

Abstract
HIV-Associated Neurocognitive disorder (HAND) affects nearly half of infected patients. The HIV envelope protein gp120 is shed by infected cells and is a potent neurotoxin in vitro that reproduces many aspects of HAND when expressed in vivo. Here, we show that HIV gp120 increases the amplitude of a tonic current mediated by γ-aminobutyric acid type-A receptors (GABAARs).
Treating rat hippocampal cultures with 600 pM gp120IIIB for 4 h increased a tonic bicuculline-sensitive current, which remained elevated for 24 h. The increased current resulted from upregulation of extrasynaptic α5-containing GABAARs, as indicated by inhibition with the selective inverse agonist basmisanil. Treatment with gp120 increased α5-GABAAR immunoreactivity on the cell surface without new protein synthesis. The increase in tonic inhibition was prevented by a C-X-C chemokine receptor type 4 (CXCR4) antagonist or elimination of microglia from the culture. Treatment with interleukin-1β (IL-1β) increased the tonic current and an IL-1 receptor antagonist blocked the gp120-evoked response. Pharmacological or genetic inhibition of p38 mitogen-activated protein kinase (MAPK) prevented the gp120-evoked increase in tonic current and direct activation of a mutant form of p38 MAPK expressed in neurons increased the current.
Collectively, these data show that gp120 activates CXCR4 to stimulate microglia to release IL-1β. Subsequent stimulation of IL-1 receptors activates p38 MAPK in neurons leading to the upregulation of α5-containing GABAARs. Increased tonic inhibition impairs neuroplasticity and inhibition of α5-containing GABAARs improves cognitive function in disease models. Thus, gp120-induced upregulation of α5-containing GABAARs presents a novel therapeutic target for HAND.
Keywords: HIV gp120, HAND, α5 GABAAR subunit, basmisanil
Graphical Abstract

1. Introduction
There is no effective treatment for HIV-associated neurocognitive disorder (HAND) which affects about half of the over 36 million people infected (Saylor et al., 2016). HIV can remain in the brain during combination antiretroviral therapy (Ellis et al., 2007; Saylor et al., 2016) producing chronic neuroinflammation that is a major component of HAND pathogenesis (Chen et al., 2014; Gill and Kolson, 2014; Hong and Banks, 2015; Walsh et al., 2014). HIV-infected cells in the brain release viral proteins and inflammatory mediators that produce synaptodendritic damage and impair cognitive function (Ellis et al., 2007; Saylor et al., 2016).
Numerous studies have described changes in cellular and network excitability that occur under inflammatory conditions (Ransohoff, 2016), including the upregulation of GABAergic signaling (Serantes et al., 2006). GABA-mediated inhibition is critical to maintain network function and control excitability. Extrasynaptic GABAARs mediate tonic inhibition that is produced by ambient GABA and continuous activation of high affinity receptors (Farrant and Nusser, 2005; Luscher and Keller, 2004). Tonic inhibition can control network excitability by having a modulatory effect on excitatory input through shunting inhibition. For example, in cerebellar granule neurons tonic inhibition controls the gain of rate-coded sensory input (Mitchell and Silver, 2003), whereas in the hippocampus tonic inhibition regulates spike timing dependent plasticity by setting the threshold for back propagating action potentials (Groen et al., 2014). Thus, proper levels of tonic inhibition are critical for neuronal function.
Upregulation of tonic GABAergic signaling in response to inflammatory and excitotoxic stress may overcompensate, resulting in impaired function. For example, in transgenic models of Alzheimer’s disease increased tonic inhibition is associated with cognitive decline (Wu et al., 2014). The inflammatory cytokine interleukin-1β (IL-1β) increases tonic inhibition mediated by α5 subunit-containing GABAARs, contributing to memory deficits (Wang et al., 2012). Inhibitors selective for α5-containing GABAARs restore cognitive function in these models providing a basis for clinical trials. These drugs are well tolerated and are currently being evaluated for their ability to improve cognitive function in patients with schizophrenia (NCT02953639; http://www.clinicaltrials.gov). During prolonged exposure to HIV neurotoxins markers for GABA signaling are elevated, suggesting that excess inhibitory tone may contribute to HAND (Fitting et al., 2013; Hargus and Thayer, 2013). At present, whether tonic inhibition is increased during HIV-induced neuroinflammation is not known.
Because HIV does not infect neurons, HAND pathogenesis results from the release of toxic agents, such as the HIV envelope protein gp120 that is shed by infected cells (Kaul et al., 2001). HIV gp120 is a potent neurotoxin (Kim et al., 2011; Meucci and Miller, 1996; Zhou et al., 2017) that has been measured in the brains of patients with HAND (Jones et al., 2000). HIV gp120 evokes synaptic and behavioral deficits in vivo that mimic significant aspects of HAND (Thaney et al., 2018; Toggas et al., 1994). In this study, we used a cell culture model to study changes in tonic inhibition during exposure to the neuroinflammatory stimulus gp120. HIV gp120 increased the amplitude of tonic currents mediated by extrasynaptic GABAARs. This increase in GABAergic signaling was dependent on microglial release of IL-1β. Activation of IL-1Rs stimulated the p38 MAPK pathway to increase tonic inhibition mediated by α5-containing GABAARs, for the first time linking upregulation of these receptors to a model of HAND.
2. Materials and Methods
2.1. Materials
Materials were obtained from the following sources: IL-1β and IL-1ra were from R&D Systems (Minneapolis, MN, USA); 1,1-Dioxidothiomorpholino)(6-((3-(4-fluorophenyl)-5-methylisoxazol-4-yl)methoxy)pyridin-3-yl)methanone (basmisanil) was from MedChemExpress (Monmouth Junction, NJ, USA); 4,5,6,7-Tetrahydroisoxazolo[5,4-c]pyridin-3-ol hydrochloride (THIP hydrochloride), cycloheximide, 4-[5-(4-Fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-imidazol-4-yl]pyridine (SB203580), 6-Cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX), bicuculline methiodide, and tetrodotoxin (TTX) were from Tocris Bioscience (Bristol, UK); 4-Ethyl-2(p-methoxyphenyl)-5-(4ʹ-pyridyl)-IH-imidazole (SB202474) was from Millipore Sigma (St. Louis, MO, USA); Dulbecco’s modified Eagle’s medium (DMEM), Hanks’ balanced salt solution (HBSS), fetal bovine serum, horse serum and penicillin/streptomycin were from Invitrogen (Carlsbad, CA, USA). The plasmid encoding RapR-p38 (pCMV5-Flag-RapR-p38) was generated in the laboratory of Klaus Hahn and obtained from Addgene (catalog number: 25935). HIV-1 gp120IIIB was obtained through the National Institutes of Health (NIH) AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH (catalog number:11784).
