

Abstract
We expose significant changes in the emission color of carbazole-based thermally activated delayed fluorescence (TADF) emitters that arise from the presence of persistent dimer states in thin films and organic light-emitting diodes (OLEDs). Direct photoexcitation of this dimer state in 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) reveals the significant influence of dimer species on the color purity of its photoluminescence and electroluminescence. The dimer species is sensitive to the sample preparation method, and its enduring presence contributes to the widely reported concentration-mediated red shift in the photoluminescence and electroluminescence of evaporated thin films. This discovery has implications on the usability of these, and similar, molecules for OLEDs and explains disparate electroluminescence spectra presented in the literature for these compounds. The dimerization-controlled changes observed in the TADF process and photoluminescence efficiency mean that careful consideration of dimer states is imperative in the design of future TADF emitters and the interpretation of previously reported studies of carbazole-based TADF materials.
Introduction
Thermally activated delayed fluorescence (TADF) has become a topic of intense interest to improve the performance of organic light-emitting diodes (OLEDs) through the utilization of the nonemissive triplet manifold.1−3 The triplet states formed in the emitter molecules during device operation are converted back into emissive singlet states through the reverse intersystem crossing (rISC) mechanism. Recent experimental and theoretical evidence has shown that rISC in these organic emitters is not simply reliant on a small singlet–triplet gap between two states, but that spin-vibronic coupling plays a significant role.4,5
A typical molecular scheme to achieve TADF is a donor–acceptor (D–A) system that can form intramolecular charge-transfer (CT) states with a very small singlet–triplet energy gap. However, there is significant evidence to suggest that a close-in-energy locally excited triplet state (3LE) is required to mediate rISC between the charge-transfer triplet (3CT) and charge-transfer singlet (1CT) states.4,5 This mediation of rISC by 3LE is the basis of the spin-vibronic coupling mechanism. Due to the sensitivity of these CT states to their local environment (i.e., solvent or solid-state host), the spin-vibronic model explains the ability to tune the rISC and TADF by changing the polarity of the host, and thus minimizing the energy gap between the CT manifold and 3LE.6−8 As a result, the choice of emitter host has become a crucial factor during OLED fabrication.
Going beyond the polarity of the host, concentration-induced shifts of the photoluminescence9−11 and electroluminescence12−17 maxima in samples that employ 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) as an emitter are commonly reported in the literature. A study by Kim et al.9 showed that increasing the concentration of 4CzIPN in a nonpolar host caused a significant shift in the CT state emission of 4CzIPN. They attributed this shift to the polarity of the dopant 4CzIPN molecules (ground-state dipole moment, 3.95 D18) influencing other 4CzIPN molecules indirectly, just as solvent molecules would do in solution. Due to its similarity to the polarity effect of solvents on CT state emission, the shift of CT emission in films is known as the solid-state solvation effect (SSSE). It implies that consideration of the concentration of the emitter is also an important variable for fabrication of TADF OLEDs.
However, recent work by Northey et al.19 has shown that SSSE is not as effective as the analogous phenomenon in liquid solvents in changing the emission of CT states when comparing solvents and hosts of similar polarity. The difference arises due to the inability of the solid-state host to reorganize around the emitter molecule, which is the manifold that gives rise to the solvatochromism in solution. In this work, we use 4CzIPN (Figure 1a) to demonstrate that, rather than SSSE, the formation of aggregated/dimer states is instead the predominant reason for the significant emission shifts observed with concentration for multi-carbazole TADF emitters. This explanation is based on the wealth of literature relating to carbazole-based compounds, e.g., poly(N-vinylcarbazole) (PVK) and 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) as well as carbazole’s known propensity for intermolecular interactions through either fully overlapped (FO) or partially overlapped (PO) co-facial dimers (Figure 1b).20−24 This aggregate/dimer interpretation is built on recent work performed on several multifunctional materials that exhibit aggregation-induced emission, TADF, and mechanochromic luminescence (MCL)25−30 while enabling further discussion on how to tackle the problem of color purity for TADF molecules. On this basis, we expect that future studies of TADF-active emitters will benefit from the use of a broader range of analytical techniques to fully interpret novel molecules’ properties.
Figure 1.

(a) Structural formulas of TADF-active compounds. (b) Fully overlapped (FO) and partially overlapped (PO) dimers formed by two carbazole units.
Results and Discussion
In this study, 4CzIPN is used as the model system. However, to provide supporting evidence for the conclusions and to expand the generality of our findings, four other similar molecules were also investigated (Figure 1a). Synthetic procedures and NMR spectroscopic characterization can be found in Schemes S1–S3 and Figures S1–S8 in the Supporting Information.
Photoluminescence of Dilute Solutions and Crystals
Differences in the emission energy between dilute solutions and crystals are not conclusive evidence for aggregation/dimerization and could be explained by SSSE. However, the significant emission shifts we observed for all molecules (Figures 2a,b and S9) would be difficult to explain by SSSE alone. The energies of the peaks and onsets of the emission spectra of the molecules in solution and crystal states are shown in Table 1.
Figure 2.
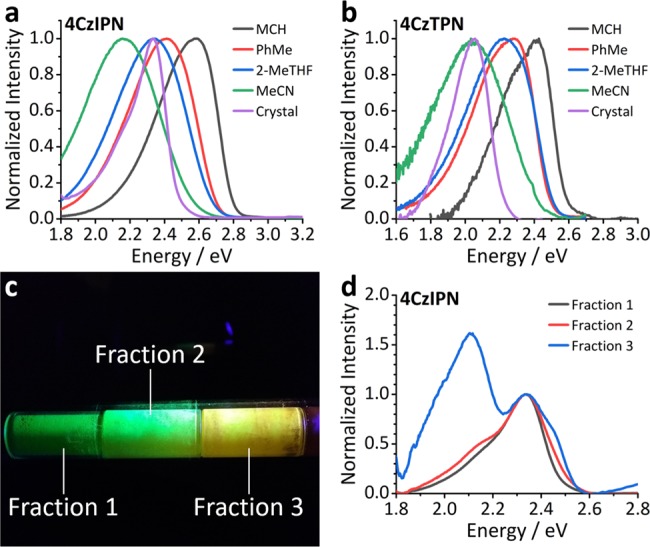
(a) Steady-state photoluminescence of 4CzIPN in a series of solvents (20 μm) and in crystal form (Fraction 1 from sublimation). (b) Solvent series for 4CzTPN (10 μm, due to low solubility) and as a crystal (Fraction 1 from sublimation). (c) Photograph of 4CzIPN after sublimation. The sublimation tube is illuminated by an ultraviolet torch and the observed colors are indicative of crystal polymorphs with different emission properties. The regions referred to as Fractions 1, 2, and 3 are labelled. (d) The steady-state fluorescence spectra of Fractions 1, 2, and 3 of 4CzIPN.
