

Abstract
Oculocerebrocutaneous syndrome (OCCS) is a rare disorder characterized primarily by congenital skin, eye and brain anomalies. The most distinctive findings are hypoplastic or aplastic skin defects; pedunculated, typically hamartomatous, or nodular skin appendages; cystic microphthalmia; and a combination of forebrain anomalies and a specific mid-hindbrain malformation. Based on a review of 40 patients with OCCS, existing clinical criteria have been revised. Because of the asymmetric and patchy distribution of features, lack of recurrence in families, male preponderance and completely skewed X-inactivation in one female, OCCS is hypothesized to result from postzygotic mosaic variants in an X-linked gene. Whole exome and genome sequencing on blood DNA in two patients failed to identify pathogenic variants so far. In view of the overlapping features, in particular of the brain, of OCCS and Aicardi syndrome, both may be pathogenetically related or even result from different variants in the same gene. For the elucidation of the cause of OCCS, exome or genome sequencing on multiple lesional tissues is the primary goal.
Keywords: Delleman-Oorthuys syndrome, oculocerebrocutaneous syndrome, OCCS, cystic microphthalmia, orbital cyst, polymicrogyria, giant tectum absent vermis
1. INTRODUCTION
Oculocerebrocutaneous syndrome (OCCS), also known as Delleman-Oorthuys syndrome, is characterized by a triad of congenital eye, brain and skin malformations (OMIM 164180). The first reports of OCCS appeared in 1981-84, although a few older and less complete reports had been published earlier under other headings [Delleman and Oorthuys, 1981; Delleman et al., 1984; Dollfus et al., 1968; Ladenheim and Metrick, 1956; Renard et al., 1964]. Considering only patients with sufficient information to establish a reasonable diagnosis of OCCS, at least 40 patients with OCCS are known to date, including 35 previously published (Supporting Information Tables S1–S2 and References). OCCS thus belongs to the group of ultra-rare syndromes. In its complete form the phenotypic hallmarks are obvious and unlikely to be missed, although incomplete forms could be more common.
2. CLINICAL FEATURES
2.1. Skin Features
The most common skin features consist of focal hypoplastic or aplastic skin defects (aplasia cutis congenita) and unusual skin appendages (Supporting Information Table S1). The hypo- or aplastic defects can be small (several mm) and punch-like or large (several cm, Figure 1A-C). They are located primarily on the face or neck, or on the scalp within areas of alopecia, and rarely extend to the trunk. No particular pattern has been defined. A postauricular crescent-shaped lesion (Fig. 1C) has been observed in 13 of 40 patients and may be unique to this syndrome [Clericuzio, 1989]. Linear skin defects have been seen in only a few patients. Most skin appendages are located in the face, especially around the orbit, and only rarely extend to the trunk. The most characteristic appendages are pedunculated, and may be finger-like and able to move (Figure 1D-E-F). These have been histologically classified as striated muscle hamartomas (SMH). Other appendages are small and nodular, often occurring in small clusters (Figure 1G).
Figure 1.
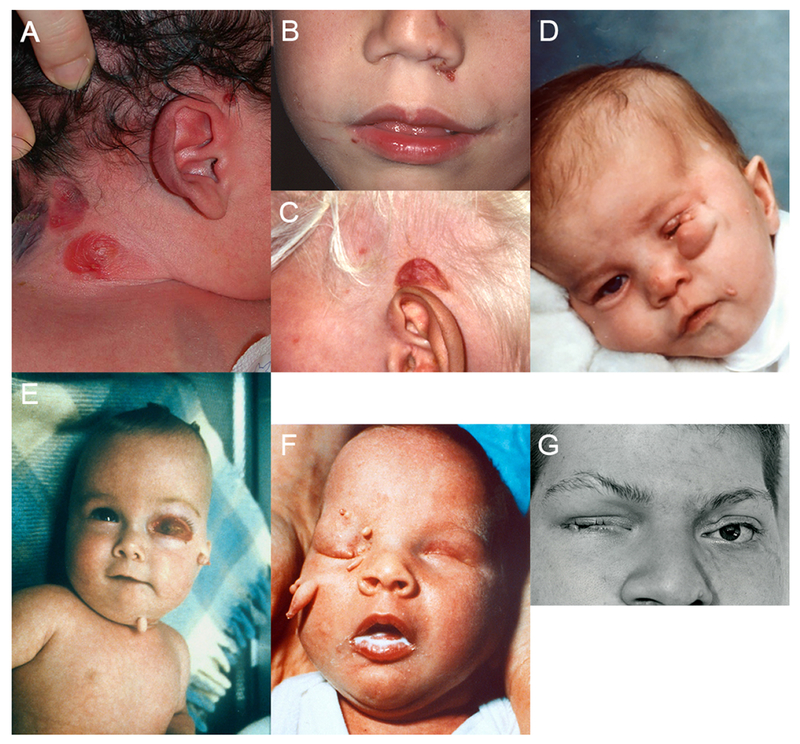
Skin and eye lesions in OCCS (color figure can be viewed online). (A) large focal skin defects on the right neck; (B) small punched-out focal skin defects on the left side of the nose; (C) typical postauricular crescent-shaped lesion above the left ear; (D, E, F) pedunculated, finger-like skin appendages over the face in three subjects; (D) microphthalmia; (E) orbital cyst; and (G) small, nodular skin tags, eyelid coloboma and microphthalmia on the right. Informed consent was obtained for publication of all photographs. Figure 1 A [Moog et al., 2005], 1 E, F [Moog et al., 1997], and 1 G [Moog et al., 1996] reproduced with permission.
