

Abstract
Range expansions are crucibles for rapid evolution, acting via both selective and neutral mechanisms. While selection on traits such as dispersal and fecundity may increase expansion speed, neutral mechanisms arising from repeated bottlenecks and genetic drift in edge populations (i.e. gene surfing) could slow spread or make it less predictable. Thus, it is necessary to disentangle the effects of selection from neutral mechanisms to robustly predict expansion dynamics. This is difficult to do with expansions in nature, as replicated expansions are required to distinguish selective and neutral processes in the genome. Using replicated microcosms of the red flour beetle (Tribolium castaneum), we identify a robust signature of stochastic, neutral mechanisms in genomic changes arising over only eight generations of expansion and assess the role of standing variation and de novo mutations in driving these changes. Average genetic diversity was reduced within edge populations, but with substantial among-replicate variability in the changes at specific genomic windows. Such variability in genomic changes is consistent with a large role for stochastic, neutral processes. This increased genomic divergence among populations was mirrored by heightened variation in population size and expansion speed, suggesting that stochastic variation in the genome could increase unpredictability of range expansions.
Keywords: range expansion, rapid evolution, gene surfing, genetic drift, pool-seq
1. Introduction
Range expansion, or the spatial spread of a species into a previously unoccupied geographical area, has long been studied as a purely ecological phenomenon [1,2], due in part to the importance of range expansion to the dynamics of invasive species [3], and in part to the assumption that evolutionary processes are too slow to influence expansion dynamics [4]. Recently, however, research has demonstrated that evolutionary changes can significantly alter the dynamics of range expansions, even over only a handful of generations [5–9]. In particular, rapid evolutionary changes at the expansion edge can lead to dramatic increases in the intrinsic variability of expansion speeds [6,7], thus increasing uncertainty in prediction of range expansions. Given the fundamental role of range expansions for invasive species [2] and populations shifting in response to climate change [10], it is important to understand the mechanisms by which evolutionary dynamics alter the speed and variance of range expansions. However, this is not a trivial task due to the combination of evolutionary mechanisms that can affect range expansions via both selective and neutral processes [11] and the complexities of disentangling their effects [12].
Evolution during range expansions occurs via several distinct, yet potentially interacting mechanisms. Selective processes can be delimited into three mechanisms. First, not unique to range expansions, is the evolutionary response to novel selection pressures a population can encounter when expanding into a new habitat [9,13]. Second, the spatial population structure formed during range expansion can impose selection on the population. Spatial selection, or the assortative mating among highly dispersive individuals at the expanding edge combined with increased fitness at the edge due to a release from intraspecific competition, can lead to the evolution of heightened dispersal in edge populations [5–8,14–16]. Third, the gradient in population density formed across the expanding range can lead to differential evolution of density-sensitive traits, such as competitive ability, across the population [17,18]. In contrast to such selection-driven mechanisms of evolution, the spatial genetic and demographic dynamics of range expansion also provide a neutral mechanism, gene surfing, in which random alleles ‘surf’ to high frequencies at the expanding edge, while other alleles are lost [19]. Gene surfing results from the repeated founding events and small population sizes characteristic of the expanding range edge [11,20], thus altering allele frequencies in an unbiased stochastic manner, leading to an overall reduction in genetic diversity in edge populations as random alleles become fixed or are lost from the expansion wave [20–23]. As gene surfing can result in stochastic fluctuations in allele frequencies regardless of the adaptive value of a given allele, it has the potential to lead to increased variability in demographic rates at the population level. Under some circumstances, gene surfing can lead to a systematic reduction in fitness at the expansion edge due to an accumulation of deleterious alleles [24–26].
While driven by fundamentally distinct processes (selection versus neutral allele fluctuations), both mechanisms acting via selection and gene surfing are expected to rapidly reduce genetic diversity through the loss and fixation of alleles. Thus, in data from a single range expansion, as is typical in the field, it can be extremely difficult to differentiate between selection and gene surfing [27]. As with any stochastic process, a single range expansion provides only one realization from a distribution of possible outcomes [28]. Only by experimentally replicating range expansions can multiple realizations from the distribution of possible outcomes be sampled, as we do here.
Selection and gene surfing are expected to produce distinct genomic patterns across such a distribution of outcomes. Evolutionary mechanisms acting via selection, such as spatial selection, would be expected to lead to consistent, though not necessarily identical, genomic changes in repetitions of a range expansion starting from the same source population [29]. In contrast, gene surfing, as a neutral mechanism, would be expected to manifest uniquely in each repetition due to the unbiased, stochastic nature of the underlying processes (i.e. founder effects and genetic drift in small populations). Thus, replicated expansions provide a method to distinguish selection from gene surfing.
Empirical evidence for gene surfing remains sparse, typically limited to inference from a single, unreplicated expansion [26,30–32] or to phenotypic rather than genetic data in bacteria [33,34]. In all of these studies, expansions took place over long timescales of hundreds of generations or more and genomic data were only analysed for single realizations of expansion [26,30–34]. Here, we use replicated range expansions of the red flour beetle (Tribolium castaneum) in laboratory microcosms to experimentally test the importance of gene surfing in shaping evolutionary patterns across the genome in sexually reproducing, diploid populations over short timescales (eight generations). We previously reported results from this experiment focused on phenotypic outcomes [6], showing that spatially evolving (structured) populations expanded faster on average but with significantly more variability in expansion speed among replicates compared to populations in which spatial evolution was prevented (shuffled; see Methods for treatment descriptions). Additionally, we observed evolution of heightened dispersal phenotypes in edge populations of structured landscapes relative to the core, implicating spatial selection as a significant evolutionary mechanism in these landscapes [6]. Importantly, we saw no evidence of a response to selection due to the density gradient across the range and our experimental design intentionally controlled for selection from novel environmental conditions. Thus, spatial selection was the key selective process acting on the structured populations. Here, we use genomic data from 37 replicated experimental range expansions, assessing allele frequencies across the genome in 96 distinct populations. We evaluate genomic signatures of gene surfing and the relative contribution of both standing variation and de novo mutations to observed changes. We find that gene surfing plays a strong role in shaping genomic diversity in range expansions, despite ongoing spatial selection, suggesting that increased variability in genomic outcomes driven by stochastic evolutionary processes could explain the increased variance in the dynamics of range expansion at the population level observed in this [6] and other recent experiments [7,28,35].
