

Abstract
Thiopeptide natural products have gained interest recently for their diverse pharmacological properties, including antibacterial, antifungal, anticancer, and antimalarial activities. Due to their inherent poor solubility and uptake, there is interest in developing new thiopeptides that mimic these unique structures, but which exhibit better pharmacokinetic properties. One strategy is to exploit the biosynthetic pathways using a chemoenzymatic approach to make analogs. However, a complete understanding of thiopeptide biosynthesis is not available, especially for those molecules that contain a large number of modifications to the thiopeptide core. This gap in knowledge and the lack of a facile method for generating a variety of thiopeptide intermediates makes studying particular enzymatic steps difficult. We developed a method to produce thiopeptide mimics based on established synthetic procedures to study the reaction catalyzed by NosN, the class C radical S-adenosylmethionine methylase involved in carbon transfer to C4 of 3-methylindolic acid and completion of the side-ring system in nosiheptide. Herein, we detail strategies for overproducing and isolating NosN, as well as procedures for synthesizing substrate mimics to study the formation of the side-ring system of nosiheptide.
1. INTRODUCTION
Thiopeptide natural products are ribosomally synthesized and post-translationally modified peptides (RiPPs) that exhibit a variety of biologically activities (Ortega & van der Donk, 2016). Most notably, these activities include inhibition of protein synthesis either by binding to the 50S ribosome or by interacting with elongation factor thermal unstable (EF-tu), and thus can produce antibiotic effects (Bagley, Dale, Merritt, & Xiong, 2005). These natural products originate from a precursor peptide before undergoing a multistep transformation to achieve, among other modifications, multiple thiazole and/or oxazole rings, dehydrated alanine (Dha) and dehydrated threonine (Dht) side chains, and a central nitrogen-containing six-membered ring of different oxidation states (Ortega & van der Donk, 2016; Zheng, Fang, & Liu, 2017). The core macrocycle comprises a 26- or 29-membered ring, which is related to the specific biological target. The larger thiopeptides include thiostrepton and nosiheptide (NOS), which have been cocrystallized with the large subunit of the ribosome (Harms et al., 2008). Such thiopeptides interact with H44 and L11 in the A-site, thus confirming the observation that thiostrepton prevents elongation factor G (EF-G) crosslinking (Cameron, Thompson, March, & Dahlberg, 2002).
NOS from Streptomyces actuosus 40,037 (NRRL 2954) was one of the first thiopeptides to be identified and characterized and has been used as a feed additive to promote the growth of poultry livestock (Benazet & Cartier, 1980). NOS exhibits potent in vitro activity against bacteria of urgent clinical relevance, such as methicillin-resistant Staphylococcus aureus, penicillin-resistant Streptococcus pneumoniae, and vancomycin-resistant-enterococci (Benazet et al., 1980). Although it exhibits a minimum inhibitory concentration in the range of 10−3 μg/mL against a list of Gram-positive bacteria, NOS is inactive when used on experimentally infected mice (Benazet et al., 1980). The inability to act as an antibiotic in vivo is due to a large extent to its poor solubility in aqueous solution and gastrointestinal absorption ( Just-Baringo, Albericio, & Alvarez, 2014). Remarkably, the total synthesis of NOS has been achieved, but with poor yields (Wojtas et al., 2016). One strategy for optimizing the yields and for generating analogs that are better suited as therapeutic agents is to use a chemoenzymatic approach ( Just-Baringo et al., 2014; Lin, He, & Liu, 2017).
Prior to determining the complete structure of NOS by X-ray crystallography, NMR spectroscopy and chemical hydrolysis helped to identify key fragments of the molecule (Depaire, Thomas, Brun, Hull, et al., 1977; Depaire, Thomas, Brun, & Lukacs, 1977; Depaire, Thomas, Brun, Olesker, & Lukacs, 1977; Pascard, Ducruix, Lunel, & Prange, 1977; Prange, Ducruix, Pascard, & Lunel, 1977). Notably, NOS carries a 3,4-dimethyl-2-indolic acid (DMIA) bridge that connects the 4-methyl carbon of DMIA to Glu6 of the peptide framework and the carboxylic acid group of DMIA to Cys8 by ester and thioester linkages, respectively, to form a second macrocycle, termed the side-ring (Fig. 1). The presence of the DMIA moiety can be confirmed by hydrolysis, which generates (4-hydroxymethyl)-3-methyl-2-indolic acid (Depaire, Thomas, Brun, & Lukacs, 1977). The biosynthesis of the side-ring system in NOS has been of general interest because of the complex and unique chemistry that it entails.
Fig. 1.

Chemical structure of nosiheptide. The side-ring system formed by DMIA was highlighted in red color.
Pioneering studies by Wen Liu and coworkers led to the identification of the nosiheptide biosynthetic operon (nosA–nosP), and encoded gene products were assigned preliminary functions based on amino acid sequence similarities to proteins of known function and gene knockout studies in S. actuosus (Yu et al., 2009). A working model was proposed wherein NosA, B, and C are responsible for terminal posttranslational modifications to the peptide core (Liu et al., 2015; Wang et al., 2015), and NosD, E, F, G, H, M, O are responsible for generating the core thiopeptide (Yu et al., 2009). NosG contains a YcaO-like domain, which is found in Ser/Thr cyclodehydratases involved in natural product biosynthesis (Dunbar, Melby, & Mitchell, 2012). Recent work by Wen Liu and coworkers showed that NosGHF modify Cys39, 42, 44, 46, and 48 to form thiazole rings on the linear precursor peptide while selectively leaving Cys47 for incorporation of 3-methylindolic acid (MIA) by NosK (Qiu et al., 2017). Dehydrated amino acids are presumably formed by NosD and NosE, which together are homologous to NisB, a dehydratase involved in nisin biosynthesis. This dehydration requires activation of the Ser or Thr residue by glutamylation using tRNAGlu, and subsequent elimination to form Dha and Dht, respectively (Garg, Salazar-Ocampo, & van der Donk, 2013). Despite our understanding of these enzymes, the timing in which these modifications are installed is unclear. In thiomuracin biosynthesis, thiazole rings are generated before dehydrated amino acids (Hudson, Zhang, Tietz, Mitchell, & van der Donk, 2015), but the sequence of events may be pathway specific. NosO is annotated as a Diels–Alderase that catalyzes a [4 + 2] aza-cycloaddition and two subsequent eliminations to remove water and the leader peptide to afford the central pyridine ring of NOS (Wever et al., 2015).
NosI, J, K, L, and N are involved in the preparation and installation of the 3,4-dimethylindolic acid moiety to form the side-ring of NOS. Biosynthesis of the side-ring begins with NosL, which catalyzes the Cα–Cβ fragmentation–recombination of L-Trp to afford MIA (Bhandari, Fedoseyenko, & Begley, 2016; Sicoli et al., 2016; Zhang et al., 2011). AMP is then attached to the carboxyl group of MIA by NosI to allow for transfer of MIA to the 4′-phosphopantetheine prosthetic group on NosJ (Badding et al., 2017; Ding, Ji, et al., 2017; Qiu et al., 2017). NosJ then shuttles MIA to NosK, an α/β hydrolase that attaches MIA onto a seryl residue within its active site. The structure of NosK revealed that the active site contains a cleft that is large enough to accommodate the structural peptide (Badding et al., 2017). Further studies showed that NosK transfers MIA to Cys45 of NosM at some stage during NOS maturation after the installation of the thiazole rings (Qiu et al., 2017).