2.2. Ocular Features
The most common eye anomalies are congenital orbital cysts with cystic microphthalmia (MOC, Figure 1E) seen in 27 of 40 patients, while a few have either orbital cysts or microphthalmia (Supporting Information Table S2). Colobomas of the eyelid were seen in 18 of 40 patients, including one with an iris coloboma as well. Less common eye anomalies, sometimes seen in the contralateral eye, include persistent hyperplastic primary vitreous and cataracts.
2.3. Brain Features
Most if not all individuals with OCCS have a complex pattern of brain malformations that is provisionally unique and easily recognized when all components are present, defined from review of brain imaging in 18 patients (Table 1). Given the rarity of OCCS, many studies were older (with lower resolution) or consisted of only selected images still available for review. In all, we have reviewed high-resolution MRI for 3 patients, limited MRI for another 6, head CT scans for 4, and a few published images for another 5 patients. The key features consist of a complex malformation of cortical development with both cortical and subcortical components, a striking brainstem-cerebellar malformation, and constant and usually striking asymmetry.
Table 1.
Clinical features in OCCS – brain imaging
| Individual | Study | MCD | LV | Midline | BS and CBL | Other | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | DB# | Sex | SYM | PMG | SUB | PNH | VMEG | ACC | IHEM | GTAV | CBVH | CBLH | PF cyst | ||
| 1 | LR01-191 | M | MRI* | L>R* | FL>TL-PL | L FL | L FH | L>R | T | + | ++ | + | L>R | + | DWML |
| 2 | LR04-005 | M | MRI* | L>R* | FL>TL-PL | L FL | L body | L>R | T | − | ++ | + | L>R | + | DWML |
| 3 | LR04-010 | F | MRI | L>R* | FL>TL-PL | L FL | … | L>R | P | − | + | + | L>R | + | DWML |
| 4 | LR01-135 | M | MRI | R>L | FL>TL-PL | R>L FL | R>L FHB | R>L | T | + | + | + | L=R | + | DWML |
| 5 | LR01-031 | M | CT/MRI | L>R | Diffuse | Diffuse | L FH | L>R | P | + | + | + | L>R | + | HYD, DWML |
| 6 | LR01-136 | M | CT | R>L | Diffuse | … | R FHB | R>L | P | + | + | + | R>L | + | HYD, CEPH |
| 7 | LR01-221 | M | CT | L>R* | … | … | … | R>L | P | − | (+) | + | R>L | + | HYD, DWML |
| 9 | LR01-253 | M | CT/MRI | L>R* | PL-OL>FL | L PL | L | L>R | P | − | − | − | − | − | LV cysts |
| 23 | F | CT | L>R* | FL>TL-PL | (L FL) | … | L>R | (P) | + | … | + | L | − | ||
| 24 | M | CT/MRI | L>R* | FL>TL-PL | (L FL) | (L) | L>R | T | … | − | + | … | − | MEGT | |
| 28 | M | MRI | L>R | (L FL) | L FL | L>R | + | P | + | … | (+) | (+) | … | ||
| 29 | LR05-138 | F | MRI | R>L* | R FL | R FL | R FHB | R>L | T | − | − | ++ | (R>L) | + | DWML |
| 33 | LR07-002 | M | MRI | R>L | FL-PL>OL | R FL | R FHB | R>L | T | + | + | ++ | R>L | ++ | DWML |
| 34 | LR01-190 | M | CT | L>R | FL>TL-PL | L FL | L FHB | L>R | P | + | + | + | + | − | |
| 35 | LR05-235 | M | MRI* | L>R | FL>TL-PL | L FL/OL | L FHB | L>R | T | + | − | + | + | − | MEGT |
| 36 | LR01-275 | M | CT | L>R | FL>TL-PL | … | … | R>L | P | − | … | + | L>R | … | |
| 37 | M | CT | L>R | FL>TL-PL | … | … | R>L | … | − | − | − | − | − | ||
| 39 | M | MRI | R>L | PL-OL>FL | (R OL) | … | L>R | (P) | + | … | … | − | … | ||
