

Abstract
Metastatic disease is a major cause of mortality in cancer patients. While many drug delivery strategies for anticancer therapeutics have been developed in preclinical studies of primary tumors, the drug delivery properties of metastatic tumors have not been sufficiently investigated. Therapeutic efficacy hinges on efficient drug permeation into the tumor microenvironment, which is known to be heterogeneous thus potentially making drug permeation heterogeneous, also. In this study, we have identified that 4T1 liver metastases, treated with pegylated liposomal doxorubicin, have unfavorable and heterogeneous transport of doxorubicin. Our drug extravasation results differ greatly from analogous studies with 4T1 tumors growing in the primary site. A probabilistic tumor population model was developed to estimate drug permeation efficiency and drug kinetics of liver metastases by integrating the transport and structural properties of tumors and delivered drugs. The results demonstrate significant heterogeneity in metastases with regard to transport properties of doxorubicin within the same animal model, and even within the same organ. These results also suggest that the degree of heterogeneity depends on the stage of tumor progression and that differences in transport properties can define transport-based tumor phenotypes. These findings may have valuable clinical implications by illustrating that therapeutic agents can permeate and eliminate metastases of “less resistant” transport phenotypes, while sparing tumors with more “resistant” transport properties. We anticipate that these results could challenge the current paradigm of drug delivery into metastases, highlight potential caveats for therapies that may alter tumor perfusion, and deepen our understanding of the emergence of drug transport-based therapeutic resistance.
Introduction
Current estimates regarding the global incidence of cancer predict that, by the year 2020, the number of new cancer cases diagnosed will increase to 15 million annually and that cancer will be responsible for more than 12 million deaths [1]. Metastasis, the spread of cancer cells from the primary tumor to secondary tumors at distant sites, is currently one of the greatest challenges in cancer treatment. For many patients, by the time a tumor is clinically detected, metastasis has already occurred [2]. Across all cancer types, only one in five patients diagnosed with metastatic cancer will survive more than 5 years [3]. The lungs and liver are among the most common sites for metastases from tumors originating in other organs, including breast, kidney, colorectal, bladder, kidney, and skin (melanoma). The lack of effective treatments for metastatic tumors negatively impacts patient survival. Although recent medical advances have resulted in many new drugs, the survival of patients with metastatic cancer has not significantly improved for more than a decade across many cancer types [4]. While resistance mechanisms in cancer cells, based on genetic and non-genetic studies, have been characterized, poor drug delivery and limited tumor uptake are often neglected as mechanisms of drug resistance in cancer [5]. In contrast, systemic drug pharmacokinetics has been historically accepted as the key determinant of a drug’s clinical utility and efficacy; yet, clinical evidences indicate that systemic pharmacokinetics alone may not be sufficient to account for therapeutic efficacy, as it does not reflect the heterogeneity of intra- and inter-tumor pharmacokinetics [6, 7]. This quandary has been illustrated by one study demonstrating an excellent association between tumor-associated pharmacokinetics and response to 5-fluorouracil [8], while others indicated that tumor-associated pharmacokinetics should be emphasized when considering delivery of drugs such as doxorubicin (DOX) or PEGylated liposomal doxorubicin (PLD) [9–11]. Using tumor-associated pharmacokinetics, the authors were able to identify a difference in therapeutic efficacy between 3LL and 4T1 tumor models, while systemic pharmacokinetics could detect any differences between these animal models [12]. The pharmacokinetics of PLD involves dramatically extended circulation times and reduced volume of distribution compared to free DOX [13], but the efficacy gains with PLD are minimal in clinical application [12, 14, 15].
The enhanced permeation and retention (EPR) effect can also enhance the local pharmacokinetic properties of free drugs or drug vectors. However, EPR benefits vary among tumors in different locations because of differences in the tissue microenvironment of a tumor or an organ [16, 17]. Despite differences in drug formulations and their systemic pharmacokinetics, the transport properties of the tumor microenvironment is the absolute determinant of the efficiency of drug delivery to cancer cells, and ultimately therapeutic efficacy.
While drug delivery has been widely investigated in preclinical studies using primary or subcutaneous tumor models, drug delivery to metastases and the heterogeneity of drug delivery are minimal at best. Based on growing evidence that the tumor microenvironment can affect drug transport, we investigated drug transport properties in a mouse model of murine breast cancer (4T1) liver metastases. Our study provides insight into the origins of drug transport-based therapeutic resistance in metastases and proposes strategies to overcome this resistance.
Materials and Methods
In vivo experiments.
Female Balb/C mice, 6–8 weeks of age, were purchased from Charles River Laboratories (Wilmington, MA, USA). To establish experimental liver metastasis, 4T1 breast cancer cells (1 × 105/100 μL) were injected into the spleen bodies of mice [18]. The cells were injected into the spleen and disseminated to the liver through the portal vein, producing multiple experimental liver metastases.