2. Methods
(a). Range expansion experiment
Full details of the range expansion experiment are described elsewhere [6]. We provide a brief reiteration here. Experimental landscapes consisted of 4 cm × 4 cm × 6 cm acrylic boxes placed in a linear arrangement with holes drilled between neighbouring boxes that could be blocked to allow controlled dispersal events. Each box contained standard medium (95% wheat flour and 5% brewer's yeast) to serve as both habitat and a food source. Tribolium castaneum life cycles were manipulated to produce non-overlapping generations of 35 days each, consisting of discrete growth, dispersal, and reproductive phases [28,36].
To begin the experiment, 20 beetles from a large, well-mixed source population of T. castaneum (called SF, after their origin from Schlegel farm near Bloomington, IN) were placed in the first patch of each of 60 empty landscapes. This procedure mimicked the dynamics of many naturally occurring range expansions beginning from small founding populations (e.g. invasive or reintroduced species). These landscapes were then randomly divided between two treatments, which were designed to test the role of spatial evolution in range expansions. One treatment, which we refer to as shuffled, prevented spatial evolution by randomizing the location of individual beetles within the landscape once per generation, while keeping local population abundances constant. In effect, the shuffled treatment decoupled an individual's genotype from its location. In the other treatment, referred to as structured, beetles remained in the location in which they were censused, thus allowing the formation of genetic spatial structure necessary for spatial evolution during range expansions. These two treatments allowed for a direct quantification of the effect of genetic spatial structure during range expansion, as the shuffled landscapes resulted in populations increasing in abundance at similar rates to the structured landscapes and with similar demographic spatial structure, but without the genetic spatial structure that naturally arises from the expansion process in the structured populations. All 60 replicates were further divided into three temporal blocks. Three replicates were lost over the course of the experiment due to laboratory mishaps, resulting in 29 replicates of the shuffled treatment and 28 replicates of the structured treatment. Range expansions proceeded for eight generations, after which phenotypic assays were performed to test for evolution in dispersal, fitness, and competitive ability among beetles from the edge and core of replicate structured and shuffled populations, confirming that both dispersal and fitness had evolved in response to the spatial structure imposed by range expansion, as we reported elsewhere [6].
For each replicate, all 20 founding beetles (generation 0) were collected and stored at −80°C after reproducing. For structured replicates, 20 beetles randomly sampled from the range core (defined as patch 1 of the landscape) and the 20 furthest-spreading beetles (defining the expansion edge) were collected and stored at −80°C after the 8th generation of expansion. In most replicates, the 20 edge beetles represented two, three or four patches (16, 6 and 1 replicate respectively), as the last patch contained less than 20 beetles. One replicate contained exactly 20 beetles in the last patch and four replicates contained over 20 beetles in the furthest patch (23, 37, 40 and 52). For those four landscapes the 20 edge beetles represent a random subset of the population in the last patch. Similarly, 20 beetles from the shuffled treatment were also stored at −80°C after the 8th generation of the experiment. The 20 beetles collected from shuffled landscapes were randomly selected since spatial location has no biological meaning in that treatment, and any 20 individuals are representative of allele frequencies across the population. In summary, collected beetles represent entire populations (founders), entire or almost entire subpopulations (edge), or random samples of populations (core and shuffled). Henceforth, we will refer to these observation units as populations.
(b). DNA pooling, extraction and sequencing
To pool DNA, we combined all 20 beetles from a population with 180 µl phosphate-buffered saline and homogenized the mixture with a disposable pestle. We then extracted total genomic DNA from the mixture using a DNeasy Blood and Tissue Kit (Qiagen). We used fluorometric quantification (Qubit) and a NanoDrop spectrophotometer (Thermo Scientific) to check each extracted DNA sample for adequate quantity and quality respectively.
We selected 22 landscapes (out of a total of 28) from the structured treatment and 15 landscapes (out of a total of 29) from the shuffled treatment for sequencing. To select landscapes, we first excluded any landscapes with subpar quantities or qualities of extracted DNA (two structured landscapes and three shuffled landscapes) and then randomly selected the designated number of landscapes. The number of landscapes was chosen to yield 96 total populations for sequencing (the number in a single 96 well plate for library construction). Each structured landscape provided three populations comprising two time points for analysis: (i) the founding population (generation 0), (ii) the core population (generation 8), and (iii) the edge population (generation 8). Each shuffled landscape provided two populations comprising two time points: (i) the founding population (generation 0) and (ii) the population from generation 8. Taken together, this yielded 66 populations from structured landscapes and 30 populations from shuffled landscapes for sequencing.
Paired end Illumina libraries were prepared for each pool using the Nextera DNA Sample Preparation Kit (Illumina) by the Next-Generation Sequencing Facility at the Biofrontiers Institute of the University of Colorado, Boulder and subsequently sequenced on an Illumina NextSeq V2 machine. Sequencing reads were trimmed using Trimmomatic (v. 0.36) [37] and aligned to the reference genome for T. castaneum [38] using bwa mem (v. 0.7.5a-r405) [39], with default settings for both. After the initial bwa alignment, we used the CleanSam utility (v. 2.8.1) from Picard (http://broadinstitute.github.io/picard) to soft-clip alignments beyond the end of reference sequences and to set the mapping quality to 0 for unmapped reads. We used the RealignerTargetCreator and IndelRealigner utilities from the Genome Analysis Toolkit (v. 3.7-0-gcfedb67) to target and realign reads around indels [40]. We used samtools (v. 0.1.19-96b5f2294a) view and mpileup under default settings to exclude aligned reads with a mapping quality below 20 and to extract multi-sequence pileups, respectively [41]. Quality scores, read lengths, and sequencing depths, averaged over all replicates and broken down by population, are reported in the electronic supplementary material and confirm high quality and sufficient depth of aligned reads among all experimental populations.
