

Abstract
Background:
22q11.2 deletion syndrome (22q11.2DS) is caused by recurrent, chromosome specific, low copy repeat mediated copy number losses of chromosome 22q11. The Children’s Hospital of Philadelphia has been involved in the clinical care of individuals with what is now known as 22q11.2DS since our initial report of the association with DiGeorge syndrome in 1982.
Methods:
We reviewed the medical records on our continuously growing longitudinal cohort of 1,421 patients with molecularly confirmed 22q11.2DS from 1992 to 2018.
Results:
Most individuals are Caucasian and older than eight years old. The median age at diagnosis was 360 days. The majority of patients (85%) had typical LCR22A-LCR22D deletions, and only 7% of these typical deletions were inherited from a parent harboring the deletion constitutionally. However, 6% of individuals harbored other nested deletions that would not be identified by traditional 22q11.2 FISH, thus requiring an orthogonal technology to diagnose. Major medical problems included immune dysfunction or allergies (77%), palatal abnormalities (67%), congenital heart disease (64%), gastrointestinal difficulties (65%), endocrine dysfunction (>50%), scoliosis (50%), renal anomalies (16%), and airway abnormalities. Median full-scale IQ was 76, with no significant difference between individuals with and without congenital heart disease or hypocalcemia. Characteristic dysmorphic facial features were present in most, but dermatoglyphic patterns of our cohort are similar to normal controls.
Conclusions:
This is the largest longitudinal study of patients with 22q11.2DS, helping to further describe the condition and aid in diagnosis and management. Further surveillance will likely elucidate additional clinically relevant findings as they age.
Introduction
Deletions of sub-band 11.2 of the long arm of chromosome 22 are estimated to be as common as 1 in 3000 – 6000 live births (Carey et al., 1992; Wilson et al., 1992; Devriendt et al., 1998; Goodship et al., 1998; Scambler, 2000; Botto et al., 2003; Oskarsdóttir et al., 2004) and approximately 1 in 1000 fetuses (Grati et al., 2015). These deletions are pathogenic variants causing a range of phenotypes previously classified as DiGeorge syndrome (la Chapelle et al., 1981; Kelley et al., 1982; Scambler et al., 1991; Driscoll et al., 1992a), velocardiofacial syndrome (Driscoll et al., 1992b; 1993), conotruncal anomaly face syndrome (Burn et al., 1993; Matsuoka et al., 1994), autosomal dominant Opitz G/BBB syndrome (McDonald-McGinn et al., 1995; Fryburg et al., 1996; Lacassie and Arriaza, 1996) and Cayler Cardio-facial syndrome (Giannotti et al., 1994; Bawle et al., 1998). The 22q11.2 deletion syndrome (22q11.2DS) owes this number of previously described entities to the diverse medical subspecialists who originally classified the genetic condition, each focusing on their specific areas of interest and signs and symptoms related to these subspecialties. However, the development and widespread use of fluorescence in situ hybridization (FISH) allowed for these conditions to be collectively referred to by their underlying molecular etiology, the 22q11.2DS (McDonald-McGinn et al., 1996). The range of associated clinical features is broad and variably expressive. These features include immunodeficiency, congenital heart disease, palatal defects, hypocalcemia, dysphagia, renal anomalies, and developmental disabilities (McDonald-McGinn et al., 2015).
Previous studies have revealed that most affected individuals with 22q11.2DS have a recurrent 3 megabase deletion, which includes ~50 genes. Of those with a nonstandard deletion, a majority have a smaller nested deletion that also results in haploinsufficiency of the “DiGeorge critical region” (Gong et al., 1996; McDonald-McGinn et al., 2015). Some of the genes located within the standard deletion have major clinical impact, such as UFD1L, COMT, and particularly TBX1. The T-box 1 (TBX1) gene is part of the larger family of T-box genes, which help to regulate tissue and organ formation during development (Baldini, 2005). However, a different minority of patients harbor nested distal deletions but retain two copies of the TBX1 gene (Kurahashi et al., 1996; O’Donnell et al., 1997; Yamagishi et al., 1999; Amati et al., 1999; McQuade et al., 1999; Garcia-Miñaur et al., 2002; McDonald-McGinn et al., 2015). Nonetheless, these individuals are haploinsufficient for other apparently developmentally important genes such as CRKL (Racedo et al., 2015). Some patients who exhibit overlapping features of 22q11.2DS but without copy number variation in the expected region have been shown to have both gain and loss of function variants of TBX1 (Yagi et al., 2003; Zweier et al., 2007).
Previous studies indicate that a substantial majority of patients harbor de novo deletions of 22q11.2 (McDonald-McGinn et al., 2001; 2015). The recurrent nature of the deletion is a result of non-allelic homologous recombination that occurs during meiosis between several segmental duplications or low copy repeats (LCR22A, LCR22B, LCR22C, and LCR22D) flanking the critical region. High sequence identity among modular elements within LCRs make the region particularly vulnerable to genomic rearrangements due to non-allelic homologous recombination (Edelmann et al., 1999; Shaikh et al., 2000; Baumer et al., 2004; Saitta et al., 2004; McDonald-McGinn et al., 2015). Previous studies suggest that individuals with the deletion have an approximately 50% risk of having an affected child with each offspring.
There has been much advancement into the treatment of children with this syndrome since 1992. However, findings from a single sizeable cohort remain relevant to the field. Here, we report our observations on 1,421 patients with 22q11.2DS, including demographics, physical examination findings, molecular genetics, inheritance, medical comorbidity, and mortality.
Methods
The Institutional Review Board at the Children’s Hospital of Philadelphia (CHOP) approved this longitudinal study. We enrolled 1,421 individuals with a diagnosis of 22q11.2DS diagnosed using FISH, multiplex ligation-dependent probe amplification (MLPA) or chromosomal microarray between 1992 and 2018. The medical records of these patients were obtained and manually reviewed to provide the clinical data presented here. Not all individuals were assessed for all phenotypes; in most cases, results are reported as percentages of total individuals with available data. Congenital heart disease was classified based on a previously reported standard by Billett et al (Billett et al., 2008) as complex, moderate, or simple. Data analysis and statistical testing was performed using the R Statistical Programming Language.
Results
Males and females were equally likely to reach clinical attention; 51% of this cohort was male while 49% was female (Table 1). Our cohort is primarily Caucasian, with individuals of African ancestry being the second largest group represented. Our cohort was also aging; at the time of analysis in 2018, 53 % was greater than 18 years of age. This age represented the current age of the patients, not necessarily the age of most recent evaluation.
