treatment. (b) Crystal staining showing the deposition area of CaOx crystals in kidney tissues of the rats treated with PHTPP or vehicle control. CaOx crystal formation was detected by Pizzolato staining and imaged by polarized light optical microphotography. (c) The PHTPP-treated rats showed increased urine oxalate excretion compared to vehicle-treated control rats. Prior to sacrifice, we collected and detected 24 hr oxalate excretion in urine of rats treated with PHTPP or vehicle control. (d) IHC staining of AGT1 in liver tissues of the rats treated with PHTPP or vehicle. The PHTPP-treated rats showed decreased AGT1 expression. (e) Detection of 24 hr urine H2O2 levels in the rats treated with PHTPP or vehicle. The PHTPP-treated rats showed higher H2O2 levels. (f) IHC staining of 8-OHdG in kidney tissues of the rats treated with PHTPP or vehicle. The PHTPP-treated rats showed increased 8-OHdG expression. (g) IHC staining of NOX2 in kidney tissues of the rats treated with PHTPP or vehicle. The PHTPP-treated rats showed increased NOX2 expression as compared to control rats. For (b)–(g), data are presented as mean ± SD. ∗ P < 0.05 and ∗∗ P < 0.01.
Prior to sacrifice, 24 hr urine samples were collected from each mouse (using metabolism cages). Results from the 24 hr urinary oxalate assay also revealed that rats treated with PHTPP have significantly higher urinary oxalate than the controls (Figure 6(c)). In addition, there was lower expression of AGT1 in the liver of rats treated with PHTPP (Figure 6(d)).
We also examined the ROS production by detecting H2O2 concentrations in the urine samples and found a much higher H2O2 concentration in the urine of rats treated with PHTPP than in urine of the control rats (Figure 6(e)). The consequences of the ERβ-suppressed, oxalate-mediated, oxidative stress-induced renal injury were also demonstrated by the increased 8-hydroxy-2′-deoxyguanosine (8-OHdG) levels, a critical biomarker of oxidative stress [15], in the kidney tissues of rats in the PHTPP group compared with the control group (Figure 6(f)). IHC staining also showed a higher NOX2 expression in renal tubular cells of rats treated with PHTPP as compared with the control rats (Figure 6(g)).
Taken together, the results from HLP-induced CaOx crystal deposition rat model (Figures 6(a)–6(g)) are in agreement with all above in vitro cell lines and in vivo mouse model studies showing that ERβ can suppress the renal CaOx crystal deposition via altering the liver oxalate biosynthesis and inhibiting renal oxidative stress-induced cell injury.
3. Discussion
CaOx crystal formation is one of the metabolic disorders in which multiple factors, such as obesity, hypertension, and diabetes, might be involved. Furthermore, dysfunctions of the gut, liver, and kidney are related to CaOx crystal formation [15].
Hyperoxaluria is a key risk factor for the development of kidney stones [35], and urinary oxalate excretion is a biomarker for the development of kidney stones [36–38]. Reduction of urinary oxalate is associated with a lower recurrent rate of CaOx kidney stones [39]. Urinary oxalate is derived from two major sources, 80% to 90% comes from endogenous production in the liver with the rest obtained from dietary oxalate [40, 41]. In the liver, AGT1 is a key enzyme in glyoxylate detoxification [19] and is necessary to avoid oxalate formation from glyoxylate. Under normal circumstances, AGT1 metabolizes oxalate precursors into the harmless amino acid glycine, which is then used by the body or is excreted. When AGT1 expression or function decreases, oxalate production is elevated in the liver, consequently resulting in an increased renal CaOx crystal deposition [19]. Primary hyperoxaluria type 1, a rare inherited metabolic disorder, results from a deficiency of AGT1 and leads to increased oxalate synthesis. In the present study, our findings indicate that ERβ could enhance hepatic AGT1 expression via direct binding to the AGT1 gene 5′ promoter to decrease endogenous oxalate production, leading to reduction in urinary oxalate excretion, and then further suppress the renal CaOx crystal deposition (Figure 7).
Figure 7.
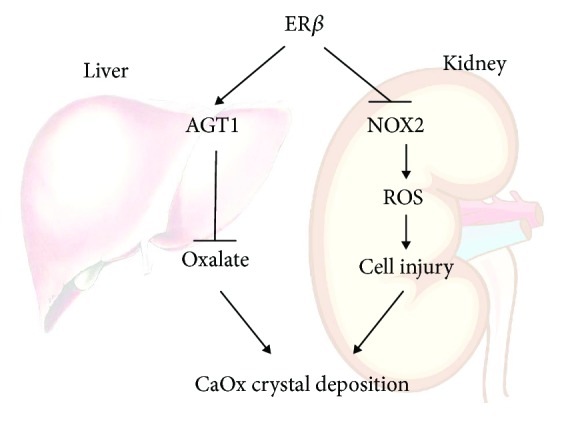
Scheme. The schematic illustration of the ERβ signal regulation of CaOx crystal formation in the liver and kidney. For the liver part, ERβ signaling promotes activity of AGT1, which decreases the biosynthesis of oxalate by converting glyoxylate into glycine decreasing CaOx crystal formation. For the kidney part, ERβ has a protective effect in human renal tubular cells against oxalate-induced oxidative stress via suppression of NOX2 (a NADPH oxidase), which finally leads to decreasing CaOx crystal deposition on the damaged cell surface.