(c). Analysis
We used Popoolation [42] (v. 1.2.2) to calculate nucleotide diversity (π) across the T. castaneum genome, dividing the genome into non-overlapping windows. We explored a range of window sizes from 5000 to 15 000 bp. Results from all analyses were invariant to these different window sizes, so we report results using windows of 10 000 bp. We used a pool size of 40, corresponding to the number of chromosome copies in our pooled samples of 20 diploid individuals. We restricted Popoolation to only consider bi-allelic sites with coverage between 4 and 22 (chosen according to the distribution of average depths across populations; electronic supplementary material, figures S1 and S2) and a site quality score of at least 20. Although polyallelic loci are expected to occur at a low frequency for organisms with large population sizes, these sites are quite rare [43], contributing little to the overall genetic variation that is of interest here. Furthermore, apparent polyallelic sites detected from low-depth, pooled sequencing data could be caused by sequencing errors, leading to overestimates of genetic diversity. To quantify overall change in nucleotide diversity in each population, we first calculated the mean across all windows from autosomes and the X chromosome separately. Using these data, we first tested for a difference in average nucleotide diversity among founding populations with a linear mixed effects model assessing the fixed effect of experimental treatment and accounting for temporal block as a random effect. We then calculated the change in autosomal and X chromosome means between the founding and generation 8 populations for each landscape to determine the effects of eight generations of range expansion and population growth. We analysed these changes with a linear mixed model assessing the fixed effect of spatial structure (core, edge or shuffled) on the change in mean π from founding populations to the 8th generation with temporal block as a random effect.
To quantify how spatial population structure during range expansion altered genetic patterns at a finer genomic scale, we considered the change in nucleotide diversity (Δπ) at each individual window. We defined the change in nucleotide diversity at a given window as the nucleotide diversity of that window from an 8th generation population minus the nucleotide diversity of the same window from the corresponding founding population. Using only windows for which all populations had estimates of diversity, we first calculated the mean change among replicates in nucleotide diversity at each window (). This quantity reveals the genomic windows most consistently impacted by evolution during the expansion process. As we know spatial selection acted to increase dispersal phenotypes in edge populations [6], we expect edge populations to display reductions compared to core and shuffled populations, but those reductions are likely concentrated on particular chromosomes under selection. Gene surfing can also contribute to the reduction of nucleotide diversity through the fixation and loss of random alleles [20], but those random reductions in diversity should be found across all chromosomes. To directly test for a signature of gene surfing, we then calculated the standard deviation of the changes in nucleotide diversity among replicates (). While selection should mostly affect the same loci in all replicates (resulting in a low ), gene surfing is expected to manifest differently in replicated range expansions, driving down nucleotide diversity in different genomic windows in each replicate. Thus, the standard deviation of changes in nucleotide diversity among replicates at each genomic window () should be higher in populations experiencing gene surfing. For both of these metrics ( and ), we analysed the data using a linear mixed model incorporating the fixed effects of spatial structure, chromosome, and their interaction and autocorrelated random effects associated with window position along the genome (assuming an exponential correlation structure). In contrast to the analyses above, as these metrics were calculated across temporal blocks, there was not a random effect of temporal block.
We then examined the end result of eight generations of selection and gene surfing by calculating the Pearson's correlation coefficient of nucleotide diversity across windows of the genome for all pair-wise combinations of replicates from the eighth generation. A high correlation indicates that replicates converged on similar patterns of genomic diversity, as would be expected if replicates were driven purely by selection. Similarly, a low correlation indicates greater divergence among replicates, in terms of genomic diversity, and would be the expected outcome if gene surfing were a dominant mechanism during expansion. Since pairwise correlations are not independent, we used a randomization test to examine the effect of spatial structure (core, edge, shuffled) on this correlation. We used the mean absolute difference between group means of correlation coefficients as the test statistic, formed a null distribution for the statistic by randomizing the assignment of spatial structure (core, edge, shuffled) among replicates (10 000 simulations), and calculated the one-tailed p-value from the simulated null distribution. We further calculated 95% confidence intervals for the differences between founders and the other group means of correlation coefficients using non-parametric bootstrap with 10 000 simulations.
While gene surfing acting on standing genetic variation is expected to create variable patterns of neutral genomic change among replicate populations, such variable patterns could also arise due to selection in at least two ways: (i) selection acting on de novo mutations arising in individual replicates during the expansion process or (ii) selection on rare alleles from the source population that were only sampled in some of the founding populations. However, we expected these effects to be negligible, which we confirmed by further analyses (electronic supplementary material).
To further clarify the metrics we examined, we provide concise descriptions along with predictions for the effects of selection and gene surfing for each in table 1. All model analyses and post-processing of the data were performed in R (v. 3.3.2) [44] except where otherwise indicated. Mixed effects models with autocorrelated random effects were fit and analysed using the package nlme (v. 3.1-128) [45]. For linear models, plots of residuals, quantiles, and influence were used to check relevant model assumptions where appropriate.
Table 1.