10 days after the inoculation with 4T1 cells, mice were intravenously injected with 6 mg/kg PLD (Doxoves-Liposomal Doxorubicin HCl; FormuMax Scientific Inc., Sunnyvale, CA, USA) and sacrificed 24 hours later. Then, the liver was excised, frozen, and processed to image endothelial cells, nuclei, and PLD accumulation. In another cohort of mice, intravital microscopic imaging was performed to evaluate transport properties in the liver as described below. The aforementioned surgical procedures have been approved by the Institutional Animal Care and Use Committee (IACUC) of the Houston Methodist Research Institute.
Immunofluorescent staining of endothelial cells (CD31), macrophages (CD204), nuclei (DAPI) and imaging of PLD in liver.
After fixation of the frozen sections in cold acetone and protein block, immunofluorescent staining of endothelial cells, macrophages and nuclei was performed using antibodies to CD31 (BD Biosciences) followed by staining with Alexa 647 labeled goat anti rat IgG (Invitrogen), CD204 labeled with Alexa 647 (Bio-Rad) followed by staining with Alexa 647 labeled goat anti rat IgG (Invitrogen), CD204 labeled with Alexa 647 (Bio-Rad) and DAPI (Thermo Fisher Scientific), respectively [19]. The images were captured using a confocal laser-scanning microscope (Nikon Inc., Tokyo, Japan). The ruby red fluorescence of anthracyclines in the liver was also imaged using confocal microscopy at the excitation and emission wavelengths of 488 and 590 nm, respectively [9, 10].
Intravital microscopy imaging.
Dextran (molecular weight of 10 kDa) conjugated with Alexa Fluor 488 and 70 kDa dextran conjugated with Alexa Fluor 647 (Thermo Fisher Scientific) were injected intravenously into the retro-orbital space of tumor-bearing mice during intravital microscopy (IVM) recording for visualization of blood flow dynamics. Laser wave length and detection ranges are the following: 488nm/500–550nm and 640nm/663–738nm. Mice were anesthetized with isoflurane and mounted on a heated microscope stage during IVM imaging under a Nikon A1R multiphoton microscope (Nikon Inc.). The liver was exposed by a midline incision through the abdominal wall. The images were processed to assess fluorescent intensities of tracers inside and outside the lesions at different times using NIS-Elements Image Processing Software (Nikon) [18, 20].
Cell culture.
4T1 murine breast cancer cells were obtained from ATCC. The cells were maintained in complete minimal essential medium supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, L-glutamine, vitamin solution (Life Technologies, Inc., Grand Island, NY), and a penicillin–streptomycin mixture (Flow Laboratories, Rockville, MD) [18].
Quantification.
ImageJ software [21] was used to size metastatic lesions manually after immunohistochemistry (IHC). The vascular fraction (rV) and the area fraction of DOX fluorescence inside the metastases were calculated by thresholding corresponding channels based on background intensity, and then area fractions were automatically calculated. The size distributions were fitted using gamma distributions. IHC data of metastases were collected as follows: 1) at 10 days, two non-treated mice with two whole liver lobes from each mouse (254 tumors total); 2) at 14 days, non-treated, three mice with four liver lobes total (198 tumors total); and 3) at 14 days, treated, three mice with one liver lobe from each mouse (220 tumors total).
The diffusion coefficient (D) of 10 kDa and 70 kDa dextrans was estimated from dextran tracer intensity kinetics recorded in IVM based on three tumors. The transport model was developed by using the composite Smeared Finite Element method [22] (see Supplemental Material) and used to estimate D values by fitting tumor intensity profiles. The D values for 10 kDa and 70 kDa dextrans were calculated as 7.3 ± 2.1 and 0.2 ± 0.1 μm2/s (7.3∙10−7 and 0.2∙10−9 cm2/s), respectively.
Simulation of the permeation time (τ).
The model was executed in Python. Probabilistic τ calculations were accomplished by using probability tables of tumor sizes and rV based on quantified in vivo results. Diffusion coefficients of dextran tracers were modeled with a normal distribution characterized by their estimated value and dispersion, while other compounds were assigned with general characteristic values and with dispersions equal to the coefficient of variation of 20%.
Results
Metastases have progression-dependent vascularization and drug permeation
Whole liver lobes were imaged prior to PLD injection (10 days after tumor inoculation) and again 4 days later. The size of metastatic tumors was quantitated. Metastatic tumors, easily detectible by DAPI staining and DOX fluorescence, were observed extensively in the sinusoidal spaces of the liver (Figure 1a). DOX fluorescence colocalized with Kupffer cells suggesting PLD was uptaken by macrophages in healthy liver but not by tumor-associated macrophages. The size distribution at day 10 was relatively narrow and centered around 250 μm, while the untreated group had a substantially wider distribution and was centered close to 500 μm after 4 days of progression (Figure 1b). The sizes of the metastases in treated and untreated mice were nearly identical, suggesting that there was no therapeutic effect after PLD injection, which was consistent with the absence of DOX fluorescence within metastatic tumors, except in a few limited cases (Figure 1a, inset). These results were discordant with DOX extravasation data in subcutaneous 4T1 tumors after systemic injection of PLD, in which PLD accumulated inside the primary tumors [13, 14].