2.2. Cell culture
All animal care and experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health. Ethical approval was granted by the Institutional Animal Care and Use Committee of the University of Minnesota (protocol 1612–34372A). Primary hippocampal cultures were prepared from fetal tissue collected from pregnant Sprague Dawley rats as described previously with minor modifications (Zhang and Thayer, 2018). Dams were killed by CO2 inhalation in an institutionally approved and calibrated CO2 chamber. Embryonic day 17 fetuses of both sexes were rapidly decapitated with sharp scissors then hippocampi dissected and placed in Ca2+ and Mg2+ free HBSS. Hippocampi were suspended in DMEM without glutamine, supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U mL−1 and 100 mg mL−1, respectively) and dissociated by trituration through flame-narrowed Pasteur pipettes of decreasing aperture. Dissociated cells were then plated onto a 25 mm round cover glass pre-coated with Matrigel (150 µL, 0.2 mg mL−1) (BD Biosciences, Billerica, MA, USA). Cells were grown in a humidified atmosphere of 10% CO2 and 90% air (pH 7.4) at 37o C. On day 1 and day 8 in vitro, cells were fed by exchanging 75% of the media with DMEM, supplemented with 10% horse serum and penicillin/streptomycin. Cells used in this study were grown for 11–15 days in vitro without mitotic inhibitors, resulting in a mixed glial-neuronal culture. Using previously described immunocytochemistry methods (Kim et al., 2011), we found that these cultures are composed of 24 ± 4% neurons, 55 ± 4% astrocytes and 13 ± 6% microglia.
2.3. Transfection
Transfection of cultured rat hippocampal neurons was conducted between 9 and 11 days in vitro using a previously described protocol with minor modifications (Kim et al., 2011). Briefly, a DNA/calcium phosphate precipitate containing 1 µg of total plasmid DNA per well was prepared and allowed to form for 90 min at room temperature. The media (conditioned media) was exchanged with DMEM supplemented with 1 mM kynurenic acid, 10 mM MgCl2, and 5 mM HEPES to reduce neurotoxicity. The DNA/calcium phosphate precipitate was added dropwise to the cells and allowed to incubate for 60 min. After the incubation, cells were washed twice with DMEM supplemented with 10 mM MgCl2 and 5 mM HEPES to remove leftover precipitate. After washing, conditioned media that had been saved at the beginning of the procedure was returned to the cells. Experiments were started 48–72 h after transfection.
2.4. Electrophysiology
Electrodes were pulled using a horizontal micropipette puller (P-87; Sutter Instruments) from glass capillaries (Narishige). Pipette resistance was 3–5 MΩ. Bicuculline, basmisanil, and THIP-sensitive currents were recorded with the following extracellular solution (in mM): 140 NaCl, 2 CaCl, 1 MgCl, 5.4 KCl, 25 HEPES, and 28 glucose, pH adjusted to 7.4 with NaOH. The intracellular recording solution was composed of (in mM): 140 CsCl, 10 HEPES, 11 EGTA, 4 KATP, 2 MgCl, 1 CaCl, and 2 TEA, pH adjusted to 7.3 with CsOH. Whole-cell voltages were amplified with an AxoPatch 200B (Molecular Devices), low-pass filtered at 2 kHz, and digitized at 10 kHz with a Digidata 1322A digitizer and pClamp software (Molecular Devices). Cells with series resistance over 25 MΩ were excluded from analysis. Whole-cell capacitance was measured after break-in. To record tonic currents, cells were voltage clamped at −60 mV in the presence of 0.5 µM GABA. The change in holding current was measured from a 10 s average before and after superfusion of 100 uM bicuculline. The difference between steady state currents before and after bicuculline were divided by whole-cell capacitance and reported as the bicuculline-sensitive current density. Basmisanil and THIP-sensitive currents were measured in the same manner, but recorded with a local perfusion apparatus to evoke faster changes in current due to their smaller shifts. Data were analyzed using ClampFit (Molecular Devices) and Origin software (OriginLab).
2.5. Immunocytochemistry
Hippocampal cultures were prepared as described above and maintained for at least 11 d in culture. Cells were washed with PBS and then fixed with 4% PFA for 10 min. The cells were washed with PBS and then blocked in 10% BSA in PBS (blocking buffer) for 30 min. Cells were then incubated with either mouse anti-OX-42 antibody (ab1211, 1:200 abcam) or rabbit anti-GABAAR α5 antibody (ab10098, 1:200; abcam) and mouse anti-MAP2 antibody (M1406, 1:200; Millipore Sigma) or rabbit anti-MAP2 antibody (ab32454, 1:200 abcam) in blocking buffer for 16 h at 4°C. Cells were washed with PBS and labeled with either fluorescein isothiocyanate (FITC) goat anti-rabbit antibody (F2765, 1:500; Thermo Fisher Scientific) and AlexaFluor 594 goat anti-mouse antibody (a11005, 1:500; Thermo Fisher Scientific) or Alexa Fluor 488 goat anti-mouse antibody (a32723, 1:500 Thermo Fisher Scientific) or Alexa Fluor 594 goat anti-rabbit (a11012, 1:500 Thermo Fisher Scientific) in blocking buffer for 1 h at room temperature. Cells were imaged using an inverted laser scanning confocal microscope (Nikon A1, Melville, NY, USA) using a 60 x (1.4 numerical aperture) oil-immersion objective. AlexaFluor594 was excited at 561 nm and emission collected from 570 to 620 nm. FITC and Alexa Fluor 488 were excited at 488 nm and emission collected from 500 to 550 nm.