Oxidative stress is the condition in which the production of ROS is greater than the protective capacity of antioxidant enzymes, such as superoxide dismutase. The role of oxidative stress in kidney stone formation has received increasing attention in recent years [17, 26, 42, 43]. Increasing evidence suggested that oxidative stress-induced renal injury might play key roles to promote the renal CaOx crystal deposition under higher urinary oxalate concentrations, a condition seen in patients with hyperoxaluria [44], which may involve inducing cell apoptosis through mitochondrial destruction with microvilli being injured and disintegrated. These resulting materials may then appear in the renal tubular lumen and become part of the nuclei of Ca-phosphate (CaP) that is gradually wrapped/coated by CaOx (under hyperoxaluria) and some inflammatory or fibrotic proteins, for example OPN, to finally become the major renal crystal matrix. The movement of these renal CaP-CaOx crystal matrices into the kidney tubular lumen may then cause some mechanical injuries leading to local bleeding. Consequences of these renal injuries may then induce these crystal matrices to become exposed/attached to the renal parenchyma [17, 43]. Another report demonstrated that antioxidant treatment can reduce renal CaOx crystal deposition in vivo [45].
There are at least three distinct ERs expressed in the kidney. Two ERs, ERα and ERβ, belong to the steroid hormone receptor superfamily and function as ligand-activated transcription factors. The third one, GPR30, has been recently studied and belongs to the G-protein-coupled receptor superfamily and may promote nongenomic signaling events by estrogen. The potential role of E2 in regulating renal function is evident from the observation that the kidney expresses the classical ERα and ERβ. In human fetal kidneys, ERβ is the prominent renal expressed ER, whereas ERα is only marginally expressed [46]. Recent studies also found that ERs (ERα or ERβ) might play protective roles against oxidative stress-induced cell injury in several tissues, including the brain [12], skin [47], kidney [48–50], and myocardium [51]. However, the mechanisms by which ERs can suppress the oxidative stress-induced renal injury in the presence of oxalate remain unclear [49]. Because ERα and ERβ can respond differently to E2 in mediating its transcriptional activity, it is possible that the differential expression of ER subtypes may be of physiological relevance. Kim et al. [52] reported that ERα function is involved in the regulation of renal fibroblast activation via the TGF-β1/Smad signaling pathway. Aufhauser et al. [53] found that ERα mediated female protection from renal ischemia-reperfusion injury through mechanisms extrinsic to the kidney. Interestingly and importantly, the current study provides evidence that ERβ, but not ERα, can protect against oxidative stress-induced renal injury in the presence of oxalate, which may then lead to suppressing the renal CaOx crystal deposition.
NADPH oxidase is a major source of ROS in the kidneys [54]. NOX-derived ROS are implicated in physiological processes of the kidney, including gluconeogenesis, glucose transport, tubuloglomerular feedback, hemodynamics, and electrolyte transport. NOX2 is the first described NADPH oxidase with a sole function of producing ROS. Of the seven NOX isoforms, NOX2 might be the best characterized in renal pathology. We identified NOX2, a NADPH oxidase, as a source of ROS induced by oxalate. Increased oxalate concentration could dramatically stimulate NOX2 expression in renal cells within a short time, and NOX2 depletion not only diminished knockdown of ERβ-induced ROS formation but also reduced cell injury. This response was confirmed in vivo in mice treated with apocynin, a NADPH oxidase inhibitor, showing diminished ROS production and crystal formation in ERβKO mouse kidneys. Our results confirmed that ERβ protects the kidney against oxidative stress-induced renal injury via suppressing the NOX2 expression (Figure 7). This finding also points to a novel strategy via targeting NOX2 to reduce the renal CaOx crystal deposition in the presence of oxalate, a condition seen in patients with hyperoxaluria.
In summary, this study is the first report showing that E2/ERβ signaling suppresses kidney CaOx crystal deposition via reducing hepatic biosynthesis of oxalate and inhibiting oxidative stress-induced renal injury.
4. Materials and Methods
4.1. Cell Lines
The human HK-2, HepG2, and HEK-293 cell lines were purchased from the American Type Culture Collection (ATCC) (Rockville, MD). The HKC-8 cell line was kindly provided by Dr. Syed Khundmiri of the University of Louisville (Louisville, Kentucky). All the cells were maintained in DMEM media with 8% fetal bovine serum and 1% penicillin/streptomycin.