Figure 1. Heterogeneity of transport and structural properties of 4T1 breast cancer metastases in mouse liver.
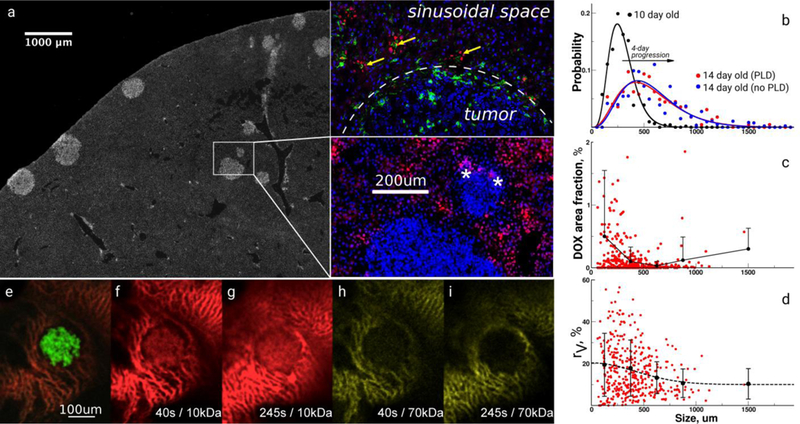
a, Immunohistochemical image of a whole liver lobe scan containing metastases stained with 4’,6-diamidino-2-phenylindole. The inset shows several magnified metastases with different sizes and the red fluorescence of extravasated doxorubicin (DOX) delivered by PEGylated liposomal DOX (PLD) and colocalizing (yellow arrows) with Kupffer cells (green) outside tumors; stars denote DOX fluorescence in tumors, white-dashed line –tumor boundary. b, Size distribution of metastatic tumors in liver lobes before and after PLD as measured by imaging analysis. c, The area fraction of DOX inside tumors. Circles represent an individual tumor measurement followed by the average and standard deviation as a function of the sizes of metastases. d, The area fraction (%) of vasculature inside tumors stained with CD31. Circles represent an individual tumor measurement followed by the average and standard deviation as a function of the sizes of metastases. e, Intravital image (IVM) of the 4T1 tumor tagged with green fluorescent protein. IVM imaging of the extravasation of red 10 kDa (f, g) and yellow 70 kDa (h, i) dextran tracers.
Quantitation revealed that the DOX extravasation area was only 0%–2% of metastases that were < 300 μm in size, while the DOX fluorescence area fraction was < 0.5% for larger tumors (Figure 1c). The DOX area fraction was highly variable for the smallest tumors. We hypothesized that the PLD did not enter tumor-associated capillaries because our previous studies of 4T1 liver metastases suggested that capillaries inside metastases were partially or fully dysfunctional [23]. It is also possible that tumors could have been permeated by DOX diffusion from the surrounding sinusoidal space, which was saturated with free DOX and could function as a PLD/DOX depot (Figure 1a, inset). However, our imaging results did not support this hypothesis.
To further characterize the DOX extravasation results, the capillary content of metastases was quantified with CD31 staining, and the capillary area fraction (rV) was calculated. Notably, the highest values and heterogeneity of rV were found for the tumors < 500 μm in size (Figure 1d). The average rV was 20% in smaller tumors but decreased to 9% for tumors > 500 μm in size. Importantly, the largest heterogeneity of rV was found in the smallest tumors. We hypothesized that such heterogeneity could be caused by differences resulting from angiogenesis and that metastases could engulf the existing liver vasculature. Taken together, the results demonstrate that metastatic 4T1 liver tumors are heterogeneous not only in vascularity but also DOX extravasation, and these heterogeneities are dependent upon tumor size.
To assess the permeability of 4T1 liver metastases, we imaged and quantified the diffusion of 10 kDa and 70 kDa dextran tracers by intravital microscopy. The 10 kDa tracer extravasated easily into metastases, while the 70 kDa tracer was confined to liver sinusoids that surrounded tumors (Figure 1e–i). The estimated diffusion coefficients were approximately 7.3 ± 2.1 and 0.2 ± 0.1 μm2/s, respectively, and suggested diffusion is inversely related to tracer size decreased diffusion based on the size of the tracer [24, 25]. Furthermore, because our IVM imaging results suggested that the perfusion through tumor-associated vessels was largely compromised, we concluded that diffusion was the prevailing transport mechanism. Thus, any anti-permeability effect of therapeutics could damage the remaining fragile opportunities for efficient drug delivery to tumors. Also, it is reasonable to think that the local heterogeneity of the diffusion properties could be an important aspect deserving separate dedicated studies.
