

Abstract
Moderate recurrent hypoglycemia (RH) is frequent in Type 1 diabetes mellitus (TIDM) patients who are under intensive insulin therapy increasing the risk for severe hypoglycemia (SH). The consequences of RH are not well understood and its repercussions on neuronal damage and cognitive function after a subsequent episode of SH have been poorly investigated. In the current study, we have addressed this question and observed that previous RH during seven consecutive days exacerbated oxidative damage and neuronal death induced by a subsequent episode of SH accompanied by a short period of coma, in the parietal cortex, the striatum and mainly in the hippocampus. These changes correlated with a severe decrease in reduced glutathione content (GSH), and a significant spatial and contextual memory deficit. Administration of the antioxidant, N-acetyl-L-cysteine, (NAC) reduced neuronal death and prevented cognitive impairment. These results demonstrate that previous RH enhances brain vulnerability to acute hypoglycemia and suggests that this effect is mediated by the decline in the antioxidant defense and oxidative damage. The present results highlight the importance of an adequate control of moderate hypoglycemic episodes in TIDM.
Keywords: Diabetes, antioxidants, cognitive impairment/decline, glucose, hippocampus, hypoglycemia, risk factors, selective neuronal death
Introduction
Glucose is the major metabolic substrate in brain and its continuous supply from blood is mandatory for its correct functioning. Repeated episodes of moderate hypoglycemia are frequently observed in 70–80% of Type-1 (T1DM) and 40% of Type-2 (T2DM) diabetes mellitus patients due to insulin therapy.1,2 Recurrent moderate hypoglycemia (RH; <70 − >55 mg/dl blood glucose) increases the risk for severe hypoglycemia (SH; <55 mg/dl), which has been reported in 36% and 7% of TIDM and T2DM patients, respectively, with an incidence of one episode per patient per year.3–5 Hypoglycemia is alleviated by the hormonal autonomic counter-regulatory response, which stimulates glycogenolysis and glyconeogenesis.6,7 This response is blunted in T1DM patients, who are unable to respond to the autonomic signs of hypoglycemia (hypoglycemia unawareness).4,7,8 A single episode of hypoglycemia induces defective glucose counter-regulation, favoring recurrent hypoglycemia (RH) and increasing the patient’s vulnerability to SH.6,9 Conversely, patients treated for SH are at risk for RH.10 SH can progress to seizures and the hypoglycemic coma in approximately 25% of the episodes.11 A prolonged period of hypoglycemic coma induces oxidative stress and neuronal death12,13 and associates with cognitive dysfunction in rodents and humans.10,14–16 However, the effect of previous episodes of RH on brain damage induced by SH, is poorly understood.
Moderate hypoglycemia is more common than SH in T1DM patients, particularly in children and adolescents. Although not an immediate threat, a repeated history of RH might cause cognitive dysfunction.17,18 Studies show that RH in healthy animals induces synaptic alterations, oxidative stress and limited neuronal death in the cerebral cortex13,19,20 and even improvement of spatial memory.20–22 In diabetic animals, RH associates with increased cognitive deficit and oxidative damage, suggesting that diabetes exacerbates the damaging effects of hypoglycemia.13,23–25 Conversely, diabetes is associated with oxidative stress26,27 and cognitive decline, which might be enhanced by hypoglycemia.28–31
The repercussions of RH on brain function after a subsequent episode of SH are not well understood. In the present study, we have addressed this question in healthy animals, precluding the influence of diabetic pathology. Results indicate that animals exposed to RH do not show neuronal death, oxidative damage or cognitive decline despite a partial decrease in the content of reduced glutathione (GSH) and increased lipoperoxidation (LPO). However, RH notably augmented oxidative damage and the number of dead cells in the cortex, the striatum and mainly in the hippocampus of animals exposed to a subsequent short period of hypoglycemic coma. These animals showed a large decrease in GSH content and severe cognitive impairment. Treatment with the antioxidant, N-acetyl-L-cysteine (NAC), significantly reduced neuronal death and prevented cognitive decline. The present results demonstrate that in non-diabetic animals, previous RH can enhance brain vulnerability to SH and suggest that this effect is mediated by a decrease in the antioxidant defense and oxidative damage.
Material and methods
Three-month-old male Wistar rats (280–300 g) obtained from Instituto de Fisiología Celular (IFC), Universidad Nacional Autónoma de México were treated according to The National Institute of Health Guide for the care and use of laboratory animals (NIH publication No.80-23 revised 1996), and experimental protocols were approved by the Committee for the Care and Use of Laboratory Animals (CICUAL) of the IFC (LMT101-16). All efforts were made to minimize the number of animals used. Animals were housed in individual cages under standard dark/light cycle and temperature conditions with food and water at libitum. Experiments are reported in compliance with the ARRIVE guidelines (Animal Research: Reporting in Vivo Experiments). A sample size of six to seven animals per group was used to achieve reliable statistical power and minimize the number of animals used.
Induction of recurrent moderate hypoglycemia
RH was induced (in the home cage) in non-anesthetized non-fasted animals receiving a daily intraperitoneal (i.p.) injection of 6.5 insulin units (IU, Humulin 70/30, Eli Lilly, Indianapolis, USA) between 11 and 12:00 a.m. during seven consecutive days, while control animals were injected with vehicle solution (0.1% acetic acid) (Supplementary Figure 1(a)). Moderate hypoglycemia was defined as blood glucose between 40 and 55 mg/dl, without loss of consciousness or seizures. Blood glucose was measured from the tail vein using a One Touch Ultra meter, every 60 min after insulin injection, and was maintained at 2.2–3.0 mM (40–55 mg/dl) for 2.5–3 h (Supplementary Figure 1(a)). Rats spontaneously recovered euglycemia after food addition. Twenty-four hours after the last hypoglycemic episode, animals were euthanized under pentobarbital overdose anesthesia and brains were prepared for histology (see below). Independent groups were used for behavioral tests (see below).