NosN is annotated as a class C radical SAM methylase, a subgroup of the radical SAM superfamily that is largely understudied (Bauerle, Schwalm, & Booker, 2015; Zhang, van der Donk, & Liu, 2012). Other reported class C radical SAM methylases appear to be involved in the biosynthesis of complex natural products or the degradation of heme in pathogenic bacteria (Hein, Klimmek, Polly, Kern, & Simon, 2017; LaMattina, Nix, & Lanzilotta, 2016). We proposed that upon attachment of MIA to the pre-cursor peptide, NosN attaches a carbon unit at C4 of MIA to form an electrophilic exocyclic methylene intermediate. A glutamyl (Glu6) residue on NosM then adds to this electrophilic species, forming the ester linkage in NOS and completing the side-ring (Fig. 2). Previous studies of NosN showed that MIA containing an N-acetylcysteamine group (MIA-SNAC) is recognized as a substrate to form DMIA. However, considering the lack of additional gene products that could transform DMIA into a closed side-ring system, we posited that DMIA is formed as a side product when NosN cannot complete its catalytic cycle (Ding, Li, et al., 2017; Ding, Wu, et al., 2017; LaMattina et al., 2017). Only when using substrate analogs that better mimic the predicted physiological one could we probe the full reaction catalyzed by NosN. With the increased interest in thiopeptides and other RiPPs, we thought that providing a synthetic strategy to generate good substrate mimics might be useful to the community.
Fig. 2.

Proposed mechanism of NosN.
2. OVERPRODUCTION AND PURIFICATION OF NosN
Our strategy to purify NosN focused on using similar overproduction and purification methods previously shown to be successful for other radical SAM enzymes (Lanz et al., 2012). Below is a detailed description of the methods used to overproduce NosN and purify it to homogeneity.
2.1. Gene Cloning and Expression Strategy
The nosN gene from S. actuosus was synthesized by Geneart (Thermo Fisher) with codon optimization for Escherichia coli to aid in optimal protein production. The synthesized gene incorporated NheI and XhoI restriction sites at its N- and C-termini, respectively, and was delivered in a pMK DNA production plasmid. The plasmid is digested with NheI and XhoI, and the fragment containing the nosN gene is ligated into a pET28a expression plasmid digested with the same restriction enzymes. The resulting construct encodes NosN with an N-terminal hexahistidine (His6) tag linked by a thrombin cleavage site for removal of the tag (LaMattina et al., 2017). The pET-based cloning system, coupled with the strong T7 promoter, allows for facile cloning and robust overproduction of proteins (Panavas, Sanders, & Butt, 2009). The pET28a vector also contains a kanamycin (Kan) resistance marker for in vivo selection, which, in addition, allows for coexpression of genes contained on plasmid pDB1282, because this plasmid confers ampicillin (Amp) resistance. pDB1282 harbors the isc operon from Azotobacter vinelandii, which encodes proteins involved in Fe—S cluster assembly (Cicchillo et al., 2004; Zheng, Cash, Flint, & Dean, 1998). The BL21(DE3) strain of E. coli is transformed both with pDB1282 and with pET28a-nosN, giving rise to a strain that confers resistance to Kan and Amp.
2.2. Protocol for Expression of the nosN Gene
Transform E. coli BL21 (DE3) with pDB1282 and pET28a-NosN using standard procedures and at least 1 μg of each plasmid. Select transformants on Luria–Bertani (LB) plates containing Kan and Amp at final concentrations of 50 and 100 μg/mL, respectively.
Use a single colony to inoculate 5 mL of LB medium containing Kan and Amp at 50 and 100 μg/mL, respectively. Incubate at 37°C and shake at 250 rpm for ~12 h. In addition, place four 6-L flasks, each containing 4 L of M9 media (Table 1), in an incubator-shaker to allow them to equilibrate at 37°C.
Add 50 μg/mL Kan and 100 μg/mL Amp to each flask just prior to inoculation.
Inoculate each 6-L flask with ~200 μL of the 5 mL starter culture grown to an OD600 of 2.0.
At an OD600 of 0.3 (~15 h), add L-arabinose to a final concentration of 0.1% (w/v) to induce the expression of the isc operon.
At an OD600 of 0.6, cool flasks to 20°C in an ice bath and add ferric ammonium citrate to a final concentration of 25 μM before adjusting the temperature in the incubator to 18°C. Once the medium is equilibrated (about 30 min), return the flasks to the shaker and induce expression of the nosN gene by adding isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 200 μM.
Allow cultures to incubate for ~16 h at 18°C while shaking at 180 rpm.
Harvest the bacteria by centrifugation and freeze the resulting cell paste (~1.5 g/L) in liquid nitrogen and store in a liquid nitrogen dewar.
Table 1.
Composition of M9 Minimal Medium
| 1 L of 20 × M9 Saltsa | g |
|---|---|
| Na2HPO4 (Anhydrous) | 136 |
| KH2PO4 | 60 |
| NaCl | 10 |
| NH4Cl | 20 |
| Volumes for 4 L of M9 Minimal Medium | |
| 20 × M9 Saltsb | 200 mL |
| 40% w/v Glucosec | 40 mL |
| 1 M CaCl2c | 0.4 mL |
| 1 M MgSO4c | 8 mL |
| ddH2Ob | 3700 mL |
After salts dissolve, adjust the pH of the medium to 7.4 with HCl and KOH; adjust volume to 4 L with water.
Medium should be autoclaved after addition to ddH2O in a 6-L Erlenmeyer flask.
Solutions should be sterilized by filtration and added just before inoculation.
2.3. Purification of NosN
One of the greatest challenges of isolating radical SAM enzymes is their instability upon exposure to oxygen. To address this issue, purification of NosN and all subsequent manipulations of the protein are performed in a Coy anaerobic chamber under an atmosphere of 95% nitrogen and 5% hydrogen, as has been previously described (Lanz et al., 2012). Additionally, the purification process is completed without interruption while keeping the protein chilled during each step. The following steps detail the protocol that is used to isolate homogeneous NosN with an N-terminal hexahistidine (His6)-tag.
In an anaerobic chamber, prepare 500 mL each of lysis buffer (50 mM HEPES, pH 7.5, 400 mM KCl, 10% glycerol, 10 mM BME, 10 mM imidazole), wash buffer (50 mM HEPES, pH 7.5, 400 mM KCl, 10% glycerol, 10 mM BME, 20 mM imidazole), elution buffer (50 mM HEPES, pH 7.5, 400 mM KCl, 10% glycerol, 10 mM BME, 250 mM imidazole), and storage buffer (50 mM HEPES, pH 7.5, 400 mM KCl, 10% glycerol, 2 mM DTT) with water that is oxygen free. Except for cen-trifugation, all subsequent steps are performed in an anaerobic chamber.
In the anaerobic chamber, resuspend 40 g of frozen cell paste in 120 mL lysis buffer containing lysozyme (1 mg/mL) and DNase I (0.1 mg/mL) by stirring in a 250 mL metal sonication cup inserted into a crystallizing dish packed with ice.
Once the cell paste is resuspended, sonicate the mixture for 45 s at 60% output using a Fisher 1550 sonic dismembrator. Repeat for at least six cycles with an 8 min interval between each cycle to allow for temperature reequilibration ( 6°C).