2.6. Statistics
All data are presented as mean ± SEM. Data collection was randomized. The homogeneity of variances was determined using Bartlett’s test; all statistical tests were performed on data of equal variance. For two-group comparisons a Student’s t-test (unpaired two-tailed) was used for statistical analysis of normally distributed data and a Mann-Whitney U-test was used for non-normally distributed data. For comparison among multiple groups, one-way or two-way ANOVAs were performed with Tukey’s post hoc test. Statistical significance was defined as p<0.05. Sample sizes were not statistically predetermined but conform to similar studies. For electrophysiological experiments, a single cell was considered as n = 1. No more than one cell was taken per coverslip and all experiments included cells from at least three different cultures. For immunocytochemistry experiments to detect α5 GABAA subunits, 1–4 cells were captured per field and each cell soma was considered as n=1. Intensities for each cell were normalized to the average intensity from untreated cells processed in parallel from the same culture. 3 different cultures were used for data collection. Statistical analyses were performed using Prism, GraphPad 7 (La Jolla, CA 92037 USA).
3. Results
3.1. gp120IIIB increases the amplitude of tonic inhibitory currents
We studied the effect of gp120IIIB on tonic GABA-evoked currents recorded from hippocampal neurons in primary culture. The shift in holding current induced by 100 µM bicuculline was recorded in the presence of a low concentration of GABA (0.5 µM) using the whole-cell configuration of the patch-clamp technique (Wang et al., 2012) (Fig. 1A). Following 4 h treatment with 600 pM gp120, the bicuculline-sensitive shift in holding current increased by 46 ± 12 % relative to untreated neurons. The increase in current was sustained for 24 h (Fig. 1B). Because this form of tonic inhibition is mediated primarily by extrasynaptic GABAA receptors (Belelli et al., 2009), these results suggest that gp120 either increases the expression of extrasynaptic GABAARs on the cell surface or increases their activity levels.
Figure 1: gp120 increases tonic GABAAR current.
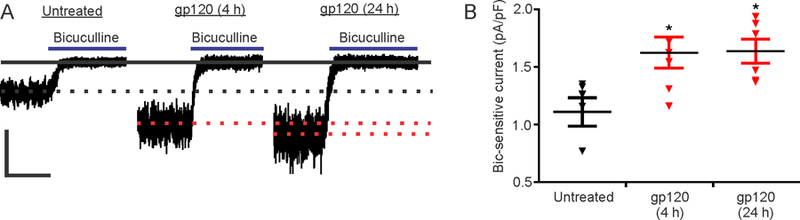
(A) Representative traces show gp120 treatment (600 pM for 4 or 24 h) increases the amplitude of the bicuculline-sensitive shift in holding current compared to untreated neurons (scale bar: 50 pA, 10 s). (B) Summary plot showing effect of gp120 on bicuculline-sensitive currents normalized to whole cell capacitance (One-way ANOVA; F(2,18)=6.21, p=0.009). Data are expressed as individual data points with mean ± SEM. One-way ANOVA was followed with Tukey’s post hoc test, *p<0.05 compared to untreated group.
3.2. Gp120 increases tonic inhibition through α5-containing GABAARs
Bicuculline is a non-selective GABAAR antagonist, therefore the gp120-induced tonic current may result from multiple subtypes of GABAARs. The predominant subtypes of extrasynaptic GABAARs in the hippocampus contain either α5 subunits or δ subunits (Brickley and Mody, 2012; Wu et al., 2013). To determine whether GABAARs were upregulated by gp120 in a subtype specific manner, we used either a selective inverse agonist for the benzodiazepine binding site on the α5 subunit (basmisanil, 10 nM) or a selective agonist for GABAARs containing the δ subunit (THIP, 1 µM). Basmisanil-sensitive currents were increased by 87 ± 20 % following treatment with gp120 (4 h) (Fig. 2A,B) suggesting up-regulation of α5-containing GABAARs. THIP-sensitive currents were not significantly changed following treatment with gp120 (4 h) (Fig. 2C,D). We next tested whether gp120 increased α5-containing GABARs on the cell surface using immunocytochemistry with an α5-specific antibody. When immunocytochemistry was performed following paraformaldehyde (PFA) fixation without a permeabilization step, neurons treated with gp120 (4 h) exhibited a 21% increase in α5-immunoreactive fluorescent intensity relative to untreated neurons (Fig. 2E,F). These data are consistent with a previous report showing increased trafficking of α5-containing GABAARs to the cell surface induced by IL-1β (Wang et al., 2012) and suggest that the surface expression of α5 subunits is selectively upregulated following treatment with gp120.
Figure 2: Upregulation of tonic inhibition is mediated by α5-containing GABAARs.
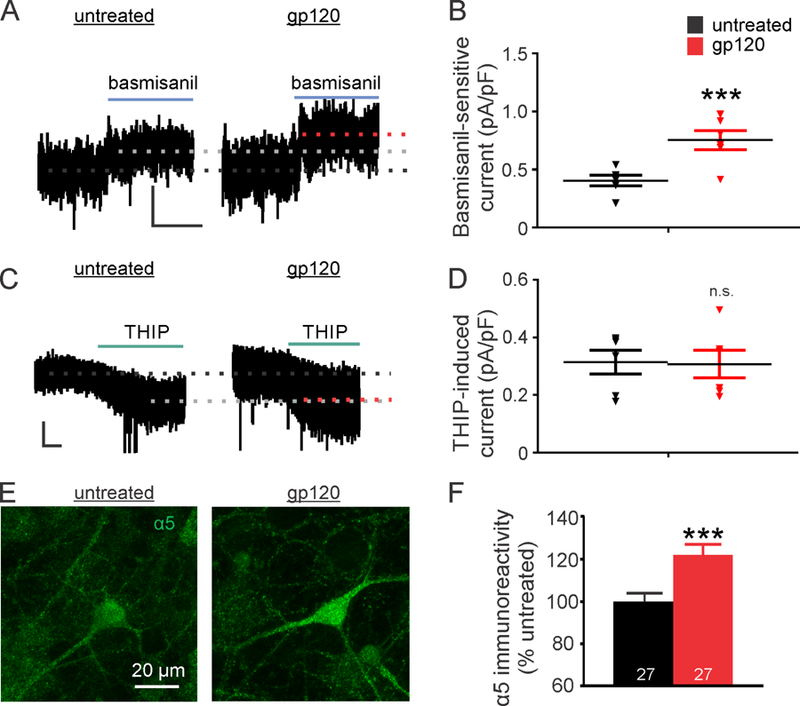
(A) Representative traces show currents sensitive to basmisanil, an α5-containing GABAAR inverse agonist (10 nM) in untreated and gp120-treated cells (scale bar: 40 pA, 10 s). (B) Basimisanil-sensitive currents normalized to whole-cell capacitance from gp120-treated and untreated cells are compared (Unpaired two-tailed t-test; t(10)=3.77, p=0.004). (C) Representative traces show gp120 did not change the amplitude of currents evoked by the agonist for δ-containing GABAARs, THIP (1 µM), compared to untreated controls (scale bar: 10 pA, 10 s). (D) THIP evoked current densities were similar in control and gp120-treated cells (Unpaired two-tailed t-test; t(10)=0.12, p=0.9). (E) Representative images show surface expression of α5-immunoreactivity increased in gp120 treated relative to untreated cells. (F) The average immunofluorescence intensity from the soma of gp120-treated cells increased relative to untreated somata (Unpaired two-tailed t-test; t(52)=3.417, p=0.001). Data are shown as individual data points with mean ± SEM (B,D) or expressed as mean ± SEM (F). ***p<0.001 compared to untreated group.