Induction of hypoglycemic coma (SH)
One week before the induction of hypoglycemia, animals were implanted with epidural electrodes under 2.5–3.0% isofluorane anesthesia for two channel (right and left hemispheres) electroencephalographic recordings (EEG). Meloxicam (1 mg/kg i.p) was used as post-surgery anesthesia. Before SH induction, food was restricted to four pellets overnight. For EEG recording, animals were maintained in acrylic special cages. The experimental animals were randomly distributed among the different experimental groups as follows: (1) SH: animals received an i.p injection of 32 IU (Humulin 70/30, Eli Lilly, Indianapolis, USA) and after 2–3 h of SH (blood glucose levels <1.0 mM), they lost their righting reflex and hypoglycemia was left to progresses to EEG isoelectricity (coma) for 7–10 min. Animals were immediately rescued with an i.p. (300 μl) bolus of 25% glucose in Krebs-Henseleit Buffer followed by a continuous intravenous perfusion at a rate of 1.5 ml/h during 3 h through the tail vein using a perfusion pump (Harvard Apparatus 22, South Natik, MA, USA) (Supplementary Figure 1(b)). (2) RH/SH: animals were treated with moderate RH during seven days and 24 h after the last episode SH was induced and left to progress to 7–10 min coma. Immediately afterwards, rats were rescued with glucose as described above (Supplementary Figure 1(c)). (3) NAC: animals were identically treated to those of the RH/SH group, except that they received one subcutaneous administrations of 100 mg/kg NAC in distilled water (Sigma–Aldrich St. Louis MO, USA) 1 h after glucose reperfusion and one daily injection during the following four days (five total administrations at 2:00 p.m.). Experimental protocols are described in Supplementary Figure 1. Twenty-four hours after all treatments, animals were anesthetized (pentobarbital overdose) and intracardially perfused with 0.9% saline solution followed by 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.3; brains were extracted and fixed during 24 h in paraformaldehyde and transferred to a 20–30% sucrose gradient (24 and 72 h, respectively) and 25 µm coronal slides were obtained in a cryostat for histological analysis. Independent groups were treated and randomly distributed for behavioral tests; they were euthanized at the indicated times. Animals showing seizures (less than 10%) were discarded. One animal from the NAC group was discarded due to the lack of exploratory activity in the open field test.
Evaluation of neuronal death
Neuronal death (n = 68 total animals, distributed as follows: n = 7 per group evaluated at 24 h; n = 6 per group evaluated at 5 days and n = 7 per group evaluated at 16 days after the different treatments) was evaluated using Fluoro Jade B (FJB) for the identification of degenerating neurons, as described.32 Sections were imaged under a fluorescence microscope (Leica DM1000) using a FITC filter (395–590 nm). The total number of FJB-positive cells in the parietal cortex and the striatum was quantified in 18 coronal sections of 25 µm (3.20 to −4.20 from bregma), while 4 sections were used for the hippocampus. In this case, cells were counted in an area of 200 µm2 of the different hippocampal subregions (CA1, CA3, dentate gyrus (DG) and hilus) using the Image J program. Data are expressed as the total number of degenerating cells in these four sections. Cell damage was confirmed by Nissl staining at 24 h (n = 7 per group) and five days (n = 4 per group), and the number of condensed darkly stained cells or pyknotic nuclei were counted in consecutive brain sections to those labeled with FJB. In the case of the NAC experiment, FJB and Nissl-positive cells were counted in three areas of 200 µm2 of the DG comprising the inferior and superior blades and the crest of the DG. To monitor DNA fragmentation, the TUNEL Roche In Situ Cell Death Detection Kit was used following the manufacturer instructions (Roche Applied Science, USA). Briefly, adjacent sections were blocked with 3% peroxide in methanol for 10 min at room temperature. Slides were incubated in a mixture containing sodium citrate 0.1%/triton 0.1% for 10 min followed by TUNEL reaction mixture (5 μl TdT enzyme, 5 μl Biotin-dUTP, 90 μl TdT buffer) for 90 min at 37℃. Slides were washed and counterstained with Hoechst (0.001% in PBS) for 10 min at room temperature to facilitate the identification of TUNEL-positive cells.
Immunohistochemistry
Twenty-four hours after the treatments (n = 7 per group), oxidative protein modification was detected using polyclonal anti-4-2-hydroxy-2-nonenal (4-HNE) (Alpha Diagnostic International, San Antonio, Texas, USA) and monoclonal anti-3-Nitrotyrosine (Cayman Company, Ann Arbor Michigan, USA) antibodies. Coronal 25 µm sections were incubated with anti-4-HNE or anti-3-NT antibodies (1:200 dilution) for 72 h at 4℃ and anti-mouse DyLight 594 or anti-rabbit DyLight 488 (Jackson ImmunoResearch Lab. Inc. West Grove, Pennsylvania, USA, 1:250 dilution) secondary antibodies, for 2 h at room temperature. Three sections per animal were examined and representative images were obtained by confocal microscopy (Leica TCS SP5) using a scanning mode for lasser 488 and 546 nm. As a control for antibody specificity, brain sections were incubated with the secondary antibody in the absence of the primary antibody and immunoreactivity was observed.
LPO levels
Twenty-four hours after the treatments (n = 7 per experimental group and n = 10 control group), brain homogenates from parietal cortex, hippocampus and striatum were obtained to determine malondialdehyde (MDA) levels using the thiobarbituric acid-reactive substances (TBARS) method as described in Haces et al.33 Briefly brain tissues were homogenized in 0.5 ml of 1.15% KCL/0.4 mM sodium azide and incubated at 37℃ for 20 min. Suspensions were centrifuged at 14,000 rpm, mixed with thiobarbituric acid at 0.75% (Merck, Darmstadt Germany) and incubated at 50℃ for 20 min. The protein concentration was determined by Bradford’s method and data are expressed as nmol MDA/mg of protein.