Definitions of the key metrics investigated in the paper, along with predictions for each due to selection and gene surfing. Specifically, we provide predictions for the patterns expected in edge populations compared to core and shuffled populations.
| predictions |
||
|---|---|---|
| metric | selection | gene surfing |
| Reduction in mean nucleotide diversity, genome wide, after eight generations of range expansion (figure 1). | Greater reductions in edge populations. | Greater reductions in edge populations. |
| Mean change in nucleotide diversity among replicates at each genomic window (; figure 2). Changes are calculated from founders to the 8th generation populations. | The consistency of selection acting on the same loci in replicate populations will lead to large reductions in certain windows of edge populations, but idiosyncratic effects across chromosomes as not all windows contain loci targeted by selection. | Gene surfing manifesting in different windows in each replicate population will lead to small, average reductions in edge populations, distributed evenly across all chromosomes. |
| Standard deviation of changes in nucleotide diversity among replicates at each genomic window (; figure 3). | Low values, as selection is expected to typically act on the same loci across replicates. | High values, as gene surfing will manifest in random windows that are likely to differ across replicates. |
| Correlation coefficient of nucleotide diversity across windows for all pair-wise combinations of replicates (ρπ; figure 4). | High values, as the consistency of selection maintains similarity among replicates. | Low values, as the stochastic nature of gene surfing drives increased divergence among replicates. |
3. Results
On average, nucleotide diversity was similar for founding populations of both structured and shuffled treatments in both autosomal and sex chromosomes (figure 1a and electronic supplementary material, figure S3a; parametric bootstrap for the effect of treatment: p = 0.58 and p = 0.43 respectively), confirming that mean differences among experimental treatments arising over the eight generations of range expansion were due to the treatments rather than initial genetic status. Among the three population types from generation eight (core and edge populations from structured landscapes and populations from shuffled landscapes), autosomal nucleotide diversity was reduced on average compared to the founding populations (figure 1b). This reduction was especially pronounced in edge populations (figure 1b; parametric bootstrap for the effect of spatial structure: p = 0.03), which experienced reductions on average 1.23 times greater than core populations (95% confidence interval (CI): 0.11–15.75). These results were repeated for the X chromosome (electronic supplementary material, figure S3; parametric bootstrap for the effect of spatial structure: p < 0.001) with 1.21 times the reduction in nucleotide diversity in edge versus core populations (95% CI: 0.50–2.73).
Figure 1.

Autosomal nucleotide diversity contained in founding and generation eight populations. Mean nucleotide diversity (π) across autosomes for all experimental populations is shown in (a). The derived data (mean π across windows) are plotted as individual points and distributions among replicates are shown with standard Tukey box plots. Differences in mean autosomal π from founders to generation-8 populations of the same landscapes are shown in (b). Differences were calculated as the generation-8 value minus the founding population from the same landscape, so a negative value indicates a loss of diversity. No change is indicated by a horizontal dashed line at 0. Derived data are shown as points and model estimated means and 95% confidence intervals are shown by the solid points and error bars. Edge populations show significantly greater reductions in diversity from the founders compared to core and shuffled populations (parametric bootstrap for the effect of spatial structure: p = 0.03). Sample sizes are 22 for structured landscapes (encompassing the structured founders and core and edge populations from generation-8) and 15 for both generations of shuffled landscapes. (Online version in colour.)
To assess the impact of evolution during range expansion at a finer genomic scale and identify a signature of gene surfing, we calculated the change in diversity at individual windows along the genome and calculated both the mean () and standard deviation () of these changes among replicates at each window as described in the methods. Analysis of these quantities revealed four important patterns. First, genomic windows in edge populations experienced greater mean reductions in nucleotide diversity in all chromosomes (likelihood ratio test for the effect of spatial structure: p < 0.001; figure 2) compared with core and shuffled populations. For selection to be the only mechanism behind this pattern, it would need to have acted on at least several loci on each chromosome, which is unlikely though certainly not impossible. Neutral processes, however, would be expected to affect all areas of the genome, and hence all chromosomes, with equal probability, with the potential to cause reductions in diversity across all chromosomes. Second, the degree to which diversity was reduced in edge populations differed idiosyncratically among chromosomes (figure 2). This idiosyncratic pattern may be a signature of selection and its differential effect on different areas of the genome. Third, genomic windows across all chromosomes had higher standard deviations in the changes in nucleotide diversity among replicates in edge populations compared to core and shuffled populations (likelihood ratio test for the effect of spatial structure: p < 0.001; figure 3). The higher among-replicate variation at individual genomic windows suggests a greater role for neutral evolutionary processes at the edge of a range expansion compared to the core. Fourth, while the effect of spatial structure on was highly variable throughout the genome (figure 2), the effect of spatial structure on was relatively consistent (figure 3). Across chromosomes, in edge populations ranged from 1.70 to 2.63 times the observed in core populations (figure 2). In contrast, the variance in Δπ among replicates (rather than which is not additive and hence not ideal for such comparisons) in edge populations only ranged from 1.30 to 1.48 times that observed in core populations. This relatively consistent effect size on the variance of changes in nucleotide diversity (related to shown in figure 3) is expected from neutral processes, such as gene surfing, that affect different loci with equal probability. Further, the mismatch between the effect sizes of the variance and implies the observed trends in the variability of changes in nucleotide diversity were not driven by a correlation with the mean.
Figure 2.
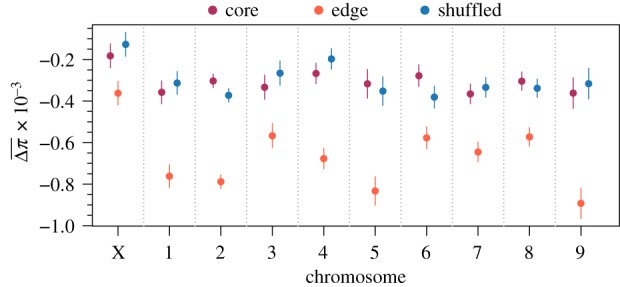
Model estimated mean changes in nucleotide diversity among replicates at individual windows in different chromosomes along the genome. Lower values (i.e. greater reductions) indicate a larger role for selection, gene surfing, or both. Estimates were generated from a linear mixed model with fixed effects for spatial structure, chromosome, and their interaction and random effects capturing the spatial autocorrelation of individual windows along the genome. Only windows with complete sets of observations across all replicates were used for this analysis (n = 6602 windows for each level of spatial structure). Points are model estimates and error bars are 95% confidence intervals. Likelihood ratio tests found all fixed effects significant with p < 0.0001.