Table 1. Onset and Peak Emission Energies of the Compounds in Solution and Crystal Statesa.
| compound | MCH | PhMe | 2-MeTHF | MeCN | crystal |
|---|---|---|---|---|---|
| 4CzIPN | 2.80 (2.58) | 2.71 (2.41) | 2.68 (2.34) | 2.57 (2.16) | 2.47 (2.33) |
| 4CzTPN | 2.58 (2.42) | 2.49 (2.28) | 2.52 (2.22) | 2.44 (2.04) | 2.23 (2.06) |
| 4CzIPN-tBu8 | 2.68 (2.47) | 2.60 (2.33) | 2.58 (2.25) | 2.44 (2.05) | 2.52 (2.35) |
| 4CzTPN-tBu8 | 2.48 (2.34) | 2.41 (2.21) | 2.41 (2.16) | 2.21 (2.04) | |
| 4CzIPN-Br1 | 2.80 (2.60) | 2.72 (2.43) | 2.71 (2.35) | 2.57 (2.16) | 2.50 (2.27) |
Peak energies are in parentheses. All values are in eV.
The crystals of both 4CzIPN and 4CzTPN have an emission onset lower in energy than equivalent solutions in acetonitrile. If the shifts in emission energies were solely a result of SSSE, considering the findings reported by Northey et al., it would imply that these small molecules have the same solvation effect as acetonitrile (dielectric constant (ε) = 37.5), even though they lack the ability to reorganize. Furthermore, while the full-width at half-maximum (FWHM) of a molecule’s CT emission profile typically increases with increasing solvent polarity,31−33 the emission spectra of the crystals feature relatively sharp peaks. Therefore, their narrower FWHM contradicts the hypothesis that the solid-state emission energies are influenced significantly by SSSE. We suggest that the red shift in emission is instead a result of direct intermolecular interactions.
The need to go beyond SSSE as an explanation for these emission shifts is further emphasized in Figure S10 which shows the difference between polycrystalline 4CzTPN and 4CzTPN in dimethylsulfoxide (DMSO). Upon allowing the crystals to take up DMSO solvent, we observe that the fluorescence of the polycrystalline sample changes from orange to yellow (emission blue shift). This would suggest that 4CzTPN in the crystal has a polarity effect greater than DMSO (ε = 47.2)! Based on the findings by Northey et al.,19 we consider it implausible that such a solid-state shift arises from SSSE, thus we must consider that the breakup of the intermolecular interactions (dimerization/aggregation) is the cause for such a large perturbation of the emission. Figure S9 also reveals that the bulky tert-butyl groups of 4CzIPN-tBu8 and 4CzTPN-tBu8 do very little to stop dimer formation in these molecules, as they behave like their non-tert-butylated analogues.
The spectra attributed to the crystal form for these compounds relate to the first fraction obtained by sublimation, “Fraction 1”, which is identified in the photograph (Figure 2c) using 4CzIPN as an example. It is also apparent from this photograph and Figure 2d that the emission color of 4CzIPN changes as a function of recrystallization temperature along the sublimation tube. These differences in the emission spectra of the sublimation “Fractions” of 4CzIPN will be explained in more detail later.
Photoluminescence of Doped Zeonex Thin Films
These intermolecular interactions not only dominate in the crystalline form but are also present in doped thin films. Figure 3 shows that the emission spectra of 4CzIPN-doped zeonex films red-shift as a function of increasing concentration of the emitter. At 10 weight percent (wt %), the emission profile resembles that observed by Kim et al.9 in neat film. Due to the high viscosity of zeonex, we ruled out large rotational movement of the carbazole donor moieties in the excited state as the cause of this emission shifts—an assumption supported by studying the variable-temperature NMR (VT NMR) spectra of 4CzIPN-Br1 (Figure S11). The ground-state barrier to 180° rotation around the carbazole N–C bond was found to be >87 kJ/mol in solution (see the VT NMR Spectroscopy section of the Supporting Information). Furthermore, there is a red shift in the absorption band of the films with increasing 4CzIPN concentration, which cannot be explained by SSSE and instead we attribute to the presence of ground-state dimers. This new absorption band allows for direct excitation of a charge-transfer dimer state (1CTD).
Figure 3.
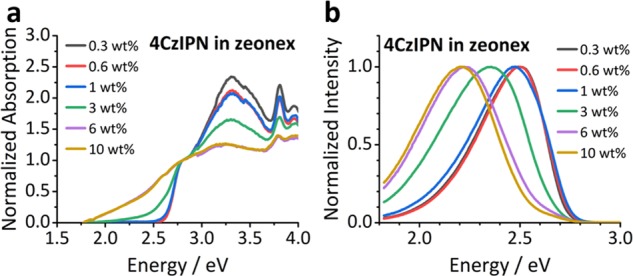
(a) Normalized absorption of 4CzIPN in a zeonex host at different doping concentrations. The band edge begins to red-shift at 1 wt %, with the shift saturating between 6 and 10 wt %. (b) The photoluminescence of the same films showing the same red shift in emission with increasing concentration. The shift occurs at the same concentrations as for the absorption and again saturates between 6 and 10 wt %. The high-energy tail in the high concentration films (3 wt % and above) is attributed to inhomogeneous dimer/aggregate formation, leaving regions of relatively isolated 4CzIPN.
The time-resolved photoluminescence of 1CTD in 4CzIPN (Figure 4) was measured by exciting the 10 wt % film at 2.33 eV [532 nm], which is significantly below the 4CzIPN monomer charge-transfer (1CTM) absorption (2.5 eV). The emission spectrum of 1CTD remains constant as a function of time and is identical to that observed in both the high concentration films in Figure 3 and the emission spectra of the neat films reported in the literature.9,111CTD is also found to be TADF active, meaning that it is not just the monomer species of 4CzIPN that undergoes rISC in this material. The delayed fluorescence (DF) lifetime of 1CTD is 2.7 μs at room temperature (RT), which increases to 4.8 μs at 80 K. The presence of delayed emission at 80 K suggests an efficient rISC process and a small singlet–triplet energy gap for 1CTD. The full kinetic fittings for the room temperature and 80 K data are shown in Table S1. All of the data include a short lifetime (4–6 ns) contribution, arising from the tail of the high-energy laser pulse (>200 μJ) required to achieve reasonable signal-to-noise using direct 1CTD excitation.
Figure 4.