Table 1:
Cohort Demographics
| Demographic | Percentage |
|---|---|
| Male | 51 % |
| Female | 49 % |
| Caucasian | 75 % |
| African American | 9 % |
| Asian | 2 % |
| Hispanic | 5 % |
| Pacific Islander | 0.1% |
| Middle Eastern | 0.1% |
| Mixed Race | 3 % |
| “Other” | 3 % |
| Did not report | 3 % |
| ≤ 8 years of age | 11 % |
| > 8 and ≤ 18 years of age | 35 % |
| > 18 years of age | 53 % |
Physical Features
The majority of individuals in our pediatric cohort were examined by a single medical geneticist (EHZ). Table 2 lists the frequency of various dysmorphic facial features. Prominent features included abnormalities of the auricular helix, such as over-folded or thick helices, eye hooding, protuberant ears, epicanthal folds and micrognathia. The available dermatoglyphic patterns (Preus and Fraser, 1972) of individuals were analyzed. Ulnar loops were the prominent dermatoglyph (Figure 1). The patterns observed in patient with 22q11.2DS were not substantially different from previously reported healthy controls (Plato et al., 1975).
Table 2.
Dysmorphic Features Among Individuals with 22q11.2 Deletion Syndrome
| Dysmorphic Feature | Percentage |
|---|---|
| At least one abnormal ear helix | 63 % |
| At least one protuberant ear | 26 % |
| At least one posteriorly rotated ear | 10 % |
| At least one low set ear | 6 % |
| At least one ear tag | 2 % |
| At least one ear pit | 0.7 % |
| Bulbous Nasal Tip | 64 % |
| Hypoplastic Alae Nasae | 27 % |
| Broad Nasal Bridge | 13 % |
| Anteverted Nares | 1 % |
| Eye Hooding | 42 % |
| Epicanthal folds | 16 % |
| Up-slanting palpebral fissures | 11 % |
| Down-slanting palpebral fissures | 4 % |
| Deep Set Eyes | 3 % |
| Micrognathia | 16 % |
| Asymmetric crying facies | 8 % |
| Down-turned corners of the mouth | 6 % |
| Thin Upper Lip | 5 % |
Figure 1.
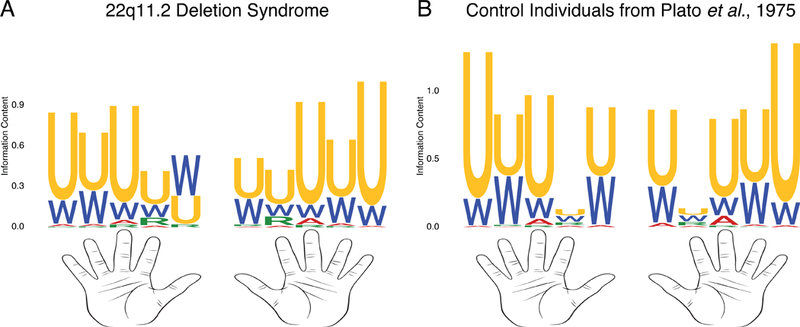
Dermatoglyphics of Patients with 22q11.2 Deletion Syndrome.
U = ulnar loop. W = whorl. A = arch. R = radial arch. The proportion of each letter indicates the fraction of individuals exhibiting that dermatoglyphic for a given finger position. The height of the Y axis indicates the information content of a given dermatoglyphic at a given position. A. Dermatoglyphics of 529 patients with 22q11.2 deletion syndrome. B. Dermatoglyphics of 720 healthy Caucasian individuals from Plato et al, 1975.
Analysis of physical features by race noted differences in either the presence of said features or of them rising to clinical attention to the examiner. Helical abnormalities, broad nasal bridge, epicanthal folds, and micrognathia were not significantly different between Caucasians and non-Caucasians (p=1, Fisher exact test, Bonferroni corrected). However, ascertainment of both bulbous nasal tip and eye hooding were significantly greater in Caucasians than non- Caucasians (p=0.00005, p=0.001, respectively, Fisher exact test, Bonferroni corrected). Identification of both protuberant ears and hypoplastic alae nasae were also greater in Caucasians, but neither was significantly different following correction for multiple testing (p=0.22 and p=0.13, respectively, Fisher exact test).
Diagnosis
The median age of diagnosis was 360 days (Figure 2). Arriving at a diagnosis was significantly easier for individuals with cardiac disease, with a median age at diagnosis of 2.6 months compared to those without cardiac issues of 3.1 years (p < 0.0001, Wilcoxon signed rank test). Among those without heart disease, the diagnosis remained challenging with common features such as ear abnormalities, hooding of the eyes, and nose differences or even seemingly more specific abnormalities such as asymmetric crying facies not helping to hasten the time to diagnosis (p > 0.05). For individuals with available data, referral information revealed that practitioners at the Children’s Hospital of Philadelphia diagnosed 93.5% of our cohort; of those, the majority (63.8%) were referred to the 22q and You Center by the General Genetics service. Meanwhile, 19.6% were diagnosed by Cardiology and 5.0% by the craniofacial specialists of Plastic Surgery.
Figure 2.
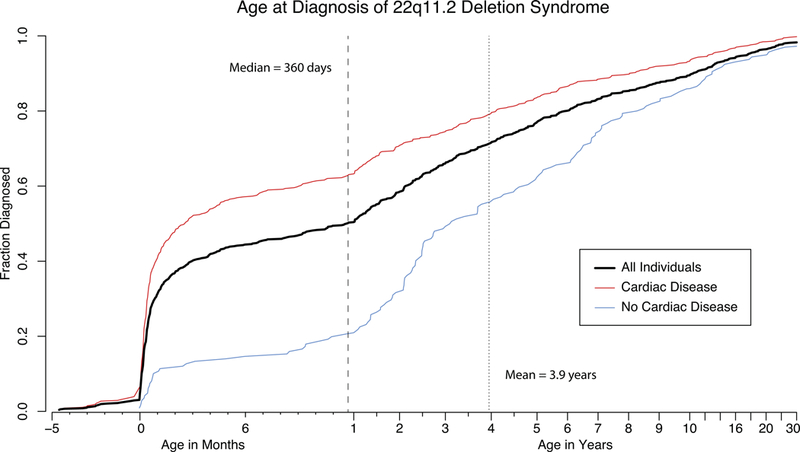
Age at Diagnosis of 22q11.2 Deletion Syndrome
The black line indicates the age at diagnosis for all individuals with available data in the cohort. The red line indicates the same data for the subset of individuals with a cardiac diagnosis. The blue line indicates the time course for individuals without cardiac disease.