Pour the lysate into centrifuge tubes. Cap the tubes tightly and seal the edges with vinyl tape before removing the tubes from the anaerobic chamber.
Centrifuge the lysate at 50,000 g for 1 h at 4°C. During centrifugation, equilibrate a column of Ni-NTA resin with the chilled lysis buffer.
After centrifugation, reintroduce the centrifuge tubes into the anaerobic chamber and carefully decant the supernatant into a clean 250 mL bottle. Remove a 100 μL sample of the supernatant and a sample of the pellet for SDS-PAGE analysis.
Load the supernatant onto the column. Proceed to wash with 150 mL of prechilled wash buffer. Take 100 μL samples of the flow-through both from the loaded lysate and from the wash for SDS-PAGE analysis.
Elute the protein from the column with prechilled elution buffer. Once the colored band begins to elute, collect fractions until all of the protein (brown/black band) elutes from the column. Concentrate the protein to a final volume of 2.5 mL by centrifugal ultrafiltration at 3500 g at 4°C using an Amicon centrifugal filter device (10 kDa molecular weight cutoff ). While the protein is concentrating, equili-brate a PD-10 column with prechilled storage buffer.
Collect a 20 μL sample of the concentrated protein for SDS-PAGE analysis. Exchange the protein into prechilled cleavage buffer using the preequilibrated PD-10 column. Collect the flow-through and concentrate the protein by centrifugal ultrafiltration if desired. Aliquot and flash-freeze the protein in liquid N2 before storing in a liquid N2dewar.
Typical expression and purification gels for NosN are shown in Fig. 3A and B, respectively.
Fig. 3.
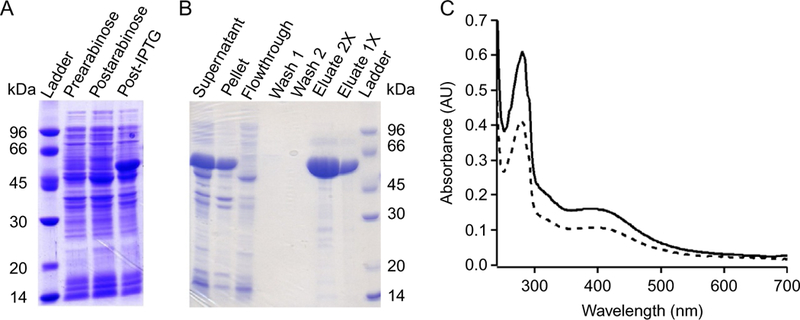
(A) NosN expression on SDS-page gel. (B) NosN purification on SDS-page gel. (C) UV–visible spectra for as-isolated (dashed line) and reconstituted (solid line) NosN.
2.4. Chemical Reconstitution of as-Isolated NosN
Although the overproduction and purification methods described above result in NosN that contains 4.6 and 3.2 mol of iron and sulfide, respectively, reconstitution can enhance FedS cluster incorporation.
Introduce the following into the anaerobic chamber: 27 mg of FeCl3 6H2O (270.3 g/mol), 24 mg of Na2S 9H2O (240.2 g/mol), 17.5 μL of stock BME (14.3 M), and a 50-mL Erlenmeyer flask.
Add NosN and 40 mL of Storage Buffer to the Erlenmeyer flask and let incubate on ice for 10 min.
Dissolve the FeCl3 6H2O and Na2S 9H2O in 1 mL of water. Add a 4:1 M equivalent of the FeCl3 solution to NosN and place mixture on ice for 30 min.
While incubating on ice, add 1 M equivalent of the Na2S solution to the reconstitution mixture every 20 min until reaching a 4:1 M ratio of sulfide to NosN. Let the mixture sit on ice overnight.
Concentrate the reconstitution mixture to ≤2.5 mL by centrifugal ultra-filtration as described above.
Exchange the concentrated protein into Gel-Filtration Buffer using a PD-10 column.
Concentrate the flow-through to ≤2 mL by centrifugal ultrafiltration as described above.
Load the protein onto an S-200 column equilibrated in Gel-Filtration Buffer and connected to an AKTA fast-protein liquid chromatography system (housed in an anaerobic chamber) for further purification by size exclusion chromatography.
UV–visible spectra for as-isolated (dashed line) and reconstituted (solid line) NosN are shown in Fig. 3C.
2.5. Determining the Stoichiometry and Forms of Iron–Sulfur Clusters
Determining the correct number of FedS clusters per polypeptide is critical for accurate characterization of radical SAM enzymes, which is typically performed in concert with Mössbauer and electron paramagnetic resonance (EPR) spectroscopies (Pandelia, Lanz, Booker, & Krebs, 2015). We follow protocols that have been previously described for determining the amount of iron and sulfide per protein in other RS enzymes (Lanz et al., 2012). A brief description of these methods follows.
Following purification, the concentration of enzyme is determined by the Bradford assay using bovine serum albumin, fraction V (BSA) as a standard (Bradford, 1976).
Amino acid analysis is conducted to determine a correction factor for NosN when BSA is used as a standard for Bradford analysis.
Quantification of the amount of iron and acid-labile sulfide per protein is done using established procedures (Beinert, 1978, 1983).
Although determining the amount of iron and sulfide per protein is useful, further characterization is required to determine the types of FedS clusters bound to an enzyme, which is done using EPR and 57Fe-Mossbauer€ spec-troscopies. Preparation of samples for Mossbauer€ spectroscopy is carried out using methods that we have previously described (Lanz et al., 2012).
3. PREPARATION OF PEPTIDE SUBSTRATES
3.1. Design and Synthesis of Peptide Mimics to Study NosN
In our proposed mechanism, NosN transfers a carbon unit to the C-4 posi-tion of MIA attached to the precursor peptide through a thioester bond. The resulting electrophilic methide intermediate is attacked by the γ-carboxylate from a glutamyl residue on the thiazolyl peptide to form a lactone bond (Fig. 2). Therefore, we designed the peptide mimic, t3NosM, as shown in Fig. 4A to validate the proposed mechanism of NosN. In our design, we maintained the thiazolylcysteine residue (structure in red) to carry the MIA (structure in black), as well as the thiazolyl glutamic acid residue (structure in blue) to act as the nucleophile for ring closure. The C-terminus of the peptide was capped with an amide to mimic a peptide bond and to avoid potential competition with the glutamyl residue. Capping a free carboxylic acid with an amide increases the stability of the entire molecule and provides a facile method for synthesis and purification. The N-terminus was capped with a thiazole-4-carboxylic acid (structure in pink) to extend the length of the peptide mimic.
Fig. 4.
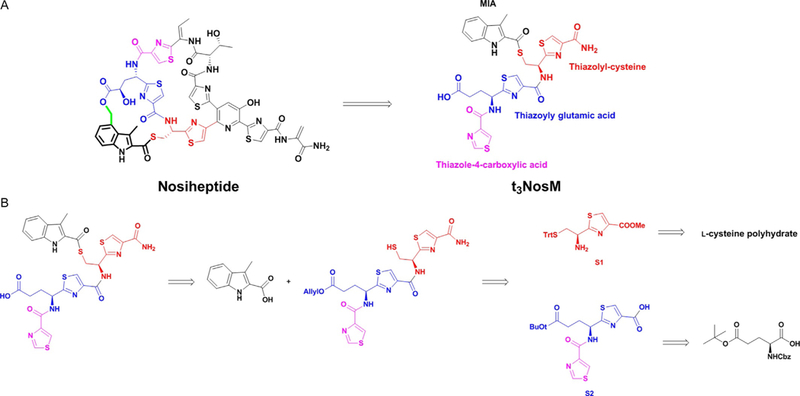
(A) Chemical structure of nosiheptide mimic (t3NosM) used for mechanistic studies of NosN. (B) Retro-synthetic analysis of nosiheptide mimic.