The time needed to permeate the entire population of metastases provides a substantial challenge
The heterogeneity of metastatic tumor size and rV resulted in additional complexity in the analysis of drug delivery to metastatic tumors, which is mitigated best by using a single property describing an individual tumor’s propensity for drug permeation. We therefore introduced the tumor permeation time (τ) as a parameter, which can be compared to drug circulation times in systemic circulation, i.e., systemic pharmacokinetics (Figure 2a). The τ was expressed through fundamental principles of diffusion and convection transports as:
| (1) |
where τD was the diffusion time of drugs through the extravascular tumor space; τV , the perfusion times of tumor capillaries; rV, the vascular fraction area of a tumor; f, the fraction of functioning tumor capillaries; A(R), the area of a tumor with the radius R; dV, the characteristic tumor capillary size; v, the characteristic blood flow velocity; D, the apparent diffusion (transport) coefficient of a drug in the extravascular domain of a tumor; and LP, the perimeter length of the tumor. The diffusion component transformed the two-dimensional extravascular space from immunohistochemistry imaging into a one-dimensional diffusion problem by using the diffusing length, where drug permeation into a tumor could occur through the tumor boundary and/or through the tumor-associated capillaries (Figure 2b). From Equation 1, τ integrated structural and transport properties of the tumor and drug. The τ was simulated in a probabilistic fashion by randomizing tumor properties based on quantitated probability distributions of tumor properties. The value of the parameter, f, is unknown, except that f << 1; therefore, we explored different f values in our analysis. The rV probabilities were coupled to tumor size based on experimental data (Figure 1d). D was assumed as a Gaussian probability distribution defined by the mean and standard deviation estimated from IVM measurements. The population of tumors may contain tumor entities possessing more or less favorable combinations of transport parameters affecting the permeation of drugs because of probabilistic nature of the parameters R, rV, and D.
Figure 2. Estimation of tumor perfusion times with different molecules.
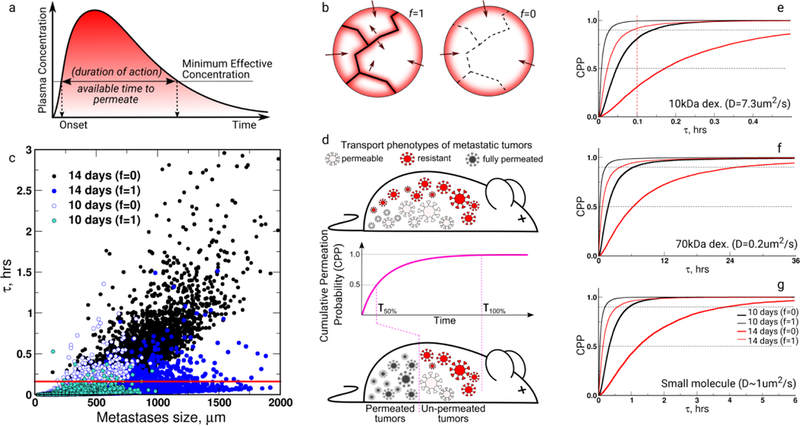
a, Drug permeation is estimated assuming concentrations that were above the minimum effective concentration. b, Drugs could permeate through the outer boundary of the tumor and through its capillaries; however, some metastases may contain a fraction of their capillaries (f) partially (0 < f <1) or fully (f = 0) dysfunctional. Molecules may extravasate (diffuse) into tumors from the tumor perimeter length (LP) or their own vasculature length (LV) (see the main text). c, An in vivo result-based probabilistic model (Equation 1) estimated the τ scattered distribution of 10 kDa dextran in the population of 10,000 4T1 tumors in liver by accounting for tumor size and transport properties (Figure 1) for 10- and 14-day-old tumors with a fully functional (f = 1) and fully dysfunctional (f = 0) tumor vasculature. The red line denotes the plasma half-life of the 10 kDa dextran. d, Cumulative permeation probability (CPP) is a cumulative metastatic tumor fraction characterized by full permeation, and CPP is a function of the time (τ). e, f, and g, Cumulative permeation probabilities in the 10,000 4T1 tumor population for 10 kDa dextran, 70 kDa dextran, and small molecules. The τ50%, τ90%, and τ100%, respectively, estimated times when the population of tumors is perfused 50%, 90%, or with total perfusion, respectively (d). The characteristic diffusion transport coefficients are provided for each molecule in e-g. Vertical red dashed lines denote the half-lives of specific molecules.