Molecular Genetics
Although some patients in our cohort have only had molecular testing by FISH (29%), most had additional, more detailed testing by MLPA or chromosomal microarray. Among individuals with better characterized pathogenic variants (60%), the vast majority (84%) had the standard LCR22A-LCR22D deletion, with 14% having other LCR-mediated deletions (Table 2). Notably, LCR22B-LCR22D, LCR22C-LCR22D and other atypical deletions would not be identified by FISH testing, as they result in normal copy number of the region interrogated by the probes used to conduct the test. We also observed a few individuals (2%) with deletions that do not appear to be LCR-mediated.
Although patients with the standard LCR22A-LCR22D deletion most frequently exhibit de novo variants, not detected in either parent, cases with nested deletions were more likely to be familial. In fact, 60% of LCR22B-LCR22D or LCR22C-LCR22D index cases have family members harboring the deletion, in contradistinction to just 7% of the overall 22q11.2 deletion cohort. Additionally, we observed one somatically mosaic patient, one presumed germline mosaic patient, and three families with a proband with a 22q11.2 deletion and a parent with a mosaic 22q11.2 duplication.
Developmental Milestones
Individuals in our cohort sat at a median age of 8 months and walked alone at a median age of 16 months (Figure 3A). Our patients spoke their first words at a median age of 19 months and were using simple phrases by a median age of 34.5 months (Figure 3B). Among those who spoke words, 95% had done so by 42 months, whereas, 95% had made simple phrases by 58 months.
Figure 3.
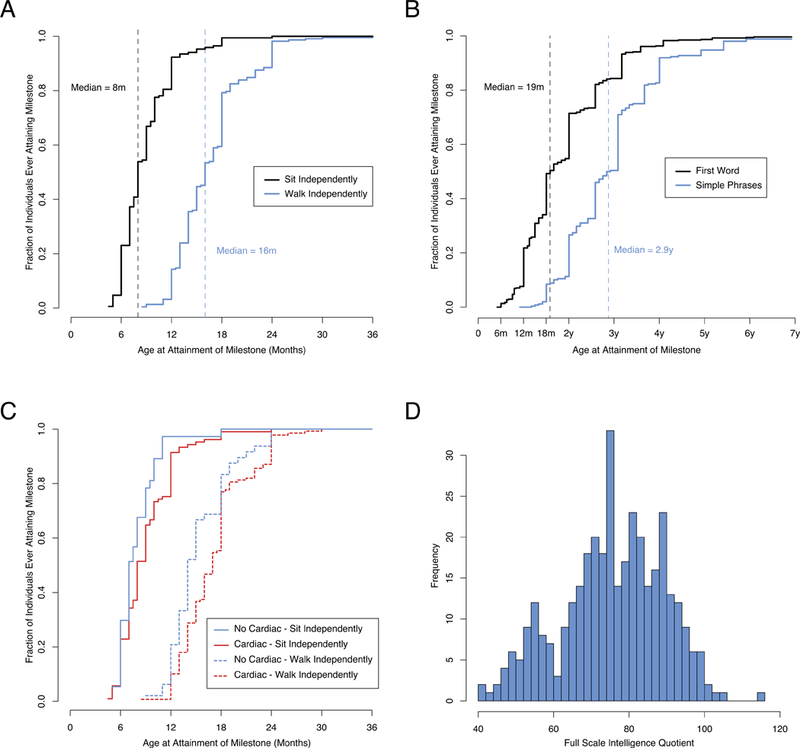
Developmental Trajectory of Individuals with 22q11.2 Deletion Syndrome.
A. Age at Attainment of Gross Motor Milestones. The black solid line indicates the time course of attainment of sitting independently by our cohort. The dashed black line indicates the median age of sitting independently. The blue solid line indicates the time course of walking independently. The dashed blue line indicates the median age of walking independently. B. Age at Attainment of Language Milestones. The black solid line indicates the time course of attainment of first words. The dashed black line indicates the median age of first words. The blue solid line indicates the time course of making simple phrases. The dashed blue line indicates the median age of attainment of simple phrases. C. Gross Motor Milestone Delays Associated with Congenital Heart Disease. The solid lines indicate the time course of attainment of sitting independently stratified by the presence (blue line) or absence (red line) of congenital heart disease. Individuals with heart disease sat a median of 1 month slower. The dashed lines indicate the age of walking independently. Individuals with heart disease walked independently a median of 3 months slower. D. Intelligence Quotient Distribution of Individuals with 22q11.2 Deletion Syndrome (N=361).
The median age for sitting and walking independently by individuals with cardiac disease was 1 month slower and 3 months slower, respectively, compared to individuals without heart issues (p=0.045, p=0.005, respectively, Wilcoxon signed rank test, Figure 3C). However, there was no significant difference between the time of first words nor full sentences between individuals with and without cardiac disease (p=0.30, p=0.25, respectively, Wilcoxon signed rank test). Interestingly, there was no significant difference between the time of first words nor speaking in full sentences among individuals with or without overt or submucous cleft palate (Supplemental Figure 1, p=0.87, p=0.17, respectively, Wilcoxon signed rank test), and they were not significantly more likely to receive speech therapy (p=0.26, Fisher exact test). However our cohort is not powered well to identify these effects, as overt or submucous cleft palate was rare in our population.
Growth
A previous study including a subset of our cohort revealed that patients with 22q11.2DS experience growth faltering from 6 to 9 months of age, followed by a catch-up period. Individuals’ weight ultimately recovered close to the reference population (mean of −0.22 standard deviation (SD), CDC growth chart). There continued to be a reduced height velocity resulting in a below average final height. At 17 years old the mean height for males was 172 ± 8 cm, (−0.72 SD, CDC growth chart), and 16 years old the mean height for girls was 158 ± 6 cm (−0.89 SD, CDC growth chart). Microcephaly was present in 30% of boys and 24% of girls before 1 year old. The final 22q11.2DS 50 percentile for head circumference was equivalent to the 9 percentile (−1.33 SD, WHO‐UK Chart) (Habel et al., 2012).
Intelligence Quotient
Full scale IQ testing was available for a subset of our patients. The median IQ among our cohort was 76 with an interquartile range of 18 (Figure 3D). A FSIQ below 69 (within the range for intellectual disabilities) was seen in 28%. There was no significant correlation between the severity of cardiac disease and IQ (p=0.14, Kendall’s tau correlation), nor did the presence of documented hypocalcemia have an effect on IQ (Wilcoxon signed rank test, p=0.70). Almost all (93%) school-aged children in our cohort had an active individualized educational plan (IEP) (American Academy of Pediatrics Council on Children With Disabilities and Cartwright, 2007), underscoring the prominent role of the Neuropsychiatric disciplines.