Legend: ACC, agenesis of the corpus callosum (P, partial; T, total); BS, brainstem; CBL, cerebellum; CBLH, cerebellar hypoplasia; CBVH, cerebellar vermis hypoplasia; CEPH, occipital cephalocele with probable DWML; DWML, Dandy-Walker like malformation with absent vermis and enlarged posterior fossa; FHB, frontal horn and body of LV; FH, frontal horn; FL, frontal lobe; HYD, hydrocephalus; IHEM: interhemispheric (midline) cyst; L=R, left same as right; L>R, left more severe than right; LV, lateral ventricle(s); MCD, malformation of cortical development; MEGT, mildly enlarged tectum suggesting variant GTAV (both subjects also have mild brainstem hypoplasia); OL, occipital lobe; PF, posterior fossa; PF cyst, enlarged posterior fossa with cystic appearance); PL, parietal lobe; PNH, periventricular nodular heterotopia; R>L, right more severe than left; SUB, subcortical malformation or heterotopia; SYM, symmetry of cortical malformation (*less severe side has only subtle malformation); TL, temporal lobe. Any entry in parenthesis is probable but not certain due to low resolution or limited images available for review. The shaded studies were reviewed based only on a few published images.
The cortical malformation consists of polymicrogyria, subcortical infolding or heterotopia found underneath the most severe areas of polymicrogyria which was not emphasized in our prior report [Moog et al., 2005], and scattered periventricular nodular heterotopia, which are sometimes connected to overlying subcortical heterotopia and sometimes consist of single or a few non-contiguous isolated heterotopia (Figures 2–3). The malformation was always asymmetric and often highly asymmetric. In the 18 patients we reviewed, the cortical malformation was more severe over the frontal lobes in 13, more severe over posterior regions in only 2, and indeterminate or diffuse in the last 3 patients; it was also more severe on the left in 13 and more severe on the right in 5 patients (the latter an interesting trend, but not statistically significant). Other supratentorial abnormalities include complete or partial agenesis of the corpus callosum, interhemispheric and less often intraventricular cysts, and enlarged dysplastic lateral ventricles.
Figure 2.

The classical brain malformation of the oculocerebrocutaneous syndrome (OCCS). Brain magnetic resonance imaging from patient 1 providing details of the OCCS brain malformation. Images through the cerebral hemispheres show an irregular surface, reduced sulcation, thick 10-15 mm cortex, and reduced white matter typical of polymicrogyria involving the left frontal, temporal, and parietal lobes (panels F–H, J–P, and white arrows in panels G, K, and L). The lateral ventricles are mildly enlarged, especially on the left, and the corpus callosum is absent (panels E, I, and J; black arrows in panels I and J). Images through the posterior fossa show a massively enlarged tectum and absent cerebellar vermis. The midbrain tegmentum is flexed forward but otherwise normal. The aqueduct is short and nearly horizontal (horizontal white arrow in panel E), and enlarges into the fourth ventricle behind the upper midbrain. The fourth ventricle is continuous with a large posterior fossa fluid collection. The midbrain tectum is greatly enlarged (black arrows in panels E, H, and O) and rotated upward; it appears to form an arch over the enlarged aqueduct (black arrows in panels G, L, and N). The cerebellar vermis is completely absent (panels B–G and L–P; black arrows in panels B, F, K, and P). The cerebellar hemispheres are nearly normal in size and seen in the midline because of the missing vermis (white arrow in panel E). The superior cerebellar peduncles are thick and dysplastic, descending vertically from the dysplastic midbrain to the cerebellar hemispheres (horizontal black arrow in panel K). The lower brain stem and spinal cord appear normal. Reproduced from Moog et al. with permission [Moog et al., 2005].
Figure 3.