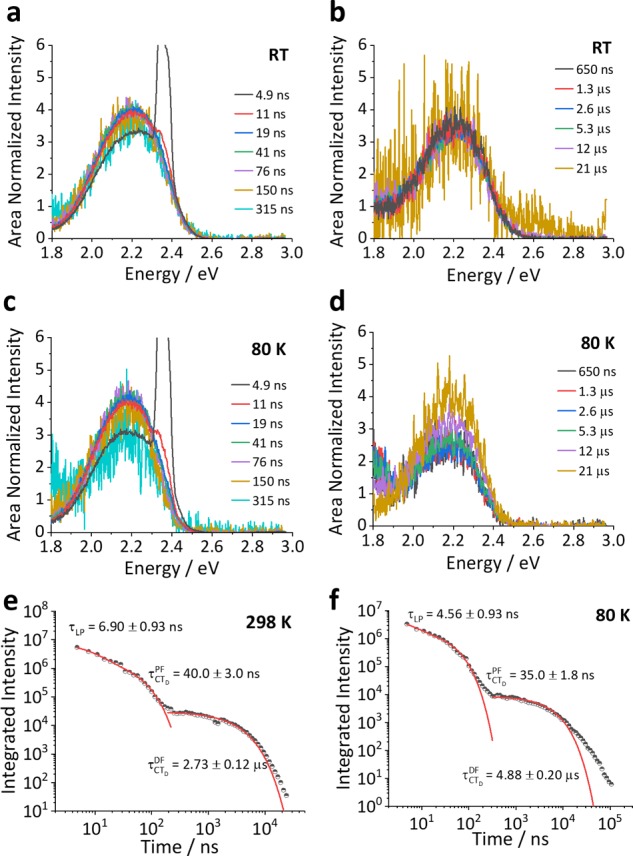
Time-resolved emission spectra and decays of 10 wt % doped 4CzIPN:zeonex film excited at 532 nm. (a, b) The room temperature spectra of the film, showing the consistent line shape maintained throughout the prompt and delayed time regions. (c, d) The spectra also remain unchanged at 80 K, and TADF is still observed, suggesting a very small singlet–triplet gap. (e) Time-resolved emission decay at RT and (f) at 80 K.
Overlaying the prompt emission spectra of 1CTD with the time-resolved photoluminescence spectra of the 0.3, 1, and 10 wt % films (excited at 355 nm i.e., into the monomer absorption band) allows the significant influence of this dimer state to be fully uncovered (Figure 5).
Figure 5.
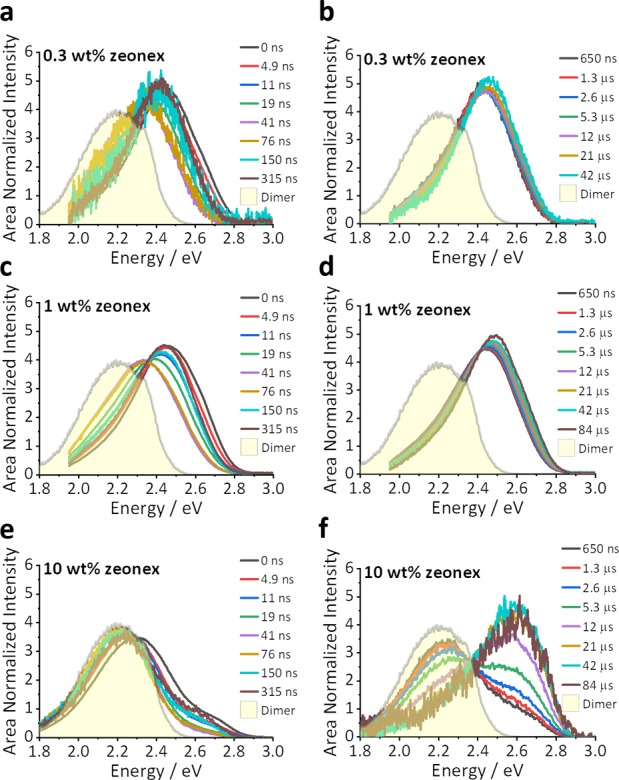
Time-resolved emission spectra of the doped 4CzIPN:zeonex films at room temperature. (a, b) The prompt and delayed emission of the 0.3 wt % doped film, which is dominated by monomer emission in a nonpolar environment (onset 2.78 eV [446 nm]). (c, d) The 1 wt % film has a small red shift at intermediate times of 50–100 ns because of the dimeric species and returns to full monomer TADF at longer times. (e, f) The 10 wt % film is dominated by the dimer emission but has contributions from the monomer species at prompt and long times, a result of the slower rISC rate for the CTM in a nonpolar environment. The filled yellow peak overlaid in each panel is the dimer emission observed after a 11 ns delay time from a 10 wt % zeonex film of 4CzIPN at RT using 532 nm excitation (as shown in Figure 4a).
While the 10 wt % film emission is predominantly from 1CTD (shaded area), even in the 1 wt % film there is also a small red shift at intermediate times as a result of the dimer. The observation of dimer contribution in such a dilute film is very surprising and conflicts with the assumption (often tacit) in previous studies of 4CzIPN that emission from “low” concentration films (<10%) comes exclusively from the monomer. In the 10 wt % films, a small contribution from 1CTM is present, which accounts for the high-energy onset of 2.78 eV [446 nm] in the prompt spectra. This high-energy component is absent with 532 nm excitation, giving further support to the assertion that the 532 nm laser excites 1CTD directly, rather than through donor 1LE excitation. The formation of 1CTM by 355 nm excitation accounts for the emergence of monomer TADF line shape in the 10 wt % film at longer times. The full shifts of the emission peaks of these doped films as a function of concentration and temperature are shown in Figure S12.
The prompt lifetimes of 1CTM in these films is approximately 5–7 ns, which is shorter than the 8 ns lifetime observed for 1CTM in methylcyclohexane (MCH) solution (Figure S13 and Table S2). This small reduction in lifetime is attributed to quenching of 1CTM to form 1CTD states (lifetime 20–30 ns) in the solid state. The 10 wt % film at room temperature has a shorter DF lifetime (2.05 μs) than the MCH solution and 0.3 and 1 wt % films, which is attributed to the faster rISC in 1CTD. The full decay profiles and fittings are shown in Figure S14 and Tables S3–S5.
The photoluminescence quantum yields (PLQYs) of the 10 and 1 wt % films were measured to identify if quenching processes shorten the measured DF lifetimes. The natural lifetimes for the DF are shown in Table S6. Even considering the significant reduction in PLQY with increasing concentration, the natural lifetime of 1CTD is still shorter than 1CTM, consistent with the smaller 1CTD–3LED energy gap (ΔEST for the dimer).