Comorbidities and Medical Management
Depending on the age of the child and their pertinent medical issues, the multidisciplinary evaluation provided by our center generally included the following subspecialties: Genetics, Allergy/Immunology, Cardiology, Gastroenterology and Feeding Clinic, Plastic Surgery, Speech-Language Pathology, Otolaryngology, Audiology, Ophthalmology, Dental, Developmental Pediatrics, Behavioral Health, Neuropsychiatry, Neurology, and General Pediatrics (Table 4). Some patients required Hematology, Oncology, Urology, Pulmonary, Allergy, or General Surgery consultation. Table 3 lists selected common medical conditions.
Table 4.
Medical comorbidities among patients with 22q11.2 deletion syndrome.
| Medical Comorbidities | Percentage of Patients |
|---|---|
| Immune Dysfunction or Allergies | |
| T-cell Dysfunction | 50% |
| Humoral Dysfunction | 17% |
| Cardiac | 64% |
| Craniofacial | |
| Velopharyngeal Dysfunction | 52% |
| Submucous Cleft Palate | 21% |
| Overt Cleft Palate | 6% |
| Gastrointestinal | |
| Constipation | 35% |
| Dysphagia | 30% |
| Tube Feeding | 21% |
| G Tube Placement | 16% |
| Endocrine | |
| Hypocalcemia | 55% |
| Musculoskeletal | |
| Scoliosis | 50% |
| Cervical Spine Abnormalities | 46% |
| Genitourinary | |
| Renal Anomalies | 16% |
| Cryptorchidism | 4% |
| Hypospadias | 4% |
| Neurologic/Psychiatric | |
| ADHD | 52% |
| Autism Spectrum Disorder | 19% |
| Seizures | 15% |
| Psychotic Disorder | 15% |
| Malignancy | 6% |
Table 3.
Molecular Characteristics of 22q11.2 Deletions in patients with deletions characterized by MLPA or chromosomal microarray. The vast majority (84%) had the standard LCR22A-LCR22D deletion, with 14% having other LCR-mediated deletions. A small proportion (2%) had non-LCR mediated deletions.
| Molecular Diagnosis | Percentage of Characterized Deletions |
|---|---|
| LCR22A-LCR22D | 84 % |
| LCR22A-LCR22B | 5 % |
| LCR22A-LCR22C | 2 % |
| LCR22B-LCR22D | 4 % |
| LCR22C-LCR22D | 1% |
| Other LCR-mediated (C-E, D-E, D-F, D-H, E-F) | 2% |
| Non-LCR mediated | 2 % |
Immune dysfunction or Allergies
The most common medical issue in our cohort was immune dysfunction or allergies, with approximately 77% displaying some issue. Among those with available data, 50% had abnormal T-cell populations. Meanwhile, 17% had some form of abnormality of humoral immunity. Allergies were common in our cohort. Autoimmune disorders were also noted. Five individuals were diagnosed with idiopathic thrombocytopenic purpura, four with juvenile rheumatoid arthritis, four with Hashimoto’s thyroiditis, two with Grave’s disease, and one with vitiligo.
Cardiac
Some form of congenital heart disease was present in 64% of patients, 42% of whom have complex or moderate defects (Billett et al., 2008). Table 5 describes the common types of heart defects seen in our cohort. Ventricular septal defects were the most common abnormality identified on echocardiography. Tetralogy of Fallot was seen in 18% of our cohort with a cardiac anomaly. Interestingly, patients with LCR22A-LCR22D deletions have a higher incidence of cardiac anomalies compared with patients with other nested LCR-mediated deletions (Supplemental Figure 2, p=0.041, Pearson chi-squared test).
Table 5.
Cardiac abnormalities observed in our cohort.
| Abnormality | Percentage |
|---|---|
| Ventricular Septal Defect | 23% |
| Tetralogy of Fallot | 18% |
| TOF with Pulmonary Atresia | 5% |
| TOF without Pulmonary Atresia | 12% |
| Aortic Arch Anomalies | 14% |
| Right Aortic Arch | 7% |
| Vascular Ring | 6% |
| Double Aortic Arch | 1% |
| Left Aortic Arch with Aberrant Right Subclavian Artery | 3% |
| Interrupted Aortic Arch | 11% |
| Atrial Septal Defect | 10% |
| Pulmonary Atresia | 6% |
| Truncus Arteriosus | 4% |
| Patent Ductus Arteriosus | 6% |
| Bicuspid Aortic Valve | 3% |
| Pulmonary Stenosis | 2% |
| Other | 1% |
Note that individual features of well-described complex defects are not repeated, for example Tetralogy of Fallot does not create a second data point for ventricular septal defect. Values are expressed as percentages of the entire cohort with available data. Many individuals had multiple findings. Other findings include double outlet right ventricle, hypoplastic left heart and complete atrioventricular canal.
Craniofacial
Palatal anomalies were present in 67% of the patients studied. The vast majority of abnormalities were not overt clefts, but rather submucous cleft palate (21%) or velopharyngeal dysfunction (VPD) (52%). VPD can result from structural issues such as velopharygeal disproportion, functional issues such as hypotonia of the velopharyngeal musculature, or a combination of these factors (McDonald-McGinn et al., 1997). Overt cleft palate was noted in only 6% of patients studied.
Gastrointestinal
Gastrointestinal disease was noted in 65% of patients. Previous studies on our cohort revealed that dysphagia may be suggested by polyhydramnios prenatally and is overall seen in 30% of individuals. Dysphagia often leads to supplemental enteral feeding strategies such as temporary nasogastric tube feeding (5%) or subsequent gastronomy tube placement (16%). If individuals are able to transition to oral only feeding regimens, 95% do so by 4.5 years of age. Previous specialty studies suggest that dysmotility in the pharyngoesophageal area appears to be the major underlying feeding issue in most children (Eicher et al., 2000). Thus, we suggest that when a patient presents in respiratory distress or is noted to have recurrent pulmonary infections or reactive airway disease, aspiration should be considered as a potential etiology.
Chronic constipation is also a common feature, with 35% affected. Structural bowel anomalies such as intestinal malrotation, intestinal nonrotation, and feeding difficulties secondary to a vascular ring have also been identified. Hirschsprung disease, imperforate anus, and congenital diaphragmatic hernia are other visceral anomalies that occur more commonly among individuals with 22qDS compared to the general population.
Endocrine
Over half of the patients in our cohort had an endocrinopathy, such as hypocalcemia, thyroid disease, and/or growth hormone deficiency. Hypocalcemia was seen in 55% of patients at some point in their life regardless of perioperative period. There was a strong correlation with the severity of cardiac disease and presence of hypocalcemia (p < 0.00001, τ = 0.31, Kendall’s tau correlation). This suggests that hypocalcemia observed among individuals with 22q11.2DS may be partially related to critical illness in the setting of heart disease. Nonetheless, 38% of individuals without heart disease also exhibited hypocalcemia sometime during life. A separate study showed 80% of adults with 22q11.2DS experienced hypocalcemia during their lifetime, and that hypoparathyroidism, hypothyroidism and hypomagnesaemia may contribute to hypocalcemia (Cheung et al., 2014).