4.2. Reagents and Materials
GAPDH (6c5) and ERα (MC-20) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ERβ (N2C2) antibody was purchased from GeneTex (Irvine, CA). NOX2 antibody was purchased from BosterBio (PA1667, Pleasanton, CA). Anti-mouse/rabbit second antibody for Western Blot was from Invitrogen (Grand Island, NY). Normal rabbit IgG was also from Santa Cruz Biotechnology.
4.3. Lentiviral Transduction of Targeted Mammalian Cells
The pLVTHM-shNOX2, pLKO.1-shERα, pLKO.1-shERβ, pLKO.1-shLuc, pWPI-vector, pWPI-ERα, or pWPI-ERβ as well as the psPAX2 packaging plasmid and pMD2G envelope plasmid were transfected into HEK-293 cells using the standard calcium chloride transfection method for 48 hr to get the lentivirus soups. The lentivirus soups were collected and concentrated by density gradient centrifugation. The collected virus were added to the target cells in the presence of polybrene (2 μg/ml) to incubate for 24 hr or frozen at -80°C for later use. Cells were refreshed with culture media and cultured for another 3 days to allow target protein expression.
4.4. Oxalate Treatments and ROS Detections Using Hydrogen Peroxide Assay and DHE Staining
Sodium oxalate (Sigma) stock solution (10 mM) in PBS was diluted in media to achieve a final concentration of 0.75 mM. HK-2 or HKC-8 cells with/without knockdown of ERα or ERβ were seeded in 12-well plates (20,000 cells/1 ml regular media per well) incubated overnight. The next day, the cell media were changed to media with 0.75 mM oxalate, and after 6 hr we then collected the cultured media to detect H2O2 amounts. The H2O2 concentration in the media, or in the mouse urine samples, was detected using Sigma Fluorimetric Hydrogen Peroxide Assay Kit (MAK165) following the manufacturer's protocol. Dihydroethidium (DHE) (Cayman, No. 12013) was diluted in PBS to the final concentration of 10 μM, and 200 μl of DHE-PBS solution was added onto cell monolayers or frozen renal tissue sections (5 μm) by dropping, and the reaction was incubated in the dark at 37°C for 30 min. Excess DHE was washed away by 1x PBS twice, and cells/tissues on the slides were fixed and stained. Images were captured immediately with a fluorescent microscope at excitation and emission wavelengths of 520 and 610 nm, respectively. For quantitative assessments, the images were analyzed by using ImageJ software.
4.5. Oxalate Measurement
Oxalate measurements in the HepG2 cell culture media and 24 hr mouse/rat urine collections were determined using an Oxalate Kit (Trinity Biotech) according to the manufacturer's instructions. The oxalate concentration in cells was measured by ion chromatography following Baker et al.'s procedure [18]. Briefly, cell monolayers were washed three times by PBS and extracted with ice-cold 10% trichloroacetic acid (TCA) for 30 min. The TCA was removed from extracts by vigorously vortexing with an equal volume of 1,1,2-trichlorotrifluoroethane (Freon)-trioctylamine (3 : 1, vol/vol), centrifuging at 4°C to promote phase separation, and collecting the upper aqueous layer for analysis.
4.6. Lactate Dehydrogenase (LDH) Release Assay (Cytotoxicity Assay)
After cells were seeded in 96-well plates at the density of 104 cells/well and cultured overnight, the confluent monolayers of cells were treated with DMEM containing 0.75 mM oxalate for 6 hr. After centrifugation, the LDH amounts in supernatants were determined according to the manufacturer's instructions (Thermo Fisher Scientific, Rochester, NY). The optical products were read at 490 nm. Values were normalized to those for control shLuc group samples.
4.7. RNA Extraction and Quantitative Real-Time PCR Analysis
Total RNA was extracted by TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNAs (1 μg) were subjected to reverse transcription using Superscript III transcriptase (Invitrogen). Quantitative real-time PCR (qRT-PCR) was conducted using a Bio-Rad CFX96 system with SYBR Green to determine the mRNA expression level of a gene of interest. RNA expression levels were normalized to the expression of GAPDH. The sequences of primers used are in Supplementary Table 1.
4.8. Western Blot Assay
Total protein was extracted by RIPA buffer containing 1% protease inhibitors (Amresco, Cochran, CA). Proteins (30-50 μg) were separated on 10% SDS/PAGE gel and then transferred onto PVDF membranes (Millipore). After blocking the membranes, they were incubated with appropriate dilutions (1 : 1000) of specific primary antibodies, and the immunopositive bands were visualized with an ECL chemiluminescent detection system (Thermo Scientific).