3.3. gp120 activates CXCR4 to release IL-1β from microglia to upregulate GABAergic signaling
The IIIB strain of the HIV-1 envelope protein gp120 binds to its co-receptor C-X-C chemokine receptor type 4 (CXCR4) and CD4 to initiate viral entry. The interaction of gp120 with CXCR4 activates microglia to release inflammatory cytokines that affect neurons (Kim et al., 2011; Viviani et al., 2006; Zhang and Thayer, 2018). To determine whether this receptor was involved in the gp120-induced increase in bicuculline-sensitive current, we co-treated neuronal cultures with the CXCR4 antagonist, AMD3100 (1 µM) and gp120. AMD3100 prevented the increase in bicuculline-sensitive current (4 h) (Fig. 3A). CXCR4 is expressed in microglia, astrocytes, and neurons (Bonavia et al., 2003), which are all present in our primary hippocampal cultures. To test whether microglia were necessary for the effects of gp120, we eliminated microglia from the culture by treating with Leucine-Methyl-Ester (LME; 25 mM for 1 h) 24 h prior to treating with gp120. LME treatment reduced the number of microglia by 95% compared to DMEM wash controls, as determined by immunocytochemistry with the Ox-42 antibody (Fig. 3B). Elimination of microglia prior to treatment with gp120 prevented the increase in bicuculline-sensitive current (4 h) compared to DMEM wash controls (Fig. 3C).
Figure 3: gp120 activates CXCR4 to stimulate microglia and increase tonic inhibition.
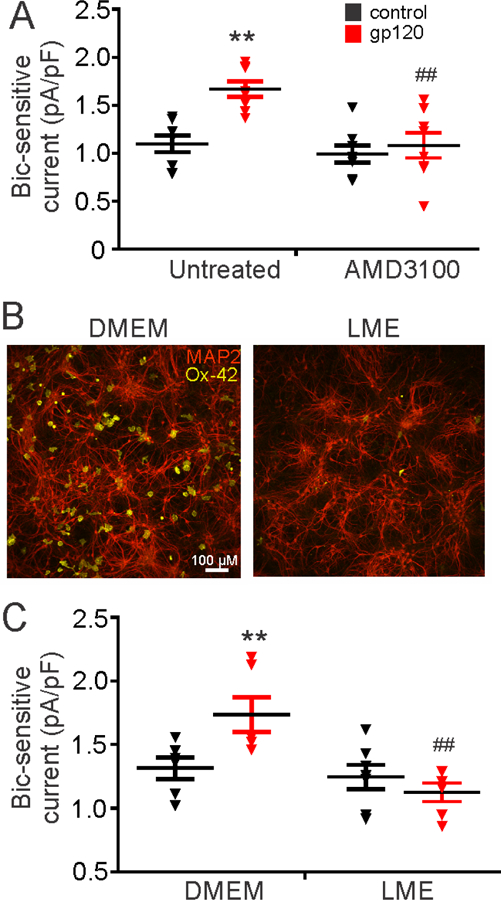
(A) The CXCR4 antagonist, AMD3100 (1 µM), inhibits gp120-induced (600 pM) increases in bicuculline-sensitive holding current normalized to whole-cell capacitance (two-way ANOVA; F(1,28)=6.00, p=0.02). (B) Representative images showing microglia labeled by Ox-42 antibody (yellow) were depleted relative to MAP2 immunoreactive neurons (red) 24 h after treatment with LME (25 mM for 1 h) compared to DMEM wash controls. (C) Removal of microglia with LME blocks gp120-induced increases in tonic current (two-way ANOVA; F(1,21)=7.63, p=0.01) compared to DMEM wash. Data are expressed as individual data points with mean ± SEM. Two-way ANOVAs were followed by Tukey’s post hoc test, **p<0.01 compared to untreated or DMEM control. ##p<0.01 compared to gp120 alone (untreated or DMEM).
The interaction of gp120 with CXCR4 on microglia can evoke the release of inflammatory cytokines such as IL-1β. IL-1β acts directly on neurons to increase currents mediated by extrasynaptic GABAARs (Wang et al., 2012). Here we examined the role of IL-1β in the gp120-induced up-regulation of GABAergic signaling. Treatment with IL-1β (20 ng/mL) for 20 min significantly increased the bicuculline-sensitive shift in holding current by 56 ± 10% (Fig. 4A,B), in close agreement to a previous study (Wang et al., 2012). Thus, IL-1β mimics the effects of gp120 on tonic inhibitory currents. To determine whether the gp120-induced increase in inhibition is mediated by IL-1β, cultures were co-treated with 1 µg/ml IL-1 receptor antagonist (IL-1ra) and gp120 (600 pM). IL-1ra prevented the gp120-induced increase in bicuculline-sensitive currents (Fig. 4C,D). Collectively, these results indicate that gp120 activates CXCR4 on microglia to evoke the release of IL-1β that acts on its receptor to alter tonic inhibition.