We simulated τ for the ensemble of 10,000 of 10- and 14-day-old 4T1 liver metastases by using D of the 10 kDa tracer at 10 and 14 days (Figure 2c). We also considered fully functional (f= 1) or fully dysfunctional (f = 0) tumor capillaries. The probabilistic model showed that 4T1 tumors with f = 1 could be fully permeated by a 10 kDa tracer within 0.5 hours, but the time triples for larger 14-day-old metastases with fully dysfunctional capillaries. Moreover, this estimation was consistent with tracer permeation kinetics in our IVM measurements (Figure 1f–i, Figure 2c–e). The majority of τ values were distributed at the bottom of the plot with scattered outliers possessing high τ values (Figure 2c). A Cumulative Permeation Probability (CPP) was calculated by integrating the τ histogram to estimate the population fraction of 4T1 tumors in an animal that could be fully permeated by the tracer (Figure 2d). CPP could be perceived as a measure of drug delivery success that was characterized by the time, τ, wherein the desired CPP was achieved. We also calculated CPP for the 70 kDa dextran tracer, for a generally small molecule drug (without strong absorption effects) by assigning to D, the characteristic values of those substances (Figure 2e–g). The CPP results point to the fact that these diffusing substances had very different time windows that varied from a few to many tens of hours, while for nanoparticles it could be even months (Supplementary Material). While the example of nanoparticles was extreme (many liposome-size nanoparticles do not extravasate far from capillaries), the example illustrates how the nature of the drug and the permeability of the tumor extravascular space may determine the time needed for successful drug delivery. The τ of DOX could be analyzed by the example of a small molecule (Figure 2g), but DOX is strongly absorbed by cell nuclei, which limits its permeation depth and makes it difficult to estimate the true D value of DOX in tissues using image analysis [9]. The absence of DOX fluorescence in tumors suggested that D was substantially smaller, hence the better example could be the 70 kDa dextran tracer with very limited permeation (Figure 2f). We compared times needed to permeate the entire population of metastases (100%), 90% of the population, or 50% of the population, which could be denoted by τ100%, τ90%, and τ50% in the CPP plots. Permeation of all metastases required more time for larger compounds. However, 50% permeation of any tumor population was achievable with current delivery approaches that could provide efficacy if dosing was maintained, because τ did not exceed 6 hours in the case of small molecules or dextran tracers (Figure 2e–g).
Anti-vascular strategies may be detrimental to efficient delivery of drugs to metastases
For the τ estimations in a practical perspective, we systematically estimated τ for the range of D values that were characteristic for different molecules and compared these estimates with the half-lives of current free and vector-based therapeutics (Figure 3a). Free, small molecule drugs usually have a half-life of a few hours in systemic circulation [26], and the D value of molecules < 1 kDa can be safely assumed to be approximately 10−7 cm2/s in in vitro buffers. However, because of molecular crowding and the dense tissue environment, D could be reduced by one or two orders of magnitude. By navigating the plot in Figure 3a, we found that 4T1 metastatic tumors could be 50% permeated by a drug within τ of a few hours with fully functioning capillaries. However, complete (100%) permeation might require tens of hours, which was difficult to achieve for free small molecule drugs. The situation is even more challenging for larger therapeutic molecules with lower D values. Drug vectors can alleviate this challenge by increasing the systemic circulation time and extending tumor exposure to drugs. Despite the ability of drug vectors to extend the systemic circulation time, D presents one of the main barriers to tumor permeation. There are limited options in improving D; either smaller therapeutic molecules must be chosen, or the extravascular space must be permeabilized. Both strategies have their own limitations, including a limited drug selection, or a chance to enhance dissemination of cancer cells.
Figure 3. The impact of transport properties and their heterogeneity on drug delivery to 4T1 metastases in liver.
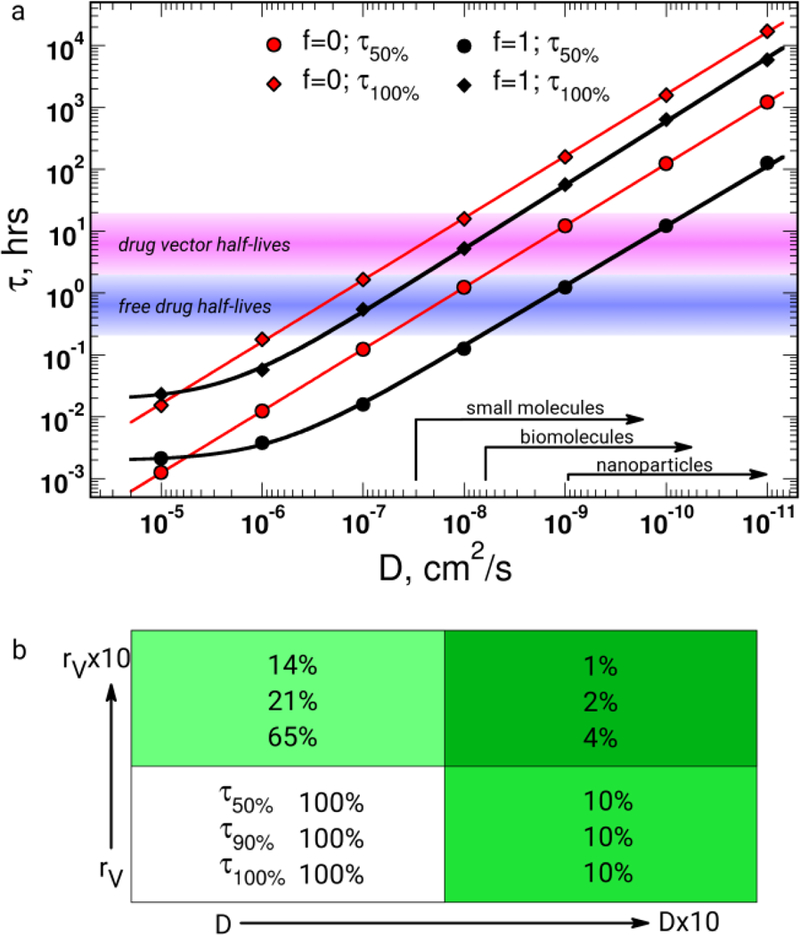
a, τ is a function of the diffusion coefficient (D) of the extravascular tumor space, where different D ranges can be associated with specific families of therapeutic substances. The characteristic half-lives of current free or delivery vector-based therapeutics are overlaid for comparison with τ and D. b, The effect of a ten-fold increase in rV and D on τ.
