

SUMMARY
Protein- and cell-based immunotherapeutic agents have revolutionized cancer treatment. However, small molecule immunomodulators with favorable pharmacological properties to reach intracellular targets remain to be developed. To explore the vast chemical space, a robust method that recapitulates the complex cancer-immune microenvironment in a high throughput format is essential. To address this critical gap, we developed a High Throughput immunomodulator Phenotypic screening platform, HTiP, which integrates the immune- and cancer-cell co-culture system with imaging- and biochemical-based multiplexed readouts. Using the HTiP platform, we have demonstrated its capability in modeling oncogenic KRAS mutation-driven immunosuppressive phenotype. From a bioactive chemical library, multiple structurally distinct compounds were identified, all of which target the same class of proteins, Inhibitor of Apoptosis Protein (IAP). IAP has demonstrated roles in cancer immunity. Identification of IAP antagonists as potent anti-tumor immune enhancers provides strong validation evidence for the use of the HTiP platform to discover small molecule immunomodulators.
Keywords: small-molecule immunomodulator, inhibitor of apoptosis protein (IAP), IAP antagonist, tumor and immune cell co-culture, KRAS mutation
Graphical Abstract
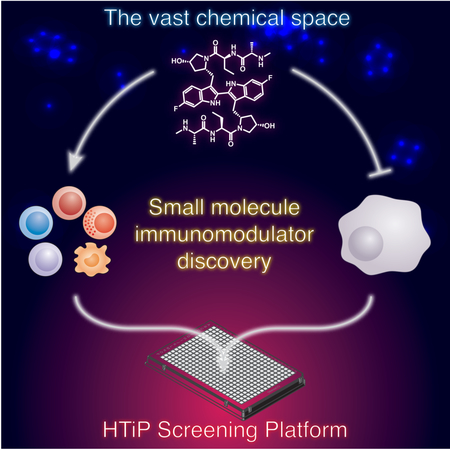
In Brief
Exploring vast chemical space for immunotherapeutic agent discovery requires robust technologies that recapitulate the tumor-immune microenvironment. Mo et al. developed an HTiP platform that models KRAS mutation-driven immunosuppressive phenotype. The identification of IAP inhibitors with known antitumor immunity activity supports the utility of HTiP to uncover small molecule anticancer immunomodulators.
INTRODUCTION
Protein- and cell-based cancer immunotherapy has led to a paradigm shift in cancer treatment through modulating the immune system using immune checkpoint blocking antibodies and engineered Chimeric Antigen Receptor T-cells (Fesnak, et al., 2016; Postow, et al., 2015). Despite the clinical success of current immunotherapies for some cancers, the limitations of these therapies have come to light such as the emerging clinical observation of limited response, the enormous economic burden in production and delivery, the complexity of pharmacokinetics, and the potential safety issue of immunogenicity (Chames, et al., 2009; Fesnak, et al., 2016; Sadelain, et al., 2017).
To complement and potentially synergize with the immunotherapeutic antibodies and engineered immune cells, alternative therapeutic agents such as small molecule immunomodulators remain to be developed (Dhanak, et al., 2017). Small molecules offer a number of advantages, including the improved bioavailability, enhanced tissue penetration, and the capability to reach intracellular targets from both immune and cancer cells. Moreover, small molecules could also serve as chemical probes for investigating mechanisms involved in anti-tumor immunity. Although there is emerging effort in target-based screenings to identify small molecules that modulate a specific protein target (Dhanak, et al., 2017; Huxley, et al., 2004; Skalniak, et al., 2017), phenotypic screenings reflecting the complex immune response network for large-scale high-throughput small molecule immunomodulator discovery are highly challenging and remain to be established.
To address this critical gap in chemical immunomodulator discovery, we report a High Throughput immunomodulator Phenotypic screening platform, HTiP, which integrates the immune- and cancer cell co-culture system with imaging- and cell viability-based multiplexed readouts in a miniaturized format. As a proof of concept, we screened a focused chemical library of clinical and pre-clinical bioactive compounds and identified a group of IAP antagonists as potent inducers of anti-tumor immunity that selectively suppress the growth of cancer cells with oncogenic KRAS mutation.
RESULTS
Design and Development of the HTiP Screening Platform
To accelerate the discovery of small molecule immunomodulators, a sensitive and scalable high throughput technology platform is essential that models the tumor microenvironment with human immune components in a high-density plate format. Towards the goal of modeling the human cancer-immune interactions for high throughput screening, we examined the feasibility of an in vitro co-culture system with both immune- and cancer-cells and tested it in a miniaturized 384-well plate format (Fig. 1A). The co-culture system consists of label-free native human peripheral blood mononuclear cells (PBMCs) and cancer cells with oncogenic alterations. Human PBMCs containing a mixture of lymphocytes, monocytes and dendritic cells were used to recapitulate the complexity of immune system. The growth phenotype of cancer cells was monitored by an imaging system based on their differential sizes (Fig. 1B). For the same well, cell viability was measured using a biochemical readout of fluorescence intensity of resofurin that produced in viable cells (Fig. 1C). These dual readouts from the same well provide orthogonal cancer cell growth status that are designed to help triage potential false positives due to intrinsic artifacts of each method. For example, the artifacts from uneven cell distribution or clustering induced biased image acquisition, the shade-off and halo hindered automated phase-contrast image segmentation, and fluorescent compound interference may lead to false discovery for image-based high-content and fluorescence-based high-throughput screenings, respectively. The simplicity and dual readouts of the design enables the screening of large chemical libraries to identify compounds that stimulate or potentiate immune cells to attack cancer cells (Fig. 1D).