Figure 4: gp120-induced release of IL-1β increases tonic inhibition.

(A) Representative traces show IL-1β (20 ng/mL for 20 min) alone increases the bicuculline sensitive current (scale bar: 50 pA, 30 s). (B) IL-1β significantly increases bicuculline-sensitive currents normalized to whole-cell capacitance compared to untreated cells (Mann-Whitney U-test; p=0.002). (C) Representative traces show that treatment with IL-1ra (1 µg/mL) blocks the gp120-induced increase in bicuculline-sensitive current. (D) IL-1ra significantly blocks the gp120-induced increase in bicuculline-sensitive current normalized to whole cell capacitance (two-way ANOVA; F (1,19)=8.19, p=0.01). Data are expressed as individual data points with mean ± SEM. Two-way ANOVA was followed by Tukey’s post hoc test, *p<0.05, **p<0.01 compared to untreated control. #p<0.05 compared to gp120 alone (untreated).
3.4. The gp120-induced increase in tonic GABA-evoked current requires activation of neuronal p38 MAPK but not gene regulation.
Activation of the p38 MAPK pathway was previously shown to increase tonic GABAAR current (Wang et al., 2012). Therefore, we tested whether inhibition of p38 affected the gp120-induced increase in bicuculline-sensitive current. Treatment with the p38 inhibitor SB203,580 (10 µM) prevented the gp120-induced increase in tonic inhibition (Fig. 5A). The gp120-evoked increase was not affected by the inactive analog SB202474 (10 µM) (Fig. 5B). HIV gp120 can activate p38 MAPK in microglia and in neurons as a result of IL-1β release and (Medders et al., 2010; Schieven, 2005; Srinivasan et al., 2004). Thus, we employed a genetic approach to complement the pharmacological approach and to examine p38 signaling specifically within neurons. RapR-p38 is a genetically modified p38 kinase that acts as a dominant negative when expressed in the presence of wild type p38. Because CaPO4 transfection has a low efficiency (Kim et al., 2011) we could limit patch-clamp experiments to a neuron that was the only transfected cell in the field (0.085 mm2). Thus, the effects of RapR-p38 expression were limited to the neuron studied. In neurons expressing RapR-p38, gp120 failed to increase the amplitude of the bicuculline-sensitive current (Fig. 5C). RapR-p38 is activated by rapamycin, enabling regulation of p38 activity independent of its upstream physiological signaling pathway (Karginov et al., 2010). In cells expressing RapR-p38 and tdTomato, 0.2 µM rapamycin increased the bicuculline-sensitive current by 32 ± 11 % compared to rapamycin-treated cells expressing tdTomato (Fig. 5D). These results show that tonic inhibition is upregulated by gp120-induced activation of p38 MAPK in neurons. P38 MAPK has a large number of downstream phosphorylation targets, many of which modify gene expression (Whitmarsh, 2007). To determine whether the gp120-induced increase in inhibition requires translation, we pre-treated the culture with the protein synthesis inhibitor, cycloheximide (CHX) (10 µM). CHX did not prevent the gp120-induced increase in bicuculline-sensitive current (Fig. 5E). These data show that GABAAR-mediated tonic inhibition is upregulated by gp120-induced activation of p38 MAPK but not through its actions on gene expression pathways.
Figure 5: Neuronal p38 MAPK activation is required for gp120-induced increases in tonic inhibition.

(A) The p38 antagonist SB203,580 (10 µM) inhibits gp120-induced increases in bicuculline-sensitive current normalized to whole-cell capacitance (two-way ANOVA; F (1,21)=9.14, p=0.006). (B) SB202,474 (10 µM), an inactive analog of SB203580, does not affect the gp120-induced increase in bicuculline-sensitive current (two-way ANOVA; F(1,17)=1.09, p=0.3). (C) RapRp38 expression prevents the gp120-induced increase in tonic inhibition (two-way ANOVA; F(1,27)=5.02, p=0.03). (D) Treatment with rapamycin (200 nM) increases tonic inhibition in RapRp38 expressing cells significantly more than cells expressing tdTomato alone (two-way ANOVA; F(1,22)=6.08, p=0.02). (E) Cycloheximide (CHX) (10 µM) does not affect the gp120-induced increase in tonic inhibition (two-way ANOVA, F(1,24)=0.003, p=0.9). Data are expressed as individual data points with mean ± SEM. Two-way ANOVAs were followed by Tukey’s post hoc test, *p<0.05, **p<0.01, ***p<.001 compared to untreated control. # p<0.05, ##p<0.01, compared to gp120 (A,C) or rapamycin (D) alone (untreated or TdTomato).
4. Discussion
Excess GABA-mediated inhibition contributes to the cognitive impairment observed in many neurodegenerative conditions (Fernandez et al., 2007; Hines et al., 2012; Wu et al., 2014) and, based on the results described here, we suggest that it could exacerbate cognitive decline in HAND patients. We used an in vitro model that captures many of the synaptic changes observed in HAND to study the effects of the HIV envelope protein gp120 on GABAergic signaling. We found that in addition to its known actions on synaptic function (Green et al., 2018), gp120 activates the p38 MAPK pathway to increase tonic inhibition mediated by extrasynaptic α5-containing GABAARs in hippocampal neurons (Fig. 6). Excess tonic inhibition has been linked to impaired cognitive function (Farrant and Nusser, 2005) and thus, targeting these receptors may provide a novel approach for the treatment of HAND.
Figure 6: Hypothetical signaling pathway: HIV gp120 activates microglial and neuronal signaling pathways to upregulate α5-containing GABAARs.

Inhibition of CXCR4 with AMD3100 prevents gp120-induced changes in GABAAR signaling. Because gp120IIIB binds to CXCR4 (Islam et al., 2013; Moore et al., 1997) we propose that the signaling cascade starts with this receptor. While multiple cell types including neurons, astrocytes, and microglia express CXCR4, LME treatment to remove microglia prevented gp120-induced changes in GABAAR signaling in neurons. Thus, gp120 does not act directly on neurons to evoke the inhibitory current and microglia are necessary and upstream from the neuronal response. Additionally, because IL-1ra blocked the gp120-induced increase in tonic inhibition and microglia release IL-1β in response to gp120 (Lisi et al., 2011), we placed microglia upstream from the neuron. Finally, p38 signaling in neurons is necessary for the gp120-induced increases in tonic inhibition as shown by RapRp38 expression in neurons. P38 activation is a canonical downstream effector of the IL-1R (Weber et al., 2010), consistent with IL1Rs on neurons mediating the response. P38 activation in other cell types may contribute to upstream steps in the pathways such as IL-1β release from microglia (Schieven, 2005), although these roles were not tested in this study. Activation of p38 increased α5-containing GABAARs on the neuronal cell surface without the need for protein synthesis; the final steps through which p38 regulates GABAAR trafficking are not known. Selective inhibitors are shown in red.