Musculoskeletal
Data about musculoskeletal abnormalities were relatively poorly available in our cohort, but among those with available data, anomalies were common. Scoliosis was noted in 50% of the cohort. Cervical spine anomalies were noted in 46% of individuals, which is substantially lower than previous estimates, suggesting ascertainment bias due to a smaller number of patients being evaluated by Orthopedics (Homans et al., 2017). Additional skeletal abnormalities included vertebral anomalies, talipes equinovarus, polydactyly, camptodactyly, patellar dislocation, hammer toe (Ming et al., 1997; Homans et al., 2017). These findings underscore the importance of evaluation by an orthopedic surgeon. Diagnosis may result in activity changes such as avoiding contact sports as well as timely initiation of treatment.
Genitourinary
Renal anomalies, mostly single hydronephrosis, renal agenesis, and multicystic dysplastic kidney, were seen in 16% our cohort. A recent study identified several genes that appear to be critical to the renal and urinary tract anomalies seen in 22q11.2DS patients, with haploinsufficiency of CRKL appearing to be a primary genetic driver behind the phenotype (Lopez-Rivera et al., 2017). However, there was no significant difference in the number of renal abnormalities, including renal agenesis, between individuals with deletions involving the CRKL gene and those without (p = 1, Fisher exact test).
A separate genitourinary analysis on a subset of our cohort found that while hydronephrosis was the most common upper tract finding, the majority (63%) had isolated upper tract dilation without any additional diagnoses (Van Batavia et al. to be cited in special issue). In terms of genital anomalies, boys were significantly more likely to be diagnosed with an abnormality than girls (8% vs. 0.5%, p < 0.001, Fisher exact test). Among males, 4% had cryptorchidism and 4% had hypospadias, which was noted to be mild (megameatus or glanular) in all except 1 boy. The only female genital anomalies noted were vaginal agenesis in 2 patients and uterine agenesis in 1 patient. Findings of hydronephrosis, unilateral renal agenesis, and multicystic dysplastic kidney occured at higher rates than expected in the general population; although in the majority of cases, no surgical intervention was necessary. Despite the higher than expected rate of hypospadias, most are mild distal hypospadias. The renal and genitourinary anomalies seen in this population emphasize the importance of a screening renal bladder ultrasound performed at the time of diagnosis of 22q11.2DS.
Neurologic / Psychiatric
Neurologic and psychiatric comorbidities were common in our cohort. 52% carried a diagnosis of attention deficit hyperactivity disorder (ADHD). Autism spectrum disorder was seen in 19% of patients. Anxiety, behavior disorders, and psychotic disorders were also seen frequently. CNS abnormalities included idiopathic seizures (15%), neural tube defects, polymicrogyria, microcephaly, hypotonia, and dystonia. A previous study including a subset of our cohort revealed that a psychotic disorder was diagnosed in 13% of patients with 22q11.2DS (Vorstman et al., 2015).
Ear, Nose, and Throat, and Respiratory
Chronic otitis media and chronic sinusitis are commonly reported in our cohort. These risks appear to be secondary to underlying immune deficiencies and palatal differences in patients with 22q11.2DS. Previous studies showed that these issues may result in conductive hearing loss, although sensorineural hearing loss and mixed hearing loss were also seen. A previous study of 104 individuals in our cohort, ranging from 5 months to 37 years, who underwent an otolaryngology procedure (microlaryngoscopy or bronchoscopy) demonstrated that 71% had airway abnormalities. Airway abnormalities included tracheomalacia (36%), subglottic stenosis (28%), laryngomalacia (26%), glottic web (21%), and bronchomalacia (16%). Additional issues observed in our cohort include vocal cord paralysis, choanal atresia, tracheoesophageal fistula, tracheal atresia, and microtia. Tracheostomy was required in 30% of these patients (Sacca et al., 2017). The high proportion of airway abnormalities in those undergoing airway evaluation, shows the importance of prompt referral to Otolaryngology when issues are suspected.
Ophthalmology
Patients exhibited a number of ocular issues, including sclerocornea, posterior embryotoxon, tortuous retinal vessels, eyelid hooding, strabismus, ptosis, amblyopia, tilted optic nerves, cataracts and/or colobomas (Forbes et al., 2007; Binenbaum et al., 2008). The number of patients with astigmatism, myopia, and hyperopia is comparable to the number of people affected by those conditions in the general population. The high proportion of abnormalities identified shows the importance of an initial Ophthalmology examination, with follow up as dictated by the initial visit.
Hematology and Oncology
Patients with 22q11.2DS may have macrothrombocytopenia (baseline thrombocytopenia with increased MPV) as well as an increased risk of autoimmune cytopenias, which tend to be recurrent. Management recommendations are the same as for patient without 22q11.2DS (Lambert et al., 2017).
Although rare, 6% of patients had malignancies including hepatoblastoma, Wilms tumor, renal cell carcinoma, thyroid carcinoma, melanoma, and leukemia. Previous studies have suggested that the overall malignancy rate is higher among our cohort than age-adjusted population estimates (Lambert et al., 2017); however, larger studies will be required to definitively identify specific tumors that are more common among individuals with 22q11.2 deletions.
Transition to Adult Care
As is evident by the aging population, there is a large need for transitional care. We have created a transitional care program with the Division of Translational Medicine and Human Genetics at the University of Pennsylvania and have evaluated 13 patients. These patients were evaluated by a single medical geneticist (SK). Patients were frequently diagnosed in pregnancy due to an affected fetus (5 patients). Similar medical issues were seen as our pediatric cohort. Neuropsychiatric issues were also prevalent; 5 patients had anxiety, 4 had ADHD, and 1 had psychosis. About half (6 patients) are living independently with their own partners/children, while the remaining 7 patients reside within their childhood home. Of these 13 adults, 8 graduated from high school, 1 graduated from college, 2 are enrolled in college courses, and 6 are employed. We believe this program will be important for the continued management as well as evaluation of the natural history of our cohort.
Dual Diagnoses
There were also a number of patients in our cohort with another significant syndrome/condition, in addition to the 22q11.2 deletion. These included severe combined immunodeficiency, Marfan syndrome, two patients with CHARGE (OMIM #214800, due to CHD7 pathogenic variants), cystic fibrosis (OMIM #21970), Ehlers-Danlos syndrome (OMIM #130000), Trisomy 8 mosaicism, and several with an additional potentially pathogenic CNV. Six patients also had autosomal recessive conditions unmasked by a 22q11.2 deletion, Bernard-Soulier syndrome (OMIM #231200, GP1BB pathogenic variant), and CEDNIK syndrome (OMIM #609528, SNAP29 pathogenic variant) (Cohen et al cite special issue).