The classical brain malformation of the oculocerebrocutaneous syndrome (OCCS). Magnetic resonance imaging from patients 1 to 4 also demonstrates the typical OCCS brain malformation. Views of the cortex show polymicrogyria (horizontal white arrows in panels B–D, G–H, K–L, and N–P), which is asymmetrical in all four patients, with more severe changes on the left (the right side of the images) in patients 1 to 3 (panels A–L) and on the right in patient 4 (panels M–P). Several periventricular nodular heterotopia are seen adjacent to the frontal horns and anterior bodies of the lateral ventricles (black arrows inside the ventricles in panels B, H, and P). The white matter is poorly myelinated in patients 2 to 4 (panels E–P) (patient 1 is older). The corpus callosum is absent in patients 1, 2, and 4 (panels A, E, and M), and dysplastic in patient 3 (black arrow in panel I). Images through the posterior fossa show a massively enlarged and upwardly rotated tectum (long white arrows in panels A, E, I, and M) and absent vermis (vertical arrows in panels J and N) in all four patients. In all, the midbrain is angled more forward than normal, leading to a short, horizontal aqueduct, enlarging prematurely into the fourth ventricle (panels A, I, and M) or appearing to form an abnormal extra ventricle behind the midbrain (panel E); the fourth ventricle is continuous with a large posterior fossa fluid collection. The cerebellar hemispheres are small and have a dysplastic foliar pattern in patients 3 and 4 (panels J and N), and are seen in the midline because of the absent vermis (short white arrows in panels A, E, I, and M). Reproduced from Moog et al. with permission [Moog et al., 2005].
The mid-hindbrain (brainstem-cerebellar) malformation in its complete form is striking and consists of variable brainstem hypoplasia, the rare “giant tectum absent vermis” (GTAV) malformation, and variable but usually asymmetric cerebellar hemisphere hypoplasia (Figures 2–3). The GTAV consists of a dramatically enlarged, nodular and upturned tectum (not vermis) that forms the roof of the aqueduct and connects with long, vertical superior cerebellar peduncles that extend down to form the only connection with the cerebellar hemispheres. Axial images through the tectum usually have a donut-like shape (Figure 4). The cerebellar malformation sometimes resembles classic Dandy-Walker malformation, except that the vermis is absent and the very dysplastic tectum is rotated upward along with the elevated tentorium.
Figure 4.
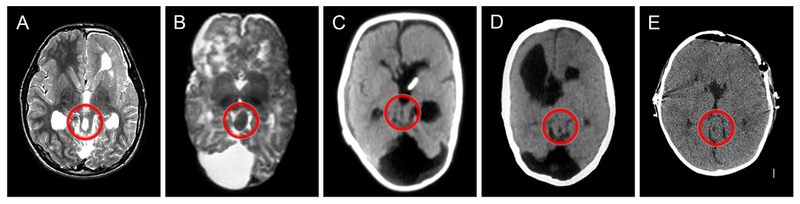
Axial T2-weighted MRI (A-B) or CT scan (C-E) images at the level of the midbrain show an unusual oval-shaped midbrain (within the red circles) with a donut-like hole in the center (in all but B). These images were taken from patients 1, 4, 5, 6 and 34.
In its complete form with polymicrogyria, subcortical heterotopia, agenesis of the corpus callosum and GTAV, the OCCS-associated brain malformation is probably pathognomonic. Incomplete forms occur and are more difficult to recognize. Limited descriptions and a few published figures for another 22 patients were generally supportive.
2.4. Development
Developmental data is available for 31 patients. Most (24 of 31) had developmental delay (DD) and/or intellectual disability (ID), which was severe in 7 and moderate or unspecified in 11 patients (Supporting Information Table S2). Another 7 patients developed normally up to a reported age of 2 months to 3 years. The last 6 patients died from causes related to the syndrome or were affected fetuses. About half of the patients had epilepsy or an abnormal EEG, with seizures well controlled in most individuals. Of the remaining patients, 7 were specifically noted to have no history of seizures.
2.5. Infrequent Features
In addition to the key features reviewed above, skull, rib and vertebral defects are commonly seen in OCCS. Craniofacial clefts were seen in 8 patients, including small unilateral defects in the nasal ala in 5 of 40 patients that take the form of a small notch or distinct cleft. All five involved the left side of the nose, consistent with the predominant left-sided brain malformations. Also, cryptorchidism has been reported in 8 patients.
2.6. Diagnostic Criteria
We have adapted the diagnostic criteria first proposed by Hunter [Hunter, 2006], still taking into account the most typical features in the three major systems but placing more weight on the striking GTAV malformation of the brain, redefining other brain features, and leaving additional systems out of consideration (Table 2).
Table 2.