Reduced GSH determination
Reduced GSH (n = 3–7) was determined using the fluorometric o-phthalaldehyde (OPA) method as described by Senft et al.34 12 and 24 h after the treatments. Briefly, parietal cortex, hippocampus and striatum were dissected, weighed and homogenized in detection buffer containing 20 mM HCl, 5 mM DTPA and 10 mM ascorbic acid; samples were rapidly centrifuged at 20,000 rpm for 20 min. For GSH determination, 5% trichloroacetic acid, 7.5 mM N-ethylmaleimide (Sigma-Aldrich, Steinheim, Germany) and 100 µl o-phthaldialdehyde (Sigma-Aldrich, Steinheim, Germany) (used as derivatizing agent to react with GSH) were added to 5 µl supernatant. After mixing, samples were incubated at room temperature for 30 min. Fluorescence was read at 365 nm excitation and 430 nm emission. GSH concentrations were calculated and expressed as nmol/mg tissue based on standard curve.
Behavioral tests
Water maze
Forty-eight hours after the treatments, animals were habituated in an isolated testing room for 30 min. Rats (n = 7 per group, 28 total animals) were trained (9:00 a.m.) during five days (five trials per day) in a circular pool divided in four quadrants where a platform was hidden 1 cm under the water at constant temperature (21℃) as described in Delint-Ramírez et al.35 Twenty-four hours after the last day of training (7 days and 15 days after the SH), the memory test was performed. The platform was removed and animals were allowed to swim during 60 s. The latency and number of crossings through the quadrant where the platform was initially placed were recorded with the Ethovision video tracking.
Contextual fear conditioning
At 48 h after the treatments, different series of animals (treated and non-treated with NAC, n = 6–9 per group, 33 total animals) were trained for contextual fear conditioning (9:00 a.m.) and they were tested 24 h later. This was carried out in standard operant chambers (Coulbourn Instruments) located inside sound-attenuating boxes (Med Associates) in an isolated testing room. The floor of the chambers consisted of stainless steel bars that delivered scrambled electric footshock. Between experiments, shock grids and floor trays were cleaned with soap and water, and the walls with wet paper towels as described in Sotres-Bayon et al.36 On day 1, rats habituated during 5 min to the chamber were immediately followed by fear conditioning to the context consisting of five 2 s, 0.8 mA foot shocks. The interval between successive foot shocks was variable with an average of 2 min. The next day (day 2), rats were tested for contextual fear conditioning memory during 10 min. Behavior was recorded with digital video cameras. Freezing was used as a measure of conditioned fear. Freezing was defined as the absence of all movement except for those related to breathing. The amount of time spent freezing to the context was expressed as a percentage of the total time in the chamber.
Open field
Twenty-four hours after contextual fear conditioning memory test (10:00 a.m.), rats were placed at the center of a wooden open arena (90 cm length × 90 cm width × 60 cm height) with a textured floor illuminated by 22 lux of intensity. Grid lines drawn on the floor of the arena (30 × 30 cm) divided it into a peripheral region (within 60 cm of the walls) and a central region (30 × 30 cm) of approximately equal area. Behavior was recorded for 10 min with a digital video camera and analyzed with commercially available video tracking software (ANY-maze, Stoelting). The total distance traveled (cm) was taken as an index of the locomotor activity and the number of entries to the center of open field as an anxiety index.
Statistical analysis
Data are expressed as mean ± SEM (Standard Error of the mean) and analyzed by one-way ANOVA followed by a Fisher’s least multiple comparison test, except for neuronal damage comparisons between NAC-treated and non-treated groups, which were compared by Student’s t test. Behavioral tests were analyzed by the Kruskal–Wallis non-parametric test for Water Maze and Tukey for fear conditioning.
Results
Changes in blood glucose and EEG recordings
No significant differences were found in mean basal blood glucose concentration (99.66 ± 1.20 mg/dl) among the different animal groups. In animals exposed to RH blood, glucose declined to 40 mg/dl 30 min after insulin administration and remained between 40 and 45 mg/dl during the following 3 h. Glucose was recovered after food addition (Supplementary Figure 2(a)). The daily mean blood glucose concentration reached during the 3-h period of moderate hypoglycemia was 40 mg/dl (Supplementary Figure 2(b)). In animals exposed to SH alone and to RH/SH, glucose concentration rapidly declined after 30 min insulin administration and remained ≤ 20 mg/dl during the next 2.5–3.0 h up to isoelectricity. It partially recovered 1 h after glucose reperfusion and at 24 h it reached control values in all animals groups (Supplementary Figure 2(a)). Basal cerebral activity was recorded for 30 min before insulin administration and no differences were observed between the groups. After 2–3 h, the EEG characteristic changes of hypoglycemia reported in previous studies were observed.33 When glucose was close to 20 mg/dl, electrical brain activity slowed and was completely absent during the coma period. Normal brain activity was recovered 1 h after glucose reperfusion (Supplementary Figure 2(c)).
Neuronal death induced by the hypoglycemic coma is exacerbated by previous RH
Figures 1 to 2 show the number of degenerating cells at 24 h and 16 days after the induction of coma. No degenerating cells were observed in animals from the control and RH groups in any of the regions studied (Figures 1(a) and (c) and 2(a)). Twenty-four hours after the animals were exposed to the hypoglycemic coma (9.5 ± 0.54 min) alone (SH), degenerating cells were observed in the parietal cortex and the dorsolateral striatum (Figure 1), while in the hippocampus, degenerating cells were observed only in the crest of DG in 50% of these animals (Figure 2(a), (b) and (d)). Animals examined 16 days after the coma (8.0 ± 0.36 min), showed a lower number of FJB-positive cells in the parietal cortex and the striatum (Figure 1(b) and (d)), while in the crest of the DG, FJB-positive cells were no longer visible (Figure 2(c)).
Figure 1.

Neuronal death induced by the hypoglycemic coma in the parietal cortex and the striatum is exacerbated by antecedent RH. Neuronal death was evaluated 24 h and 16 days after the treatments and the number of FJB-positive cells was counted. Representative micrographs showing no FJB-positive in control and RH-treated animals and degenerating cells in the parietal cortex (a) and the striatum (c) of animals exposed to SH and RH/SH. (b–d) Cell death was exacerbated in animals exposed to RH/SH at 24 h and 16 days after the treatments. Data are expressed as mean ± SEM from seven animals per group. *p < 0.0001 relative to control and RH, #p < 0.0001 relative to SH.
Figure 2.