4.9. Chromatin Immunoprecipitation Assay (ChIP)
Cell lysates were precleared sequentially with normal rabbit IgG and protein A-agarose. Anti-ERβ antibody (5.0 μg) was added to the cell lysates and incubated at 4°C overnight. For the negative control, IgG was used in the reaction. Specific primer sets were designed to amplify a target sequence within human AGT1 or NOX2 promoters, and PCR products were identified by agarose gel electrophoresis.
4.10. Luciferase Reporter Assays
For luciferase assays, 3954 bp fragments of AGT1 promoters or 1345 bp fragments of NOX2 promoters containing wild-type or mutant ERE-binding sites were constructed into the pGL3 basic vector (Promega). Cells were plated in 24-well plates and transfected with the above promoter luciferase pGL3 plasmid and pRL-TK luciferase plasmid using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. After the indicated treatments, cells were lysed and the luciferase activity was detected by the dual-luciferase Assay (Promega).
4.11. Breeding ERβKO Mice and Development of the CaOx Crystal Mouse Model
Animals were housed in a vivarium at the University of Rochester Medical Center, School of Medicine and Dentistry. All protocols related to animals were overseen and approved by the University Committee for Animal Research, and all animals were treated in accordance with National Institutes of Health guidelines.
The ERβ knockout (ERβKO, 129P2-Esr2tm1Unc/J) and the background wild-type (WT) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred to heterozygote ERβKO (ERβ +/-) male and female mice. And the ERβ +/- male and ERβ −/− female mice were mated to obtain homozygote ERβKO (ERβ −/−) female mice for experiments. The genotypes of the female pups were confirmed by polymerase chain reaction (PCR) [55].
We established the CaOx crystal mouse model following the reported protocol [28]. The 8-week-old female mice were given daily intraperitoneal injections of 80 mg/kg glyoxylate (G1134, Sigma, St. Louis, MO) for 7 days. All animals had free access to chow and water. The 24 hr urine samples were collected 1 day before sacrifice using metabolism cages to control the diet and excretion of each animal, and mouse liver and kidney tissues were collected after sacrifice. As we are testing the ERβ gene knockout effects in our animal models, those mice were not ovariectomized.
For the in vivo rescue experiment, apocynin (10 mg/kg/day) or saline was injected ip into groups of mice every day for 7 days, followed by coinjection with glyoxylate (80 mg/kg/day) for 7 days. After treatments, the mice were sacrificed and kidneys were obtained for further analysis.
4.12. Development of Hyperoxaluria-Induced CaOx Crystal Formation in Rats and Test the Effects of ERβ Antagonist PHTPP
Eight-week-old Sprague-Dawley female rats, from the Guangdong Medical Laboratory Animal Center, were divided into 2 groups (6 rats/group), and both were given chow mixed with 5% HLP (weight/weight HLP/chow) under either DMSO or PHTPP (850 μg/kg per rat) intraperitoneal injection every other day for a total of 8 weeks. As we are testing the antiestrogen effects in our animal model, those female rats were not ovariectomized. The 24 hr urine samples were collected 1 day before sacrifice, and rat liver and kidney tissues were collected after sacrifice.
4.13. H&E and Immunohistochemical (IHC) Staining
Tissues were fixed in 10% (v/v) formaldehyde in PBS, embedded in paraffin, and cut into 5 μm sections and used for H&E staining and IHC staining with specific primary antibodies against AGT1, NOX2, and 8-OHdG. To enhance antigen exposure, the slides were treated with 10 mM sodium citrate (pH = 6.0) at 98°C for 10 minutes for antigen retrieval. The slides were incubated with endogenous peroxidase blocking solution and then were incubated with the primary antibody at 4°C overnight. After rinsing with PBS, the slides were incubated for 45 minutes with biotin-conjugated secondary antibody, washed, and then incubated with enzyme conjugate horseradish peroxidase (HRP) streptavidin. Freshly prepared DAB (Zymed, South San Francisco, CA) was used as substrate to detect HRP. Finally, slides were counterstained with hematoxylin and mounted with aqueous mounting media. Positive cells were calculated as the number of immunopositive cells × 100% divided by the total number of cells/field in 10 random fields at 400x magnification.
4.14. Statistics
All statistical analyses were carried out with SPSS 13.0 (SPSS Inc., Chicago, IL). The data values were presented as mean ± SD. All experiments were performed with triplicate data points and at least 3 times. Differences in mean values between two groups were analyzed by two-tailed Student's t test, and the mean values of more than two groups were compared with one-way ANOVA. p ≤ 0.05 was considered statistically significant.
Acknowledgments
This work was supported by NIH grants (CA155477 and CA156700), George Whipple Professorship Endowment, and by grants from National Natural Science Foundation, China (No. 81670643 and No. 81800625), Natural Science Foundation of Guangdong Province (2018A030310296), China's Ministry of Health Project (No. 1802030). We confirmed that the mentioned received funding did not lead to any conflict of interests regarding the publication of this manuscript. We thank Karen Wolf for help in preparing the manuscript.