Revised diagnostic criteria for OCCS
| Eye | Major criteria • Congenital orbital cyst or microphthalmia with cyst (MOC) Minor criteria • Isolated microphthalmia/anophthalmia • Any other colobomatous defect, ocular or eyelid |
| Skin | Major criteria • Crescent-shaped skin defect above or behind the ear • Pedunculated skin appendage, finger-like and moving, or proven striated muscle hamartoma (SMH) • Pedunculated skin appendage (possible SMH) plus one of minor criteria 2-4 below Minor criteria • Pedunculated skin appendage, possible SMH • Focal hypo- or aplastic lesions • Small punched-out lesions • Three or more small nodular tags |
| Brain | Major criteria • Novel mid-hindbrain malformation consisting of giant, dysplastic tectum rotated upwards, and absent or severely malformed vermis Often associated with nearly horizontal orientation of aqueduct Minor criteria • Patchy polymicrogyria, most often frontal predominant • Subcortical or periventricular nodular heterotopia • Agenesis of corpus callosum, complete or partial, and often associated with interhemispheric cysts • Enlarged lateral ventricles, often asymmetric, or hydrocephalus • Porencephalic cysts unspecified • Cerebellar vermis and hemisphere hypoplasia • Posterior fossa fluid collection sometimes with enlarged posterior fossa |
| Application to diagnosis of OCCS | Definite OCCS • 3 systems involved with major criterium in at least one system, or • 2 systems involved with major criterium in both Probable OCCS • 3 systems involved with no major criterium • 2 systems involved with major criterium in only one system |
3. DIFFERENTIAL DIAGNOSES
3.1. OCCS Overlap with Aicardi Syndrome
The typical brain malformation associated with OCCS overlaps substantially with the brain malformation seen in Aicardi syndrome (OMIM 304050), another rare pattern recognition syndrome that is also thought to be X-linked with embryonic lethality in males and is also reviewed in this issue of the Journal. Most of the brain malformations seen in OCCS (ACC, PMG, heterotopia, enlarged ventricles and cerebellar hypoplasia) are relatively common individually, but occur together in only a few syndromes, including both OCCS and Aicardi syndrome. Other overlapping features between OCCS and Aicardi syndrome include orbital cysts, microphthalmia, optic nerve colobomas, intracranial cysts especially midline cysts, cleft lip and palate, and costovertebral defects. Further, all of these features are almost uniformly asymmetric in both OCCS and Aicardi syndrome. However, the striking GTAV malformation seen in OCCS is unknown in Aicardi syndrome, while the chorioretinal lacunae in Aicardi syndrome are unknown in OCCS. Skin tags are occasionally observed in Aicardi syndrome, but the focal hypoplastic or aplastic skin defects and pedunculated skin appendages of OCCS have not been seen in Aicardi syndrome. Finally, OCCS occurs predominately in males, while Aicardi syndrome occurs only in females. Still, in our view the similarities outweigh the differences leading us to hypothesize that OCCS and Aicardi syndrome are both genetic and caused by mosaic mutations of the same gene or genes in the same functional pathway.
3.2. Further Differential Diagnosis
Several other rare disorders have been repeatedly included in the differential diagnosis of OCCS due to phenotypic overlap. However, they are not difficult to distinguish with full expression of key features, as summarized below.
3.2.1. Microphthalmia with linear skin defects syndrome (MLS, OMIM 309801).
MLS is a rare X-linked disorder affecting only females. It is most often caused by segmental deletions of Xp22.2 that include the HCCS gene, and in smaller subgroups of individuals by heterozygous mutation in the X-linked genes HCCS, COX7B or NDUFB11, all encoding components of the mitochondrial respiratory chain [van Rahden et al., 2015]. The disorder is characterized by anophthalmia or microphthalmia plus a variety of other ocular defects including sclerocorneae, and by aplastic, typically linear, skin lesions that primarily affect the head and neck region and heal to become hyperpigmented areas with age. Other less common abnormalities include congenital heart defects, microcephaly, agenesis of the corpus callosum or other unspecific brain anomalies, growth retardation, seizures, or ID [van Rahden et al., 2014].
3.2.2. Focal dermal hypoplasia (FDH; OMIM 305600).
FDH or Goltz syndrome is another X-linked disorder that affects predominantly females that is characterised by a combination of cutaneous, ocular, neurological and skeletal features. It is caused by mutations in PORCN, a modulator of Wnt signaling. The prominent cutaneous features include focal hypoplastic or aplastic skin lesions often with herniation of fatty tissue, and linear streaks of pigmentation that can resemble OCCS [Bostwick et al., 2016]. In contrast to OCCS, the skin lesions commonly follow the lines of Blaschko and skin tags are papillomas, not hamartomatous appendages. Ocular features are diverse, include anophthalmia or microphthalmia, and iris or chorioretinal coloboma, which most often affect the anterior chamber. Digital anomalies are common and include syndactyly, polydactyly and reduction defects. X-rays show striation of the bones in most patients. Dental findings, CLP, and typical facial features have been reported. A minority of individuals show intellectual impairment or behavioral problems. Brain malformations have rarely been described and do not correspond to the brain malformations seen in OCCS.