Neuronal death induced by the hypoglycemic coma in the hippocampus is exacerbated by antecedent RH. Neuronal death was evaluated 24 h or 16 days after the different treatments in brain sections stained with Nissl or FJB. (a) Representative micrographs showing degenerating cells only in the crest of the DG of animals exposed to SH and extensive neuronal death in animals from the RH/SH group. (b–c) Quantitative data showing exacerbated neuronal death in animals exposed to RH/SH at 24 h or 16 days. (d) Representative micrographs showing FJB-positive and pyknotic cells in the different hippocampal subregions of rats exposed to the different treatments. Data are expressed as mean ± SEM from seven animals per group. *p < 0.0001 relative to control and RH, #p < 0.0001 relative to SH.
Animals exposed to coma (9.5 ± 0.20 min) and previously treated with RH (RH/SH), exhibited a significant increase in the number of degenerating cells in the parietal cortex and the striatum as compared to those treated with SH alone at 24 h. This increase was also observed when animals were examined 16 days after RH/SH (7.28 ± 0.42 min of coma) (Figure 1(b) and (d)). Representative micrographs showing FJB-positive cells of animals exposed to the different treatments are shown in Figure 1(a) and (c). Similarly, the hippocampus showed a dramatic elevation in the number of degenerating neurons, which were present not only in the crest of the DG but also in the superior and inferior blades of the DG, the hilus, CA1 and CA3 (Figure 2(a)). This increase was statistically different relative to the SH group at 24 h and 16 days in all hippocampal regions (Figure 2(b) and (c)). These results were corroborated by the quantification of pyknotic cells in Nissl-stained sections (Supplementary Figure 3). Representative micrographs showing degenerating neurons in animals exposed to the different treatments are shown in Figure 2(a) and (d). The presence of pyknotic cells correlated with FJB labeling in the parietal cortex, the striatum (not shown) and the hippocampus (Figure 2(d)). Similarly, TUNEL-positive nuclei, indicative of DNA fragmentation were found in the parietal cortex, the striatum (Supplementary Figure 4) and the hippocampus (Figure 3(a)) of animals of the RH/SH group, while in animals exposed to coma alone, TUNEL-positive nuclei were observed only in the DG (Figure 3(b)). In control and RH groups, no TUNEL-positive nuclei were observed in any region (Supplementary Figure 4). Altogether, these observations indicate that RH increases the vulnerability to neuronal death in the parietal cortex, the striatum and mainly in the hippocampus of animals exposed to a subsequent short period of coma.
Figure 3.

DNA fragmentation induced by the hypoglycemic coma is exacerbated by RH. The presence of DNA fragmentation was examined by the TUNEL assay and Hoechst was used as counterstain. (a) Representative micrographs showing TUNEL-positive cells in CA1 and the DG of rats treated with RH/SH. (b) In rats exposed to SH alone TUNEL-positive were detected only in the crest of the DG.
RH aggravates cognitive impairment induced by the hypoglycemic coma
According to the above-described results, RH notably enhanced neuronal death induced by the hypoglycemic coma in the hippocampus. Therefore, we aimed to test whether animals exposed to RH/SH showed memory impairment using two hippocampal-dependent memory tests. Figure 4(a) shows that during the five days of training in the water maze, the latency to locate the platform decreased similarly in all experimental groups indicating no difference in learning acquisition. At seven days after the coma, the SH and the RH/SH groups exhibited a significant decrease in number of crossings relative to controls, while animals exposed to RH alone showed no significant decrease (Figure 4(b)). At 15 days after the coma, only animals from the RH/SH showed a significant decrease in the number of crossings suggesting a long-term change in spatial memory (Figure 4(c)).
Figure 4.

Previous RH aggravates cognitive impairment induced by the hypoglycemic coma. Spatial memory test (Water maze) was performed 7 and 15 days after the treatments. (a) Learning curve of animals exposed to the different treatments. (b) At day 7, animals of the SH and RH/SH groups showed a decrease in the number of crossings. (c) At 15 days, only animals of the RH/SH group showed a significant decrease in the number of crossings. Data are expressed as mean ± SEM from seven animals per group. *p < 0.01 relative to control #p < 0.05 relative to SH. Contextual fear conditioning test was performed 48 h after the treatments. (d) No statistical differences between the groups were observed in the acquisition phase (day 1). The RH/SH group showed a significant decrease, while the NAC group showed no change in the percent freezing relative to control and RH groups during retrieval (day 2). Data are expressed as mean ± SEM from six to nine animals per group. *p < 0.0001 relative to control and RH &p < 0.05 relative RH/SH. (e) No significant changes were observed between groups in (e) locomotor activity and (f) anxiety tests. (g) Number of damaged cells (FJB-positive group and pyknotic Nissl stained) in the dentate gyrus (DG) and CA1 of animals of the RS/SH and the NAC groups, 24 h after the fear-conditioning test. Data are expressed as mean ± SEM from four to six animals per group. &p < 0.002 relative to RH/SH). (h) Representative images showing the protective effect induced by NAC administration against cell damage (FJB-positive cell and pyknotic cells in Nissl stained section) induced in the DG and CA1 after exposure to RH/SH.
Figure 4(d) shows the performance of animals on the contextual fear conditioning. No statistical differences were found during contextual fear acquisition (day 1) since a similar percent freezing was observed in all groups. In the retrieval test (day 2), only the RH/SH group showed a significant decrease in the percent freezing relative to the control and the RH groups (Figure 4(d) right panel). No changes in locomotor activity and anxiety were found in any of the groups (Figure 4(e) and (f)). These data are in agreement with the spatial memory test and suggest that RH preceding the hypoglycemic coma exacerbates cognitive decline, which correlates with the presence of degenerating neurons in the hippocampus.