Figure 3.
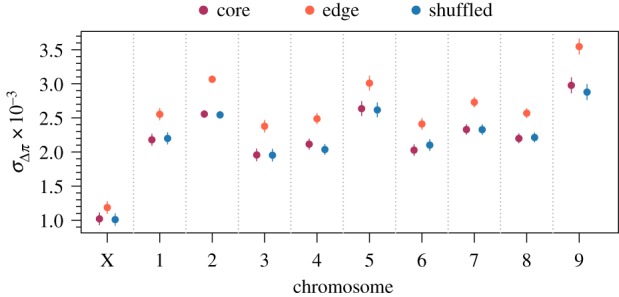
Model estimated standard deviation of changes in nucleotide diversity among replicates at individual windows in different chromosomes along the genome. Higher values of indicate a larger role for neutral processes like gene surfing. Estimates were generated from a linear mixed model with fixed effects for spatial structure, chromosome, and their interaction and random effects capturing the spatial autocorrelation of individual windows along the genome. Only windows with complete sets of observations across all replicates were used for this analysis (n = 6602 windows for each level of spatial structure). Points are model estimates and error bars are 95% confidence intervals. Likelihood ratio tests found all fixed effects significant with p < 0.0001.
The correlations of nucleotide diversity in genomic windows among replicate populations revealed a dramatic effect of gene surfing after only eight generations of range expansion. As expected, nucleotide diversities of founding populations were highly correlated among replicates, reflecting their shared history as samples from a large, well-mixed laboratory population kept in constant environmental conditions for many generations prior to the start of the experiment. If selection were the only evolutionary mechanism acting during range expansion, all experimental treatments should have highly correlated nucleotide diversity after eight generations similar to the founding populations, as selection acted either to maintain founding genotypes or caused populations to diverge from the founding populations in repeatable ways. Both types of selection would lead to high correlations among experimental populations. However, all experimental treatments had reduced correlations after eight generations compared to the founders (randomization test: p = 0.0002; figure 4). Reduced correlations were most pronounced for edge populations for which nucleotide diversity was 11.9% (95% CI: 11.6–12.3%) less correlated among replicate populations compared to the correlations among founding populations (figure 4). Core and shuffled populations were also less correlated than the founders: 5.0% (95% CI: 4.8–5.3%) and 4.7% (95% CI: 4.3–5.1%) respectively. These reductions in correlation among replicates compared to the founding populations suggest an important role for neutral stochasticity in the evolutionary trajectories of these landscapes, particularly so for the edge populations, which experienced about double the reduction in correlation compared to core and shuffled populations. This enhanced role for neutral stochasticity in edge populations is consistent with gene surfing acting during range expansion to drive population genetic differentiation at range margins.
Figure 4.
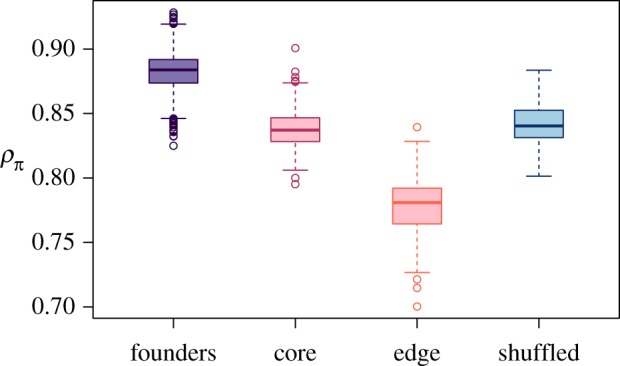
Correlations in nucleotide diversity among replicate populations. Pearson's correlation coefficient calculated for the nucleotide diversity of genomic windows between each pair-wise comparison of replicate populations within founders, shuffled, core, and edge populations (n = 666, 105, 231, 231 pair-wise comparisons respectively). Standard Tukey boxplots are shown to visualize the distribution of correlation coefficients. Edge populations had lower correlation in nucleotide diversity among replicates compared to founders, core, and shuffled populations (randomization test for the effect of experimental group: p = 0.002), suggesting that gene surfing results in increased variability among edge populations. (Online version in colour.)
We identified 66 de novo mutations in our data and constructed two probabilistic models (the sampling model and the detection model) to assess the role of selection on de novo mutations and rare alleles in driving increased genomic variation among edge populations (electronic supplementary material). None of the mutations occurred within protein-coding sequences. The plurality of de novo mutations occurred within introns (45%), with one occurring in the 3′ untranslated region of a protein coding gene. The remaining variants occurred in intergenic (7%), upstream (22%) or downstream (25%) regions. Windows containing de novo mutations did not significantly increase or decrease in nucleotide diversity in core, edge or shuffled populations (electronic supplementary material, figure S4), suggesting selection on de novo mutations was not a driver of the genome-wide reductions in diversity of edge populations. Further, analysis of the sampling model revealed the probability of even a small number of landscapes failing to sample a rare allele in the source population to be extremely low (electronic supplementary material, figure S5). It is therefore unlikely that selection on rare alleles or de novo mutations was a significant driver of the variable reductions in diversity among edge populations.
4. Discussion
Both neutral processes and selection are known to be important in range expansions, but it can be difficult to disentangle the contributions of each in field populations, where expansions are not replicated [12]. Nevertheless, it is important to understand the role of stochastic neutral processes to improve predictions of range expansion. If gene surfing (stochastic neutral processes at the range edge) plays a large role then, due to its stochastic nature, it could contribute to the large intrinsic variance observed in the speed and population dynamics of range expansions [6,7,28]. By comparing changes in nucleotide diversity across the genome for replicate populations experiencing identical conditions during experimental range expansion, we identify distinct signatures of gene surfing over a remarkably short timescale. This heightened role of neutral stochastic evolutionary processes in edge populations provides a potential explanation for the increased variance in expansion speed observed in structured populations [6].