Figure 1. Development of phenotypic small molecule immunomodulator discovery platform.

(A) 384-well plate based immune- and cancer-cell co-culture system to model human cancer-immune microenvironment. (B) Image-based readout for monitoring cancer cell proliferation. (C) Biochemical-based readout for measuring cell viability using fluorescence signal of resofurin from viable cells. (D) principle for screening of small molecules with capability to induce immune cell-mediated killing of cancer cell. (E) PBMC-dose dependent killing curves for KRAS WT and G12V isogenic SW48 cell lines. The data are presented as mean ± SEM from four independent experiments. (F) AUC analysis of PBMC-dose dependent killing curves of 3 pairs of KRAS WT and G12V isogenic cell lines. Each dot represents one cell line and the line indicate the mean. **p<0.01, *p<0.05. See also Figure S1 and S2.
The HTiP platform captures the oncogenic KRAS-induced immune suppression phenotype
To examine whether the in vitro co-culture system can model the in vivo cancer immune surveillance mechanism, we first tested its capability to capture the oncogene induced immunosuppressive phenotypes. As a proof of principle, we selected cancer cell lines with an engineered KRAS G12V mutation to examine their growth phenotype under immune surveillance conditions using the in vitro co-culture system. Cancer cells carrying KRAS mutations have been reported to exert immunosuppression phenotype via various molecular mechanisms (Chen, et al., 2017; Kortlever, et al., 2017; Zdanov, et al., 2016).
Three pairs of isogenic cell lines, parental SW48, LIM1215 and NCI-H838 cells with KRAS WT and their corresponding genetically engineered KRAS G12V knock-in cells with defined and matched genetic background, were subjected to the test. Native non-labeled human PBMCs were added to reconstitute the immune component, and the growth phenotypes of cancer cells were monitored. With the increasing number of immune cells, a dose-dependent PBMC-induced growth inhibition of KRAS WT cells was observed, whereas the effect was significantly attenuated in cells with KRAS G12V (Fig. 1E-F), capturing the reported immunosuppressive phenotype of cancer cells with a KRAS mutation. In agreement with the image-based cell proliferation readout, the biochemical-based orthogonal cell viability measurement showed significantly more viable cells with G12V than those of the WT KRAS with the same PBMCs treatment (Fig. 1E-F). These results altogether indicate that the oncogenic KRAS mutation-induced immune escape phenotype could be recapitulated using both readouts.
HTiP screening reveals potential anti-tumor inducers
To examine the utility of the co-culture system to discover chemical immunomodulators, we collected and screened the Emory Enriched Bioactive Library (EEBL), a library of 2036 bioactive compounds including FDA-approved drugs with the potential for repurposing the existing drugs. For the primary screening, we utilized the SW48-G12V cell line and selected the EC90 condition with PBMC/cancer cell ratio of 5, which was based on our assessment of their assay window and robustness in both image-based cell growth measurement and biochemical-based cell viability readout (Fig. S1).
Using this established condition for SW48-G12V, we performed primary parallel screenings in the presence or absence of PBMCs to assess the immune-mediated anti-cancer effect of small molecule compounds. The parallel screenings exhibited robust performance with S/B > 10 and Z’ values > 0.5 from both readouts (Fig. S1). The immune killing selectivity index, which was defined as the ratio of percentage inhibition of cancer cell growth with immune cells to that without immune cells, was established for each compound to prioritize potential hits based on the potency of immune cell-dependent inhibition of cancer cell growth (Fig. 2A). Based on the ranking of “immune killing selectivity” index of > 1.5-fold and the inhibitory effect of < 20% on SW48-G12V cancer cells in the absence of PBMC from both readouts, top ranked ten hit compounds with double positive readouts were prioritized for dose-response studies (Fig. 2B).
Figure 2. Identification of IAP inhibitors as potential small molecule immunomodulators.

(A) Selectivity of 2036 compounds in immune cell-dependent killing of SW48-G12V cells. (B) Venn diagram of top ranked compounds identified from image-based cell proliferation (in black) and biochemical-based cell viability (in red) readouts. (C) Chemical structure of three small molecule IAP inhibitors. (D-F) Dose-dependent curves of birinapant (D), BV-6 (E) and GDC0152 (F) in inhibiting SW48-G12V cells growth with or without PBMC. (G) AUC analysis of six IAP inhibitors-dose dependent growth and viability inhibition curves. (H-I) PBMC-dose dependent killing curves for KRAS WT and G12V isogenic SW48 cell lines with or without birinapant (1 nM) from image-based cell proliferation (H) and biochemical-based cell viability (I) readouts. The data in D-I are presented as mean ± SEM from four independent experiments. *p<0.05. (J) AUC analysis of birinapant-induced PBMC-dependent killing curves of six patient-derived colorectal cancer cell lines from cell viability readout. WT: Caco-2, LIM1215 and SW48; G12V: SW403, SW480 and SW620. Each dot represents one cell line and the data are presented as mean ± SD. *p<0.05. (K) Time dependent SW48-G12V cell proliferation curves and (L) Time lapse images of SW48-G12V cell growth at the conditions as indicated. The data are presented as mean ± SEM from four replicates from a representative experiment. See also Figure S1, S2 and S3.
Through the dose-response confirmatory assay with cherry-picked and newly ordered compounds, three out of ten primary hits were validated with significant and reproducible effect in inducing immune cell-dependent inhibition of cell growth and viability (Fig. 2C-F). The other seven primary hits were triaged due to their low potency or lack of consistent effect (data not shown).