We have compared the effects of ten-fold increases in rV and D on the CPP in the 4T1 metastasis population (Figure 3b). The CPP effects on changes of rV and D values were expressed as the remaining τ from the original values of τ50%, τ90%, and τ100%. The results suggested that D and rV provided similar effects on τ50%, meaning that improved drug delivery can be equally observed with each drug delivery enhancement strategy, which hopefully directly translates into pharmacodynamically relevant responses. The permeation of 50% of the metastatic population is an achievable aim of current therapeutic strategies (Figure 3a). The problem becomes evident analyzing τ90% or τ100%, because D shows its largest effect on τ90% or τ100 after a ten-fold increase in rV and D. Eventually, the cumulative effect is the most promising with 1%, 2%, and 4% of initial τ50%, τ90%, and τ100% values (Figure 3b). It is worth noting the interplay between the effect of increasing vascularization and extravascular diffusion: the increase in the functioning capillary fraction not only provides a larger source of drugs, but also decreases the extravascular space and thus decreases the time needed to diffuse through the extravascular space. Therefore, these results did not favor anti-vascular endothelial growth factor (VEGF) or other anti-vascular therapies, because these therapies could reduce the rV and increase the diffusion burden through the remaining functioning tumor-associated capillaries or from tumor-surrounding tissues. This may be one of the reasons why clinical anti-VEGF results are still controversial [27].
Insufficiently efficient drug delivery may create a misleading perception of efficacy
Drug permeation analyses have revealed tumor phenotypes showing different transport-based therapeutic resistances that can originate from the heterogeneity of transport properties among 4T1 liver metastases (Figure 4a). We have generated 30 simulations by using our probabilistic 4T1 tumor model, i.e., n = 30 animal groups, based on the 10 kDa tracer imaging results. Each individual simulation represents an individual animal carrying 200 14-day-old 4T1 metastatic tumors. Figure 4b shows the CPP dependence on τ, where τ started deviating from the τ group average a CPP > 80%. The results demonstrated that heterogeneity of drug delivery increased while trying to achieve higher CPP and that the number of animals deviating from the group average, or outliers, could be increasing in experiments seeking to achieve high efficiency of drug delivery.
Figure 4. Transport property-based phenotypes.

a, Subjects having different composition of transport phenotypes will have different CPP dependence on τ. b, The probability of metastases being permeated with 10 kDa dextran in the simulated delivery experiment in mice (n = 30) bearing 200 4T1 metastases based on our in vivo study. The inset shows the average and standard deviation of cumulative permeation probability (CPP) as a function of τ. c, Two groups (n = 5 each) of the best and worst τ were ensembled from the group of 30 mice and the individual statistics of each group were plotted. The inset shows t-tests for data comparisons between the two groups, suggesting that they are statistically diverging into two groups with CPPs above 0.8.
We can elucidate this effect further by selecting two groups of animals, i.e., simulations from the 30-animal group mentioned above with the shortest and the longest τ at CPP values > 80%, i.e., creating two animal groups with n = 5 each. Figure 4c shows that both groups statistically behaved as a single entity with CPP < 70%, but through heterogeneity, these two groups eventually diverged with a CPP of 80%. This effect of divergence was the most notable for animals having f << 1 or f = 0 (data not shown). The CPP analyses suggested that with insufficient drug delivery, (pre)clinical evidence of treatment responses may conceal underlying resistant transport phenotypes in the remaining population of metastases. These underlying resistant phenotypes become evident once efficient drug delivery has eliminated the most susceptible transport phenotypes. Insufficient drug delivery may therefore mask transport-based resistance in cancer treatments and obscure conclusions regarding resistance, which is frequently discussed within biological context only.