RH preceding the hypoglycemic coma induces oxidative damage
Oxidative stress has been highly implicated in neuronal death induced by the hypoglycemic coma.12,32,33,37 Therefore, we aimed to evaluate the presence of the LPO product 4-HNE and 3-NT residues in proteins by immunohistochemistry. 4-HNE positive cells were observed in the parietal cortex, the striatum and all hippocampal subregions of animals of the RH/SH group, while in the control, RH and SH groups, 4-HNE inmmunoreactivity was not observed (Figure 5). As shown in the Figure 6, 3-NT-positive cells were observed mainly in the parietal cortex, DG and hilus of animals exposed to RH/SH. In contrast, no 3-NT immunoreactivity was observed in control, and RH groups, and only a few cells were observed in the parietal cortex of some animals of the SH group (Figure 6). Results suggest that RH facilitates oxidative damage to proteins in animals exposed to a subsequent short period of coma, which correlates with the presence of cell death.
Figure 5.

Immunoreactivity to 4-HNE in the brain of animals exposed to RH/SH. Representative fluorescent images showing the presence of 4-HNE-positive cells in (a) parietal cortex, (b) striatum and (c) hippocampus of animals treated with RH/SH. No 4-HNE-positive cells were detected in control, RH and SH groups.
Figure 6.

Immunoreactivity to 3-NT in the brain of animals exposed to RH/SH. Representative fluorescent images showing the presence of 3-NT-positive cells in the parietal cortex and the hippocampus of animals treated with RH/SH. No 3-NT-positive cells were detected in control and RH groups and only a few cells were observed in the parietal cortex of animals of the SH groups.
Then we aimed to investigate whether a decrease in reduced GSH, the major antioxidant defense in brain, was involved in oxidative damage observed in animals treated with RH/SH. As observed in Figure 7, RH induced a partial (from 23 to 30%) but significant decrease in GSH, while animals of the RH/SH group showed a further decrease (from 49 to 58%) in GSH in all brain regions, the largest change was observed in the hippocampus (58%). The changes in GSH content were observed at 12 h after the end of treatments and were maintained at 24 h in all regions. These results suggest that a reduction in GSH contributes to enhanced oxidative damage in animals from the RH/SH group. Supporting this hypothesis, we observed that treatment with the antioxidant NAC, completely prevented cognitive decline in the contextual fear conditioning test (Figure 4(d)) and reduced hippocampal neuronal damage (Figure 4(g) and (h)). The number of pyknotic neurons and neurons positive to FJB was significantly reduced in the DG, CA1 (Figure 4(g) and (h)), CA3 and hilus (not shown). In the RH group, a significant increase in LPO was observed in all brain regions studied (Supplementary Figure 5); however, as in the case of GSH, increased LPO did not correlate with oxidative damage in these animals.
Figure 7.
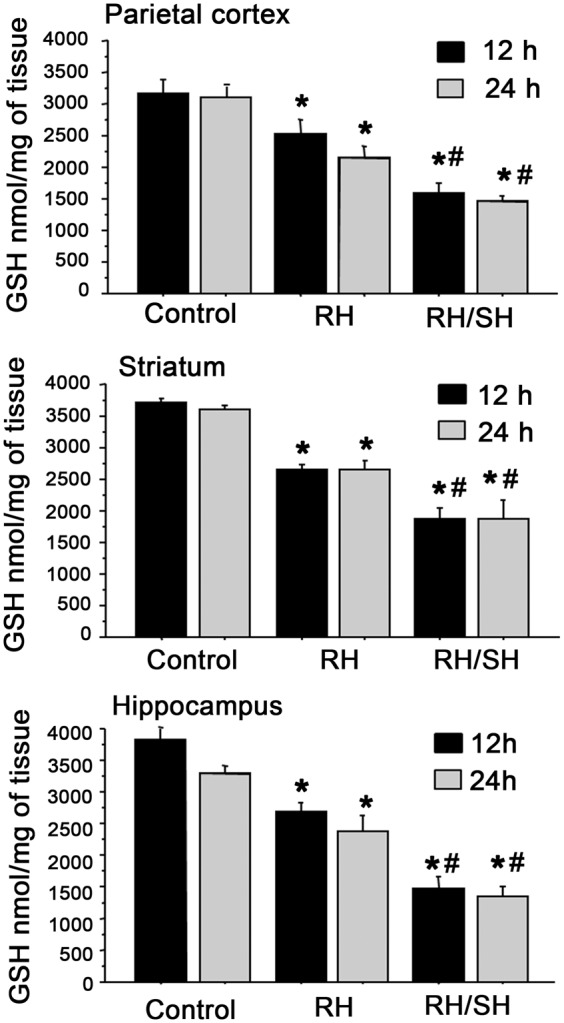
Changes in GSH content after RH and RH/SH. Animals were exposed to the different treatments and 12 and 24 h later reduced GSH levels were measured in different brain regions. Animals exposed to RH show a significant moderate decrease in reduced GSH in all brain regions, which is further decreased in animals from the RH/SH group (n = 3–7 animals per group). Data are expressed in means ± SEM. *p < 0.01 relative to control; #p < 0.01 relative to RH.
Discussion
TIDM is a long-term disease frequently diagnosed during infancy and a 3–4% annual increase rate in children and adolescents has been estimated in Europe.3,18 TIDM patients under strict insulin treatment can show one to two episodes per week of moderate hypoglycemia, which increases the risk for SH (one episode per year) due to a blunted counter-regulatory hormonal response and hypoglycemia unawareness.1–5 Mild to moderate hypoglycemia has been described as blood glucose < 70 mg/dl and > 55 mg/dl without loss of consciousness, while SH is considered when blood glucose falls below 50 mg/dl, is accompanied by seizures, altered consciousness or coma and requires external assistance for treatment with carbohydrates or glucagon.3,37,38 Hypoglycemic episodes can last for several hours (from 1 to 5 h), as those reported during nocturnal hypoglycemia.39 TIDM patients are exposed to mild RH for a life-time, and although at moderate levels, hypoglycemia has not been considered a potential threat, its consequences on brain function either alone or when it is followed by an episode of SH, are unknown so far. In the present study, we have addressed this question in a short-term period and as such, the frequency of moderate hypoglycemia was increased to seven consecutive episodes. To mimic T1DM patient’s condition, moderate hypoglycemia periods were induced by decreasing blood glucose to 55–40 mg/dl during 2.5–3 h, without loss of consciousness or seizures and spontaneous recovery of euglycemia after food addition. SH was induced by the fall in blood glucose to 20 mg/dl or below accompanied by 7–10 min of EEG isoelectricity (coma state).