Spectra and decays obtained at 80 K (Figure S15) show that the 1CTM TADF in the 0.3 and 1 wt % films is significantly reduced, although delayed emission from 1CTD remains. This smaller temperature dependence in the 1CTD delayed emission intensity is further evidence that 1CTD has a much smaller singlet–triplet energy gap—with its associated dimeric locally excited triplet (3LED)—than the monomer. This phenomenon is also seen in the 10 wt % film, where the 1CTD exhibits significant TADF even at 80 K. For all films at long times, the phosphorescence of the monomer species (3LEM) is observed (onset 2.69 eV [460 nm]), which is a direct result of the excitation wavelength (355 nm) exciting the 1LEM state of the molecule.
Excitation fluence dependence measurements of the delayed emission (Figures S16 and S17) confirm that emission arises from TADF and not any bimolecular process occurring in the aggregate regions, even at low temperature where rISC is inhibited.
Evaporated Films and Device Physics
The photophysical analysis of the doped zeonex films demonstrates that dimer formation for 4CzIPN and related molecules is critically dependent on the conditions of sample preparation. In Figure 6, the photoluminescence spectra of evaporated films of 4CzIPN in 3,3′-bis(N-carbazolyl)-1,1′-biphenyl (mCBP) show the same trend as that observed in zeonex films. This surprising presence of dimer species even in evaporated films also explains the observed behavior of 4CzIPN and related materials in OLED devices.
Figure 6.
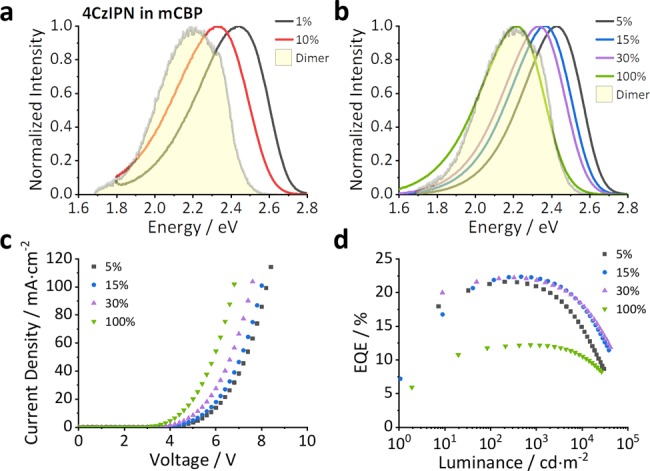
Photoluminescence from evaporated films and device data from a 4CzIPN OLED. (a) Emission from 100 nm evaporated films of 4CzIPN doped into mCBP. (b) The electroluminescence from a device with a 4CzIPN-doped mCBP emissive layer of different concentrations (X = 5, 15, 30, 100). (c) The current density voltage curves and (d) the external quantum efficiency (EQE) versus luminance curves of the same series of devices. The device structure used for these measurements was indium tin oxide (ITO) (20 nm)/buffer (20 nm)/hole injection layer (20 nm)/hole transport layer (30 nm)/electron blocking layer (10 nm)/mCBP:4CzIPN (X%, 15 nm)/hole blocking layer (10 nm)/electron transport layer (40 nm)/aluminum (100 nm).
This red shift in emission is also replicated in the electroluminescence spectra from model devices with an emissive layer of 4CzIPN co-doped in mCBP at different concentrations. A full series of concentration measurements is shown in Figure S18, and the device structure is detailed in the caption of Figure 6. The changes in the electroluminescence spectra are consistent with the photoluminescence emission9−11 and are thus independent of the device structure and transport layers. The phenomena are related to the emissive layer only and are consistent with the emission red shifts reported in solution-processed and evaporated films by others.9,10,12−16 Device data shown in Figure 6c,d demonstrate that the monomer and dimer concentrations within the emissive layer play a dominant role in all aspects of the device performance, both electrical and optical. The current–voltage (J–V) and EQE curves for the full device series are shown in Figure S18. The data demonstrate that dimer formation in 4CzIPN and related compounds is present and influences the emission color at typical concentrations used in device fabrication.
Pristine and Annealed Neat Films
The dimer state in 4CzIPN can be controlled through thermal annealing. This was demonstrated by measuring the emission of a neat dropcast film before and after heating to 250 °C. Upon heating, the emission onset of the film blue-shifted from 2.55 eV [486 nm] to 2.66 eV [466 nm] (Figure S19). This was attributed to the breakup of dimer species in the pristine film and is a property shared by 4CzIPN and 4CzIPN-tBu8 as shown in Videos S1 and S2. This further implies that the addition of tert-butyl groups to the compounds is insufficient to prevent these intermolecular interactions. The process is also completely reversible by dissolving the annealed film in solvent.
Interestingly, the emission onset of the annealed film is lower in energy than that of the dilute (0.3 and 1 wt %) films even though it is most likely the monomer state of 4CzIPN. The comparison of the onset and peak energies of the emission for the pristine, annealed, and 1 wt % films is shown in Table 2, alongside the singlet–triplet gap and empirical rISC rate. The difference between the annealed and 1 wt % emission is attributed to SSSE and the mechanism proposed by Kim et al.;9 however, the further shift between the annealed and pristine films arises from the intermolecular interactions.
Table 2. Measured Energies of 1CT and 3LE, Respective ΔES-T, and Reverse Intersystem Crossing Rates of 4CzIPN Samplesa.
| sample | 1CT/eV | 3LE/eV | ΔES-T/eV | krISC (×105 s–1) |
|---|---|---|---|---|
| 1 wt % zeonex film | 2.78 (2.48) | 2.69 (2.43) | 0.09 | 3.8 |
| pristine | 2.48 (2.26) | 2.50 (2.28) | 0.02 | 1.2 |
| annealed | 2.66 (2.43) | 2.69 (2.43)b, 2.50 (2.23)c | 0.03b, 0.16c | 28 |
Peak energies are in parentheses.
Monomer 3LE.
Dimer 3LE.
Emission from 1CTD still dominates the time-resolved spectra of the pristine neat film at room temperature and 80 K, whereas the annealed film retains the higher energy contribution from 1CTM (Figures S20 and S21). At longer times in the 80 K spectra, both films tend toward the dimer phosphorescence (3LED), which is almost isoenergetic to the 1CTD emission. The lifetime fits are shown in Figure S22 and Tables S7, S8.
By comparing the natural lifetimes (see Table S6) of the films, 1CTM in a neat film is found to have a shorter DF lifetime than 1CTD, even though they have very similar energy gaps. The rISC rate of 1CTM is slow in zeonex and fast in the annealed film due to the change in the 1CTM–3LEM energy gap from 0.09 eV to approximately 0.03 eV due to SSSE. The experimentally calculated rISC rate of 1CTD is quite low considering its small singlet–triplet energy gap. However, this is attributed to the low PLQY of the dimer state.