Multiple small molecule anticancer immune enhancers target IAP proteins
Among positive hit compounds, birinapant (Fig. 2C), a SMAC mimetic, small molecule antagonist of IAP (Inhibitor of Apoptosis Protein) family proteins, showed high “immune killing selectivity” index of >3 from the primary screening. In the dose-response study, birinapant exhibited potent effect on immune cell-dependent inhibition of cell growth and viability (Fig. 2D) with desired selectivity. Birinapant showed little effect on the proliferation of SW48-G12V cells in the absence of PBMC while exhibited highly potent inhibitory effect on these cells in the presence of PBMC with IC50 of 0.7±1.2 nM (Fig. 2D). In agreement with the image-based readout, the biochemical-based cell viability readout recaptured the specific immune cell-dependent inhibition induced by birinapant with IC50 of 0.5±0.2 nM. Interestingly, two structurally distinct positive hits identified from the screening, BV-6 and GDC0152, also belong to the class of IAP antagonists (Fig. 2C). Both BV-6 and GDC-0152 were confirmed in dose-response experiments, showing potent effects and selectivity in suppression of SW48-G12V cells with IC50 in low nanomolar range (IC50 of 2.1 ±1.3 and 7.0±1.3 nM for BV-6 and GDC-0152, respectively, in image-based assay; IC50 of 0.7±1.1, and 2.5±1.3 nM for BV-6 and GDC-0152, respectively, in viability assay) (Fig. 2E-F). These data support a general immune modulation function of these IAP antagonists, demonstrating the utility of our in vitro co-culture system in high-throughput discovery of small molecule immunomodulators.
To further support the specific impact of targeting IAP for immunomodulation, we purchased three additional IAP inhibitors, AT406, AZD5582 and LCL161, and tested their effect using HTiP platform. In support of this notion, we found that these three IAP inhibitors induced immune cell-dependent selective killing of cancer cells with potency in the nanomolar range (Fig. 2D-G and Fig. S2). In contrast, embelin, a promiscuous compound with IAP inhibitory effect at micromolar range (Nikolovska-Coleska, et al., 2004), failed to induce cancer cell death regardless of the presence of immune cells at 2 μM in the primary HTiP screening. Such convergent immunoregulatory effect from structurally divergent IAP inhibitors further reinforces the function of IAP in shaping the anti-tumor immunity.
To evaluate the immune cell-dependency of cancer cell selectivity for the identified IAP antagonists, we first used the isogenic WT and G12V SW48 cell lines to test their response to varying levels of immune cells upon compound treatment. Birinapant was used due to its potent immunomodulatory effect (Fig. 2D). The PBMC dose-dependent cell growth inhibition curves were profiled in the presence of birinapant or with DMSO. As shown in Fig. 1E-F, SW48-G12V showed reduced sensitivity to PBMC as compared to SW48 WT. Birinapant treatment reversed such a resistance phenotype, showing significantly enhanced PBMC-dependent growth inhibition of KRAS G12V mutation cells comparing to DMSO control (Fig. 2H-I). In contrast, this compound failed to show enhanced immune cell killing of KRAS WT cells, exhibiting its cancer cell selectivity. In addition to the human engineered exogenous KRAS isogenic cell line model, patient-derived cancer cell lines with differential KRAS mutation status were tested for their sensitivity to birinapant in the HTiP assay (Fig. S3). As shown in Fig. 2J, human colorectal carcinoma cell lines carrying oncogenic KRAS mutations exhibited increased sensitivity to birinapant-triggered immune-dependent killing compared to those cells without KRAS mutations. Birinapant appears to synergize with immune cells to suppress the growth of KRAS mutation-carrying cells.
To visualize the compound-induced immune cell-dependent inhibition of cancer cell growth over time, time-lapse videos of SW48-G12V cell growth were recorded in the presence of birinapant. Phase contrast images revealed that cancer cells exhibit regular polygonal epithelial-like shape with comparable proliferation rates among control groups with PBMC or birinapant alone, or that under untreated cell culture conditions (Fig. 2K-L; Supplementary Video). On the contrary, cancer cells underwent morphological change with rounding, shrinkage and loss of adherent phenotypes upon attack by immune cells in the presence of birinapant within the initial 30 hours, and thereafter no significant cell proliferation was observed (Fig. 2K-L; Supplementary Video). These results demonstrate the effect of IAP inhibitor-induced immune cell-dependent killing of cancer cells, and support the potential action of birinapant on immune cells to enhance their anti-cancer activity.
T and natural killer (NK) cells are involved in birinapant-induced immune-dependent killing effect
To determine the potential mechanism underlying the immunomodulatory effect of IAP inhibitors, we began to deconvolute the effective immune cell subtypes that might be involved in the birinapant-induced immune-dependent inhibition of cancer cell growth. Since IL-2 and anti-CD3 antibody was used in HTiP assay as generic immune activator (Tsoukas, et al., 1985), we first tested the immune selective killing effect induced by birinapant in the absence of these activators. Under the condition which immune cells were left unstimulated, birinapant exhibited significantly reduced effect in inducing immune-dependent killing of cancer cells comparing to the condition with IL-2 and anti-CD3 stimulation (Fig. 3A). Given the well documented role of IL-2 and anti-CD3 in activating T cells and NK cells (Bryceson, et al., 2006; Smith-Garvin, et al., 2009) and the potential role of IAPs in modulating T and NK cell activity (Dougan, et al., 2010; Fischer, et al., 2017), it is possible that T and NK cells might contribute to the IAP inhibitor-mediated anti-tumor immunity. Thus, we next examined the requirement of immune cell subtypes from the PBMC for mediating birinapant-induced immunomodulation. The potency of birinapant was evaluated in the HTiP co-culture assay using the PBMCs with certain immune cell subtype depleted by magnetic beads coated with corresponding anti-CD marker antibody. The depletion of CD3+ T cells and CD56+ NK cells, but not CD19+ B cells, significantly increased the IC50 of birinapant comparing to the control with intact PBMCs (Fig. 3B). These results demonstrate that T and NK cells are required for the birinapant-induced effect in the immune-cancer cell interaction system.