Contributor Information
Guohua Zeng, Email: gzgyzgh@vip.sina.com.
Shuyuan Yeh, Email: shuyuan_yeh@urmc.rochester.edu.
Data Availability
All data used to support the findings of this study are available from the corresponding author (Shuyuan Yeh, shuyuan_yeh@urmc.rochester.edu) upon request.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
W.Z and F.C designed and performed the experiments and wrote the manuscript. Z.Z and Z.L provided suggestions and participated in the experiments and data analysis. W.Z, F.C, and T.L contributed to the development of the methodology and the mouse model experiment. D.B and C.C. provided intellectual inputs and helped with the writing of the manuscript. GZ. and S.Y conceived the study, designed the experiments, and wrote, edited, and approved the final version for publication. Wei Zhu, Zhijian Zhao, Fu-Ju Chou, and Li Zuo contributed equally to this work.
Supplementary Materials
Supplementary Table 1: sequences of primers targeting genes for Q-PCR.
Supplementary Figure 1: depletion of renal ERβ with 2nd shRNA made renal epithelial cells more vulnerable to oxalate-induced ROS production and cell injury. Figure 1S: A, the second shERβ target sequence. B, qRT-PCR shows the ERβ knockdown efficiency of the second shERβ (shERβ#2) in HK-2 cells. C, HK-2 cells were transduced with lentiviral shLuc (control) or shERβ#2 and treated with 8% FBS-DMEM media or 8% FBS-DMEM media containing 0.75 mM oxalate for 6 hr; the ROS production in the cells was detected by dihydroethidium (DHE) staining under a fluorescence microscope. Representative DHE staining images are presented. Digital scans of DHE-stained cells were quantified using ImageJ software; t-test, compared to the shLuc group. D, detection of H2O2 levels in culture media of the shERβ#2 or control (shLuc) renal tubular epithelial cells after challenge with 0.75 mM oxalate for 6 hr; Student's t tests, compared to the shLuc group. E, LDH release measurement in the ERβ knocked-down HK-2 cells treated with 0.75 mM oxalate for 6 hr. For B-E, data are presented as mean ± SD. ∗ P < 0.05 and ∗∗ P < 0.01.
Supplementary Figure 2: generation and confirmation of the ERβKO mice. Figure 2S: generation and confirmation of the ERβKO mice. A, ERβKO mouse breeding scheme. B, tail genomic DNA was isolated for genotyping by PCR.
References
- 1.Scales C. D., Jr, Smith A. C., Hanley J. M., Saigal C. S., Urologic Diseases in America Project Prevalence of kidney stones in the United States. European Urology. 2012;62(1):160–165. doi: 10.1016/j.eururo.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyajima J., Hayashi T., Saito K., Iida S., Matsuoka K. The interaction between female sex hormone receptors and osteopontin in a rat hyperoxaluric model. The Kurume Medical Journal. 2010;57(3):73–80. doi: 10.2739/kurumemedj.57.73. [DOI] [PubMed] [Google Scholar]
- 3.YAGISAWA T., ITO F., OSAKA Y., AMANO H., KOBAYASHI C., TOMA H. The influence of sex hormones on renal osteopontin expression and urinary constituents in experimental urolithiasis. The Journal of Urology. 2001;166(3):1078–1082. doi: 10.1016/S0022-5347(05)65925-3. [DOI] [PubMed] [Google Scholar]
- 4.Fan J., Chandhoke P. S., Grampsas S. A. Role of sex hormones in experimental calcium oxalate nephrolithiasis. Journal of the American Society of Nephrology: JASN. 1999;10(Supplement 14):S376–S380. [PubMed] [Google Scholar]
- 5.Heller H. J., Sakhaee K., Moe O. W., Pak C. Y. C. Etiological role of estrogen status in renal stone formation. Journal of Urology. 2002;168(5):1923–1927. doi: 10.1016/S0022-5347(05)64264-4. [DOI] [PubMed] [Google Scholar]
- 6.Yoshioka I., Tsujihata M., Momohara C., Akanae W., Nonomura N., Okuyama A. Effect of sex hormones on crystal formation in a stone-forming rat model. Urology. 2010;75(4):907–913. doi: 10.1016/j.urology.2009.09.094. [DOI] [PubMed] [Google Scholar]
- 7.Mattix Kramer H. J., Grodstein F., Stampfer M. J., Curhan G. C. Menopause and postmenopausal hormone use and risk of incident kidney stones. Journal of the American Society of Nephrology. 2003;14(5):1272–1277. doi: 10.1097/01.ASN.0000060682.25472.C3. [DOI] [PubMed] [Google Scholar]
- 8.Maalouf N. M., Sato A. H., Welch B. J., et al. Postmenopausal hormone use and the risk of nephrolithiasis: results from the women’s health initiative hormone therapy trials. Archives of Internal Medicine. 2010;170(18):1678–1685. doi: 10.1001/archinternmed.2010.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner R. J., Kerber I. J. Renal stones, timing hypothesis, and eu-estrogenemia. Menopause: The Journal of The North American Menopause Society. 2012;19(1):104–108. doi: 10.1097/gme.0b013e318221be9b. [DOI] [PubMed] [Google Scholar]