The most important finding of the present study is that previous RH exacerbates oxidative damage and neuronal death, leading to memory impairment in animals subsequently exposed to a short period of coma. In previous studies, we have demonstrated that short 5–10 min coma episodes induce neuronal death mainly in the parietal cortex but not in the hippocampus except for the crest of the DG.32,33 In the present study, we demonstrate that animals exposed to a brief period of coma, which were previously treated with RH, show enhanced neuronal death in the parietal cortex and the striatum, but notably in the hippocampus, where degenerating and pyknotic cells are present in all hippocampal subregions. Degenerating neurons were observed even 16 days after the coma and correlated with deficient spatial memory. Cognitive decline was confirmed by defective performance in the contextual fear-conditioning retrieval test and interestingly we observed that in both spatial and contextual memory tests, mainly the consolidation of memory was impaired. When animals were exposed to coma alone (SH), only a transitory memory decline was present, consistent with the lack of degenerating neurons in the hippocampus, except for a small population of cells in the crest of the DG. Previous studies have implicated oxidative stress in neuronal death after a prolonged period of hypoglycemic coma.12,13,40,41 In agreement, we observed immunoreactive cells to 4-HNE and 3-NT in brain regions showing dead cells in animals treated with RH/SH. 4-HNE and 3-NT react with proteins altering their function.42,43 Thus, neuronal death correlates with oxidative alteration of proteins, which might be caused by a deficient antioxidant defense, based on the significant reduction in GSH content in all brain regions. Supporting this conclusion, it was observed that animals treated with NAC showed a significant reduction in the number of degenerating neurons and pyknotic cells in the hippocampus and no memory deficit in the contextual fear-conditioning test. GSH is the main antioxidant defense in the nervous system and its decrease is associated with oxidative stress and cell damage.44,45 In addition, NAC has been demonstrated to have antioxidant actions and to restore GSH levels in rodents’ brain after systemic administration.46 Furthermore, it was recently reported that treatment with NAC prevents neuronal damage induced by a prolonged period of hypoglycemic coma.41 Besides a deficient antioxidant defense, decreased glucose brain metabolism might also contribute to exacerbated hypoglycemic neuronal death induced by antecedent RH. A previous study has shown that three consecutive episodes of antecedent hypoglycemia result in decreased glucose oxidation in the brain of animals tested under acute hypoglycemia.47
The observed enhancement of hypoglycemic neuronal damage induced by antecedent RH, is in agreement with previous studies showing that exposure to RH in diabetic rats exacerbates brain injury induced by a short period of acute ischemia. This effect correlates with mitochondrial superoxide production implicating oxidative stress.48 In contrast, a previous study showed that previous RH during three days elicited a preconditioning effect, reducing the number of degenerating neurons and preventing cognitive impairment induced by acute hypoglycemia in non-diabetic animals.49 However, in that study, the presence of seizures possibly highly contributed to neuronal death, masking the effect of RH on hypoglycemic damage. In addition, a lower number of moderate hypoglycemic periods was used.
Rats treated with RH alone showed a partial decrease in GSH, which correlated with increased LPO but no oxidative damage, cell death or cognitive decline. The absence of neuronal death in this group of animals might result from the lack of glucose infusion since they were allowed to recover euglycemia after food addition. It is well known that glucose reperfusion plays a major role in neuronal death, mainly when blood glucose exceeds physiological levels resulting in hyperglycemia.40,50,51 Previous data have reported a decrease in GSH content even after a single episode of moderate hypoglycemia,52 and some studies have shown that RH in healthy animals produces an increase in LPO and 4-HNE immunoreactivity, without leading to significant neuronal death or cognitive decline.13,19,25 Furthermore, studies have reported that RH in healthy animals can even improve cognitive performance due to synaptic changes and brain metabolic adaptations.20,21,22,53
In summary, the present results demonstrate that previous RH can have an adverse effect on subsequent SH in healthy animals, and suggests that this effect is mediated by the decline in the antioxidant defense and oxidative damage. It is expected that the consequences of RH treatment used in the present conditions could be aggravated under diabetic conditions. Recent studies have demonstrated that the effects of RH on oxidative stress and the antioxidant defense are exacerbated in diabetic animals, enhancing cognitive decline.13,24,25,54 Furthermore, depletion of Nrf2, a major transcription factor regulating the antioxidant defense, exacerbates RH-induced oxidative damage to proteins and cognitive decline in control and diabetic animals.25
The present observations demonstrate that RH renders the brain and particularly the hippocampus, more vulnerable to oxidative damage and neuronal death induced by a subsequent episode of hypoglycemic coma, ultimately leading to cognitive dysfunction. These observations highlight the relevance of the history of hypoglycemic episodes in TIDM patients and help us to estimate the consequences of RH on a subsequent episode of SH.