Our strategy involves connecting individual amino acid building blocks through amide coupling reactions to create the desired substrate. The synthetic route is illustrated briefly in Fig. 4B. Due to the presence of a labile thioester bond, the protecting group should be chosen carefully. The thioester linkage is more sensitive than its oxygen counterpart (ester). Therefore, harsh conditions, such as the use of strong acids/bases and intense heat should be avoided to prevent hydrolysis of the MIA thioester bond. Benzyl ester/ether and Cbz protection groups, which are usually removed by catalytic hydrogenation, are also not compatible, because the molecule is sulfur rich, which is poisonous to the palladium catalyst used for catalytic hydrogenation. Hence, we use an allyl ester as the protecting group on the carboxylic acid, because it can be removed by (PPh3)4Pd(0) under very mild conditions following MIA incorporation.
3.2. Protocol to Synthesize Thiazolyl Amino Acid Building Blocks
For the synthesis of the NOS mimic, two thiazolyl amino acid building blocks are needed, a thiazolyl cysteine residue (S1) and a thiazolyl glutamic acid residue (S2); however, the methods we describe can be applied to the synthesis of any other thiazolyl amino acid analogs. The method starts from amino acids with proper protecting groups on their side chains and amino groups, most of which are commercially available from Chem-Impex Inter-national Inc. Our three-step method includes amidation of the carboxylic acid (step a), thioamide synthesis (step b), and cyclization/dehydration (step c), as shown in Fig. 5.
Fig. 5.

General procedures for the synthesis of thiazolyl amino acid building blocks.
3.2.1. Amidation of the Carboxylic Acid
In a round-bottom flask, add the substrate, DMF (5 mL/1 mmol), and a magnetic stir bar. Stir the solution and add CDI (1.5 mmol/1 mmol substrate) in three portions. Let react for 1 h.
Bubble ammonia gas into the reaction slowly and monitor the reaction by silica TLC until disappearance of the starting material. For separation, use chloroform:methanol (5:1 v/v) as the mobile phase.
After completion, dilute the reaction mixture with ethyl acetate (20 mL/1 mmol substrate) and transfer to a properly sized separatory funnel.
Wash the organic phase thoroughly with 0.5 M HCl, aqueous saturated NaHCO3, and brine. Dry the organic layer with anhydrous sodium sulfate and concentrate by rotary evaporation with the bath water set no higher than 45°C. The resulting residue is used for the next step without further purification.
Caution:
The reaction conditions of step a tolerates most of the protecting groups for commercially available starting material, including Fmoc, Boc, Cbz, Alloc for the amino group, as well as tBu and benzyl for side chains of amino acids. However, excessive ammonia gas can cleave Fmoc. There-fore, when using such a protecting group, silica TLC monitoring should be done frequently and, once the starting material is gone, the bubbling of ammonia gas should be ceased immediately.
3.2.2. Thioamide Synthesis
Weigh the resulting material from Section 3.2.1 in a round-bottom flask with a magnetic stir bar. Add 50 mL of THF per 1 mmol of the sub-strate and stir before the addition of Lawesson’s reagent (MW = 404, 0.5 mmol per 1 mmol substrate). Stir the resulting reaction mixture at room temperature overnight.
After completion, concentrate the reaction by rotary evaporation in a fume hood and resuspend the resulting residue in ethyl acetate (20 mL/1 mmol of substrate). Wash the organic phase thoroughly with 0.5 M HCl, aqueous saturated NaHCO3, and brine.
Dry the organic layer with anhydrous sodium sulfate and concentrate by rotary evaporation. The resulting residue is used for the next step without further purification.
Caution:
Lawesson’s reagent gives a strong unpleasant odor. Use in a fume hood that functions well. Deodorization of glassware used for this step can be done by rinsing with 5% potassium permanganate in water.
3.2.3. Cyclization/Dehydration
Weigh the resulting material from Section 3.2.2 in a round-bottom flask with a magnetic stir bar and add THF (30 mL/1 mmol substrate).
While stirring, add potassium bicarbonate (MW = 100, 10 mmol per 1 mmol substrate) as well as methyl bromopyruvate (MW = 181, 3 mmol per 1 mmol substrate). Stir the reaction at room temperature for 3 h or until disappearance of the starting material as determined by silica TLC.
After completion, dilute the reaction with ethyl acetate (30 mL/1 mmol substrate) and wash thoroughly with 0.5 M HCl, aqueous saturated NaHCO3, and brine. Dry the organic layer over anhydrous sodium sul-fate and concentrate by rotary evaporation.
Dissolve the resulting residue in ethyl acetate (30 mL/1 mmol substrate) and pyridine (MW = 79, 10 mmol per 1 mmol substrate). After cooling thoroughly in an ice-water bath, add trifluoroacetic anhydride (MW = 210, 3 mmol per 1 mmol substrate) dropwise using a dropping funnel.
After completion, as indicated by TLC, quench the reaction by adding methanol (MW = 32, 2 mmol per 1 mmol trifluoroacetic anhydride).
Wash the reaction mixture thoroughly with 0.5 M HCl, aqueous satu-rated NaHCO3, and brine.
Dry the organic layer over anhydrous sodium sulfate and concentrate by rotary evaporation.
Purify the resulting residue using a silica gel column with hexanes and ethyl acetate as the eluent.
3.3. General Method for Protection/Deprotection and Amide Coupling
The synthesis of S1 (Fig. 6, step d), S2 (Fig. 7, step e), and the final product (Fig. 8) requires several protection/deprotection and amide coupling steps. Here, we describe the detailed procedure. To avoid repetition, each subsec-tion corresponds to a particular reaction step during the synthesis and can be referred to at different stages in the synthesis. Each of the figures has section references for each of the reaction steps to make it easier to follow.
Fig. 6.
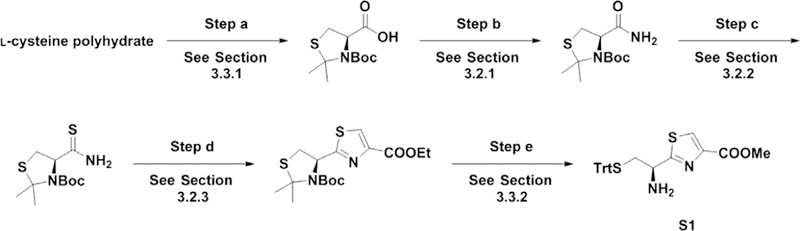
Synthesis of S1.
Fig. 7.

Synthesis of S2.
Fig. 8.

Assembly of S1 and S2, incorporation of MIA, and deprotection.
3.3.1. Protection of the Amino Group and Side Chain Thiol Group With Boc and Acetonide
Suspend L-cysteine in polyhydratein, a mixture of acetone (20 mL/1 g substrate) and 2,2-dimethoxypropane (4 mL/1 g substrate), and reflux the resulting suspension for 48 h.
After concentrating by rotary evaporation, dissolve the remaining resi-due in pyridine (12 mL/1 g substrate) and Boc2O (1.5 mmol per 1 mmol substrate).