After eight generations of range expansion, populations across all types of spatial structure (core, edge and shuffled) had reduced nucleotide diversity on average (figure 1 and electronic supplementary material, figure S3) due to a combination of initial founder events and eight generations of selection and drift. Edge populations experienced the largest reduction in diversity (figure 1b), indicating an increased role for selection, neutral mechanisms, or both at the edge of expansion relative to the core and the shuffled populations. Idiosyncratic reductions of nucleotide diversity in certain chromosomes of edge populations (; figure 2) suggest a role for spatial selection, which is consistent with the observed increases in dispersal ability among edge populations of structured landscapes [6]. However, reduced diversity throughout the genome (i.e. on all chromosomes) of edge populations (figure 2) combined with larger variation in diversity changes among replicates of edge populations (; figure 3) indicate an important role for gene surfing. Further, the low correlations of nucleotide diversity among replicate edge populations compared to core and shuffled populations (figure 4) demonstrates that gene surfing led to highly dissimilar genomic patterns among replicated range expanding populations. Importantly, our experimental treatments allow these genomic results to be directly attributed to the spatial structure formed during range expansion. The changes to nucleotide diversity were calculated relative to the founding populations of each landscape, thus accounting for the initial bottleneck of landscape colonization. Further, the shuffled treatment provides a direct comparison to the genomic patterns expected from a single, panmictic population, allowing precise quantification of the effect of spatial structure formed during range expansion.
A possible alternative explanation for our results is that spatial selection acting on de novo mutations or initially rare alleles found only in a handful of founding populations could result in the uncorrelated patterns in nucleotide diversity. If selection were primarily acting on de novo mutations, which would not be expected to occur at the same loci in replicate populations, the process of mutation and subsequent selection could also result in seemingly divergent genomic patterns among replicates. Similarly, if initial alleles in the source population were rare, they might have been sampled in only a few founding populations. If such alleles provided a fitness advantage in edge populations, then selection on them could also lead to uncorrelated changes in genomic diversity among replicates. Indeed, the expansion edge is known to produce distinct selection pressures not present in core populations (i.e. spatial selection) [14–18], potentially creating opportunities for de novo mutations or rare alleles to rise rapidly in frequency due to the small population size at the edge.
However, our results demonstrate selection on de novo mutations or rare alleles are unlikely mechanisms for five reasons. First, although simulations suggested we were able to identify de novo mutations with reasonable accuracy (electronic supplementary material, figure S6), de novo mutations only rarely rose to measurable frequencies in our data. Second, those windows with identified de novo mutations contributed little to the mean reductions in nucleotide diversity in edge populations, since the windows containing de novo mutations did not exhibit large, consistent reductions in diversity (electronic supplementary material, figure S4). Third, de novo mutations occurring in edge populations did not exhibit different trends in changes in nucleotide diversity compared to core and shuffled populations (electronic supplementary material, figure S4). Fourth, the sampling model of the probability of rare alleles being missed in some founding populations reveals that the probability of more than a single founding population missing an allele is extremely low, even for low allele frequencies (electronic supplementary material, figure S5). Given the number of alleles that would need to be involved to drive the patterns observed here, it is extremely unlikely that rare variants or de novo mutations contributed significantly to the observed genomic patterns. Fifth, no de novo mutations fell within coding regions, making them less likely targets of spatial selection. Thus, while some de novo mutations were observed, and some rare variants could have been missed in sequencing, the genomic patterns observed were highly likely to be driven by standing variation.
Taken together, our results imply an important role for stochastic neutral processes in driving the observed genomic changes during range expansion. Although spatial selection helped shape edge populations, it was unable to fully counter the divergent effects of gene surfing, which resulted in heightened among replicate variability leading to substantially reduced correlations in nucleotide diversity after eight generations. In addition to the genomic evidence, previously reported results from this experiment [6] demonstrated reduced reproductive fitness of edge populations on average, which is consistent with theoretical expectations of gene surfing [24]. These results suggest that gene surfing can be an important component of genomic evolution at the expansion edge, even over only eight generations.
However, more work is needed to fully understand the role of gene surfing in range expansions, particularly the interaction between gene surfing and selection due to variable environmental conditions. The simplified structure of our experimental landscapes intentionally removed any role for selection due to spatial or temporal environmental variation so as to quantify the importance of spatial population structure (i.e. spatial selection) alone. Theoretical work suggests, however, that environmental variation could alter the role of gene surfing in range expansions, with local adaptation to environmental gradients reducing the expansion load caused by gene surfing [46]. Recent experimental work in the T. castaneum system is consistent with this prediction and the potential role of gene surfing in driving variability in the speed of range expansions. Evolving populations expanding through a novel environment (in which theory would predict gene surfing to play a reduced role) did not display reduced fitness in the edge relative to the core, and only modestly increased variability in expansion speeds compared to non-evolving control populations [9]. More studies are needed to disentangle the complex relationship between environmentally driven selection, gene surfing, and variability in the speed of range expansions.
Understanding the potential for evolutionary changes to impact range expansions is critical, given the importance of range expansion to invasive species [2] and species' range shifts in response to climate change [10]. Previously reported results from this experiment [6] and a simultaneous study in a different model organism [7] demonstrated that spatial evolutionary mechanisms substantially increased variance in expansion rates. Our results show that the increased variation in expansion rates is mirrored by an increased role for neutral mechanisms at the expansion edge, suggesting that variation on the genomic level could be propagated to impact variance in expansion speed at the population level. In other words, stochastic neutral processes at the genomic level may increase stochasticity and uncertainty at the population level. Thus, understanding the role of gene surfing is critical to understanding and predicting the dynamics of range expansions, even over very short timescales.