- 10.Paterni I., Granchi C., Katzenellenbogen J. A., Minutolo F. Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids. 2014;90:13–29. doi: 10.1016/j.steroids.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Y., Bian Z., Lu P., et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor β . Science. 2002;295(5554):505–508. doi: 10.1126/science.1065250. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q.-G., Raz L., Wang R., et al. Estrogen attenuates ischemic oxidative damage via an estrogen receptor-mediated inhibition of NADPH oxidase activation. The Journal of Neuroscience. 2009;29(44):13823–13836. doi: 10.1523/JNEUROSCI.3574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rago V., Romeo F., Giordano F., Maggiolini M., Carpino A. Identification of the estrogen receptor GPER in neoplastic and non-neoplastic human testes. Reproductive Biology and Endocrinology. 2011;9(1):p. 135. doi: 10.1186/1477-7827-9-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng G., Mai Z., Xia S., et al. Prevalence of kidney stones in China: an ultrasonography based cross-sectional study. BJU International. 2017;120(1):109–116. doi: 10.1111/bju.13828. [DOI] [PubMed] [Google Scholar]
- 15.Liang L., Li L., Tian J., et al. Androgen receptor enhances kidney stone-CaOx crystal formation via modulation of oxalate biosynthesis & oxidative stress. Molecular Endocrinology. 2014;28(8):1291–1303. doi: 10.1210/me.2014-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin-Higueras C., Luis-Lima S., Salido E. Glycolate oxidase is a safe and efficient target for substrate reduction therapy in a mouse model of primary hyperoxaluria type I. Molecular Therapy. 2016;24(4):719–725. doi: 10.1038/mt.2015.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan S. R. Reactive oxygen species as the molecular modulators of calcium oxalate kidney stone formation: evidence from clinical and experimental investigations. The Journal of Urology. 2013;189(3):803–811. doi: 10.1016/j.juro.2012.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker P. R. S., Cramer S. D., Kennedy M., Assimos D. G., Holmes R. P. Glycolate and glyoxylate metabolism in HepG2 cells. American Journal of Physiology Cell Physiology. 2004;287(5):C1359–C1365. doi: 10.1152/ajpcell.00238.2004. [DOI] [PubMed] [Google Scholar]
- 19.Salido E. C., Li X. M., Lu Y., et al. Alanine–glyoxylate aminotransferase-deficient mice, a model for primary hyperoxaluria that responds to adenoviral gene transfer. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(48):18249–18254. doi: 10.1073/pnas.0607218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiessner J. H., Hasegawa A. T., Hung L. Y., Mandel G. S., Mandel N. S. Mechanisms of calcium oxalate crystal attachment to injured renal collecting duct cells. Kidney International. 2001;59(2):637–644. doi: 10.1046/j.1523-1755.2001.059002637.x. [DOI] [PubMed] [Google Scholar]
- 21.Li S., Wu W., Wu W., Duan X., Kong Z., Zeng G. L-carnitine protects renal tubular cells against calcium oxalate monohydrate crystals adhesion through preventing cells from dedifferentiation. Kidney & Blood Pressure Research. 2016;41(5):582–592. doi: 10.1159/000443455. [DOI] [PubMed] [Google Scholar]
- 22.Curhan G. C., Willett W. C., Speizer F. E., Stampfer M. J. Twenty-four-hour urine chemistries and the risk of kidney stones among women and men. Kidney International. 2001;59(6):2290–2298. doi: 10.1046/j.1523-1755.2001.00746.x. [DOI] [PubMed] [Google Scholar]
- 23.Coe F. L., Evan A., Worcester E. Kidney stone disease. The Journal of Clinical Investigation. 2005;115(10):2598–2608. doi: 10.1172/JCI26662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang M. E. I. Y. I., Chaturvedi L. S., Koul S., Koul H. K. Oxalate stimulates IL-6 production in HK-2 cells, a line of human renal proximal tubular epithelial cells. Kidney International. 2005;68(2):497–503. doi: 10.1111/j.1523-1755.2005.00427.x. [DOI] [PubMed] [Google Scholar]