Figure 3. Identification of effective immune subtypes and cytokines involved in birinapant induced anti-tumor immunity.

(A) Bar graph showing the IC50 values calculated from birinapant dose-response inhibition curve with (+) or without (−) immune activation using IL2 and anti-CD3 antibody, and (B) with PBMC or depletion of immune subtypes as indicated. (C) Dose- and time-dependent curves of TNFα production upon birinapant stimulation. (D) Bar graph showing the IC50 values calculated from birinapant dose-response inhibition curves with TNFα neutralization using anti-TNFα antibody. (E) Dose- and time-dependent curves of IFNγ production upon birinapant stimulation. (F) Bar graph showing the IC50 values calculated from birinapant dose-response inhibition curves under conditions as indicated. The data are presented as mean ± SD from three independent experiments. *p<0.05, **p<0.01, ***p<0.001.
TNFα and IFNγ contribute to the birinapant-induced PBMC-dependent killing effect
Various IAP antagonists (SMAC mimetics) have been shown to produce synergistic effect with Tumor Necrosis Factor alpha (TNFα) in inducing cancer cell death (Petersen, et al., 2007; Vince, et al., 2007). Since we have demonstrated that the birinapant-induced immunomodulatory effect are partially mediated through the T and NK cells, and given that these cells are able to secret TNFα (Dinarello, 2007), we next tested whether TNFα is one of the effective cytokines in this scenario. First, we measured the TNFα secretion from the immune- and cancer-cell co-culture system. Upon the birinapant treatment, we observed a dose- and time-dependent increase of TNFα concentration in the conditioned medium from the co-cultures (Fig. 3C). In addition, depletion of TNFα using 50 ng/ml of TNFα neutralizing antibody significantly attenuated the birinapant-induced PBMC-dependent inhibition of cancer cell growth (Fig. 3D). These results suggested that birinapant-induced TNFα secretion contribute to the observed immune-dependent killing of cancer cells.
Besides TNFα, another important immune cytokine that can be produced from T and NK cells is Type II Interferon (IFNγ)(Dinarello, 2007), which has also been demonstrated to cooperate with IAP antagonists in activating cell necroptosis (Cekay, et al., 2017; Tanzer, et al., 2017). Similarly, we first detected a dose- and time-dependent increase of IFNγ secretion from the co-cultures (Fig. 3E). In support of this notion, inhibition of IFNγ signaling pathway by using 5 μg/ml IFNγ neutralizing antibody significantly decreased the birinapant-induced immune cell killing effect (Fig. 3F). These results suggest the importance of IFNγ in the birinapant-induced anti-tumor immune response. In addition, depletion of both TNFα and IFNγ together further attenuate the immunomolulatory effect of birinapant (Fig. 3F). Collectively, our results suggested that TNFα and IFNγ are two of the effective cytokines that are important in mediating immune-dependent cancer cell death induced by IAP antagonist.
DISCUSSION
Vast chemical space has been extensively explored to discover inhibitors of cancer cells using a variety of molecular, cellular, or phenotypic assays with tumor population alone. It has been challenging to screen large chemical libraries using clinically relevant tumor models, particularly those with the complexity of cancer and immune system interactions. Our in vitro cancer and immune cell co-culture system provides a simple and scalable platform that enables its application for systematic profiling of large chemical libraries using immune cell-dependent inhibition of cancer cell growth as a phenotypic readout. The use of non-labeled native cancer and immune cells with the imaging system avoids artificial tagging-associated artifacts. The design of a built-in orthogonal detection step with the biochemical-based measurement from the same well is expected to enhance the screening efficiency and quality for hit prioritization. Promising results from the current HTiP platform that utilizes PBMC as a source of immune cells supports further development that will incorporate defined subtypes and engineered tumor antigen-specific T cells for directed screening campaigns.
Using the HTiP platform, we tested 2036 bioactive compounds and identified three IAP antagonists as potential immunomodulators that selectively enhance the immune-cell dependent inhibition of KRAS mutation cells. Our screening strongly supports that our co-culture HTiP platform can be applied to identify novel small molecule immunomodulators. These IAP antagonists could serve as the first-in-class immunomodulators identified from the HTS setting and as benchmark compounds for future small molecule immunomodulator campaign. Meanwhile, the streamlined assay platform could be rapidly adapted and extended to a broad range of epithelial cancer cells with a wide variety of specific immune cell subtypes, such as “exhausted” T cells, CD8+ cytotoxic T lymphocytes, or NK cells, or other cytotoxic organisms, such as bacteria or viruses.
In addition to IAP antagonists, it is possible that other hits might be identified if we relaxed the selection criteria. Indeed, ten different primary hits were revealed with selectivity index > 1.5 but with the inhibitory effect > 20% on cancer cell alone. Among these potential hits, one compound, batimastat, a matrix metalloproteinase inhibitor (Botos, et al., 1996), showed immune-selectivity in the dose-response study (Fig. S2). This compound requires further examination with additional orthogonal assays for validation.