Stir the reaction mixture overnight at room temperature.
After completion, dilute the mixture with EtOAc (50 mL/1 g substrate) and follow with a thorough washing of the organic layer with hydrochloric acid (0.5 M), saturated aqueous sodium bicarbonate, and then brine.
Concentrate the organic layer by rotary evaporation and use the resulting viscous residue for the next step without further purification.
3.3.2. Deprotection of Boc and Acetonide and Protection of the Thiol Group With Trt
Weigh the round-bottom flask to obtain the amount of starting material. Add TFA (10 mL per 1 g substrate) and water (0.35 mL per 1 g substrate). Stir the solution at 50°C overnight and then concentrate by rotary evaporation.
Resuspend the resulting residue in TFA (5 mL per 1 mmol substrate) and trityl chloride (1 mmol per 1 mmol substrate). Stir the reaction mixture at room temperature for 2 h and then concentrate by rotary evaporation.
Resuspend the resulting residue in EtOAc (75 mL/1 g substrate) and wash the organic phase thoroughly with aqueous saturated NaHCO3, and then brine.
Dry the organic layer over anhydrous Na2SO4 and concentrate by rotary evaporation. Purify the resulting residue by silica-gel flash-chromatography (hexanes:ethyl acetate ¼1:1 as the eluent).
3.3.3. Deprotection of Cbz and Reprotection With Alloc of Amino Group
Dissolve the starting material in methanol (20 mL per 1 g of substrate) and add palladium on carbon (10% palladium on carbon, 200 mg per 1 g substrate). Stir the reaction under a hydrogen atmosphere overnight.
Filter the palladium on carbon through a pad of Celite before concen-trating the filtrate by rotary evaporation. The resulting residue is used for the next step without further purification.
Add THF (20 mL per 1 g of crude product from previous step). Place the round-bottom flask in an ice-water bath and stir the solution to chill.
Once chilled, add aqueous saturated NaHCO3 (20 mL/1 g substrate).
Add allyl chloroformate (1.5 mmol per 1 mmol substrate) dropwise to the reaction while stirring.
Continue stirring the reaction at 0°C for 2 h before diluting with EtOAc (20 mL/1 g substrate). Wash the organic phase thoroughly with water and then brine.
Dry the organic layer over anhydrous Na2SO4 and concentrate by rotary evaporation. Purify the resulting residue by silica-gel flash-chromatography (hexanes:ethyl acetate ¼2:1 as the eluent).
Follow steps outlined in Sections 3.2.2 and 3.2.3 to prepare the substrate for the next step
3.3.4. Removal of Alloc
In a round-bottom flask containing the starting material, add dioxane (10 mmol per 1 mmol substrate) and water (1 mL per 1 mmol substrate).
Add (PPh3)4Pd (MW = 1155, 0.05 mmol per 1 mmol substrate) and 1,3-dimethylbarbituric acid (MW = 156, 1 mmol per 1 mmol substrate) to the reaction and stir at room temperature for 3 h. Dilute the reaction mixture with EtOAc (30 mL per 1 g substrate) and wash the organic phase thoroughly with aqueous saturated NaHCO3, and brine.
Dry the organic layer over anhydrous Na2SO4 and concentrate by rotary evaporation. The resulting residue is used in the next step without further purification.
3.3.5. Amide Coupling Reaction
Dissolve acid (1 mmol) and amine (1 mmol) in DCM (20 mL) and chill the reaction in an ice-water bath while stirring.
Add DIPEA (MW = 129, 3 mmol per 1 mmol acid) and HATU (MW = 380, 1.5 mmol per 1 mmol acid). Let the reaction gradually warm up to room temperature and continue stirring overnight.
After completion, transfer the reaction mixture to a separatory funnel and wash the reaction mixture thoroughly with 0.5 M HCl, aqueous saturated NaHCO3, and brine.
Dry the organic layer over anhydrous Na2SO4 and concentrate by rotary evaporation. Purify the resulting residue by silica-gel flash-chromatography using hexanes and acetone as the eluent.
3.3.6. Hydrolysis of the Methyl Ester
In a round-bottom flask containing the starting material, add a cosolvent of THF, methanol, and water (5 mL of each solvent per 1 mmol substrate).
Add lithium hydroxide monohydrate (MW = 42, 2 mmol per 1 mmol substrate) and stir at room temperature for 5 h.
After completion, as indicated by silica TLC, dilute the reaction with EtOAc (30 mL per 1 mmol substrate) and wash thoroughly with 0.5 M HCl and then brine.
Dry the organic layer over anhydrous Na2SO4 and concentrate by rotary evaporation. The resulting residue is used for the next step with no further purification.
3.4. Incorporation of MIA and Final Deprotection
As shown in Figs. 6 and 7, precursors S1 and S2 are prepared from the pre-vious synthetic protocols described above. The two precursors are first coupled using the amide coupling procedure in Section 3.3.5. After replace-ment of the t-butyl protecting group with an allyl group on the carboxylic acid of the glutamate, the Trt can be removed selectively to allow for the incorporation of MIA on the thiol group via an acyl chloride intermediate of MIA. Below is the detailed protocol to make the final peptide mimic used to study NosN turnover.
3.4.1. Amidation of Carboxylic Ester
Resuspend the starting material in 7 N ammonia in methanol (200 mL/1 g substrate) and stir at room temperature overnight.
After completion, as indicated by silica TLC, concentrate the reaction mixture by rotary evaporation.
Purify the resulting residue by silica-gel flash-chromatography using hexanes and acetone as the eluent.
3.4.2. Deprotection of t-Butyl Ester and Reprotection With Allyl Ester
Add 20 mL of DCM per 1 mmol substrate in a round-bottom flask.
While stirring, add TFA (1/4 in volume of DCM used) and continue stirring for 1 h at room temperature.
Concentrate the reaction mixture by rotary evaporation and use the resulting residue for the next step without further purification.
Add DMF (10 mL per 1 mmol substrate) to the residue while stirring.
Add K2CO3 (MW = 138, 5 mmol per 1 mmol substrate) in one portion and stir for 10 min.
Add allyl bromide (MW = 121, 2 mmol per 1 mmol substrate) dropwise to the stirred reaction.
Stir the reaction mixture at room temperature for 2 h before diluting with EtOAc (30 mL per 1 mmol substrate).
Wash the organic phase thoroughly with 0.5 M HCl, aqueous saturated NaHCO3, and brine.
Dry the organic layer over Na2SO4 and concentrate by rotary evaporation.
Purify the resulting residue by silica-gel flash-chromatography using chloroform and methanol as the eluent.
3.4.3. Deprotection of Trt
Add DCM (20 mL per 1 mmol substrate) in a round-bottom flask that contains the substrate from the previous step.
Add triethylsilane (4 mL per 1 mmol substrate) while stirring with a mag-netic stir bar. Continue stirring for 5 min before adding TFA (4 mL per 1 mmol substrate).
Stir the resulting clear solution for 30 min at room temperature.
Concentrate the reaction mixture by rotary evaporation and use the resulting thiol intermediate for the next step without further purification.
3.4.4. Synthesis of MIA Acyl Chloride and Thioesterification
Prepare MIA acyl chloride (2 mmol per 1 mmol of crude product from previous step) for the subsequent thioesterification as described below.
Add DCM (10 mL per 1 mmol of MIA) to a round-bottom flask with MIA and a magnetic stir bar.