- 25.Koul S., Khandrika L., Pshak T. J., et al. Oxalate upregulates expression of IL-2Rβ and activates IL-2R signaling in HK-2 cells, a line of human renal epithelial cells. American Journal of Physiology-Renal Physiology. 2014;306(9):F1039–F1046. doi: 10.1152/ajprenal.00462.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thamilselvan V., Menon M., Thamilselvan S. Selective Rac1 inhibition protects renal tubular epithelial cells from oxalate-induced NADPH oxidase-mediated oxidative cell injury. Urological Research. 2012;40(4):415–423. doi: 10.1007/s00240-011-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bedard K., Krause K.-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 28.Okada A., Nomura S., Higashibata Y., et al. Successful formation of calcium oxalate crystal deposition in mouse kidney by intraabdominal glyoxylate injection. Urological Research. 2007;35(2):89–99. doi: 10.1007/s00240-007-0082-8. [DOI] [PubMed] [Google Scholar]
- 29.Bessler W. K., Hudson F. Z., Zhang H., et al. Neurofibromin is a novel regulator of Ras-induced reactive oxygen species production in mice and humans. Free Radical Biology & Medicine. 2016;97:212–222. doi: 10.1016/j.freeradbiomed.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henríquez-Olguín C., Díaz-Vegas A., Utreras-Mendoza Y., et al. NOX2 inhibition impairs early muscle gene expression induced by a single exercise bout. Frontiers in Physiology. 2016;7 doi: 10.3389/fphys.2016.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrônio M. S., Zeraik M. L., da Fonseca L. M., Ximenes V. F. Apocynin: chemical and biophysical properties of a NADPH oxidase inhibitor. Molecules. 2013;18(3):2821–2839. doi: 10.3390/molecules18032821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan S. R. Nephrocalcinosis in animal models with and without stones. Urological Research. 2010;38(6):429–438. doi: 10.1007/s00240-010-0303-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao S., Yang R., Peng Z., et al. Metabolomics analysis for hydroxy-L-proline-induced calcium oxalate nephrolithiasis in rats based on ultra-high performance liquid chromatography quadrupole time-of-flight mass spectrometry. Scientific Reports. 2016;6(1, article 30142) doi: 10.1038/srep30142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zuo J., Khan A., Glenton P. A., Khan S. R. Effect of NADPH oxidase inhibition on the expression of kidney injury molecule and calcium oxalate crystal deposition in hydroxy-L-proline-induced hyperoxaluria in the male Sprague–Dawley rats. Nephrology Dialysis Transplantation. 2011;26(6):1785–1796. doi: 10.1093/ndt/gfr035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stieger B. Regulation of the expression of the hepatocellular sulfate–oxalate exchanger SAT-1 (SLC26A1) by glyoxylate: a metabolic link between liver and kidney? Journal of Hepatology. 2011;54(3):406–407. doi: 10.1016/j.jhep.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 36.McHarg T., Rodgers A., Charlton K. Influence of cranberry juice on the urinary risk factors for calcium oxalate kidney stone formation. BJU International. 2003;92(7):765–768. doi: 10.1046/j.1464-410X.2003.04472.x. [DOI] [PubMed] [Google Scholar]
- 37.Finkielstein V. A., Goldfarb D. S. Strategies for preventing calcium oxalate stones. CMAJ. 2006;174(10):1407–1409. doi: 10.1503/cmaj.051517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor E. N., Curhan G. C. Oxalate intake and the risk for nephrolithiasis. Journal of the American Society of Nephrology. 2007;18(7):2198–2204. doi: 10.1681/ASN.2007020219. [DOI] [PubMed] [Google Scholar]
- 39.Pearle M. S., Goldfarb D. S., Assimos D. G., et al. Medical management of kidney stones: AUA guideline. The Journal of Urology. 2014;192(2):316–324. doi: 10.1016/j.juro.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 40.Holmes R. P., Kennedy M. Estimation of the oxalate content of foods and daily oxalate intake. Kidney International. 2000;57(4):1662–1667. doi: 10.1046/j.1523-1755.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 41.Schrier R. W. Manual of Nephrology. Lippincott Williams & Wilkins; 2008. [Google Scholar]
- 42.Thamilselvan V., Menon M., Thamilselvan S. Oxalate-induced activation of PKC-α and -δ regulates NADPH oxidase-mediated oxidative injury in renal tubular epithelial cells. American Journal of Physiology-Renal Physiology. 2009;297(5):F1399–F1410. doi: 10.1152/ajprenal.00051.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan S. R. Reactive oxygen species, inflammation and calcium oxalate nephrolithiasis. Translational Andrology and Urology. 2014;3(3):256–276. doi: 10.3978/j.issn.2223-4683.2014.06.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thamilselvan S., Khan S. R., Menon M. Oxalate and calcium oxalate mediated free radical toxicity in renal epithelial cells: effect of antioxidants. Urological Research. 2003;31(1):3–9. doi: 10.1007/s00240-002-0286-x. [DOI] [PubMed] [Google Scholar]