Because the chemical library employed contains compounds with known activities, it may offer opportunities to evaluate performance of the screening, i.e., to assess the capture rate of known immunomodulatory compounds. However, it is challenging to define compounds with tumor-directed immunostimulatory activities among those known compounds at this stage due to limited knowledge in this area. Our efforts through chemoinformatics analysis did reveal (i) a known immunostimulant, and (ii) compounds that target proteins that are involved in immunomodulation. Pidotimod is a synthetic dipeptide with immunomodulatory properties and demonstrated role in both innate and adaptive immunity (Ferrario, et al., 2015). However, pidotimod showed <20% cancer cell-killing effect in our primary screening. It needs additional experimental studies to understand whether pidotimod has tumor-directed immunogenic cell death activity. To expand the definition of the expected hits, we included compounds that are primarily not classified as immunomodulators, but act on protein targets implicated in regulating immune response. The IAP family of proteins belong to this class with recently demonstrated roles in regulating both innate and adaptive immunity, in addition to its established apoptosis inhibition function(Dougan and Dougan, 2018). In this case, IAP inhibitors could be the expected hits and indeed were identified from the HTiP platform. Other protein targets, such as PI3K and mTOR, have been implicated in regulating immune cell fate (Koyasu, 2003; Powell, et al., 2012), their corresponding compounds were not identified as hits. It should be noted that PI3K and mTOR have a broad range of biological activities from the regulation of cell survival to protein translation. Thus, it is possible that the HTiP platform may allow the identification of compounds that target pathways selective for specific oncogenic lesions.
IAP antagonists, also known as second mitochondrial-derived activator of caspase (SMAC) mimetics, have been extensively studied as potential anti-cancer therapeutics in the last decade (Cheung, et al., 2009; Lu, et al., 2008; Petersen, et al., 2007; Vince, et al., 2007). However, the dramatic differences in their activities between in vitro 2-D cell culture system and in vivo human and murine cancer models when used as a single agent remain puzzling(Cheung, et al., 2010; Dineen, et al., 2010; Krepler, et al., 2013; Lecis, et al., 2013; Probst, et al., 2010). The use of IAP antagonists have indicated the importance of IAP in modulating the TNFα/NFκB-mediated inflammatory responses (Vince, et al., 2007). Moreover, the significant synergistic effects between IAP inhibitors and inflammatory cytokines, such as TNFα, IL-1β or IFNγ, in inhibiting cancer cell growth have implicated a potential combination strategy of using these compounds and immune stimuli to promote tumor death (Cekay, et al., 2017; Cheung, et al., 2010; Dineen, et al., 2010; Dougan, et al., 2010; Krepler, et al., 2013; Lecis, et al., 2013; Probst, et al., 2010; Vince, et al., 2007). In addition, several IAP inhibitors have recently been shown to target the immune system to evoke anti-tumor immunity in various cancer types (Beug, et al., 2017; Chesi, et al., 2016; Dougan, et al., 2010). Our unbiased screening independently revealed three structurally distinct IAP inhibitors as potent inducers of anti-tumor immunity from our in vitro co-culture system, in support of the reported role of IAPs in suppression of antitumor immune response pathways (Dougan, et al., 2010; Estornes and Bertrand, 2015; Sharma, et al., 2017). Unraveling of three IAP antagonists/SMAC mimetics from our HTiP screening illustrates the power of the HTiP platform in recapitulating the complex in vivo immune-cancer microenvironment for the discovery of small molecule antitumor immunity enhancers. The immune cell dependency for the cancer cell killing effect of these IAP antagonists strongly supports their action on immune cell population to reverse immune-resistance of cancer cells.
Significance
We have developed a HTiP platform that employs the in vitro immune- and cancer-cell co-culture system using an image-based phenotypic readout and a built-in orthogonal viability readout to monitor cancer cell growth status for accelerated discovery of small molecule immunomodulators. The identification of multiple IAP antagonists/SMAC mimetics, with demonstrated roles as antitumor immunity enhancers, supports the validity of the HTiP system for the accelerated discovery of small molecule anticancer immune enhancing compounds. This platform could be readily adapted to a wide range of immune and cancer cell types with various genetic background for the identification of small molecule immunomodulators or studying immune surveillance mechanisms towards personalized immunotherapy.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Haian Fu (hfu@emory.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All cells were grown at 37°C and 5% CO2. The X-MAN® KRAS isogenic cell lines, including two human colorectal adenocarcinoma cells SW48 (female) and LIM1215 (male) and one human lung adenocarcinoma cell NCI-H838 (male) and their corresponding genomic engineered counterparts with the heterozygous knockin of KRAS G12V activating mutation, were from Horizon Discovery (Cambridge, UK) and maintained according to manufacturer’s protocol. Four additional patient-derived human colorectal adenocarcinoma cell lines, including Caco-2 (male), SW403 (female), SW480 (male) and SW620 (male), were purchased from the American Type Culture Collection (ATCC). The authenticity of these cell lines was verified by western blot using KRAS-G12V specific antibody (Cell Signaling #14412S) (Figure S3). The primary Peripheral Blood Mononuclear Cells (PBMC) from healthy donor (ATCC PCS-800-011™) were recovered from liquid nitrogen and were used immediately for co-culturing with cancer cells in T cell medium, which is Roswell Park Memorial Institute (RPMI) 1640 medium containing L-glutamine (CORNING Cat# 10-040) supplemented with 10% fetal bovine serum and 100 units/ml of penicillin/streptomycin.
METHOD DETAILS
Immune and tumor cell co-culture assay
PBMCs as effector immune cells and KRAS isogenic cancer cells as target cells were used for co-culture assays. Tumor cells were seeded at specific density in 384-well cell culture plate (Corning #3764). Twenty-four hours later, PBMCs were then thawed and co-cultured in T-cell medium with tumor cells in a dose dependent manner for four days. CD3 monoclonal antibody (100 ng/mL, OKT3, ThermoFisher) and human recombinant interleukin-2 (10 ng/mL, PeproTech) were used as activation cocktail to activate immune cells.