While stirring, add oxalyl chloride (MW = 127, 3 mmol per 1 mmol MIA) and DMF (1 drop).
Stir the reaction for 1 h at room temperature.
After completion, concentrate the reaction mixture by rotary evapora-tion. The resulting acyl chloride is used in subsequent steps without fur-ther purification.
In the round-bottom flask with the thiol intermediate from Section 3.4.3, add THF (10 mL per 1 mmol of thiol intermediate).
While stirring, add DIPEA (MW = 129, 5 mmol per 1 mmol thiol intermediate).
Dissolve the MIA acyl chloride intermediate in THF (5 mL per 1 mmol MIA acyl chloride).
While stirring, add MIA acyl chloride in THF from the previous step dropwise to the round-bottom flask containing the thiol intermediate and DIPEA.
Stir the resulting reaction mixture for 30 min at room temperature.
After completion, dilute the reaction with EtOAc (50 mL/1 mmol thiol intermediate). Wash the resulting mixture thoroughly with 0.5 M HCl, 0.1 M aqueous NaOH, and brine.
Dry the organic layer over anhydrous Na2SO4 and concentrate by rotary evaporation.
Purify the resulting residue by silica-gel flash-chromatography using chloroform and methanol as the eluent.
3.4.5. Deprotection of Allyl Ester
Add dioxane (10 mL per 0.1 mmol substrate) and water (0.5 mL per 0.1 mmol substrate) in the round-bottom flask with the allyl ester starting material.
While stirring, add (PPh3)4Pd (MW = 1155, 0.005 mmol per 0.1 mmol substrate) and 1,3-dimethylbarbituric acid (MW = 156, 0.1 mmol per 0.1 mmol substrate).
Stir the reaction for 2 h at room temperature.
Once complete, dilute the reaction mixture with EtOAc (50 mL per 0.1 mmol substrate).
Wash the organic layer thoroughly with brine before drying the organic layer over anhydrous Na2SO4 and concentrating by rotary evaporation.
Purify the resulting residue by silica-gel flash-chromatography using chloroform and methanol as the eluent.
4. QUANTITATIVE ANALYSIS OF NosN TURNOVER
NosN has been stated to produce 5′-deoxyadenosine (5′-dA) and either S-adenosylhomocysteine (SAH) or thioadenosine (tA), depending on whether S-adenosylmethionine (SAM) or methylthioadenosine (MTA) acts as a methyl donor, respectively (Ding, Li, et al., 2017; LaMattina et al., 2017). SAM typically degrades to MTA during long periods of storage, which is heavily dependent on the pH of the storage solution and the temperature. When using unlabeled MTA and (13C-methyl) SAM in NosN assays, we only observe formation of labeled product, indicating that MTA is not a direct methyl donor in our reactions. These products can be quantified by LC– MS/MS using multiple reaction monitoring, as described below for a NosN-catalyzed reaction. The substrate for NosN is more difficult to quantify due to a lack of synthetic protocols to make a peptide mimic that contains the intact side-ring product. One alternative is to use MIA-SNAC as a sub-strate analog for NosN assays and quench with sodium hydroxide to form a hydroxymethyl adduct of MIA (LaMattina et al., 2017). However, such methods result in a mixture of products formed due to a lack of specificity with the poor substrate, making the quantification less meaningful.
The NosN peptide reaction products can be analyzed structurally by MS, as well as by MS/MS. For the addition of a CH2 group, the mass will only change 12 units (684–696 as shown in Fig. 9B and C), which can be a result of either the predicted exocyclic methylene intermediate or the closed side-ring. For this reason, MS/MS is used to detect the fragmented molecule and determine which product is produced. For NosN turnover, the side-ring closed product (t3NosMSR) will fragment to an m/z of 188 as shown in Fig. 9D, which corresponds to the hydroxyl adduct from the glutamate residue. This fragment is only observed when the side ring is formed and can be verified by 18O-labeling of the carboxylate oxygens of Glu6 of the peptide mimic (LaMattina et al., 2017).
Fig. 9.

LC–MS of the NosN activity assay. (A) Chromatogram displaying the total ion chromatography (TIC) for t3NosM and t3NosMSR (i, no SAM; ii, t = 1 min with 50 μM NosN; iii, t = 250 min with 50 μM NosN; iv, t = 250 min with 100 μM NosN. (B) MS scan of t3NosM. (C) MS scan of t3NosMSR. (D) MS/MS of the t3NosMSR.
4.1. NosN Enzyme Assays Using Synthesized Peptide Mimics
Assays should contain final concentrations of the following in a total volume of 200 μL: 50 mM HEPES, pH 7.5, 100 mM KCl, 5% glycerol, 1 mM SAM, 1 mM substrate, 10–50 μM NosN, 100 μM Trp (internal standard), and 2 mM sodium dithionite as a reductant. Note: We have not observed NosN activity with the E. coli flavodoxin/flavodoxin reductase reducing system.
Initiate the reaction by adding SAM, then remove 20 μL aliquots at specified times and quench the reactions with an equal volume of 100% MeOH.
Centrifuge the samples for 15 min at 14,000 × g before removing supernatants and placing them into MS vials.
4.2. Protocol for Quantification of Reaction Products by HPLC Coupled to Electrospray-Ionization Mass Spectrometry
Prepare standards of SAH, 5′-dA, and Trp using extinction coefficients for each molecule. SAH and 5′-dA display absorption features at 260 nm with extinction coefficients of 16,000 M−1 cm−1 and 15,400 M−1 cm−1, respectively, while Trp displays an absorption feature at 278 nm with an extinction coefficient of 5560 M−1 cm−1 (Wood, 1987). Using these standards, generate a standard curve containing 0.5–250 μM of each molecule and 100 μM Trp (internal standard).
Analyze samples on an Agilent Technologies Zorbax Extend-C18 RRHD column (4.6 mm × 50 mm, 1.8 μm particle size) equilibrated in 98% solvent A (0.1% formic acid, pH 2.6) and 2% solvent B (methanol). Apply a linear gradient of 2%–10% solvent B from 0 to 0.5 min, and then a linear gradient of 10%–50% solvent B from 0.5 to 2.5 min. Next, increase solvent B to 100% from 2.5 to 3.5 min and hold constant from 3.5 to 4.5 min before returning to 2% solvent B from 4.5 to 5 min. Allow the column to reequilibrate for 2.5 min under initial conditions before subsequent sample injections.
Detect products using MS/MS with electrospray ionization in positive mode. All products are detected in their +1 charge states. Table 2 displays the m/z values (parent and daughter ions), fragmentor voltages, collision energies, and retention times of all molecules that are monitored in a NosN reaction.
Quantify product formation. Shown in Fig. 10 is the time-dependent formation of SAH, 5′-dA, MTA, and NosMSR in the NosN-catalyzed reaction.
Table 2.
Parameters for LC–MS/MS Analysis of Reaction Products
| Compound | Precursor Ion | Product Ion | Fragmentor | Collision Energy |
|---|---|---|---|---|
| SAH | 385 | 136 | 92 | 17 |
| 87.9 | 92 | 61 | ||
| 5′-dA | 252 | 136 | 63 | 29 |
| 119 | 92 | 13 | ||
| MTA | 298 | 136 | 106 | 18 |
| 119 | 106 | 60 | ||
| MIA-t3NosM | 684 | 158 | 100 | 50 |
| 112 | 100 | 50 | ||
| NosMSR | 696 | 188 | 100 | 50 |
| 170 | 100 | 50 | ||
| L-Tryptophan | 205 | 188 | 63 | 9 |
| 118 | 63 | 29 |
Fig. 10.