Sublimed Crystals
To deconvolute the steady-state spectra of the 4CzIPN crystals obtained after sublimation, the time-resolved photoluminescence of these samples was analyzed (see Figures S23 and S24). While the spectra are dominated by 1CTD emission, all fractions have a small contribution from 1CTM, resulting in the high-energy onset above 2.6 eV [477 nm]. This monomer contribution is more prominent in Fractions 2 and 3, where there is also a blue shift in the TADF. The prompt and DF emission lifetimes of 1CTM and 1CTD are consistent with those observed in the neat films (Figure S25 and Tables S9–S11).
The structure observed in the time-resolved spectra of the sublimed crystals is attributed to the nature of the dimer state. We consider the dimers to be intermolecular species, and there will be a fixed distance between the carbazoles in a single crystal. Consequently, certain modes and geometries will be prevalent. This phenomenon is the origin of the vibronic structure in the spectra of the sublimed crystals.
Upon analyzing the powder X-ray diffraction (PXRD) data obtained from these samples (Figure S26a), it appears that the contribution from 1CTM in Fractions 2 and 3 is due to a reduction in the long-range intermolecular order. The diffraction patterns from the powdered samples are in good agreement with the simulated PXRD pattern based on the single crystal X-ray structure of material obtained from Fraction 1 (Figure S26b).
Solvent-Grown Crystals
Crystals formed as solvates were grown from solvent mixtures of acetone/hexanes, tetrahydrofuran (THF)/hexanes, and chloroform/hexanes. The 532 nm-excited 1CTD spectrum matches perfectly with the time-resolved emission of these solvent-grown crystals (Figures S27 and S28), as do the decay lifetimes (Figure S29 and Tables S12–S14).
Crystal Structures and Polymorphism
Single crystal X-ray diffraction data of the Fraction 1 sublimed crystals of 4CzIPN and 4CzTPN (Figure 7) reveal face-to-face contacts between the aromatic groups of the molecules, particularly in the structure of 4CzTPN. 4CzIPN also has some neighboring-molecule interactions reminiscent of PO carbazole dimers (Figure 1b), which account for the red shift compared to MCH solution in Figure 2a. The same is true for the remaining compounds in the series (Figures S30 and S31). The dimer emission in the solvent-grown crystals is also explained by these interactions (Figures S32 and S33).
Figure 7.
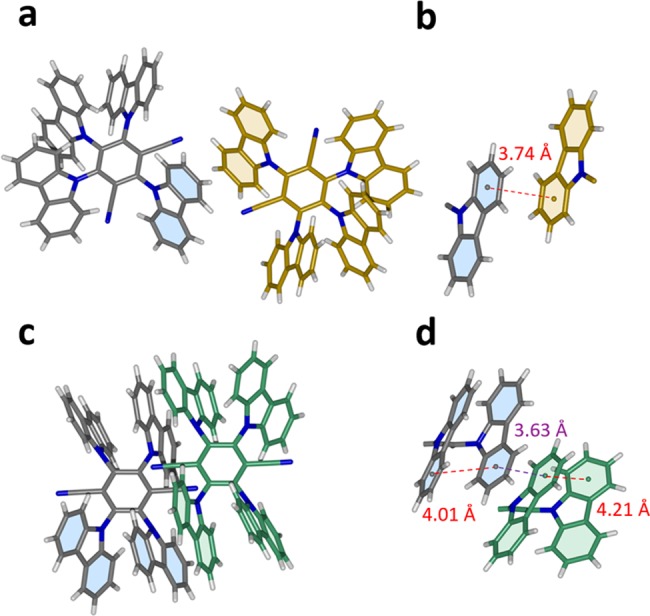
Packing in the crystal structures of 4CzIPN and 4CzTPN obtained by sublimation. (a) Two neighboring molecules of 4CzIPN with the carbazoles involved in the interaction are highlighted. (b) The measured distance between the two interacting molecules. (c) Two neighboring molecules of 4CzTPN with the carbazoles involved are highlighted. (d) The intermolecular and intramolecular distances between the involved carbazoles.
Although 4CzIPN was isolated as a crystalline solid after synthesis, the sublimation of the sample gave several visually different fractions (Figure 2c). Nonetheless, all fractions gave NMR spectra identical to the original material showing that these changes in appearance, like the color change of the annealed neat film, are not a result of decomposition. Fractions 1 and 2 were characterized by differential scanning calorimetry (DSC) (Figure S34) and thermogravimetric analysis (TGA) (Figure S35). For both fractions, the DSC data show two endothermic peaks: a sharp one at 651.0 K and a shoulder at 650.7 K that is more pronounced in Fraction 2. These observations are consistent with the presence of polymorphs of 4CzIPN in the sublimed fractions. Different temperatures of initial decomposition (676 and 685 K for Fractions 1 and 2, respectively) were observed by TGA, which also supports the presence of polymorphs.
The DSC data of the solvent-grown crystals show several possible melting peaks ranging from 650.1 to 651.0 K (Figure S34), whose prevalence depends on the preparation conditions. Upon grinding the crystals obtained from chloroform/hexanes, only one sharp melting peak at 650.1 K appears in the DSC. This observation provides evidence that mechanical force can modulate the dimer formation in solid-state samples of this compound (to be discussed in detail later).
On the other hand, 4CzTPN and 4CzTPN-tBu8 do not show polymorphism. After sublimation, both 4CzTPN and 4CzTPN-tBu8 retain their orange color both in reflectance and fluorescence, characteristics that are attributed to the dimer species of these molecules. The emission profiles also do not change during the excited-state lifetime of 4CzTPN at room temperature and 80 K (Figure S36), which is attributed to the intermolecular interactions/dimers that are seen in its crystal structure (Figure 7). The DF lifetime of the dimer state in 4CzTPN at room temperature is 1.89 μs as shown in Figure S36 and Table S15. The lifetime fit at 80 K is shown in Figure S37.
Mechanochromic Luminescence (MCL)
The MCL of 4CzIPN (Figure S38) is also related to these dimeric interactions. When crystals of sublimed 4CzIPN were ground by hand in a pestle and mortar, a significant red shift and broadening of the emission occurred. This red shift was attributed to an increase of the dimer states and the resultant emission is consistent with that of 1CTD. However, when a host molecule was included in the grinding, weaker MCL and a reduced red shift in emission were observed. Even the introduction of a polar host bis[2-(diphenylphosphino)phenyl] ether oxide (DPEPO), which has a ground-state dipole moment of 8.06 D,19,34 did not lead to the significant shifts in emission observed upon grinding of the neat crystals. The reduction in the red shift of the emission by introducing a more polar molecule than 4CzIPN once again confirms that SSSE cannot explain the reported emission shifts in 4CzIPN and related molecules.