Cell proliferation measurement
The co-culture assay plates were imaged using the IncuCyte® S3 Live-Cell Analysis System (Essen Biosciences). The cancer cell proliferation was monitored and characterized as the percentage of confluence using the IncuCyte® basic analysis module. Because of the size distinction between effector immune cells and target cancer cells, the area filter of >400 μm2 was used to select cancer cells that are larger in size.
Cell viability measurement
Cell Titer Blue (Promega, G8081) was added to each well. The plates were incubated for desired time at 37 °C to allow the generation of sufficient signal within the linear range. The fluorescence intensity of each well was read using an PHERAstar FSX multi-mode plate reader (Ex 545 nm, Em 615 nm; BMG LABTECH). Cells containing medium or immune cells alone were used as blank control for background correction.
The Emory Enriched Bioactive Library and other chemicals
The Emory Enriched Bioactive Library (EEBL) consists of a collection of 2036 diverse small molecules with validated biological and pharmacological activities, including 1018 FDA approved compounds (Selleckchem). These molecules are focused on over two hundred targets that are part of more than twenty signaling pathways, including survival and apoptosis pathways. The selected primary hits were re-ordered from Selleckchem for validation studies. Additional IAP inhibitors, AT406, AZD5582 and LCL161, were purchased from Selleckchem.
HTiP method for small molecule immunomodulator discovery
For the primary screening, SW48-G12V cells were seeded in 384-well cell culture plate (1,000 cells/well in 40 μl medium; Corning, Cat#3764) using a Multidrop Combi Reagent Dispenser (ThermoScientific). The last column was used as a medium or PBMCs only control (blk). The next day, PBMCs were dispensed at 5,000 cells/well in 10 μl media containing the activation cocktail. Subsequently, the 2036 Emory Enriched Bioactive Library (EEBL) compounds (100 nl) were added into wells in each plate using Biomek NXP Automated Workstation (Beckman) from a compound stock plate to give the final concentration of 2 μM. The final DMSO concentration was 0.2% (v/v) in samples with compound treatment. Each sample was tested with single point. A parallel screening was performed with SW48-G12V cells alone (1000 cells/well) in 50 μl medium containing the same amount of activation cocktail in the absence of PBMCs. After 4 days of incubation, image-based cell proliferation readouts followed by biochemical-based cell viability measurements were used to examine the compound effect on cancer cell growth. Z’ factor was calculated as
Where SDpositive and SDblank are the standard deviations, and Spositive and Sblank are the corresponding average of the cell confluence or fluorescence intensity for wells with DMSO containing PBMCs/medium only or plus cancer cells, respectively. A Z’ factor between 0.5 and 1.0 indicates that the assay is suitable for HTS(Zhang, et al., 1999). The percentage of control (%C) was calculated using the equation 100X(Scompound-Sblank)/(Spositive-Sblank). The immune killing selectivity index was calculated using the equation log10(%C−PBMC/%C+PBMC).
Time-lapse video for cell proliferation kinetics
SW48-G12V cells were plated in 96-well IncuCyte® ImageLock plates (Essen Biosciences, #4379) at 6,000 cells per wells. Twenty-four hours later, cells were treated with various conditions as indicated and imaged with time-lapse setting using the IncuCyte® S3 Live-Cell Analysis System (Essen Biosciences). PBMCs were added at 30,000 cells/well containing the activation cocktail. Birinapant with final concentration of 5 nM was used with the final DMSO concentration of 0.1% (v/v). The time-lapse video was composed by displaying images at 6 frames per second using Final Cut Pro X software.
Immune cell subtype and cytokine depletion assays
PBMCs depleted with CD3+ T cells, CD56+ NK cells, or CD19+ B cells were obtained by incubating the primary PBMCs with magnetic beads coated with anti-CD3, CD56 or CD19 antibody (ThermoFisher Scientific), respectively, according to the manufacturer’s instruction. The human TNFα neutralization mouse monoclonal antibody was purchased from R&D Systems (#MAB210-100). The human IFNγ neutralization mouse monoclonal antibody and mouse IgG1 isotype control were purchased from ThermoFisher. For Enzyme-linked immunosorbent assay (ELISA) assay, the conditioned medium from the co-culture assay upon the treatment with compound or controls was collected and centrifuged to remove any cells or debris. Then the supernatant was analyzed for the TNFα and IFNγ amount using the TNF alpha and IFN gamma Human ELISA Kit (ThermoFisher Scientific), respectively, according to manufacturer's protocol.
QUATIFICATION AND STATISTICAL ANALYSIS
The dose-dependent PBMC-induced or small molecule induced cancer cell growth inhibition curve was established using GraphPad Prism based on the Sigmoidal dose-response (variable slope) equation. For quantitative analysis of the dose response, the area under the curve (AUC) was computed as a measurement of cancer growth signal, as AUC integrates both the amplitude and shape of the growth curve. The student’s t-test was used for statistics and P-value of 0.05 or less were considered statistically significant.