Supplementary Material
Acknowledgements
The authors thank Dr. Ruth Rincón-Heredia for her valuable help in confocal imaging, Gianfranco Chávez Marchetta for his help in Water Maze assays and Francisco Pérez Eugenio for computer facilities. GL performed this study in partial fulfillment of the requirements of the Programa de Doctorado en Psicología at the Universidad Nacional Autónoma de México (UNAM).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by CB239607 CONACYT grant to LM, CB250870 and 176639 CONACYT grants to FB-R and FS-B. GL was supported by CONACYT fellowship 363968.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
LM and GL conceived the study, analyzed all data and wrote the manuscript. GL performed all experiments; TM performed all hypoglycemic treatments, contributed to data analysis and provided technical assistance; LR-L and FS-B participated in the contextual fear-conditioning experiments, data analysis and results interpretation; IB and FB-R participated in water maze experiments, data analysis and results interpretations; GS-C provided technical assistance and participated in manuscript revision. All authors participated in discussions and critically reviewed the manuscript.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Donelly LA, Morris AD, Frier BM, et al. Frequency and predictors of hypoglycaemia in Type 1 and insulin-treated type 2 diabetes: a population-based study. Diab Med 2005; 22: 749–755. [DOI] [PubMed] [Google Scholar]
- 2.Heller S, Colagiuri S, Vaaler S, et al. Hypoglycaemia with insulin aspart: a doubleblind randomised, crossover trial in subjects with Type 1 diabetes. Diab Med 2004; 21: 769–775. [DOI] [PubMed] [Google Scholar]
- 3.Blasetti A, Di Giulio C, Tocco AM, et al. Variables associated with severe hypoglycemia in children and adolescents with Type 1 diabetes: a population-based study. Pediatr Diab 2011; 12: 4–10. [DOI] [PubMed] [Google Scholar]
- 4.Frier BM. Hypoglycaemia in diabetes mellitus: epidemiology and clinical implications. Nat Rev Endocrinol 2014; 10: 711–722. [DOI] [PubMed] [Google Scholar]
- 5.Languren G, Montiel T, Julio-Amilpas A, et al. Neuronal damage and cognitive impairment associated with hypoglycemia: an integrated view. Neurochem Int 2013; 63: 331–343. [DOI] [PubMed] [Google Scholar]
- 6.Cryer PE. Mechanisms of sympathoadrenal failure and hypoglycemia in diabetes. J Clin Invest 2006; 116: 1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tesfaye N, Seaquist ER. Neuroendocrine responses to hypoglycemia. Ann Acad Sci 2010; 1212: 12–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beall C, Michael L, McCrimmon A, et al. The physiology and pathophysiology of the neural control of the counterregulatory response. Am J Physiol Regul Integr Comp Physiol 2012; 202: 215–223. [DOI] [PubMed] [Google Scholar]
- 9.Jensen VF, Bogh IV, Lykesfeldt J. Effect of insulin–induced hypoglycemia on the central nervous system: evidence from experimental studies. J Neuroendocrinol 2014; 26: 123–150. [DOI] [PubMed] [Google Scholar]
- 10.Lin YY, Hsu CW, Sheu W, et al. Risk factors for recurrent hypoglycemia induced in hospitalized diabetic patients admitted for severe hypoglycemia. Yonsei Med J 2010; 51: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strachan MWJ. Frequency, causes and risk factors for hypoglycaemia in type 1 diabetes. In: Frier BM, Heller SR, McCrimmon R. (eds). Hypoglycaemia in clinical diabetes, 3rd ed. eBook Chichester: John Wiley & Sons Ltd, 2014, pp. 63–95. [Google Scholar]
- 12.Suh SW, Hamby AM, Gum ET, et al. Sequential release of nitric oxide, zinc and superoxide in hypoglycemic neuronal death. J Cereb Blood Flow Metab 2008; 28: 1697–1706. [DOI] [PubMed] [Google Scholar]
- 13.Won SJ, Yoo BH, Kauppinen TM, et al. Recurrent/moderate hypoglycemia induces hippocampal dendritic injury, microglial activation, and cognitive impairment in diabetic rats. J Neuroinflammation 2012; 9: 1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suh S, Aoyama K, Matsumori Y, et al. Pyruvate administered after severe hypoglycemia reduces neuronal death and cognitive impairment. Diabetes 2005; 54: 1452–1458. [DOI] [PubMed] [Google Scholar]
- 15.Fujioka M, Okuchi K, Hiramatsu K, et al. Specific changes in human brain after hypoglycemic injury. Stroke 1997; 28: 584–587. [DOI] [PubMed] [Google Scholar]
- 16.Xu C, Yogaratman J, Lua R, et al. Persistent, severe hypoglycemia–induced organic brain syndrome with neurological sequelae: a case report. Gen Hosp Psichiatr 2011; 33: 411–412. [DOI] [PubMed] [Google Scholar]
- 17.Perantie DC, Lim A, Wu J, et al. Effects of prior hypoglycemia and hyperglycemia on cognition in children with type 1 diabetes mellitus. Pediatr Diab 2008; 9: 87–95. [DOI] [PubMed] [Google Scholar]
- 18.Franchini S, Comegna L, Prezioso G, et al. Hypoglycemia in children with type 1 diabetes: unawareness is a concrete risk. Curr Med Res Opin 2016; 32: 1487–1491. [DOI] [PubMed] [Google Scholar]
- 19.Yamada KA, Rensing N, Izumi Y, et al. Repetitive hypoglycemia in young rats impairs hippocampal long-term potentiation. Pediatr Res 2004; 55: 372–379. [DOI] [PubMed] [Google Scholar]
- 20.McNay EC, Williamson A, McCrimmon R, et al. Cognitive and neural hippocampal effects of long-term moderate recurrent hypoglycemia. Diabetes 2006; 55: 1088–1095. [DOI] [PubMed] [Google Scholar]
- 21.McNay EC, Sherwin RS. Effect of recurrent hypoglycemia on spatial cognition and cognitive metabolism in normal and diabetic rats. Diabetes 2004; 53: 418–425. [DOI] [PubMed] [Google Scholar]
- 22.Osborne DM, ÓLeary KE, Fitzgerald DP, et al. Context-dependent memory following recurrent hypoglycaemia in non-diabetic rats is mediated via glucocorticoid signaling in the dorsal hippocampus. Diabetologia 2016; 60: 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bree AJ, Puente EC, Daphna-Iken D, et al. Diabetes increases brain damage caused by severe hypoglycemia. Am J Physiol Endocrinol Metab 2009; 297: 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cardoso S, Santos RX, Correia SC, et al. Insulin-induced recurrent hypoglycemia exacerbates diabetic brain mitochondrial dysfunction and oxidative imbalance. Neurobiol Dis 2012; 49: 1–12. [DOI] [PubMed] [Google Scholar]