3.2.3. Encephalocraniocutaneous lipomatosis (ECCL, OMIM 613001).
Like OCCS, ECCL is characterized by congenital skin, eye, and brain anomalies. The dermatological hallmark is an alopecic fatty tissue nevus of the scalp (so-called nevus psiloliparus) which is unknown in OCCS. Other skin features include focal alopecia, aplastic scalp defects, nodular skin tags, and subcutaneous fatty masses. Ocular findings are mainly choristomas, but ocular and palpebral colobomas, microphthalmia and other findings can be seen. Intracranial and spinal lipomas are the most frequent CNS feature; in addition, anomalies of the meninges and vascular defects leading to asymmetric lesions, but no primary brain malformations are documented [Moog et al., 2007]. ECCL is associated with a disposition to lytic bone lesions, jaw tumors, and gliomas [Bennett et al., 2016; Moog, 2009]. In some patients recurrent postzygotic activating mutations in FGFR1 and rarely in KRAS [Bennett et al., 2016; Boppudi et al., 2016] have been reported. Whereas at a first look ECCL may easily be confused with OCCS, the typical features are all different, especially the ocular and brain defects.
3.2.4. Oculoauriculovertebral spectrum (OAVS, OMIM 164210).
OAVS (also known as hemifacial microsomia or Goldenhar syndrome) has also been discussed in the differential diagnosis of OCCS based primarily on facial asymmetry, skin tags and congenital eye anomalies including microphthalmia. However, the ophthalmologic hallmarks are different (epibulbar dermoids versus orbital cyst or MOC) and skin tags are restricted to small preauricular tags in OAVS. In OAVS, only a minority of the patients have ID. A few have hydrocephalus or various brain malformations, but no pattern has emerged. Several patients with (possible) OCCS and overlap with OAVS have been reported [Angle and Hersh, 1997; Leichtman et al., 1994; McCandless and Robin, 1998; Ming et al., 1998]. One boy had PMG over the frontal lobes, ACC and a midline cyst and so possibly had OCCS [Angle and Hersh, 1997]. Several other patients with overlapping features of OAVS and OCCS have had massive hydrocephalus without the brain anomalies characteristic of OCCS. In this group, the combination of severe hydrocephalus with anophthalmia or severe microphthalmia and clefts favours OAVS, or another entity.
4. MOLECULAR PATHOGENESIS
4.1. Inheritance and Genetic Studies
A striking male preponderance has been observed in OCCS with 30 affected males but only 10 affected females, most of the latter severely affected. In one affected female (patient 12), X-chromosome inactivation analysis on lymphocyte-derived DNA showed a completely skewed pattern, which supports X-linked inheritance. No other female patients have been studied.
Chromosome analysis has been performed on lymphocytes in many patients and on skin tags in a few patients with OCCS, always with normal results. We performed chromosome microarrays on lymphocyte-derived DNA using the Affymetric 6.0 SNP-array in four affected males (patients 1, 2, 35, 40) but found no recurrent CNVs including none on the X-chromosome [Sajan et al., 2013]. We also performed whole genome sequencing in two trios (patients 2 and 34: LR04-005, LR01-190) using fibroblast- or tissue-derived DNA as well as blood or saliva-derived DNA from the two probands. However, no de novo heterozygous, homozygous or compound heterozygous, or X-linked candidate variants were identified. We are aware of no other genome-wide sequencing studies by other groups.
4.2. Evidence for Mosaicism
The malformations observed in OCCS are uniformly asymmetric, either unilateral or bilateral but always much more severe on one side. From our review, the eye and skin abnormalities appear unilateral in half and bilateral but asymmetric in the other half, while the brain abnormalities are almost always bilateral but asymmetric. As might be expected from similar syndromes, the eye, brain and skin features are almost always more severe on the same side.
Further, no recurrence in siblings or children has been reported although no individuals with OCCS are known to have had children, likely due to severe ID and other handicaps. The family history has been negative for related malformations with the exception of patients 7 (unilateral anophthalmia in the paternal grandmother) and 18 (eye cyst in a paternal cousin and bilateral coloboma in the mother). Paternal age is not advanced, and no exposures to teratogenic or mutagenic agents during or shortly before pregnancy have been demonstrated. Consanguinity was reported in two families (patients 23 and 25). A genealogical study of 5 patients of full or partial Dutch extraction revealed no consanguinity in up to 5 generations [Bleeker-Wagemakers et al., 1990].
The asymmetric and patchy pattern of malformations combined with the absence of any reports of familial recurrence first led Happle to hypothesize that OCCS as well as several other patchy disorders results from postzygotic (mosaic) mutations of one or more genes that would be lethal in the non-mosaic state [Happle, 1987]. Remarkably, mosaicism has been proven for all of the disorders proposed by Happle except for OCCS. Still, the preponderance of available evidence suggests that OCCS is indeed caused by mosaic mutations of one or more genes, with one gene likely located on the X chromosome.