Drug delivery strategies that permeate only the least resistant (with respect to transport) tumors may show therapeutic effects, and, for example, the patient’s progression-free survival may increase. However, the burden of metastatic tumors bearing a more resistant transport phenotype will ultimately affect the patient’s overall survival. Another interesting aspect of this analysis was the perception of outliers that were frequently discarded. In view of our results, the outliers may provide insights concerning the appearance of animals dominated by specific transport phenotypes; instead of discarding the data, such outliers could be helpful to understand the metastases dominating a specific animal. It is important to remember that CPP and transport phenotypes pertain to the therapy stage before a drug can provide its pharmacological action. Often, in a purely biological approach to the treatment of cancer, concern for and knowledge of drug delivery is absent; the drug is presumed to reach the tumor cell with the focus primarily on mechanism of action.
Discussion
This study of transport properties of metastases provides insight into how fundamental transport laws may control drug delivery, and eventually therapeutic efficacy or resistance. The drug extravasation results were distinct from analogous studies with 4T1 tumors growing at the primary site [28]. The newly introduced permeation time τ enabled us to integrate morphological properties of tumors and transport properties of tissues and drugs. The results have shown that τ analysis can be a useful tool connecting pharmacokinetics, transport properties, and biology. The underlying hypothesis of our study was that metastases may possess heterogeneity in transport properties that significantly affects the delivery of drugs to tumors. However, the majority of clinical and preclinical studies focus primarily on scrutinizing mostly molecular determinants of therapeutic resistance. Frequently, the analysis concludes with the characterization of target gene mutations or expressions [29]. Interestingly, out of 57 references describing molecular origins of resistance [29], approximately 50% of the studies were in vitro cell culture studies and only 40% relied on patient biopsies. While the molecular origins of therapeutic resistance are of fundamental importance for determining pharmacological adequacy of drug selection or its affinity for a target, the common assumption is that a drug de facto arrives at the lesion site and permeates the tumor. However, this assumption can be deceiving during preclinical therapy or clinical studies because it is not known if and how much of the drug has reached the tumor. Understanding and harnessing the power of transport could be the missing link in therapeutic success and overcoming resistance. Thus, the failure to integrate transport concepts beyond systemic pharmacokinetics could explain why purely genetic approaches to therapeutic design have not proven to be successful treatment strategies [30].
Conclusions
The results of our study suggested that the drug permeation of the extravascular space, integrating cell wall permeability of the functioning metastases-associated capillaries, was the most fundamental challenge for efficient drug delivery into cancer cells. Furthermore, insufficient drug delivery, e.g. permeating only 50% of metastatic tumors, may cloud the interpretation of drug delivery and efficacy studies, because the first 50% of drug-permeated tumors in this example would be the most permeable to metastases. However, transport heterogeneity can create transport phenotypes that reveal themselves only in experiments after the population of very permeable metastases has been eliminated by drug(s). Furthermore, therapeutics can permeate and eliminate a fraction of metastases resulting in surviving tumors that possess challenging transport properties, thus rendering these tumors resistant to repeated treatments. Some of the preclinical findings discussed in this study could be translated to the clinic by using non-invasive imaging with contrast agents that can interrogate transport properties of tissues and determine the transport differential between tumors and healthy tissue. While more effort should be focused to better understand the origins of resistance in metastatic diseases in both preclinical and clinical studies, our study emphasizes the importance of ongoing and future studies of the role of transport heterogeneity in determining the therapeutic susceptibility of metastases.
Supplementary Material
Acknowledgements
This work was supported by the National Cancer Institute Physical Sciences-Oncology Network of the National Institutes of Health, under award number U54CA210181. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Breast Cancer Research Program, under award number W81XWH-17–1-0389. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. The authors also acknowledge partial support from OI 174028 of the Serbian Ministry of Education and Science (M.K.), and the Ernest Cockrell Jr. Distinguished Endowed Chair (M.F.). We thank Rolf Brekken for his valuable comments and discussions.
References
- 1.World Health Orgaization Press Release WHO/27/IARC/145. The World Cancer Report - the major findings 2003; Available from: http://www.who.int/cancer/en/.
- 2.Schroeder A, et al. , Treating metastatic cancer with nanotechnology. Nature Reviews Cancer, 2011. 12(1): p. 39–50. [DOI] [PubMed] [Google Scholar]
- 3.SEER Cancer Statistics Review, 1975–2008. SEER Cancer Statistics Review, 1975–2008 2011; Available from: http://seer.cancer.gov/csr/1975_2008/.