- 25.McNeilly AD, Gallagher JR, Dinkova-Kostova AT, et al. Nrf2-mediated neuroprotection against recurrent hypoglycemia is insufficient to prevent cognitive impairment in a rodent model of Type 1 diabetes. Diabetes 2016; 65: 3151–3160. [DOI] [PubMed] [Google Scholar]
- 26.McGrowder DA, Anderson-Jackson L and Crawford T. Biochemical evaluation of oxidative stress. In: Escher AP and Li A (eds) Type 1 diabetes . Rijeka, Croatia: InTech Open Accsess Publisher, 2013; pp.223–248. 10.5772/45927. [DOI]
- 27.Kumar P, Rao N, Bhusan Pal B, et al. Hyperglycemia-induced oxidative stress induces apoptosis by inhibiting PI3-kinase/Akt and ERK 1/2 MAPK mediated signaling pathway causing downregulation of 8-oxoG-DNA glycosylase levels in glial cells. Int J Biochem Cell Biol 2014; 53: 302–319. [DOI] [PubMed] [Google Scholar]
- 28.Gold A, Macleod K, Deary I, et al. Hypoglycemia-induced cognitive dysfunction in diabetes mellitus: effect of hypoglycemia unawareness. Physiol Behav 1995; 58: 501–511. [DOI] [PubMed] [Google Scholar]
- 29.Kirchhoff BA, Lugar HM, Smith SE, et al. Hypoglycemia-induced changes in regional brain volume and memory function. Diab Med 2013; 30: 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moheet A, Mangia S, Seaquist ER. Impact of diabetes on cognitive function and brain structure. Ann N Y Acad Sci 2015; 1353: 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirchhoff BA, Jundt DK, Doty T, et al. A longitudinal investigation of cognitive function in children and adolescents with type 1 diabetes mellitus. Pediatr Diab 2016; 12414: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Julio Amilpas A, Montiel T, Soto-Tinoco E, et al. Protection of hypoglycemia-induced neuronal death by ß-hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J Cereb Blood Flow Metab 2015; 35: 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haces ML, Montiel T, Massieu L. Selective vulnerability of brain regions to oxidative stress in a non-coma model of insulin-induced hypoglycemia. Neurosci 2010; 165: 28–38. [DOI] [PubMed] [Google Scholar]
- 34.Senft AP, Dalton TP, Shertzer HG. Determining glutathione and glutathione disulfide using the fluorescence probe o-Phthalaldehyde. Anal Biocehem 2000; 280: 80–86. [DOI] [PubMed] [Google Scholar]
- 35.Delint-Ramírez I, Salcedo-Tello P, Bermúdez-Rattoni F. Spatial memory formation induced recruitment of NMDA receptor and PSD-95 to synaptic lipid rafts. J Neurochem 2008; 106: 1658–1668. [DOI] [PubMed] [Google Scholar]
- 36.Sotres-Bayón F, Sierra-Mercado D, Pardilla-Delgado, et al. Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron 2012; 76: 804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryan C, Gurtunca N, Becker D. Hypoglycemia: a complication of diabetes therapy in children. Pediatr Clin N Am 2005; 52: 1705–1733. [DOI] [PubMed] [Google Scholar]
- 38.Festa A, Heller S, Seaquist E, et al. Association between mild and severe hypoglycemia in people with type 2 diabetes initiating insulin. J Diab Complicat 2017; 31: 1047–1052. [DOI] [PubMed] [Google Scholar]
- 39.Yale J. Nocturnal hypoglycemia in patients with insulin-treated diabetes. Diab Res Clin Pract 2004; 65S: S41–S46. [DOI] [PubMed] [Google Scholar]
- 40.Suh SW, Gum ET, Hamby AM, et al. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest 2007; 117: 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kho AR, Choi BY, Kim JH, et al. Prevention of hypoglycemia-induced hippocampal neuronal death by N-acetil-L- Cysteine (NAC). Amino Acids 2016; 49: 367–378. [DOI] [PubMed] [Google Scholar]
- 42.Ullery C, Marnett LJ. Protein modification by oxidized phospholipids and hydrolytically released lipid electrophiles: investigating cellular response. Biochim Biophys Acta 2012; 1818: 2424–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radi R. Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc Chem Res 2013; 46: 550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain. Metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem 2000; 16: 4912–4916. [DOI] [PubMed] [Google Scholar]
- 45.Baxter PS, Hardingham PE. Adaptative regulation of the brain’s antioxidant defenses by neurons and astrocytes. Free Radic Biol Med 2016; 100: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reyes R, Cittolin-Santos G, Kim J, et al. Neuronal glutathione content and antioxidant capacity can be normalized In Situ by N-acetyl Cysteine concentrations attained in human cerebrospinal fluid. Neurotherapeutics 2016; 13: 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang L, Herzog RI, Mason GF, et al. Recurrent antecedent hypoglycemia alters neuronal oxidative metabolism in vivo. Diabetes 2009; 58: 1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dave KR, Tamariz J, Desai KM, et al. Recurrent hypoglycemia exacerbates cerebral ischemic damage in streptozotocin-induced diabetic rats. Stroke 2011; 42: 1404–1411. [DOI] [PubMed] [Google Scholar]
- 49.Puente EC, Silverstein J, Bree AJ, et al. Recurrent moderate hypoglycemia ameliorates brain damage and cognitive dysfunction induced by severe hypoglycemia. Diabetes 2010; 59: 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ennis K, Dotterman H, Stein A, et al. Hyperglycemia accentuates and ketonemia attenuates hypoglycemia-induced neuronal injury in the developing rat brain. Pediatr Res 2015; 77: 84–90. [DOI] [PubMed] [Google Scholar]
- 51.Rao R, Ennis K, Mitchell P, et al. Recurrent moderate hypoglycemia suppresses brain-derived neurotrophic factor expression in the prefrontal cortex and impairs sensoriomotor gating in the post-hypoglycemia period in young rats. Dev Neurosci 2016; 38: 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rao A, Quach H, Smith E, et al. Changes in ascorbate, glutathione and α-tocopherol concentrations in the brain regions during normal development and moderate hypolgycemia in rats. Neurosci Lett 2014; 568: 67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Canada S, Weaver S, Sharpe S, et al. Brain glycogen supercompensation in the mouse after recovery from insulin-induced hypoglycemia. J Neurosci Res 2011; 4: 585–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi BY, Kim JH, Kim HJ, et al. Pyruvate administration reduces recurrent/moderate hypoglycemia-induced cortical neuron death in diabetic rats. PLoS One 2013; 8: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.