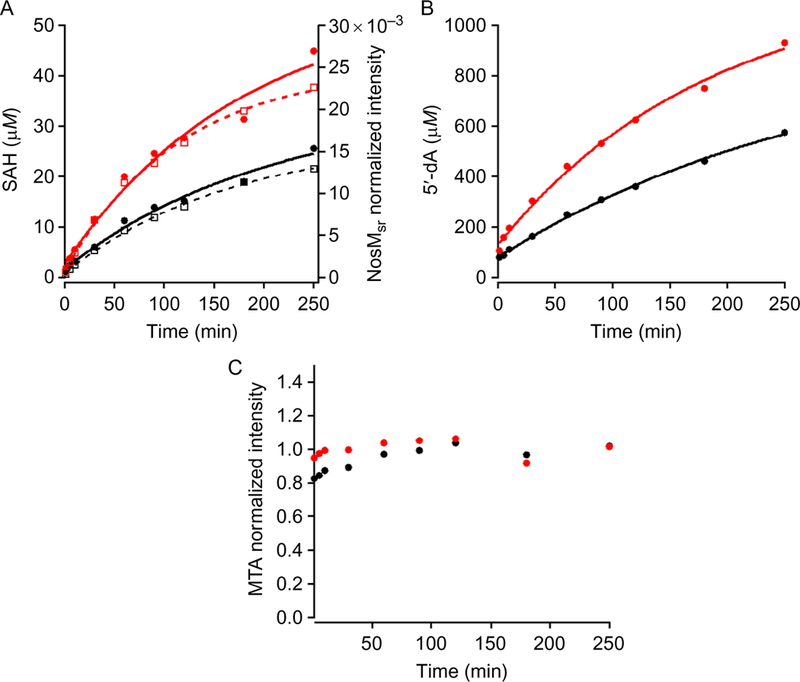
(A) Detection of SAH (closed circles) and NosMSR (open squares) production when 50 μM NosN (black) or 100 μM NosN (red) is used in the assay. Solid curves are the fit for SAH production, while dashed curves are for NosMSR. (B and C) Progress curves for 5′-dA and MTA production, respectively.
5. CONCLUSIONS
Class C radical SAM methylases catalyze the appendage of a carbon unit to sp2-hybridized carbon centers, resulting in the transfer of either a methyl group ( CH3), as in the example of TbtI for thiazole methylation, or a methylene group ( CH2), which appears to be the case for NosN and YtkT (Bauerle et al., 2015; LaMattina et al., 2017). With the role that these enzymes play in the biosynthesis of medically relevant natural prod-ucts, determining their mechanisms of catalysis can lead to producing RiPP analogs to combat antibiotic resistant bacteria. However, the previous understanding of these biosynthetic pathways and the lack of substrate syntheses have hindered the study of the class C radical SAM methylaes. This method provides a facile synthetic approach to generate similar thiopeptides as a means to probe the reactions catalyzed by some of the unique enzymes within the respective pathway.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (GM-122595 and AI-133318). S.J.B. is an investigator of the Howard Hughes Medical Institute.
REFERENCES
- Badding ED, Grove TL, Gadsby LK, LaMattina JW, Boal AK, & Booker SJ 2017). Rerouting the pathway for the biosynthesis of the side ring system of nosiheptide: The roles of NosI, NosJ, and NosK. Journal of the American Chemical Society, 139(16), 5896–5905. 10.1021/jacs.7b01497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley MC, Dale JW, Merritt EA, & Xiong X (2005). Thiopeptide antibiotics. Chemical Reviews, 105(2), 685–714. 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- Bauerle MR, Schwalm EL, & Booker SJ (2015). Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. The Journal of Biological Chemistry, 290(7), 3995–4002. 10.1074/jbc.R114.607044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinert H (1978). Micro methods for the quantitative determination of iron and copper in biological material. Methods in Enzymology, 54, 435–445. [DOI] [PubMed] [Google Scholar]
- Beinert H (1983). Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Analytical Biochemistry, 131(2), 373–378. [DOI] [PubMed] [Google Scholar]
- Benazet F, & Cartier JR (1980). Effect of nosiheptide as a feed additive in chicks on the quantity, duration, prevalence of excretion, and resistance to antibacterial agents of Salmonella typhimurium; on the proportion of Escherichia coli and other coliforms resistant to antibacterial agents; and on their degree and spectrum of resistance. Poultry Science, 59(7), 1405–1415. [DOI] [PubMed] [Google Scholar]
- Benazet F, Cartier M, Florent J, Godard C, Jung G, Lunel J, et al. (1980). Nosiheptide, a sulfur-containing peptide antibiotic isolated from Streptomyces actuosus 40037. Experientia, 36(4), 414–416. [DOI] [PubMed] [Google Scholar]
- Bhandari DM, Fedoseyenko D, & Begley TP (2016). Tryptophan Lyase (NosL): A cornucopia of 50-deoxyadenosyl radical mediated transformations. Journal of the American Chemical Society, 138(50), 16184–16187. 10.1021/jacs.6b06139. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemis-try, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Cameron DM, Thompson J, March PE, & Dahlberg AE (2002). Initiation factor IF2, thiostrepton and micrococcin prevent the binding of elongation factor G to the Escherichia coli ribosome. Journal of Molecular Biology, 319(1), 27–35. 10.1016/S0022-2836(02)00235-8. [DOI] [PubMed] [Google Scholar]
- Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, et al. (2004). Lipoyl synthase requires two equivalents of S-adenosyl-L-methionine to synthesize one equivalent of lipoic acid. Biochemistry, 43(21), 6378–6386. 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- Depaire H, Thomas JP, Brun A, Hull WE, Olesker A, & Lukacs G (1977). N-15 NMR-spectroscopy of nosiheptide—Determination of elemental formula and molecular-weight of antibiotic. Tetrahedron Letters, 16, 1401–1402. [Google Scholar]
- Depaire H, Thomas JP, Brun A, & Lukacs G (1977). Acid and alkaline-hydrolysis of antibiotic nosiheptide—Structure elucidation of 5 fragments. Tetrahedron Letters, 16, 1395–1396. [Google Scholar]
- Depaire H, Thomas JP, Brun A, Olesker A, & Lukacs G (1977). C-13 NMR-spectroscopy of nosiheptide. Tetrahedron Letters, 16, 1397–1400. [Google Scholar]
- Ding W, Ji W, Wu Y, Wu R, Liu WQ, Mo T, et al. (2017). Biosynthesis of the nosiheptide indole side ring centers on a cryptic carrier protein NosJ. Nature Communications, 8(1), 437 10.1038/s41467-017-00439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Li Y, Zhao J, Ji X, Mo T, Qianzhu H, et al. (2017). The catalytic mech-anism of the class C radical S-Adenosylmethionine methyltransferase NosN. Angewandte Chemie (International Ed. in English), 56(14), 3857–3861. 10.1002/anie.201609948. [DOI] [PubMed] [Google Scholar]
- Ding W, Wu Y, Ji X, Qianzhu H, Chen F, Deng Z, et al. (2017). Nucleoside-linked shunt products in the reaction catalyzed by the class C radical S-adenosylmethionine methyltransferase NosN. Chemical communications (Cambridge), 53(37), 5235–5238. 10.1039/c7cc02162c. [DOI] [PubMed] [Google Scholar]
- Dunbar KL, Melby JO, & Mitchell DA (2012). YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations. Nature Chemical Biology, 8(6), 569–575. 10.1038/nchembio.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg N, Salazar-Ocampo LM, & van der Donk WA (2013). In vitro activity of the nisin dehydratase NisB. Proceedings of the National Academy of Sciences of the United States of America, 110(18), 7258–7263. 10.1073/pnas.1222488110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms JM, Wilson DN, Schluenzen F, Connell SR, Stachelhaus T, Zaborowska Z, et al. (2008). Translational regulation via L11: Molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Molecular Cell, 30(1), 26–38. 10.1016/j.molcel.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Hein S, Klimmek O, Polly M, Kern M, & Simon J (2017). A class C radical S-adenosylmethionine methyltransferase synthesizes 8-methylmenaquinone. Molecular Microbiology, 104(3), 449–462. 10.1111/mmi.13638. [DOI] [PubMed] [Google Scholar]