Mortality
We observed a 4% mortality rate overall. Most of these deaths were related to complex congenital heart disease. The median age at death was 5 months. This is in contrast to the median age of death of 41.5 years from a prior study of adults with 22q11.2DS and is likely due to the age of our cohort (Bassett et al., 2009).
Discussion
To our knowledge, this is the largest longitudinal study of 22q11.2DS. The size of our patient population allowed us to more accurately characterize the demographics, dysmorphology, molecular etiologies, common medical issues, and neurodevelopmental outcomes of this disease. Notably, 74% of our cohort described themselves as Caucasian, with only 9% being of African descent, differing demographically from our general hospital population of approximately 35% African American. Analysis of physical examination findings by race suggests that some common facial features are either less common or more difficult to identify clinically in non-Caucasian patients, potentially hindering diagnosis. Alternative explanations include differences in access to our referral center or possibly population variations in a heritable predisposition to development of 22q11.2 deletions (Demaerel et al., 2017).
Our findings confirm that the range of behavioral and developmental phenotypes that can be seen in patients with the 22q11.2 deletion are considerable (Swillen and McDonald-McGinn, 2015; Gerdes et al., 1999; McDonald-McGinn et al., 2001 PMID: 28328118). Previous studies revealed that delays in reaching motor milestones and emergence of language are common in children with 22q11.2DS (Gerdes et al., 1999; Solot et al., 2001). Our findings suggest that motor delays are somewhat less severe and may be associated with congenital heart disease. Meanwhile, language delays are more prominent and do not correlate with major medical issues. We find that measured FSIQ is significantly below normal, although there is likely ascertainment bias in that individuals with apparently low IQ are more likely to have formal testing. We also found that neuropsychiatric disorders such as autism and ADHD are common in our cohort. The majority of the children in our cohort have an IEP. We suspect the remaining 7% merit an IEP, but are either still too young or attend school in poor districts. Notably, not all patients necessarily had standardized formal diagnosis according to DSM-5 criteria, which is a potential weakness of our analysis.
Similar to previously published information (Ryan et al., 1997; McDonald-McGinn et al., 2001; Repetto et al., 2014), we find that congenital heart defects are extremely common in 22q11.2DS. We find that rare congenital heart abnormalities such as interrupted aortic arch and vascular rings are frequent among our patients. We find that palate abnormalities are also common, despite that reported incidence of palatal anomalies in the literature varies greatly, from 9 to 98% (Driscoll et al., 1993; Cohen et al., 1999; McDonald-McGinn et al., 1999). These discrepancies are likely due to variations in authors’ classification of palate abnormalities.
Overall, our analysis of this cohort provides an in-depth view of the phenotypic spectrum associated with the 22q11.2DS. Although not every individual received formalized testing in each area, our data estimates the prevalence of various features. As our cohort ages, we will further identify and classify medical comorbidities that develop during adulthood.
Supplementary Material
References
- Amati F, Conti E, Novelli A, Bengala M, Diglio MC, Marino B, Giannotti A, Gabrielli O, Novelli G, Dallapiccola B 1999. Atypical deletions suggest five 22q11.2 critical regions related to the DiGeorge/velo-cardio-facial syndrome. Eur J Hum Genet 7: 903–909. [DOI] [PubMed] [Google Scholar]
- American Academy of Pediatrics Council on Children With Disabilities, Cartwright JD 2007. Provision of educationally related services for children and adolescents with chronic diseases and disabling conditions. Pediatrics 119: 1218–1223. [DOI] [PubMed] [Google Scholar]
- Baldini A 2005. Dissecting contiguous gene defects: TBX1. Curr. Opin. Genet. Dev. 15: 279–284. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Chow EWC, Husted J, Hodgkinson KA, Oechslin E, Harris L, Silversides C 2009. Premature death in adults with 22q11.2 deletion syndrome. J Med Genet 46: 324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumer A, Riegel M, Schinzel A 2004. Non-random asynchronous replication at 22q11.2 favours unequal meiotic crossovers leading to the human 22q11.2 deletion. J Med Genet 41: 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bawle EV, Conard J, Van Dyke DL, Czarnecki P, Driscoll DA. 1998. Seven new cases of Cayler cardiofacial syndrome with chromosome 22q11.2 deletion, including a familial case. Am. J. Med. Genet. 79: 406–410. [DOI] [PubMed] [Google Scholar]
- Billett J, Cowie MR, Gatzoulis MA, Vonder Muhll IF, Majeed A 2008. Comorbidity, healthcare utilisation and process of care measures in patients with congenital heart disease in the UK: cross-sectional, population-based study with case-control analysis. Heart 94: 1194–1199. [DOI] [PubMed] [Google Scholar]
- Binenbaum G, McDonald-McGinn DM, Zackai EH, Walker BM, Coleman K, Mach AM, Adam M, Manning M, Alcorn DM, Zabel C, Anderson DR, Forbes BJ. 2008. Sclerocornea associated with the chromosome 22q11.2 deletion syndrome. Am J Med Genet A 146A: 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O’Leary LA, Wong L-Y, Elixson EM, Mahle WT, Campbell RM 2003. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 112: 101–107. [DOI] [PubMed] [Google Scholar]
- Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, Scambler P, Goodship J 1993. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J Med Genet 30: 822–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey AH, Kelly D, Halford S, Wadey R, Wilson D, Goodship J, Burn J, Paul T, Sharkey A, Dumanski J 1992. Molecular genetic study of the frequency of monosomy 22q11 in DiGeorge syndrome. The American Journal of Human Genetics 51: 964–970. [PMC free article] [PubMed] [Google Scholar]