5. FUTURE DIRECTIONS
In summary, OCCS is a rare but highly recognizable disorder that consists of patches of skin aplasia or hypoplasia, unusual skin appendages that often appear finger-like, microopthalmia and orbital cysts, and multiple brain malformations including polymicrogyria, heterotopia, agenesis of the corpus callosum, cerebellar hypoplasia, and the very rare giant tectum-absent vermis malformation.
While the cause of OCCS remains unknown, few genome-wide sequencing studies have been done due to the extreme rarity of the disorder, the predicted mosaicism, and the paucity of DNA samples, especially DNA derived from affected tissues rather than blood or saliva. Despite our focus in collecting samples over more than a decade, we have not yet obtained DNA from other female patients or DNA derived from affected tissue. The obvious next step is to perform genome-wide sequencing using DNA samples derived from affected tissue in multiple trios. In addition, additional X inactivation studies need to be performed in female patients and their mothers.
Supplementary Material
CALL OUT QUOTES.
-
1
In its complete form with polymicrogyria, subcortical heterotopia, agenesis of the corpus callosum and GTAV, the OCCS-associated brain malformation is probably pathognomonic.
-
2
OCCS is hypothesized to be indeed caused by mosaic mutations of one or more genes, with one gene likely located on the X chromosome.
-
3
[We] hypothesize that OCCS and Aicardi syndrome are both genetic and caused by mosaic mutations of the same gene or genes in the same functional pathway.
-
4
OCCS is a rare but highly recognizable disorder that consists of patches of skin aplasia or hypoplasia, unusual skin appendages, microopthalmia and orbital cysts, and multiple brain malformations.
ACKNOWLEDGEMENTS
We thank the families for permission to use photographs of their children and their referring physicians for their important contribution to our ongoing work on OCCS. This study was funded by the US National Institutes of Health under NINDS grants 5R01NS050375 to WBD and 1R01NS058721 to E.H. Sherr and WBD. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding sources.
Grant numbers:
NIH (NINDS)
5R01NS050375 (to W.B. Dobyns)
1R01NS058721 (to E.H. Sherr and W. B. Dobyns)
REFERENCES
- Angle B, Hersh JH. 1997. Anophthalmia, intracerebral cysts, and cleft lip/palate: expansion of the phenotype in oculocerebrocutaneous syndrome? Am J Med Genet 68(1):39–42. [DOI] [PubMed] [Google Scholar]
- Bennett JT, Tan TY, Alcantara D, Tetrault M, Timms AE, Jensen D, Collins S, Nowaczyk MJM, Lindhurst MJ, Christensen KM, Braddock SR, Brandling-Bennett H, Hennekam RCM, Chung B, Lehman A, Su J, Ng S, Amor DJ, University of Washington Center for Mendelian G, Care4Rare Canada C, Majewski J, Biesecker LG, Boycott KM, Dobyns WB, O’Driscoll M, Moog U, McDonell LM. 2016. Mosaic Activating Mutations in FGFR1 Cause Encephalocraniocutaneous Lipomatosis. Am J Hum Genet 98(3):579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleeker-Wagemakers LM, Hamel BC, Hennekam RC, Beemer FA, Oorthuys HW. 1990. Oculocerebrocutaneous syndrome. J Med Genet 27(1):69–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boppudi S, Bogershausen N, Hove HB, Percin EF, Aslan D, Dvorsky R, Kayhan G, Li Y, Cursiefen C, Tantcheva-Poor I, Toft PB, Bartsch O, Lissewski C, Wieland I, Jakubiczka S, Wollnik B, Ahmadian MR, Heindl LM, Zenker M. 2016. Specific mosaic KRAS mutations affecting codon 146 cause oculoectodermal syndrome and encephalocraniocutaneous lipomatosis. Clin Genet 90(4):334–42. [DOI] [PubMed] [Google Scholar]
- Bostwick B, Fang P, Patel A, Sutton VR. 2016. Phenotypic and molecular characterization of focal dermal hypoplasia in 18 individuals. Am J Med Genet C Semin Med Genet 172C(1):9–20. [DOI] [PubMed] [Google Scholar]
- Clericuzio C 1989. Oculocerebrocutaneous syndromme and the family of neurodermal disorders: developmental considerations 10th Smith David W Workshop on Malformaions and Morphogenesis. Madrid, Spain. [Google Scholar]