Supplementary Material
Supplementary Video. Time-lapse videos of SW48-G12V cell proliferation, related to Figure. 2. SW48-G12V cell growth at the conditions with DMSO (A and C) or birinapant (B and D) in the absence (A and B) or the presence of PBMC (C and D). Videos were acquired with time-lapse imaging interval of 30 minutes and displayed at 6 frames per second.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-β-Actin antibody | Sigma-Aldrich | Cat# A2228 |
| Rabbit polyclonal anti-KRAS antibody | Proteintech | Cat# 12063-1-AP |
| Rabbit monoclonal anti-KRAS (G12V specific) antibody | Cell signaling | Cat# 14412S |
| Human TNFα neutralization mouse monoclonal antibody | R&D Systems | Cat# MAB201-100 |
| Human IFNγ neutralization mouse monoclonal antibody | Thermo Fisher | Cat# 14-7318-81 |
| Mouse IgG1 isotype control | Thermo Fisher | Cat# 02-6100 |
| CD3 monoclonal antibody (OKT3) | Thermo Fisher | Cat# 16-0037-81 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human recombinant IL-2 | PeproTech | Cat# 200-02 |
| Birinapant | Selleckchem | Cat# S7015 |
| BV-6 | Selleckchem | Cat# S7597 |
| GDC0152 | Selleckchem | Cat# S7010 |
| AZD5582 | Selleckchem | Cat# S7362 |
| LCL161 | Selleckchem | Cat# S7009 |
| AT406 | Selleckchem | Cat# S2754 |
| EEBL | Emory ECBDC | N/A |
| Batimastat | Selleckchem | Cat# S7155 |
| Critical Commercial Assays | ||
| CellTiter-Blue® Cell Viability Assay | Promega | Cat# G8081 |
| TNFα Human ELISA Kit | Thermo Fisher | Cat# KHC3011 |
| IFNγ Human ELISA Kit | Thermo Fisher | Cat# EHIFNG |
| Experimental Models: Cell Lines | ||
| Human KRAS (G12V/+) SW48 Cell Lines | Horizon Discovery | Cat# HD 103-007 |
| Human KRAS (G12V/+) LIM1215 Cell Lines | Horizon Discovery | Cat# HD 116-006 |
| Human KRAS (G12V/+9n) NCI-H838 Cell Line | Horizon Discovery | Cat# HD 114-003 |
| Caco-2 | ATCC | Cat# HTB-37 |
| SW403 | ATCC | Cat# CCL-230 |
| SW480 | ATCC | Cat# CCL-228 |
| SW620 | ATCC | Cat# CCL-227 |
| Peripheral Blood Mononuclear Cells, Human, Normal | ATCC | Cat# PCS-800-011 |
| Software and Algorithms | ||
| Graphpad Prism | Graphpad; v7 | https://www.graphpad.com/scientific-software/prism/ |
| IncuCyte S3 software | Essen BioScience; S3 | https://www.essenbioscience.com/en/products/software/incucyte-s3-software-v2018b/ |
| Final Cut Pro X | Apple | https://www.apple.com/final-cut-pro/ |
| Other | ||
| Dynabeads™ CD3 | Thermo Fisher | Cat# 11151D |
| MagniSort™ Human CD56 Positive Selection Kit | Thermo Fisher | Cat# 8802-6835-74 |
| Dynabeads™ CD19 Pan B | Thermo Fisher | Cat# 11143D |
Highlights.
HTiP is a miniaturized co-culture system with cancer and immune cells
HTiP integrates imaging- with biochemical-based readouts for built-in counterscreening
HTiP models oncogenic KRAS mutation-induced immunosuppression
HTiP screening identifies IAP inhibitors as anti-tumor immunity enhancers
Acknowledgements
We would like to thank Dr. Andrey A. Ivanov (Department of Pharmacology and Emory Chemical Biology Discovery Center) for chemoinformatic analysis of EEBL annotation. This research was supported in part by NIH NCI U01CA217875, NIH NCI 5U01CA199241 and a Georgia Cancer Coalition Award from Georgia Research Alliance, the Emory Chemical Biology Discovery Center, and Winship Cancer Institute (NIH 5P30CA138292).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Beug ST, Beauregard CE, Healy C, Sanda T, St-Jean M, Chabot J, Walker DE, Mohan A, Earl N, Lun X, et al. (2017). Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat Commun 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botos I, Scapozza L, Zhang D, Liotta LA, and Meyer EF (1996). Batimastat, a potent matrix mealloproteinase inhibitor, exhibits an unexpected mode of binding. Proc. Natl. Acad. Sci. U. S. A. 93, 2749–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryceson YT, March ME, Ljunggren HG, and Long EO (2006). Activation, coactivation, and costimulation of resting human natural killer cells. Immunol. Rev. 214, 73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekay MJ, Roesler S, Frank T, Knuth AK, Eckhardt I, and Fulda S (2017). Smac mimetics and type II interferon synergistically induce necroptosis in various cancer cell lines. Cancer Lett. 410, 228–237. [DOI] [PubMed] [Google Scholar]
- Chames P, Van Regenmortel M, Weiss E, and Baty D (2009). Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 157, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Fang W, Lin Z, Peng P, Wang J, Zhan J, Hong S, Huang J, Liu L, Sheng J, et al. (2017). KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 66, 1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, Akhmetzyanova I, Kosiorek HE, Calcinotto A, Riggs DL, et al. (2016). IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat. Med. 22, 1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HH, Beug ST, St Jean M, Brewster A, Kelly NL, Wang S, and Korneluk RG (2010). Smac mimetic compounds potentiate interleukin-1beta-mediated cell death. J. Biol. Chem. 285, 40612–40623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HH, Mahoney DJ, Lacasse EC, and Korneluk RG (2009). Down-regulation of c-FLIP Enhances death of cancer cells by smac mimetic compound. Cancer Res. 69, 7729–7738. [DOI] [PubMed] [Google Scholar]
- Dhanak D, Edwards JP, Nguyen A, and Tummino PJ (2017). Small-Molecule Targets in Immuno-Oncology. Cell Chem Biol 24, 1148–1160. [DOI] [PubMed] [Google Scholar]