- Hudson GA, Zhang Z, Tietz JI, Mitchell DA, & van der Donk WA (2015). In vitro biosynthesis of the core scaffold of the thiopeptide thiomuracin. Journal of the American Chemical Society, 137(51), 16012–16015. 10.1021/jacs.5b10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just-Baringo X, Albericio F, & Alvarez M (2014). Thiopeptide engineering: A multidisciplinary effort towards future drugs. Angewandte Chemie (International Ed. in English), 53(26), 6602–6616. 10.1002/anie.201307288. [DOI] [PubMed] [Google Scholar]
- LaMattina JW, Nix DB, & Lanzilotta WN (2016). Radical new paradigm for heme degradation in Escherichia coli O157:H7. Proceedings of the National Academy of Sciences of the United States of America, 113(43), 12138–12143. 10.1073/pnas.1603209113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMattina JW, Wang B, Badding ED, Gadsby LK, Grove TL, & Booker SJ (2017). NosN, a radical S-adenosylmethionine methylase, catalyzes both C1 transfer and formation of the ester linkage of the side-ring system during the biosynthesis of nosiheptide. Journal of the American Chemical Society, 139(48), 17438–17445. 10.1021/jacs.7b08492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz ND, Grove TL, Gogonea CB, Lee KH, Krebs C, & Booker SJ (2012). RlmN and AtsB as models for the overproduction and characterization of radical SAM proteins. Methods in Enzymology, 516, 125–152. 10.1016/B978-0-12-394291-3.00030-7. [DOI] [PubMed] [Google Scholar]
- Lin Z, He Q, & Liu W (2017). Bio-inspired engineering of thiopeptide antibiotics advances the expansion of molecular diversity and utility. Current Opinion in Biotechnology, 48, 210–219. 10.1016/j.copbio.2017.06.008. [DOI] [PubMed] [Google Scholar]
- Liu S, Guo H, Zhang T, Han L, Yao P, Zhang Y, et al. (2015). Structure-based mechanistic insights into terminal amide synthase in nosiheptide-represented thiopeptides biosynthesis. Scientific Reports, 5, 12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega MA, & van der Donk WA (2016). New insights into the biosynthetic logic of ribosomally synthesized and post-translationally modified peptide natural products. Cell Chemical Biology, 23(1), 31–44. 10.1016/j.chembiol.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panavas T, Sanders C, & Butt TR (2009). SUMO fusion technology for enhanced pro-tein production in prokaryotic and eukaryotic expression systems. In Ulrich HD (Ed.), SUMO Protocols (pp. 303–317). Totowa, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- Pandelia ME, Lanz ND, Booker SJ, & Krebs C (2015). Mossbauer spectroscopy of Fe/S proteins. Biochimica et Biophysica Acta, 1853(6), 1395–1405. 10.1016/j.bbamcr.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Pascard C, Ducruix A, Lunel J, & Prange T (1977). Highly modified cysteine-containing antibiotics. Chemical structure and configuration of nosiheptide. Journal of the American Chemical Society, 99(19), 6418–6423. [DOI] [PubMed] [Google Scholar]
- Prange T, Ducruix A, Pascard C, & Lunel J (1977). Structure of nosipeptide, a polythiazole-containing antibiotic. Nature, 265(5590), 189–190. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Du Y, Zhang F, Liao R, Zhou S, Peng C, et al. (2017). Thiolation protein-based transfer of indolyl to a ribosomally synthesized polythiazolyl peptide intermediate during the biosynthesis of the side-ring system of nosiheptide. Journal of the American Chemical Society 10.1021/jacs.7b11367. [DOI] [PubMed]
- Sicoli G, Mouesca JM, Zeppieri L, Amara P, Martin L, Barra AL, et al. (2016). Fine-tuning of a radical-based reaction by radical S-adenosyl-L-methionine tryptophan lyase. Science, 351(6279), 1320–1323. 10.1126/science.aad8995. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu S, Yao P, Yu Y, Zhang Y, Lan W, et al. (2015). Crystallographic analysis of NosA, which catalyzes terminal amide formation in the biosynthesis of nosiheptide. Acta Crystallographica Section F, 71, 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wever WJ, Bogart JW, Baccile JA, Chan AN, Schroeder FC, & Bowers AA (2015). Chemoenzymatic synthesis of thiazolyl peptide natural products featuring an enzyme-catalyzed formal [4 + 2] cycloaddition. Journal of the American Chemical Society, 137(10), 3494–3497. 10.1021/jacs.5b00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtas KP, Riedrich M, Lu JY, Winter P, Winkler T, Walter S, et al. (2016). Total synthesis of nosiheptide. Angewandte Chemie (International Ed. in English), 55(33), 9772–9776. 10.1002/anie.201603140. [DOI] [PubMed] [Google Scholar]
- Wood EJ (1987). Data for biochemical research (3rd ed.) by Dawson RMC, Elliott DC, Elliott WH and Jones KM, pp 580 Oxford Science Publications, OUP, Oxford, 1986. £35/$59. ISBN 0-19-855358-7. Biochemical Education, 15(2), 97. 10.1016/0307-4412(87)90110-5 [DOI] [Google Scholar]
- Yu Y, Duan L, Zhang Q, Liao R, Ding Y, Pan H, et al. (2009). Nosiheptide biosynthesis featuring a unique indole side ring formation on the characteristic thiopeptide framework. ACS Chemical Biology, 4(10), 855–864. 10.1021/cb900133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Li Y, Chen D, Yu Y, Duan L, Shen B, et al. (2011). Radical-mediated enzymatic carbon chain fragmentation-recombination. Nature Chemical Biology, 7(3), 154–160. 10.1038/nchembio.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, van der Donk WA, & Liu W (2012). Radical-mediated enzymatic methyl-ation: A tale of two SAMS. Accounts of Chemical Research, 45(4), 555–564. 10.1021/ar200202c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Cash VL, Flint DH, & Dean DR (1998). Assembly of iron-sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. The Journal of Biological Chemistry, 273(21), 13264–13272. [DOI] [PubMed] [Google Scholar]
- Zheng Q, Fang H, & Liu W (2017). Post-translational modifications involved in the biosynthesis of thiopeptide antibiotics. Organic & Biomolecular Chemistry, 15(16), 3376–3390. 10.1039/c7ob00466d. [DOI] [PubMed] [Google Scholar]