- 45.Naghii M., Jafari M., Mofid M., Eskandari E., Hedayati M., Khalagie K. The efficacy of antioxidant therapy against oxidative stress and androgen rise in ethylene glycol induced nephrolithiasis in Wistar rats. Human & Experimental Toxicology. 2015;34(7):744–754. doi: 10.1177/0960327114558889. [DOI] [PubMed] [Google Scholar]
- 46.Gava A. L., Freitas F. P. S., Meyrelles S. S., Silva I. V., Graceli J. B. Gender-dependent effects of aging on the kidney. Brazilian Journal of Medical and Biological Research. 2011;44(9):905–913. doi: 10.1590/S0100-879X2011007500101. [DOI] [PubMed] [Google Scholar]
- 47.Stevenson S., Thornton J. Effect of estrogens on skin aging and the potential role of SERMs. Clinical Interventions in Aging. 2007;2:283–297. doi: 10.2147/cia.s798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park K. M., Cho H. J., Bonventre J. V. Orchiectomy reduces susceptibility to renal ischemic injury: a role for heat shock proteins. Biochemical and Biophysical Research Communications. 2005;328(1):312–317. doi: 10.1016/j.bbrc.2004.12.177. [DOI] [PubMed] [Google Scholar]
- 49.Aufhauser D. D., Jr, Wang Z., Murken D. R., et al. Improved renal ischemia tolerance in females influences kidney transplantation outcomes. The Journal of Clinical Investigation. 2016;126(5):1968–1977. doi: 10.1172/JCI84712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park K. M., Kim J. I., Ahn Y., Bonventre A. J., Bonventre J. V. Testosterone is responsible for enhanced susceptibility of males to ischemic renal injury. The Journal of Biological Chemistry. 2004;279(50):52282–52292. doi: 10.1074/jbc.M407629200. [DOI] [PubMed] [Google Scholar]
- 51.Wang M., Crisostomo P. R., Markel T., Wang Y., Lillemoe K. D., Meldrum D. R. Estrogen receptor beta mediates acute myocardial protection following ischemia. Surgery. 2008;144(2):233–238. doi: 10.1016/j.surg.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 52.Kim D., Lee A. S., Jung Y. J., et al. Tamoxifen ameliorates renal tubulointerstitial fibrosis by modulation of estrogen receptor α-mediated transforming growth factor-β1/Smad signaling pathway. Nephrology, Dialysis, Transplantation. 2014;29(11):2043–2053. doi: 10.1093/ndt/gfu240. [DOI] [PubMed] [Google Scholar]
- 53.Aufhauser D., Murken D., Wang Z., et al. Estrogen receptor alpha mediates female protection from renal ischemia-reperfusion injury through mechanisms extrinsic to the kidney. 2017 American Transplant Congress; 2017; Chicago, IL, USA. p. p. B114. [Google Scholar]
- 54.Sedeek M., Nasrallah R., Touyz R. M., Hebert R. L. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. Journal of the American Society of Nephrology. 2013;24(10):1512–1518. doi: 10.1681/ASN.2012111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krege J. H., Hodgin J. B., Couse J. F., et al. Generation and reproductive phenotypes of mice lacking estrogen receptor β . Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15677–15682. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: sequences of primers targeting genes for Q-PCR.
Supplementary Figure 1: depletion of renal ERβ with 2nd shRNA made renal epithelial cells more vulnerable to oxalate-induced ROS production and cell injury. Figure 1S: A, the second shERβ target sequence. B, qRT-PCR shows the ERβ knockdown efficiency of the second shERβ (shERβ#2) in HK-2 cells. C, HK-2 cells were transduced with lentiviral shLuc (control) or shERβ#2 and treated with 8% FBS-DMEM media or 8% FBS-DMEM media containing 0.75 mM oxalate for 6 hr; the ROS production in the cells was detected by dihydroethidium (DHE) staining under a fluorescence microscope. Representative DHE staining images are presented. Digital scans of DHE-stained cells were quantified using ImageJ software; t-test, compared to the shLuc group. D, detection of H2O2 levels in culture media of the shERβ#2 or control (shLuc) renal tubular epithelial cells after challenge with 0.75 mM oxalate for 6 hr; Student's t tests, compared to the shLuc group. E, LDH release measurement in the ERβ knocked-down HK-2 cells treated with 0.75 mM oxalate for 6 hr. For B-E, data are presented as mean ± SD. ∗ P < 0.05 and ∗∗ P < 0.01.
Supplementary Figure 2: generation and confirmation of the ERβKO mice. Figure 2S: generation and confirmation of the ERβKO mice. A, ERβKO mouse breeding scheme. B, tail genomic DNA was isolated for genotyping by PCR.
Data Availability Statement
All data used to support the findings of this study are available from the corresponding author (Shuyuan Yeh, shuyuan_yeh@urmc.rochester.edu) upon request.