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We thank Torrey Davis for assistance with the DNA extractions and Jasmine Vidreo, Tristin Myers, Sean Race, Nicholas Richter, Frances Janz, McKenzie Gibson, Jacob Depompolo, Kabita Ghale, Adam Solon, Naomi Ochwat, Tyler Turk, Minh Duc Le, and Rajani Ghale for assistance with the range expansion experiment. We also thank Michael Wade and Doug Drury for providing SF beetles. Finally, we thank two anonymous reviewers for comments and suggestions that greatly improved the manuscript.
Data accessibility
All genomic data necessary to produce the results presented here are available from the NCBI Sequence Read Archive (see electronic supplementary material for a list of accession numbers). R scripts for all models and analyses are available from https://github.com/tpweiss06/StochasticGenomicEvolution.
Authors' contributions
C.W.-L., R.A.H., N.C.K. and B.A.M. designed the experiment. C.W.-L. conducted the experiment. C.W.-L., S.T. and B.A.M. performed the statistical analyses. All authors contributed to writing the manuscript.
Competing interests
The authors declare no competing financial interests.
Funding
Funding for this work was provided by National Science Foundation grant nos. DEB 0949595 and DEB 1457660 to B.A.M., DEB 0949619 to R.A.H., DEB 1601333 to B.A.M. and C.W.-L., and the University of Colorado Dean's Grant to C.W.-L. During this work, R.A.H. received support from the USDA NIFA Hatch project 1012868 and USDA AFRI COLO-2016-09135 and C.W.-L. was partially supported by a Graduate Research Fellowship from the National Science Foundation (grant no. 1144083).
References
- 1.Skellam JG. 1951. Random dispersal in theoretical populations. Biometrika 38, 196–218. ( 10.1093/biomet/38.1-2.196) [DOI] [PubMed] [Google Scholar]
- 2.Hastings A, et al. 2005. The spatial spread of invasions: new developments in theory and evidence. Ecol. Lett. 8, 91–101. [Google Scholar]
- 3.Elton CS. 1958. The ecology of invasions by animals and plants. Chicago, IL: University of Chicago Press. [Google Scholar]
- 4.Pelletier F, Garant D, Hendry AP. 2009. Eco-evolutionary dynamics. Phil. Trans. R. Soc. B 364, 1483–1489. ( 10.1098/rstb.2009.0027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fronhofer EA, Altermatt F. 2015. Eco-evolutionary feedbacks during experimental range expansions. Nat. Commun. 6, 6844 ( 10.1038/ncomms7844) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss-Lehman C, Hufbauer RA, Melbourne BA. 2017. Rapid trait evolution drives increased speed and variance in experimental range expansions. Nat. Commun. 8, 14303 ( 10.1038/ncomms14303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ochocki BM, Miller TEX. 2017. Rapid evolution of dispersal ability makes biological invasions faster and more variable. Nat. Commun. 8, 14315 ( 10.1038/ncomms14315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams JL, Kendall BE, Levine JM. 2016. Rapid evolution accelerates plant population spread in fragmented experimental landscapes. Science 353, 482–485. ( 10.1126/science.aaf6268) [DOI] [PubMed] [Google Scholar]
- 9.Szűcs M, Vahsen ML, Melbourne BA, Hoover C, Weiss-Lehman C, Hufbauer RA. 2017. Rapid adaptive evolution in novel environments acts as an architect of population range expansion. Proc. Natl Acad. Sci. USA 114, 13 501–13 506. ( 10.1073/pnas.1712934114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen I-C, Hill JK, Ohlemüller R, Roy DB, Thomas CD. 2011. Rapid range shifts of species associated with high levels of climate warming. Science 333, 1024–1026. ( 10.1126/science.1206432) [DOI] [PubMed] [Google Scholar]
- 11.Excoffier L, Foll M, Petit RJ. 2009. Genetic consequences of range expansions. Annu. Rev. Ecol. Evol. Syst. 40, 481–501. ( 10.1146/annurev.ecolsys.39.110707.173414) [DOI] [Google Scholar]
- 12.Hoban S, et al. 2016. Finding the genomic basis of local adaptation: pitfalls, practical solutions, and future directions. Am. Nat. 188, 379–397. ( 10.1086/688018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szűcs M, Melbourne BA, Tuff T, Weiss-Lehman C, Hufbauer RA. 2017. Genetic and demographic founder effects have long-term fitness consequences for colonising populations. Ecol. Lett. 20, 436–444. ( 10.1111/ele.12743) [DOI] [PubMed] [Google Scholar]
- 14.Shine R, Brown GP, Phillips BL.. 2011. An evolutionary process that assembles phenotypes through space rather than through time. Proc. Natl Acad. Sci. USA 108, 5708–5711. ( 10.1073/pnas.1018989108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perkins TA, Phillips BL, Baskett ML, Hastings A. 2013. Evolution of dispersal and life history interact to drive accelerating spread of an invasive species. Ecol. Lett. 16, 1079–1087. ( 10.1111/ele.12136) [DOI] [PubMed] [Google Scholar]
- 16.Phillips BL, Brown GP, Travis JMJ, Shine R. 2008. Reid's paradox revisited: the evolution of dispersal kernels during range expansion. Am. Nat. 172, S34–S48. ( 10.1086/588255) [DOI] [PubMed] [Google Scholar]
- 17.Burton OJ, Phillips BL, Travis JMJ. 2010. Trade-offs and the evolution of life-histories during range expansion. Ecol. Lett. 13, 1210–1220. ( 10.1111/j.1461-0248.2010.01505.x) [DOI] [PubMed] [Google Scholar]
- 18.Phillips BL, Brown GP, Shine R. 2010. Life-history evolution in range-shifting populations. Ecology 91, 1617–1627. ( 10.1890/09-0910.1) [DOI] [PubMed] [Google Scholar]
- 19.Edmonds CA, Lillie AS, Cavalli-Sforza LL.. 2004. Mutations arising in the wave front of an expanding population. Proc. Natl Acad. Sci. USA 101, 975–979. ( 10.1073/pnas.0308064100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallatschek O, Nelson DR. 2008. Gene surfing in expanding populations. Theor. Popul. Biol. 73, 158–170. ( 10.1016/j.tpb.2007.08.008) [DOI] [PubMed] [Google Scholar]