- Cheung ENM, George SR, Costain GA, Andrade DM, Chow EWC, Silversides CK, Bassett AS. 2014. Prevalence of hypocalcaemia and its associated features in 22q11·2 deletion syndrome. Clin. Endocrinol. (Oxf) 81: 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Chow EW, Weksberg R, Bassett AS. 1999. Phenotype of adults with the 22q11 deletion syndrome: A review. Am. J. Med. Genet. 86: 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaerel W, Hestand MS, Vergaelen E, Swillen A, López-Sánchez M, Pérez-Jurado LA, McDonald-McGinn DM, Zackai E, Emanuel BS, Morrow BE, Breckpot J, Devriendt K, Vermeesch JR, International 22q11.2 Brain and Behavior Consortium. 2017. Nested Inversion Polymorphisms Predispose Chromosome 22q11.2 to Meiotic Rearrangements. Am J Hum Genet 101: 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Devriendt K, Fryns J-P, Mortier G, van Thienen MN, Keymolen K 1998. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet 35: 789–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Budarf ML, Emanuel BS. 1992a. A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. The American Journal of Human Genetics 50: 924–933. [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, Emanuel BS. 1993. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet 30: 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Spinner NB, Budarf ML, McDonald-McGinn DM, Zackai EH, Goldberg RB, Shprintzen RJ, Saal HM, Zonana J, Jones MC. 1992b. Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome. Am. J. Med. Genet. 44: 261–268. [DOI] [PubMed] [Google Scholar]
- Edelmann L, Pandita RK, Morrow BE. 1999. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. The American Journal of Human Genetics 64: 1076–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher PS, McDonald-McGinn DM, Fox CA, Driscoll DA, Emanuel BS, Zackai EH. 2000. Dysphagia in children with a 22q11.2 deletion: unusual pattern found on modified barium swallow. J. Pediatr. 137: 158–164. [DOI] [PubMed] [Google Scholar]
- Forbes BJ, Binenbaum G, Edmond JC, DeLarato N, McDonald-McGinn DM, Zackai EH. 2007. Ocular findings in the chromosome 22q11.2 deletion syndrome. J AAPOS 11: 179–182. [DOI] [PubMed] [Google Scholar]
- Fryburg JS, Lin KY, Golden WL. 1996. Chromosome 22q11.2 deletion in a boy with Opitz (G/BBB) syndrome. Am. J. Med. Genet. 62: 274–275. [DOI] [PubMed] [Google Scholar]
- Garcia-Miñaur S, Fantes J, Murray RS, Porteous MEM, Strain L, Burns JE, Stephen J, Warner JP. 2002. A novel atypical 22q11.2 distal deletion in father and son. J Med Genet 39: E62–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes M, Solot C, Wang PP, Moss E, Larossa D, Randall P, Goldmuntz E, Clark BJ, Driscoll DA, Jawad A, Emanuel BS, McDonald-McGinn DM, Batshaw ML, Zackai EH. 1999. Cognitive and behavior profile of preschool children with chromosome 22q11.2 deletion. Am. J. Med. Genet. 85: 127–133. [PubMed] [Google Scholar]
- Giannotti A, Digilio MC, Marino B, Mingarelli R, Dallapiccola B 1994. Cayler cardiofacial syndrome and del 22q11: part of the CATCH22 phenotype. Am. J. Med. Genet. 53: 303–304. [DOI] [PubMed] [Google Scholar]
- Gong W, Emanuel BS, Collins J, Kim DH, Wang Z, Chen F, Zhang G, Roe B, Budarf ML. 1996. A transcription map of the DiGeorge and velo-cardio-facial syndrome minimal critical region on 22q11. Hum Mol Genet 5: 789–800. [DOI] [PubMed] [Google Scholar]
- Goodship J, Cross I, LiLing J, Wren C 1998. A population study of chromosome 22q11 deletions in infancy. Arch. Dis. Child. 79: 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati FR, Molina Gomes D, Ferreira JCPB, Dupont C, Alesi V, Gouas L, Horelli-Kuitunen N, Choy KW, García-Herrero S, la Vega, de AG, Piotrowski K, Genesio R, Queipo, Malvestiti B, Hervé B, Benzacken B, Novelli A, Vago P, Piippo K, Leung TY, Maggi F, Quibel T, Tabet AC, Simoni G, Vialard F 2015. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat. Diagn. 35: 801–809. [DOI] [PubMed] [Google Scholar]
- Habel A, McGinn M-J, Zackai EH, Unanue N, McDonald-McGinn DM 2012. Syndrome-specific growth charts for 22q11.2 deletion syndrome in Caucasian children. Am J Med Genet A 158A: 2665–2671. [DOI] [PubMed] [Google Scholar]
- Homans JF, Tromp IN, Colo D, Schlösser TPC, Kruyt MC, Deeney VFX, Crowley TB, McDonald-McGinn DM, Castelein RM. 2017. Orthopaedic manifestations within the 22q11.2 Deletion syndrome: A systematic review. Am J Med Genet A. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Zackai EH, Emanuel BS, Kistenmacher M, Greenberg F, Punnett HH. 1982. The association of the DiGeorge anomalad with partial monosomy of chromosome 22. J. Pediatr. 101: 197–200. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Nakayama T, Osugi Y, Tsuda E, Masuno M, Imaizumi K, Kamiya T, Sano T, Okada S, Nishisho I 1996. Deletion mapping of 22q11 in CATCH22 syndrome: identification of a second critical region. The American Journal of Human Genetics 58: 1377–1381. [PMC free article] [PubMed] [Google Scholar]
- la Chapelle, de A, Herva R, Koivisto M, Aula P 1981. A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet 57: 253–256. [DOI] [PubMed] [Google Scholar]
- Lacassie Y, Arriaza MI. 1996. Opitz GBBB syndrome and the 22q11.2 deletion. Am J Med Genet A 62: 318–318. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Arulselvan A, Schott A, Markham SJ, Crowley TB, Zackai EH, McGinn DMM. 2017. The 22q11.2 deletion syndrome: Cancer predisposition, platelet abnormalities and cytopenias. Am J Med Genet A 16: 318. [DOI] [PubMed] [Google Scholar]
- Lopez-Rivera E, Liu YP, Verbitsky M, Anderson BR, Capone VP, Otto EA, Yan Z, Mitrotti A, Martino J, Steers NJ, Fasel DA, Vukojevic K, Deng R, Racedo SE, Liu Q, Werth M, Westland R, Vivante A, Makar GS, Bodria M, Sampson MG, Gillies CE, Vega-Warner V, Maiorana M, Petrey DS, Honig B, Lozanovski VJ, Salomon R, Heidet L, Carpentier W, Gaillard D, Carrea A, Gesualdo L, Cusi D, Izzi C, Scolari F, van Wijk JAE, Arapovic A, Saraga-Babic M, Saraga M, Kunac N, Samii A, McDonald-McGinn DM, Crowley TB, Zackai EH, Drozdz D, Miklaszewska M, Tkaczyk M, Sikora P, Szczepanska M, Mizerska-Wasiak M, Krzemien G, Szmigielska A, Zaniew M, Darlow JM, Puri P, Barton D, Casolari E, Furth SL, Warady BA, Gucev Z, Hakonarson H, Flogelova H, Tasic V, Latos-Bielenska A, Materna-Kiryluk A, Allegri L, Wong CS, Drummond IA, D’Agati V, Imamoto A, Barasch JM, Hildebrandt F, Kiryluk K, Lifton RP, Morrow BE, Jeanpierre C, Papaioannou VE, Ghiggeri GM, Gharavi AG, Katsanis N, Sanna-Cherchi S 2017. Genetic Drivers of Kidney Defects in the DiGeorge Syndrome. N Engl J Med 376: 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka R, Takao A, Kimura M, Imamura S, Kondo C, Joh-o K, Ikeda K, Nishibatake M, Ando M, Momma K 1994. Confirmation that the conotruncal anomaly face syndrome is associated with a deletion within 22q11.2. Am. J. Med. Genet. 