- Delleman JW, Oorthuys JW. 1981. Orbital cyst in addition to congenital cerebral and focal dermal malformations: a new entity? Clin Genet 19(3):191–8. [DOI] [PubMed] [Google Scholar]
- Delleman JW, Oorthuys JW, Bleeker-Wagemakers EM, ter Haar BG, Ferguson JW. 1984. Orbital cyst in addition to congenital cerebral and focal dermal malformations: a new entity. Clin Genet 25(5):470–2. [DOI] [PubMed] [Google Scholar]
- Dollfus MA, Marx P, Langlois J, Clement JC, Forthomme J. 1968. Congenital cystic eyeball. Am J Ophthalmol 66(3):504–9. [DOI] [PubMed] [Google Scholar]
- Happle R 1987. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol 16(4):899–906. [DOI] [PubMed] [Google Scholar]
- Hunter AG. 2006. Oculocerebrocutaneous and encephalocraniocutaneous lipomatosis syndromes: blind men and an elephant or separate syndromes? Am J Med Genet A 140(7):709–26. [DOI] [PubMed] [Google Scholar]
- Ladenheim J, Metrick S. 1956. Congenital microphthalmos with cyst formation. Am J Ophthalmol 41(6):1059–62. [DOI] [PubMed] [Google Scholar]
- Leichtman LG, Wood B, Rohn R. 1994. Anophthalmia, cleft lip/palate, facial anomalies, and CNS anomalies and hypothalamic disorder in a newborn: a midline developmental field defect. Am J Med Genet 50(1):39–41. [DOI] [PubMed] [Google Scholar]
- McCandless SE, Robin NH. 1998. Severe oculocerebrocutaneous (Delleman) syndrome: overlap with Goldenhar anomaly. Am J Med Genet 78(3):282–5. [DOI] [PubMed] [Google Scholar]
- Ming JE, Katowitz J, McDonald-McGinn DM, Schnur RE, Hunter JV, Zackai EH. 1998. Hemifacial microsomia in a newborn with hypoplastic skin lesions, an eyelid skin tag, and microphthalmia: an unusual presentation of Delleman syndrome. Clin Dysmorphol 7(4):279–83. [DOI] [PubMed] [Google Scholar]
- Moog U 2009. Encephalocraniocutaneous lipomatosis. J Med Genet 46(11):721–9. [DOI] [PubMed] [Google Scholar]
- Moog U, de Die-Smulders C, Systermans JM, Cobben JM. 1997. Oculocerebrocutaneous syndrome: report of three additional cases and aetiological considerations. Clin Genet 52(4):219–25. [DOI] [PubMed] [Google Scholar]
- Moog U, Jones MC, Bird LM, Dobyns WB. 2005. Oculocerebrocutaneous syndrome: the brain malformation defines a core phenotype. J Med Genet 42(12):913–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moog U, Jones MC, Viskochil DH, Verloes A, Van Allen MI, Dobyns WB. 2007. Brain anomalies in encephalocraniocutaneous lipomatosis. Am J Med Genet A 143A(24):2963–72. [DOI] [PubMed] [Google Scholar]
- Moog U, Kruger G, Stengel B, De Die-Smulders C, Dykstra S, Bleeker-Wagemakers E. 1996. Oculocerebrocutaneous syndrome: a case report, a follow-up, and differential diagnostic considerations. Genet Couns 7(4):257–65. [PubMed] [Google Scholar]
- Renard G, Fontaine M, Dhermy P, Caquet N. 1964. Microphthalmie bilatérale avec kystes orbitaires associéé à des appendices faciaux surnuméraires. Bull Med Soc Fr Ophthalmol 77:297–316. [Google Scholar]
- Sajan SA, Fernandez L, Nieh SE, Rider E, Bukshpun P, Wakahiro M, Christian SL, Riviere JB, Sullivan CT, Sudi J, Herriges MJ, Paciorkowski AR, Barkovich AJ, Glessner JT, Millen KJ, Hakonarson H, Dobyns WB, Sherr EH. 2013. Both rare and de novo copy number variants are prevalent in agenesis of the corpus callosum but not in cerebellar hypoplasia or polymicrogyria. PLoS Genet 9(10):e1003823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rahden VA, Fernandez-Vizarra E, Alawi M, Brand K, Fellmann F, Horn D, Zeviani M, Kutsche K. 2015. Mutations in NDUFB11, encoding a complex I component of the mitochondrial respiratory chain, cause microphthalmia with linear skin defects syndrome. Am J Hum Genet 96(4):640–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rahden VA, Rau I, Fuchs S, Kosyna FK, de Almeida HL Jr, Fryssira H, Isidor B, Jauch A, Joubert M, Lachmeijer AM, Zweier C, Moog U, Kutsche K 2014. Clinical spectrum of females with HCCS mutation: from no clinical signs to a neonatal lethal form of the microphthalmia with linear skin defects (MLS) syndrome. Orphanet J Rare Dis 9:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.