Because HIV does not infect neurons, the neurotoxicity in HAND is thought to result from the neuroinflammation evoked by the release of viral proteins and cytokines from infected microglia and macrophages (Ellis et al., 2007; Saylor et al., 2016). Viral tropism is determined by gp120 binding to CD4 and a chemokine co-receptor. The IIIB strain of gp120 used in this study binds to CXCR4 (Islam et al., 2013; Moore et al., 1997), consistent with inhibition of gp120-evoked effects by a CXCR4 receptor antagonist. Recombinant gp120IIIB has previously been shown to induce release of inflammatory cytokines, including IL-1β, from human monocytes (Clouse et al., 1991). CCR5 tropic gp120s induce release of IL-1β from microglia (Ashraf et al., 2014; Walsh et al., 2014). Thus, CCR5-preferring strains of gp120 would also be predicted to upregulate α5-containing GABAARs. Furthermore, neuronal injury resulting from expression of gp120IIIB is prevented in CCR5 knockout mice, suggesting CCR5 could also participate in the inflammatory response evoked by CXCR4-preferring strains of HIV (Maung et al., 2014).
The effects observed were likely caused by CXCR4-mediated activation on microglia and subsequent IL-1β release. Removal of microglia using LME completely blocked the gp120-induced increase in tonic inhibition indicating microglia are essential for gp120-induced changes in GABAR signaling. Astrocytes have also been shown to play a significant role in gp120-induced toxicity (Bezzi et al., 2001) and we cannot rule out the possibility that they contributed to the increase in tonic inhibitory current. However, the signaling pathway proposed here (Fig. 6), in which microglia release of IL-1β that acts on neurons, is the most parsimonious pathway consistent with the results of this study. This model is consistent with prior studies from our laboratory and others showing that the highly potent (picomolar) effects that result from prolonged exposure to gp120 are indirect (Kim et al., 2011; Meucci and Miller, 1996; Viviani et al., 2006; Yang et al., 2013), whereas higher concentrations will elicit direct actions on neurons (Hesselgesser et al., 1998; Teodorof et al., 2014; Wenzel et al., 2017).
IL-1β alone increased tonic inhibition, as previously reported (Wang et al., 2012), effectively mimicking the actions of gp120. Neurons express IL-1 receptors (Vezzani et al., 2011) and upregulation of tonic inhibition was blocked selectively by expression of a dominant negative form of p38 in neurons, suggesting that key steps in the downstream regulatory pathway are located within neurons (Fig. 6). However, we cannot exclude contributions from IL-1 receptors and p38 signaling in other cells types such as astrocytes (Ravizza and Vezzani, 2006) and microglia (Medders et al., 2010). P38 signaling in both microglia and neurons is necessary for gp120-induced cell death (Medders et al., 2010), and p38 is required for microglial production of inflammatory cytokines such as IL-1β (Schieven, 2005). We show here that activation of p38 MAPK in neurons is required to upregulate α5-containing GABAARs, our study does not address whether p38 in other cell types also contributed to the response.
The gp120-induced increase in tonic inhibition was observed within 4 h and persisted during a 24 h exposure. This IL-1β-mediated increase in tonic current did not require protein synthesis and may be an early response to inflammation. It required activation of p38 MAPK as indicated by both pharmacological and dominant negative approaches. P38 is a known downstream effector of IL-1Rs although, how p38 regulates GABAARs is not clear. Immunocytochemistry experiments indicated that treatment with gp120 increased surface expression of α5 GABAAR subunits. Because inhibition of protein synthesis did not block the gp120-induced increase in tonic inhibition, p38 must alter the trafficking of α5-containing GABAARs. This finding is consistent with a previous study that found exposure to IL-1β increased trafficking of α5-containing GABAARs to the cell surface (Wang et al., 2012). Protein kinases such as CamKii and PKC increase trafficking of GABAARs to the cell surface by phosphorylating the β3 and α4 subunits, respectively (Abramian et al., 2014; Saliba et al., 2012). However, p38 mediated phosphorylation of GABAAR subunits has not been reported so whether p38 acts directly or via an intermediary step is not clear.
Selective pharmacological inhibition of tonic currents indicated that the effects of gp120 are predominantly mediated by α5-containing GABAARs. The other dominant subtype that contributes to tonic current in the hippocampus, the δ-containing GABAAR, was unaffected by gp120 treatment. Additionally, immunocytochemistry experiments showed an increase in surface expression of α5-containing GABAARs. While other subtypes of GABAARs may also be upregulated, the α5-containing GABAAR appears to be the predominant subtype upregulated by gp120. Mice deficient in the α5 subunit actually perform better in hippocampal-dependent cognitive tests than their wildtype counterparts (Collinson et al., 2002). Indeed, reducing tonic inhibition ameliorates cognitive impairment in animal models of Down syndrome (Vidal et al., 2018), Alzheimer’s disease (Wu et al., 2014) and stroke (Clarkson et al., 2010). Additionally, pharmacological inhibition of α5-containing GABAARs is well tolerated in rodents and humans (Rudolph and Mohler, 2014), in contrast to inhibition of synaptic GABAA receptors which produces anxiety and seizures (Baram and Snead, 1990; Sanders and Shekhar, 1995). Thus, this study has important clinical implications because the drug used to inhibit the tonic inhibitory currents, basmisanil, is also being tested as a therapeutic to improve cognitive function in schizophrenia patients (NCT02953639; http://www.clinicaltrials.gov).