Conclusions
We have discovered that 4CzIPN and related molecules form intermolecular ground-state dimers, which impact on their emission energy and TADF performance, even in evaporated films previously assumed to emit only from 4CzIPN monomers. The influence of these aggregate/dimer states on the TADF mechanism has been characterized through analysis of their fluorescence (in crystal, film, and solution), crystal structures, and MCL behavior. The crystal analysis shows that the molecules pack closely leading to this aggregate/dimer formation, which explains previously reported phenomena, including a significant emission red shift observed by Kim et al.9 Interestingly, the dimer state (1CTD) undergoes TADF with its own characteristic kinetics.
Our observations also contribute to understanding the photoinduced absorption measurements of Hosokai et al.,35 who reported TADF from “local” and “delocalized” CT triplet states. Their observations of the photoinduced absorption of these states in toluene solution of 4CzIPN would be consistent with excited-state-induced absorption from monomer and dimer states, respectively. Moreover, we find that the 1CTD–3LED gap is very small, giving efficient TADF in nonpolar hosts, consistent with the observations of Hosokai et al.35 that the best design of a TADF emitter comes from the delocalized component (or in our interpretation, the dimer). However, the low PLQY of the dimer state means that it is more likely to act as a triplet-trap state in applications.
This work highlights the ubiquity and importance of dimerization in carbazole-based materials and the effects this can have on TADF behavior and color purity. Without this knowledge, specious interpretations may be made. We suggest that the unpredictable behavior of many well-known carbazole-based TADF emitters may arise from similar dimer formation in films. As a result, observations in the literature should be reinterpreted within this model and future molecular design should be directed to the choice of bulkier sterically hindered units, which will minimize intermolecular interactions and ensure the predictable properties of the emitters in OLEDs.
Experimental Section
Sample Preparation
Zeonex films were prepared via drop-casting using a mixture of dopant and host (zeonex) at the weight percentage specified. The initial solution concentrations were 1 mg/mL for the dopant and 100 mg/mL for zeonex. The films were dropcast onto a quartz substrate at room temperature to avoid any thermal annealing. The crystal samples were prepared using either the sublimed or solvent-grown crystals fixed with vacuum grease on a quartz substrate. The 4CzIPN-doped mCBP films were evaporated at 100 nm thickness onto a sapphire substrate using a Kurt J. Lesker deposition chamber at a vacuum below 10–6 mbar.
Sublimation
The materials were sublimed using a Creaphys DSU05 organic sublimation system.
Optical Characterization
Optical measurements in solution used concentrations in the 10–5–10–2 M range. Degassed solutions were prepared via the freeze–thaw method (5 repeats). Absorption and emission spectra were collected using a UV-3600 double beam spectrophotometer (Shimadzu) and a Fluoromax fluorimeter (Jobin Yvon). The MCL measurements were performed using an Ocean Optics spectrometer and a 395 nm UV torch. The PLQY measurements were performed using 365 nm excitation from an Ocean Optics LLS-LED and an integrating sphere connected to an Ocean Optics QePro Spectrometer.
Time-Resolved Photoluminescence
Time-resolved photoluminescence spectra and decays were recorded using a nanosecond gated spectrograph-coupled iCCD (Stanford) using either an Nd:YAG laser emitting at 355 nm (EKSPLA) or an N2 laser (Lasertechnik Berlin) emitting at 337 nm.36 Values of krISC were calculated according to “approach b” reported by dos Santos et al.37 using the natural lifetime and the integrals of the prompt and delayed fluorescence.
Device Fabrication
OLED devices were fabricated using precleaned indium tin oxide (ITO)-coated glass substrates with an ITO thickness of 50 nm. A 20 nm thick PEDOT:PSS layer was spin-coated to the precleaned ITO substrates and the substrates were heated afterward for 10 min at 180 °C. The small-molecule and cathode layers were thermally evaporated using a Kurt J. Lesker deposition chamber below 10–6 mbar.
Single Crystal X-ray Diffraction
Experimental details and a detailed description of the 4CzIPN crystal dihedral angles can be found in Figures S39–S42 and Tables S16–S18.
Acknowledgments
We thank W. D. Carswell for obtaining DSC and TGA data, Gary Oswald for collecting the powder X-ray diffraction patterns, and Dr D. S. Yufit for help with the single crystal X-ray study. The Diamond Light Source (RAL) is thanked for the award of instrument time on Station I19 (MT 11145) and the instrument scientists for their kind support. Funding: M.K.E., H.F.H., and A.P.M. acknowledge the EU’s Horizon 2020 research and innovation programme for funding the PHEBE project under Grant No. 641725. M.K.E., A.D., N.H., C.P., A.M., M.R.B., and A.P.M. also acknowledge the EU’s Horizon 2020 research and innovation programme for funding the HyperOLED project under grant agreement No. 732013. A.P.M. and M.R.B. also acknowledge the EPSRC for funding under grant number EP/L02621X/1. A.N.B. is supported by an EPSRC doctoral training grant. P.R.M. and A.P.M. thank BEIS and UUKi for support through a Rutherford Strategic Partner Grant. D.R.G. thanks Merck KGaA for funding his PhD studies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcc.9b01458.
General methods; synthetic methods; Photographs of 4CzTPN in powder form and DMSO solution under room light and UV light conditions; Photoluminescence (PL) spectroscopy of zeonex films and methylcyclohexane solution of 4CzIPN; PL spectroscopy of sublimed crystals of 4CzIPN; molecular packing and interactions in the solids; mechanochromic luminescence; X-ray crystallography (PDF)
Breakup of dimer species in pristine films of 4CzIPN and 4CzIPN-tBu8 (AVI) (AVI)
CCDC 1873416–1873427 contains the supplementary crystallographic data for this paper (ZIP)
The authors declare no competing financial interest.
This paper was published ASAP on April 18, 2019, with incorrect Supporting Information due to a production error. The corrected version was reposted on April 18, 2019.