- Dinarello CA (2007). Historical insights into cytokines. Eur. J. Immunol. 37 Suppl 1, S34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineen SP, Roland CL, Greer R, Carbon JG, Toombs JE, Gupta P, Bardeesy N, Sun H, Williams N, Minna JD, et al. (2010). Smac mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer Res. 70, 2852–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan M, Dougan S, Slisz J, Firestone B, Vanneman M, Draganov D, Goyal G, Li W, Neuberg D, Blumberg R, et al. (2010). IAP inhibitors enhance costimulation to promote tumor immunity. J. Exp. Med. 207, 2195–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan SK, and Dougan M (2018). Regulation of innate and adaptive antitumor immunity by IAP antagonists. Immunotherapy 10, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estornes Y, and Bertrand M (2015). IAPs, regulators of innate immunity and inflammation. Semin. Cell Dev. Biol. 39, 106–114. [DOI] [PubMed] [Google Scholar]
- Ferrario BE, Garuti S, Braido F, and Canonica GW (2015). Pidotimod: the state of art. Clin. Mol. Allergy 13, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesnak AD, June CH, and Levine BL (2016). Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 16, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer K, Tognarelli S, Roesler S, Boedicker C, Schubert R, Steinle A, Klingebiel T, Bader P, Fulda S, and Ullrich E (2017). The Smac Mimetic BV6 Improves NK Cell-Mediated Killing of Rhabdomyosarcoma Cells by Simultaneously Targeting Tumor and Effector Cells. Front. Immunol. 8, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley P, Sutton DH, Debnam P, Matthews IR, Brewer JE, Rose J, Trickett M, Williams DD, Andersen TB, and Classon BJ (2004). High-affinity small molecule inhibitors of T cell costimulation: compounds for immunotherapy. Chem. Biol. 11, 1651–1658. [DOI] [PubMed] [Google Scholar]
- Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, Littlewood TD, and Evan GI (2017). Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 171, 1301–1315 e1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyasu S (2003). The role of PI3K in immune cells. Nat. Immunol. 4, 313–319. [DOI] [PubMed] [Google Scholar]
- Krepler C, Chunduru SK, Halloran MB, He X, Xiao M, Vultur A, Villanueva J, Mitsuuchi Y, Neiman EM, Benetatos C, et al. (2013). The novel SMAC mimetic birinapant exhibits potent activity against human melanoma cells. Clin. Cancer Res. 19, 1784–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecis D, De Cesare M, Perego P, Conti A, Corna E, Drago C, Seneci P, Walczak H, Colombo MP, Delia D, et al. (2013). Smac mimetics induce inflammation and necrotic tumour cell death by modulating macrophage activity. Cell Death Dis. 4, e920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu S, Miller RS, Yi H, Shangary S, Sun Y, et al. (2008). SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res. 68, 9384–9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolovska-Coleska Z, Xu L, Hu Z, Tomita Y, Li P, Roller PP, Wang R, Fang X, Guo R, Zhang M, et al. (2004). Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three-dimensional structure database. J. Med. Chem. 47, 2430–2440. [DOI] [PubMed] [Google Scholar]
- Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, Harran P, and Wang X (2007). Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 12, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow M, Callahan M, and Wolchok J (2015). Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 33, 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JD, Pollizzi KN, Heikamp EB, and Horton MR (2012). Regulation of immune responses by mTOR. Annu. Rev. Immunol. 30, 39–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst BL, Liu L, Ramesh V, Li L, Sun H, Minna JD, and Wang L (2010). Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-alpha-dependent manner. Cell Death Differ. 17, 1645–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Riviere I, and Riddell S (2017). Therapeutic T cell engineering. Nature 545, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kaufmann T, and Biswas S (2017). Impact of inhibitor of apoptosis proteins on immune modulation and inflammation. Immunol. Cell Biol. 95, 236–243. [DOI] [PubMed] [Google Scholar]
- Skalniak L, Zak KM, Guzik K, Magiera K, Musielak B, Pachota M, Szelazek B, Kocik J, Grudnik P, Tomala M, et al. (2017). Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 8, 72167–72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, and Jordan MS (2009). T cell activation. Annu. Rev. Immunol. 27, 591–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzer MC, Khan N, Rickard JA, Etemadi N, Lalaoui N, Spall SK, Hildebrand JM, Segal D, Miasari M, Chau D, et al. (2017). Combination of IAP antagonist and IFNgamma activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 24, 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoukas CD, Landgraf B, Bentin J, Valentine M, Lotz M, Vaughan JH, and Carson DA (1985). Activation of resting T lymphocytes by anti-CD3 (T3) antibodies in the absence of monocytes. J. Immunol. 135, 1719–1723. [PubMed] [Google Scholar]
- Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, et al. (2007). IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 131, 682–693. [DOI] [PubMed] [Google Scholar]
- Zdanov S, Mandapathil M, Abu Eid R, Adamson-Fadeyi S, Wilson W, Qian J, Carnie A, Tarasova N, Mkrtichyan M, Berzofsky JA, et al. (2016). Mutant KRAS conversion of conventional T cells into regulatory T Cells. Cancer Immunol Res 4, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, and Oldenburg KR (1999). A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 4, 67–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video. Time-lapse videos of SW48-G12V cell proliferation, related to Figure. 2. SW48-G12V cell growth at the conditions with DMSO (A and C) or birinapant (B and D) in the absence (A and B) or the presence of PBMC (C and D). Videos were acquired with time-lapse imaging interval of 30 minutes and displayed at 6 frames per second.