- 21.Austerlitz F, Jung-Muller B, Godelle B, Gouyon P-H. 1997. Evolution of coalescence times, genetic diversity, and structure during colonization. Theor. Popul. Biol. 51, 148–164. ( 10.1006/tpbi.1997.1302) [DOI] [Google Scholar]
- 22.Le Corre V, Kremer A.. 1998. Cumulative effects of founding events during colonization on genetic diversity and differentiation in an island and stepping-stone model. J. Evol. Biol. 11, 495–512. ( 10.1007/s000360050102) [DOI] [Google Scholar]
- 23.Slatkin M, Excoffier L. 2012. Serial founder effects during range expansion: a spatial analog of genetic drift. Genetics 191, 171–181. ( 10.1534/genetics.112.139022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peischl S, Dupanloup I, Kirkpatrick M, Excoffier L. 2013. On the accumulation of deleterious mutations during range expansions. Mol. Ecol. 22, 5972–5982. ( 10.1111/mec.12524) [DOI] [PubMed] [Google Scholar]
- 25.Peischl S, Kirkpatrick M, Excoffier L. 2015. Expansion load and the evolutionary dynamics of a species range. Am. Nat. 185, E81–E93. ( 10.1086/680220) [DOI] [PubMed] [Google Scholar]
- 26.Henn BM, et al. 2016. Distance from sub-Saharan Africa predicts mutational load in diverse human genomes. Proc. Natl Acad. Sci. USA 113, E440–E449. ( 10.1073/pnas.1510805112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lotterhos KE, Whitlock MC. 2015. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol. Ecol. 24, 1031–1046. ( 10.1111/mec.13100) [DOI] [PubMed] [Google Scholar]
- 28.Melbourne BA, Hastings A. 2009. Highly variable spread rates in replicated biological invasions: fundamental limits to predictability. Science 325, 1536–1539. ( 10.1126/science.1176138) [DOI] [PubMed] [Google Scholar]
- 29.Stern DL. 2013. The genetic causes of convergent evolution. Nat. Rev. Genet. 14, 751–764. ( 10.1038/nrg3483) [DOI] [PubMed] [Google Scholar]
- 30.Graciá E, Botella F, Anadón JD, Edelaar P, Harris DJ, Giménez A. 2013. Surfing in tortoises? Empirical signs of genetic structuring owing to range expansion. Biol. Lett. 9, 20121091 ( 10.1098/rsbl.2012.1091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peischl S, et al. 2018. Relaxed selection during a recent human expansion. Genetics 208, 763–777. ( 10.1534/genetics.117.300551) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bosshard L, Dupanloup I, Tenaillon O, Bruggmann R, Ackermann M, Peischl S, Excoffier L.. 2017. Accumulation of deleterious mutations during bacterial range expansions. Genetics 207, 669–684. ( 10.1534/genetics.117.300144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hallatschek O, Hersen P, Ramanathan S, Nelson DR.. 2007. Genetic drift at expanding frontiers promotes gene segregation. Proc. Natl Acad. Sci. USA 104, 19 926–19 930. ( 10.1073/pnas.0710150104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldschmidt F, Regoes RR, Johnson DR. 2017. Successive range expansion promotes diversity and accelerates evolution in spatially structured microbial populations. ISME J. 11, 2112–2123. ( 10.1038/ismej.2017.76) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips BL. 2015. Evolutionary processes make invasion speed difficult to predict. Biol. Invasions 17, 1949–1960. ( 10.1007/s10530-015-0849-8) [DOI] [Google Scholar]
- 36.Melbourne BA, Hastings A. 2008. Extinction risk depends strongly on factors contributing to stochasticity. Nature 454, 100–103. ( 10.1038/nature06922) [DOI] [PubMed] [Google Scholar]
- 37.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. ( 10.1093/bioinformatics/btu170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tribolium Genome Sequencing Consortium. 2008. The genome of the model beetle and pest Tribolium castaneum. Nature 452, 949–955. ( 10.1038/nature06784) [DOI] [PubMed] [Google Scholar]
- 39.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. ( 10.1093/bioinformatics/btp324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKenna A, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. ( 10.1101/gr.107524.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. ( 10.1093/bioinformatics/btp352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kofler R, Orozco-terWengel P, De Maio N, Pandey RV, Nolte V, Futschik A, Kosiol C, Schlötterer C. 2011. PoPoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS ONE 6, e15925 ( 10.1371/journal.pone.0015925) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodgkinson A, Eyre-Walker A. 2010. Human triallelic sites: evidence for a new mutational mechanism? Genetics 184, 233–241. ( 10.1534/genetics.109.110510) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.R Core Team. 2016. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; see https://www.R-project.org/. [Google Scholar]
- 45.Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team. 2016. nlme: linear and nonlinear mixed effects models. R package version 3.1-128; see http://CRAN.R-project.org/package=nlme.
- 46.Gilbert KJ, Sharp NP, Angert AL, Conte GL, Draghi JA, Guillaume F, Hargreaves AL, Matthey-Doret R, Whitlock MC. 2017. Local adaptation interacts with expansion load during range expansion: maladaptation reduces expansion load. Am. Nat. 189, 368–380. ( 10.1086/690673) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic data necessary to produce the results presented here are available from the NCBI Sequence Read Archive (see electronic supplementary material for a list of accession numbers). R scripts for all models and analyses are available from https://github.com/tpweiss06/StochasticGenomicEvolution.