Supplementary Material
References
- Nakagawa T.; Ku S.-Y.; Wong K.-T.; Adachi C. Electroluminescence Based on Thermally Activated Delayed Fluorescence Generated by a Spirobifluorene Donor–Acceptor Structure. Chem. Commun. 2012, 48, 9580. 10.1039/c2cc31468a. [DOI] [PubMed] [Google Scholar]
- Uoyama H.; Goushi K.; Shizu K.; Nomura H.; Adachi C. Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature 2012, 492, 234–238. 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
- Dias F. B.; Bourdakos K. N.; Jankus V.; Moss K. C.; Kamtekar K. T.; Bhalla V.; Santos J.; Bryce M. R.; Monkman A. P. Triplet Harvesting with 100% Efficiency by Way of Thermally Activated Delayed Fluorescence in Charge Transfer OLED Emitters. Adv. Mater. 2013, 25, 3707–3714. 10.1002/adma.201300753. [DOI] [PubMed] [Google Scholar]
- Etherington M. K.; Gibson J.; Higginbotham H. F.; Penfold T. J.; Monkman A. P. Revealing the Spin–Vibronic Coupling Mechanism of Thermally Activated Delayed Fluorescence. Nat. Commun. 2016, 7, 13680 10.1038/ncomms13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J.; Monkman A. P.; Penfold T. J. The Importance of Vibronic Coupling for Efficient Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Molecules. ChemPhysChem 2016, 17, 2956–2961. 10.1002/cphc.201600662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos P. L.; Ward J. S.; Bryce M. R.; Monkman A. P. Using Guest–Host Interactions To Optimize the Efficiency of TADF OLEDs. J. Phys. Chem. Lett. 2016, 7, 3341–3346. 10.1021/acs.jpclett.6b01542. [DOI] [PubMed] [Google Scholar]
- Santos P. L.; Ward J. S.; Data P.; Batsanov A. S.; Bryce M. R.; Dias F. B.; Monkman A. P. Engineering the Singlet–Triplet Energy Splitting in a TADF Molecule. J. Mater. Chem. C 2016, 4, 3815–3824. 10.1039/C5TC03849A. [DOI] [Google Scholar]
- Dos Santos P. L.; Etherington M. K.; Monkman A. P. Chemical and Conformational Control of the Energy Gaps Involved in the Thermally Activated Delayed Fluorescence Mechanism. J. Mater. Chem. C 2018, 6, 4842–4853. 10.1039/C8TC00991K. [DOI] [Google Scholar]
- Kim H. S.; Park S.-R.; Suh M. C. Concentration Quenching Behavior of Thermally Activated Delayed Fluorescence in a Solid Film. J. Phys. Chem. C 2017, 121, 13986–13997. 10.1021/acs.jpcc.7b02369. [DOI] [Google Scholar]
- Niwa A.; Haseyama S.; Kobayashi T.; Nagase T.; Goushi K.; Adachi C.; Naito H. Triplet-Triplet Annihilation in a Thermally Activated Delayed Fluorescence Emitter Lightly Doped in a Host. Appl. Phys. Lett. 2018, 113, 083301 10.1063/1.5025870. [DOI] [Google Scholar]
- Kim B. S.; Lee J. Y. Phosphine Oxide Type Bipolar Host Material for High Quantum Efficiency in Thermally Activated Delayed Fluorescent Device. ACS Appl. Mater. Interfaces 2014, 6, 8396–8400. 10.1021/am501301g. [DOI] [PubMed] [Google Scholar]
- Wang P.; Zhao S.; Xu Z.; Qiao B.; Long Z.; Huang Q. The Electroluminescence Mechanism of Solution-Processed TADF Emitter 4CzIPN Doped OLEDs Investigated by Transient Measurements. Molecules 2016, 21, 1365 10.3390/molecules21101365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F.; Zhou C.; Zhou D.; Yu J. Improving Electroluminescent Efficiency and Ultraviolet Detectivity of Optoelectronic Integrated Devices by Doping a Thermally Activated Delayed Fluorescent Material in an Aggregation-Induced Emission Material as an Active Layer. Opt. Mater. Express 2017, 7, 3538. 10.1364/OME.7.003538. [DOI] [Google Scholar]
- Nakanotani H.; Masui K.; Nishide J.; Shibata T.; Adachi C. Promising Operational Stability of High-Efficiency Organic Light-Emitting Diodes Based on Thermally Activated Delayed Fluorescence. Sci. Rep. 2013, 3, 2127 10.1038/srep02127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu R.; Sasabe H.; Inomata S.; Pu Y.-J.; Kido J. High Efficiency Solution Processed OLEDs Using a Thermally Activated Delayed Fluorescence Emitter. Synth. Met. 2015, 202, 165–168. 10.1016/j.synthmet.2015.02.009. [DOI] [Google Scholar]
- Li N.; Fang Y.; Li L.; Zhao H.; Quan Y.; Ye S.; Fan Q.; Huang W. A Universal Solution-Processable Bipolar Host Based on Triphenylamine and Pyridine for Efficient Phosphorescent and Thermally Activated Delayed Fluorescence OLEDs. J. Lumin. 2018, 199, 465–474. 10.1016/j.jlumin.2018.03.003. [DOI] [Google Scholar]
- Liu Z.; Lei Y.; Fan C.; Peng X.; Ji X.; Jabbour G. E.; Yang X. Simple-Structure Organic Light Emitting Diodes: Exploring the Use of Thermally Activated Delayed Fluorescence Host and Guest Materials. Org. Electron. 2017, 41, 237–244. 10.1016/j.orgel.2016.11.010. [DOI] [Google Scholar]
- Noguchi Y.; Kim H.-J.; Ishino R.; Goushi K.; Adachi C.; Nakayama Y.; Ishii H. Charge Carrier Dynamics and Degradation Phenomena in Organic Light-Emitting Diodes Doped by a Thermally Activated Delayed Fluorescence Emitter. Org. Electron. 2015, 17, 184–191. 10.1016/j.orgel.2014.12.009. [DOI] [Google Scholar]
- Northey T.; Stacey J. E.; Penfold T. J. The Role of Solid State Solvation on the Charge Transfer State of a Thermally Activated Delayed Fluorescence Emitter. J. Mater. Chem. C 2017, 5, 11001–11009. 10.1039/C7TC04099G. [DOI] [Google Scholar]
- Jankus V.; Monkman A. P. Is Poly(Vinylcarbazole) a Good Host for Blue Phosphorescent Dopants in PLEDs? Dimer Formation and Their Effects on the Triplet Energy Level of Poly(N-Vinylcarbazole) and Poly(N-Ethyl-2-Vinylcarbazole). Adv. Funct. Mater. 2011, 21, 3350–3356. 10.1002/adfm.201100732. [DOI] [Google Scholar]