- 4.Freyer G, et al. , Pharmacokinetic studies in cancer chemotherapy: usefulness in clinical practice. Cancer Treatment Reviews, 1997. 23(3): p. 153. [DOI] [PubMed] [Google Scholar]
- 5.Minchinton AI and Tannock IF, Drug penetration in solid tumours. Nature Reviews Cancer, 2006. 6(8): p. 583–592. [DOI] [PubMed] [Google Scholar]
- 6.Wolf W and Presant CA, Tumor-based pharmacokinetics has greater significance for anticancer drugs than does blood-based pharmacokinetics. Clinical Pharmacology & Therapeutics, 2004. 76(5): p. 508–508. [DOI] [PubMed] [Google Scholar]
- 7.Müller M, dela Peña A, and Derendorf H, Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: distribution in tissue. Antimicrobial Agents and Chemotherapy, 2004. 48(5): p. 1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Presant C, et al. , Association of intratumoral pharmacokinetics of fluorouracil with clinical response. The Lancet, 1994. 343(8907): p. 1184–1187. [DOI] [PubMed] [Google Scholar]
- 9.Yokoi K, et al. , Liposomal doxorubicin extravasation controlled by phenotype-specific transport properties of tumor microenvironment and vascular barrier. Journal of Controlled Release, 2015. 217: p. 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokoi K, et al. , Capillary-wall collagen as a biophysical marker of nanotherapeutic permeability into the tumor microenvironment. Cancer Research, 2014. 74(16): p. 4239–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziemys A, et al. , Computational analysis of drug transport in tumor microenvironment as a critical compartment for nanotherapeutic pharmacokinetics. Drug Delivery, 2016. 23(7): p. 2524–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon AN, et al. , Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. Journal of Clinical Oncology, 2001. 19(14): p. 3312–3322. [DOI] [PubMed] [Google Scholar]
- 13.Gabizon A, Shmeeda H, and Barenholz Y, Pharmacokinetics of pegylated liposomal doxorubicin. Clinical Pharmacokinetics, 2003. 42(5): p. 419–436. [DOI] [PubMed] [Google Scholar]
- 14.Jain RK and Stylianopoulos T, Delivering nanomedicine to solid tumors. Nature Reviews Clinical Oncology, 2010. 7(11): p. 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Brien M, et al. , CAELYX Breast Cancer Study Group: Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Annals of Oncology, 2004. 15(3): p. 440–449. [DOI] [PubMed] [Google Scholar]
- 16.Maeda H, et al. , Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. Journal of Controlled Release, 2000. 65(1): p. 271–284. [DOI] [PubMed] [Google Scholar]
- 17.Davis ME, Nanoparticle therapeutics: an emerging treatment modality for cancer. Nature Reviews Drug Discovery, 2008. 7(9): p. 771–782. [DOI] [PubMed] [Google Scholar]
- 18.Tanei T, et al. , Redirecting transport of nanoparticle albumin-bound paclitaxel to macrophages enhances therapeutic efficacy against liver metastases. Cancer Research, 2016. 76(2): p. 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yokoi K, et al. , Simultaneous inhibition of EGFR, VEGFR, and platelet-derived growth factor receptor signaling combined with gemcitabine produces therapy of human pancreatic carcinoma and prolongs survival in an orthotopic nude mouse model. Cancer Research, 2005. 65(22): p. 10371–10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corr SJ, et al. , A new imaging platform for visualizing biological effects of non-invasive radiofrequency electric-field cancer hyperthermia. PLoS One, 2015. 10(8): p. e0136382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abràmoff MD, Magalhães PJ, and Ram SJ, Image processing with ImageJ. Biophotonics international, 2004. 11(7): p. 36–42. [Google Scholar]
- 22.Kojic M, et al. , A composite smeared finite element for mass transport in capillary systems and biological tissue. Computer methods in applied mechanics and engineering, 2017. 324: p. 413–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiseliovas V, et al. , Tumor progression effects on drug vector access to tumor-associated capillary bed. Journal of Controlled Release, 2017. [DOI] [PubMed] [Google Scholar]
- 24.Dreher MR, et al. , Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. Journal of the National Cancer Institute, 2006. 98(5): p. 335–344. [DOI] [PubMed] [Google Scholar]
- 25.Nugent LJ and Jain RK, Extravascular diffusion in normal and neoplastic tissues. Cancer Research, 1984. 44(1): p. 238–244. [PubMed] [Google Scholar]
- 26.Norvaisas P and Ziemys A, The Role of Payload Hydrophobicity in Nanotherapeutic Pharmacokinetics. Journal of Pharmaceutical Sciences, 2014. [DOI] [PubMed] [Google Scholar]
- 27.Jain RK, et al. , Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nature clinical practice Oncology, 2006. 3(1): p. 24–40. [DOI] [PubMed] [Google Scholar]
- 28.Yokoi K, et al. , Serum biomarkers for personalization of nanotherapeutics-based therapy in different tumor and organ microenvironments. Cancer Letters, 2014. 345(1): p. 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holohan C, et al. , Cancer drug resistance: an evolving paradigm. Nature Reviews Cancer, 2013. 13(10): p. 714–726. [DOI] [PubMed] [Google Scholar]
- 30.Letai A, Functional precision cancer medicine [mdash] moving beyond pure genomics. Nature Medicine, 2017. 23(9): p. 1028–1035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.