This report shows for the first time that gp120 upregulates α5-containing GABAARs, which may provide a selective pharmacological target within the GABAergic system of importance to HAND. Future studies will determine whether gp120-induced increases in tonically active GABAARs dampen neuronal excitability in vivo and impair cognitive function. If so, drugs that inhibit α5 GABAARs might improve cognitive function in HAND patients.
Highlights.
HIV envelope protein gp120 increases tonic inhibition through extrasynaptic GABAARs
α5-containing GABAARs on the cell surface are selectively upregulated by gp120
Increased tonic inhibition occurs via gp120-evoked release of IL-1β from microglia
Stimulation of IL-1 receptors activates p38 in neurons to increase tonic inhibition
Acknowledgements
This work was supported by a grant from the National Institute on Drug Abuse, National Institutes of Health (DA07304) to S.A.T. The authors thank Adrienne Jo for assistance with immunocytochemistry.
Abbreviations:
- CNQX
6-Cyano-7-nitroquinoxaline-2,3-dione disodium
- DMEM
Dulbecco’s modified Eagle’s Medium
- FITC
fluorescein isothiocyanate
- GABAAR
γ-aminobutyric acid type A receptor
- GFP
green fluorescent protein
- HAND
HIV-associated neurocognitive disorder
- HIV-1
human immunodeficiency virus type 1
- HBSS
Hanks’ balanced salt solution
- IL-1β
interleukin-1β
- MAPK
mitogen-activated protein kinase
- NMDA
N-methyl-D-aspartate
- SB202474
4-Ethyl-2(p-methoxyphenyl)-5-(4ʹ-pyridyl)-IH-imidazole
- SB203580
4-[5-(4-Fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-imidazol-4-yl]pyridine
- TTX
tetrodotoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Chemical compounds studied in this article: basmisanil, 1,1-Dioxidothiomorpholino)(6-((3-(4-fluorophenyl)-5-methylisoxazol-4-yl)methoxy)pyridin-3-yl)methanone (PubChem CID: 57336276)
Conflict of Interest
The authors declare no conflicts of interest.
References
- Abramian AM, Comenencia-Ortiz E, Modgil A, Vien TN, Nakamura Y, Moore YE, Maguire JL, Terunuma M, Davies PA, Moss SJ, 2014. Neurosteroids promote phosphorylation and membrane insertion of extrasynaptic GABAA receptors. Proc Natl Acad Sci U S A 111, 7132–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf T, Jiang W, Hoque MT, Henderson J, Wu C, Bendayan R, 2014. Role of anti-inflammatory compounds in human immunodeficiency virus-1 glycoprotein120-mediated brain inflammation. J Neuroinflammation 11, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Snead OC 3rd, 1990. Bicuculline induced seizures in infant rats: ontogeny of behavioral and electrocortical phenomena. Brain Res Dev Brain Res 57, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW, 2009. Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci 29, 12757–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A, 2001. CXCR4-activated astrocyte glutamate release via TNFa: amplification by microglia triggers neurotoxicity. Nature Neuroscience 4, 702–710. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Bajetto A, Barbero S, Pirani P, Florio T, Schettini G, 2003. Chemokines and their receptors in the CNS: expression of CXCL12/SDF-1 and CXCR4 and their role in astrocyte proliferation. Toxicol Lett 139, 181–189. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Mody I, 2012. Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MF, Gill AJ, Kolson DL, 2014. Neuropathogenesis of HIV-associated neurocognitive disorders: roles for immune activation, HIV blipping and viral tropism. Curr Opin HIV AIDS 9, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST, 2010. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouse KA, Cosentino LM, Wein KA, Pyle SW, Robbins PB, Hochstein HD, Natarajan V, Farrar WL, 1991. The HIV-1 gp120 envelope protein has the intrinsic capacity to stimulate monokine secretion. The journal of Immunology 147, 2892–2901. [PubMed] [Google Scholar]
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, Smith A, Otu FM, Howell O, Atack JR, McKernan RM, Seabrook GR, Dawson GR, Whiting PJ, Rosahl TW, 2002. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci 22, 5572–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E, 2007. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci 8, 33–44. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z, 2005. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 6, 215–229. [DOI] [PubMed] [Google Scholar]
- Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, Garner CC, 2007. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci 10, 411–413. [DOI] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF, 2013. Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry 73, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill AJ, Kolson DL, 2014. Chronic inflammation and the role for cofactors (hepatitis C, drug abuse, antiretroviral drug toxicity, aging) in HAND persistence. Curr HIV/AIDS Rep 11, 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MV, Raybuck JD, Zhang X, Wu MM, Thayer SA, 2018. Scaling Synapses in the Presence of HIV. Neurochem Res [DOI] [PMC free article] [PubMed]
- Groen MR, Paulsen O, Perez-Garci E, Nevian T, Wortel J, Dekker MP, Mansvelder HD, van Ooyen A, Meredith RM, 2014. Development of dendritic tonic GABAergic inhibition regulates excitability and plasticity in CA1 pyramidal neurons. J Neurophysiol 112, 287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Thayer SA, 2013. Human immunodeficiency virus-1 Tat protein increases the number of inhibitory synapses between hippocampal neurons in culture. J Neurosci 33, 17908–17920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselgesser J, Taub D, Baskar P, Greenberg M, Hoxie J, Kolson DL, Horuk R, 1998. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol 8, 595–598. [DOI] [PubMed] [Google Scholar]
- Hines RM, Davies PA, Moss SJ, Maguire J, 2012. Functional regulation of GABAA receptors in nervous system pathologies. Curr Opin Neurobiol 22, 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Banks WA, 2015. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav Immun 45, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam S, Hoque SA, Adnan N, Tanaka A, Jinno-Oue A, Hoshino H, 2013. X4-tropic human immunodeficiency virus IIIB utilizes CXCR4 as coreceptor, as distinct from R5X4-tropic viruses. Microbiol Immunol 57, 437–444. [DOI] [PubMed] [Google Scholar]
- Jones MV, Bell JE, Nath A, 2000. Immunolocalization of HIV envelope gp120 in HIV encephalitis with dementia. Aids 14, 2709–2713. [DOI] [PubMed] [Google Scholar]
- Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM, 2010. Engineered allosteric activation of kinases in living cells. Nat Biotechnol 28, 743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA, 2001. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410, 988–994. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Shin AH, Thayer SA, 2011. Activation of cannabinoid type 2 receptors inhibits HIV-1 envelope glycoprotein gp120-induced synapse loss. Mol Pharmacol 80, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Keller CA, 2004. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther 102, 195–221. [DOI] [PubMed] [Google Scholar]
- Maung R, Hoefer MM, Sanchez AB, Sejbuk NE, Medders KE, Desai MK, Catalan IC, Dowling CC, de Rozieres CM, Garden GA, Russo R, Roberts AJ, Williams R, Kaul M, 2014. CCR5 knockout prevents neuronal injury and behavioral impairment induced in a transgenic mouse model by a CXCR4-using HIV-1 glycoprotein 120. J Immunol 193, 1895–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medders KE, Sejbuk NE, Maung R, Desai MK, Kaul M, 2010. Activation of p38 MAPK is required in monocytic and neuronal cells for HIV glycoprotein 120-induced neurotoxicity. J Immunol 185, 4883–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Miller RJ, 1996. Gp120-induced neurotoxicity in hippocampal pyramidal neuron cultures - protective action of tgf-beta-1. Journal of Neuroscience 16, 4080–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA, 2003. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron 38, 433–445. [DOI] [PubMed] [Google Scholar]
- Moore JP, Trkola A, Dragic T, 1997. Co-receptors for HIV-1 entry. Curr Opin Immunol 9, 551–562. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, 2016. How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Vezzani A, 2006. Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience 137, 301–308. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Mohler H, 2014. GABAA receptor subtypes: Therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu Rev Pharmacol Toxicol 54, 483–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba RS, Kretschmannova K, Moss SJ, 2012. Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. Embo J 31, 2937–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SK, Shekhar A, 1995. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav 52, 701–706. [DOI] [PubMed] [Google Scholar]
- Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ, McArthur JC, 2016. HIV-associated neurocognitive disorder - pathogenesis and prospects for treatment. Nat Rev Neurol 12, 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieven GL, 2005. The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem 5, 921–928. [DOI] [PubMed] [Google Scholar]
- Serantes R, Arnalich F, Figueroa M, Salinas M, Andres-Mateos E, Codoceo R, Renart J, Matute C, Cavada C, Cuadrado A, Montiel C, 2006. Interleukin-1beta enhances GABAA receptor cell-surface expression by a phosphatidylinositol 3-kinase/Akt pathway: relevance to sepsis-associated encephalopathy. J Biol Chem 281, 14632–14643. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Yen JH, Joseph DJ, Friedman W, 2004. Cell type-specific interleukin-1beta signaling in the CNS. J Neurosci 24, 6482–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodorof C, Divakar S, Soontornniyomkij B, Achim CL, Kaul M, Singh KK, 2014. Intracellular mannose binding lectin mediates subcellular trafficking of HIV-1 gp120 in neurons. Neurobiol Dis 69, 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaney VE, Sanchez AB, Fields JA, Minassian A, Young JW, Maung R, Kaul M, 2018. Transgenic mice expressing HIV-1 envelope protein gp120 in the brain as an animal model in neuroAIDS research. J Neurovirol 24, 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L, 1994. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 367, 188–193. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Maroso M, Balosso S, Sanchez M-A, Bartfai T, 2011. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain, Behavior, and Immunity 25, 1281–1289. [DOI] [PubMed] [Google Scholar]
- Vidal V, Garcia-Cerro S, Martinez P, Corrales A, Lantigua S, Vidal R, Rueda N, Ozmen L, Hernandez MC, Martinez-Cue C, 2018. Decreasing the Expression of GABAA alpha5 Subunit-Containing Receptors Partially Improves Cognitive, Electrophysiological, and Morphological Hippocampal Defects in the Ts65Dn Model of Down Syndrome. Mol Neurobiol 55, 4745–4762. [DOI] [PubMed] [Google Scholar]
- Viviani B, Gardoni F, Bartesaghi S, Corsini E, Facchi A, Galli CL, Di Luca M, Marinovich M, 2006. Interleukin-1 beta released by gp120 drives neural death through tyrosine phosphorylation and trafficking of NMDA receptors. J Biol Chem 281, 30212–30222. [DOI] [PubMed] [Google Scholar]
- Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG, Broadhurst DI, Power C, 2014. Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology 11, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DS, Zurek AA, Lecker I, Yu J, Abramian AM, Avramescu S, Davies PA, Moss SJ, Lu WY, Orser BA, 2012. Memory deficits induced by inflammation are regulated by alpha5-subunit-containing GABAA receptors. Cell Rep 2, 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Wasiliew P, Kracht M, 2010. Interleukin-1 (IL-1) pathway. Sci Signal 3, cm1. [DOI] [PubMed] [Google Scholar]
- Wenzel ED, Bachis A, Avdoshina V, Taraballi F, Tasciotti E, Mocchetti I, 2017. Endocytic Trafficking of HIV gp120 is Mediated by Dynamin and Plays a Role in gp120 Neurotoxicity. J Neuroimmune Pharmacol 12, 492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ, 2007. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim Biophys Acta 1773, 1285–1298. [DOI] [PubMed] [Google Scholar]
- Wu X, Huang L, Wu Z, Zhang C, Jiang D, Bai Y, Wang Y, Chen G, 2013. Homeostatic competition between phasic and tonic inhibition. J Biol Chem 288, 25053–25065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Guo Z, Gearing M, Chen G, 2014. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat Commun 5, 4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Hu D, Xia J, Liu J, Zhang G, Gendelman HE, Boukli NM, Xiong H, 2013. Enhancement of NMDA receptor-mediated excitatory postsynaptic currents by gp120-treated macrophages: implications for HIV-1-associated neuropathology. J Neuroimmune Pharmacol 8, 921–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Thayer SA, 2018. Monoacylglycerol lipase inhibitor JZL184 prevents HIV-1 gp120-induced synapse loss by altering endocannabinoid signaling. Neuropharmacology 128, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu J, Xiong H, 2017. HIV-1 Glycoprotein 120 Enhancement of N-Methyl-D-Aspartate NMDA Receptor-Mediated Excitatory Postsynaptic Currents: Implications for HIV-1-Associated Neural Injury. J Neuroimmune Pharmacol 12, 314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]