- Tani K.; Tohda Y.; Takemura H.; Ohkita H.; Ito S.; Yamamoto M. Synthesis and Photophysical Properties of [3.3](3,9)Carbazolophanes. Chem. Commun. 2001, 1914–1915. 10.1039/b104101k. [DOI] [PubMed] [Google Scholar]
- de Sainte Claire P. Molecular Simulation of Excimer Fluorescence in Polystyrene and Poly(Vinylcarbazole). J. Phys. Chem. B 2006, 110, 7334–7343. 10.1021/jp056987e. [DOI] [PubMed] [Google Scholar]
- Saiful I. S. M.; Heinze P.; Ohba Y.; Yamauchi S.; Yamamoto M.; Tohda Y.; Tani K. Interplanar Interactions in the Excited Triplet States of Carbazole Dimers by Means of Time-Resolved EPR Spectroscopy. Mol. Phys. 2006, 104, 1535–1542. 10.1080/00268970600638606. [DOI] [Google Scholar]
- Wex B.; Kaafarani B. R. Perspective on Carbazole-Based Organic Compounds as Emitters and Hosts in TADF Applications. J. Mater. Chem. C 2017, 5, 8622–8653. 10.1039/C7TC02156A. [DOI] [Google Scholar]
- Pashazadeh R.; Pander P.; Lazauskas A.; Dias F. B.; Grazulevicius J. V. Multicolor Luminescence Switching and Controllable Thermally Activated Delayed Fluorescence Turn on/Turn off in Carbazole–Quinoxaline–Carbazole Triads. J. Phys. Chem. Lett. 2018, 9, 1172–1177. 10.1021/acs.jpclett.8b00136. [DOI] [PubMed] [Google Scholar]
- Skuodis E.; Bezvikonnyi O.; Tomkeviciene A.; Volyniuk D.; Mimaite V.; Lazauskas A.; Bucinskas A.; Keruckiene R.; Sini G.; Grazulevicius J. V. Aggregation, Thermal Annealing, and Hosting Effects on Performances of an Acridan-Based TADF Emitter. Org. Electron. 2018, 63, 29–40. 10.1016/j.orgel.2018.09.002. [DOI] [Google Scholar]
- Huang B.; Chen W.-C.; Li Z.; Zhang J.; Zhao W.; Feng Y.; Tang B. Z.; Lee C.-S. Manipulation of Molecular Aggregation States to Realize Polymorphism, AIE, MCL, and TADF in a Single Molecule. Angew. Chem., Int. Ed. 2018, 57, 12473–12477. 10.1002/anie.201806800. [DOI] [PubMed] [Google Scholar]
- Hladka I.; Volyniuk D. Y.; Bezvikonnyi O.; Kinzhybalo V.; Bednarchuk T.; Danyliv Y.; Lytvyn R.; Lazauskas A.; Grazulevicius J. V. Polymorphism of Derivatives of tert-Butyl Substituted Acridan and Perfluorobiphenyl as Sky-Blue OLED Emitters Exhibiting Aggregation Induced-Active Thermally Activated Delayed Fluorescence. J. Mater. Chem. C 2018, 13179–13189. 10.1039/C8TC04867C. [DOI] [Google Scholar]
- Zeng X.; Zhou T.; Liu J.; Wu K.; Li S.; Xiao X.; Zhang Y.; Gong S.; Xie G.; Yang C. Incorporating Thermally Activated Delayed Fluorescence into Mechanochromic Luminescent Emitters: High-Performance Solution-Processed Yellow Organic Light Emitting Diodes. Adv. Opt. Mater. 2018, 1801071 10.1002/adom.201801071. [DOI] [Google Scholar]
- Sturala J.; Etherington M. K.; Bismillah A. N.; Higginbotham H. F.; Trewby W.; Aguilar J. A.; Bromley E. H. C.; Avestro A.-J.; Monkman A. P.; McGonigal P. R. Excited-State Aromatic Interactions in the Aggregation-Induced Emission of Molecular Rotors. J. Am. Chem. Soc. 2017, 139, 17882–17889. 10.1021/jacs.7b08570. [DOI] [PubMed] [Google Scholar]
- Verbeek G.; Depaemelaere S.; Van der Auweraer M.; De Schryver F. C.; Vaes A.; Terrell D.; De Meutter S. Amino Derivatives of Symmetric 1,3,5-Triphenylbenzene. Nature of the Polar Singlet Excited State. Chem. Phys. 1993, 176, 195–213. 10.1016/0301-0104(93)85017-3. [DOI] [Google Scholar]
- Laguitton-Pasquier H.; Pansu R.; Chauvet J.-P.; Collet A.; Faure J.; Lapouyade R. The Charge Transfer State of Excited Bianthryl and a Derivative: Solvatochromism, Emission CT Spectra Broadening in Homogeneous Solvents. Chem. Phys. 1996, 212, 437–455. 10.1016/S0301-0104(96)00139-5. [DOI] [Google Scholar]
- Brunschwig B. S.; Ehrenson S.; Sutin N. Solvent Reorganization in Optical and Thermal Electron-Transfer Processes: Solvatochromism and Intramolecular Electron-Transfer Barriers in Spheroidal Molecules. J. Phys. Chem. 1987, 91, 4714–4723. 10.1021/j100302a017. [DOI] [Google Scholar]
- Méhes G.; Goushi K.; Potscavage W. J.; Adachi C. Influence of Host Matrix on Thermally-Activated Delayed Fluorescence: Effects on Emission Lifetime, Photoluminescence Quantum Yield, and Device Performance. Org. Electron. 2014, 15, 2027–2037. 10.1016/j.orgel.2014.05.027. [DOI] [Google Scholar]
- Hosokai T.; Matsuzaki H.; Nakanotani H.; Tokumaru K.; Tsutsui T.; Furube A.; Nasu K.; Nomura H.; Yahiro M.; Adachi C. Evidence and Mechanism of Efficient Thermally Activated Delayed Fluorescence Promoted by Delocalized Excited States. Sci. Adv. 2017, 3, e1603282 10.1126/sciadv.1603282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankus V.; Data P.; Graves D.; McGuinness C.; Santos J.; Bryce M. R.; Dias F. B.; Monkman A. P. Highly Efficient TADF OLEDs: How the Emitter-Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve Near 100% Triplet Harvesting and High Efficiency. Adv. Funct. Mater. 2014, 24, 6178–6186. 10.1002/adfm.201400948. [DOI] [Google Scholar]
- dos Santos P. L.; Ward J. S.; Congrave D. G.; Batsanov A. S.; Eng J.; Stacey J. E.; Penfold T. J.; Monkman A. P.; Bryce M. R. Triazatruxene: A Rigid Central Donor Unit for a D-A 3 Thermally Activated Delayed Fluorescence Material Exhibiting Sub-Microsecond Reverse Intersystem Crossing and Unity Quantum Yield via Multiple Singlet-Triplet State Pairs. Adv. Sci. 2018, 5, 1700989 10.1002/advs.201700989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.