53: 285–289. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J 1995. Autosomal dominant “Opitz” GBBB syndrome due to a 22q11.2 deletion. Am. J. Med. Genet. 59: 103–113. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Driscoll DA, Emanuel BS, Goldmuntz E, Clark BJ, Solot C, Cohen M, Schultz P, Larossa D, Randall P, Zackai EH. 1997. Detection of a 22q11.2 deletion in cardiac patients suggests a risk for velopharyngeal incompetence. Pediatrics 99: E9. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Emanuel BS, Zackai EH. 1996. Autosomal dominant “Opitz” GBBB syndrome due to a 22q11.2 deletion. Am. J. Med. Genet. 64: 525–526. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Solot C, Wang P, Jacobs I, Handler S, Knightly C, Heher K, Wilson M, Ming JE, Grace K, Driscoll D, Pasquariello P, Randall P, Larossa D, Emanuel BS, Zackai EH. 1999. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genet. Couns. 10: 11–24. [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JAS, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS. 2015. 22q11.2 deletion syndrome. Nature Reviews Disease Primers 1: 15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH. 2001. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med 3: 23–29. [DOI] [PubMed] [Google Scholar]
- McQuade L, Christodoulou J, Budarf M, Sachdev R, Wilson M, Emanuel B, Colley A 1999. Patient with a 22q11.2 deletion with no overlap of the minimal DiGeorge syndrome critical region (MDGCR). Am. J. Med. Genet. 86: 27–33. [PubMed] [Google Scholar]
- Ming JE, McDonald-McGinn DM, Megerian TE, Driscoll DA, Elias ER, Russell BM, Irons M, Emanuel BS, Markowitz RI, Zackai EH. 1997. Skeletal anomalies and deformities in patients with deletions of 22q11. Am. J. Med. Genet. 72: 210–215. [DOI] [PubMed] [Google Scholar]
- O’Donnell H, McKeown C, Gould C, Morrow B, Scambler P 1997. Detection of an atypical 22q11 deletion that has no overlap with the DiGeorge syndrome critical region. The American Journal of Human Genetics 60: 1544–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsdóttir S, Vujic M, Fasth A 2004. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch. Dis. Child. 89: 148–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plato CC, Cereghino JJ, Steinberg FS. 1975. The dermatoglyphics of American Caucasians. Am. J. Phys. Anthropol. 42: 195–210. [DOI] [PubMed] [Google Scholar]
- Preus M, Fraser FC. 1972. Dermatoglyphics and syndromes. Am. J. Dis. Child. 124: 933–943. [DOI] [PubMed] [Google Scholar]
- Racedo SE, McDonald-McGinn DM, Chung JH, Goldmuntz E, Zackai E, Emanuel BS, Zhou B, Funke B, Morrow BE. 2015. Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am J Hum Genet 96: 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repetto GM, Guzmán ML, Delgado I, Loyola H, Palomares M, Lay-Son G, Vial C, Benavides F, Espinoza K, Alvarez P 2014. Case fatality rate and associated factors in patients with 22q11 microdeletion syndrome: a retrospective cohort study. BMJ Open 4: e005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brøndum-Nielsen K, Scambler PJ. 1997. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet 34: 798–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacca R, Zur KB, Crowley TB, Zackai EH, Valverde KD, McDonald-McGinn DM. 2017. Association of airway abnormalities with 22q11.2 deletion syndrome. Int. J. Pediatr. Otorhinolaryngol. 96: 11–14. [DOI] [PubMed] [Google Scholar]
- Saitta SC, Harris SE, Gaeth AP, Driscoll DA, McDonald-McGinn DM, Maisenbacher MK, Yersak JM, Chakraborty PK, Hacker AM, Zackai EH, Ashley T, Emanuel BS. 2004. Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum Mol Genet 13: 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scambler PJ. 2000. The 22q11 deletion syndromes. Hum Mol Genet 9: 2421–2426. [DOI] [PubMed] [Google Scholar]
- Scambler PJ, Carey AH, Wyse RK, Roach S, Dumanski JP, Nordenskjold M, Williamson R 1991. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics 10: 201–206. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, O’Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, Emanuel BS. 2000. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet 9: 489–501. [DOI] [PubMed] [Google Scholar]
- Solot CB, Gerdes M, Kirschner RE, McDonald-McGinn DM, Moss E, Woodin M, Aleman D, Zackai EH, Wang PP 2001. Communication issues in 22q11.2 deletion syndrome: children at risk. Genet Med 3: 67–71. [DOI] [PubMed] [Google Scholar]
- Swillen A, McDonald-McGinn D 2015. Developmental trajectories in 22q11.2 deletion. American journal of medical genetics Part C, Seminars in medical genetics 169: 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JAS, Breetvelt EJ, Duijff SN, Eliez S, Schneider M, Jalbrzikowski M, Armando M, Vicari S, Shashi V, Hooper SR, Chow EWC, Fung WLA, Butcher NJ, Young DA, McDonald-McGinn DM, Vogels A, van Amelsvoort T, Gothelf D, Weinberger R, Weizman A, Klaassen PWJ, Koops S, Kates WR, Antshel KM, Simon TJ, Ousley OY, Swillen A, Gur RE, Bearden CE, Kahn RS, Bassett AS, International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. 2015. Cognitive decline preceding the onset of psychosis in patients with 22q11.2 deletion syndrome. JAMA Psychiatry 72: 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DI, Goodship JA, Burn J, Cross IE, Scambler PJ. 1992. Deletions within chromosome 22q11 in familial congenital heart disease. Lancet 340: 573–575. [DOI] [PubMed] [Google Scholar]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S-I, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R 2003. Role of TBX1 in human del22q11.2 syndrome. Lancet 362: 1366–1373. [DOI] [PubMed] [Google Scholar]
- Yamagishi H, Garg V, Matsuoka R, Thomas T, Srivastava D 1999. A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science 283: 1158–1161. [DOI] [PubMed] [Google Scholar]
- Zweier C, Sticht H, Aydin-Yaylagul I, Campbell CE, Rauch A 2007. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am J Hum Genet 80: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.