

Abstract
We report a dynamic kinetic resolution (DKR) of chiral 4-pentenals by olefin hydroacylation. A primary amine racemizes the aldehyde substrate via enamine formation and hydrolysis. Then, a cationic Rh-catalyst promotes hydroacylation to generate α,γ-disubstituted cyclopentanones with high enantio- and diastereoselectivities.
Keywords: dynamic kinetic resolution, hydroacylation, dual catalysis, C–H activation, rhodium
Graphical Abstract
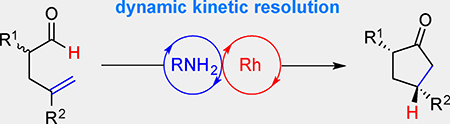
Dynamic duo: Racemic α-allyl aldehydes undergo stereoconvergent hydroacylation to generate α,γ-disubstituted cyclopentanones with high diastereo- and enantioselectivities. In this dynamic kinetic resolution, a primary amine catalyst racemizes the aldehyde substrate via enamine formation and hydrolysis, while a Rh-catalyst promotes cyclization.
By merging epimerization with asymmetric catalysis, chemists have developed powerful ways to convert racemic reagents into enantiopure precursors, including those used for making natural products and medicinal targets.[1] While most dynamic kinetic resolutions (DKR’s) feature hydrogenation[2a–c] or acylation,[2d,e] variants that exploit C–C bond formation remain rare.[3] Olefin hydroacylation is an atom-economical[4] route to ketones that achieves both C–H bond activation and C–C bond formation.[5] Herein, we disclose a DKR strategy to prepare α,γ-disubstituted cyclopentanones by intramolecular hydroacylation.
The first kinetic resolution of an α-chiral aldehyde was fortuitously discovered by James in 1983. While attempting to develop an enantioselective decarbonylation, the authors observed that 2-methyl-2-phenylpent-4-enal underwent intramolecular hydroacylation to furnish the corresponding cyclopentanone in up to 69% ee (Figure 1a).[6a,b] Fu described a parallel kinetic resolution of racemic 4-alkynals to generate a mixture of enantioenriched cyclopentenones and cyclobutanones.[6c] Most recently, Willis disclosed an kinetic resolution of β-thio aldehydes by intermolecular alkyne hydroacylation.[6d] Aldehydes bearing either α- or β-stereocenters undergo kinetic resolution. These early studies contribute to emerging kinetic resolutions that occur by C–H bond activation,[7] however, the theoretical yield for the enantiopure ketone products is limited to fifty percent. Despite the first resolution over three decades ago, the DKR of aldehydes by hydroacylation had yet to be achieved. In light of this challenge, we imagined combining aldehyde racemization with formyl C–H bond functionalization to invent DKR’s via hydroacylation.[8]
Figure 1.

Resolutions of chiral aldehydes by hydroacylation.
We propose using an amine organocatalyst and a Rh-catalyst in tandem to produce α,γ-disubstituted cyclopentanones, a motif not yet accessible by hydroacylation (Figure 1b). Given that branched aldehydes readily undergo epimerization,[9] we reasoned a DKR variant of hydroacylation would be feasible. Since the substrate and product have similar acidities, one challenge would be to identify a catalyst that would rapidly and selectively epimerize the aldehyde reagent, in preference to the ketone product. If successful, this DKR by C–C bond formation would complement Buchwald’s DKR of cyclopentenones by asymmetric reduction.[10]
To test our hypothesis, we investigated the cyclization of aldehyde 1a (Table 1). Our initial studies included various bases, such as alkoxides and tertiary amines. The use of pyridine A1 and a Segphos-derived ligand (Segphos = 5,5’-bis(diphenylphosphino)-4,4’-bi-1,3-benzodioxole) provided an early lead (Table 1a), where bulkier phosphine substituents afforded higher reactivity (L2 and L3), presumably due to increased dispersive interactions.[11] The combination of L3 and A1 led to cyclopentanone 2a in 33% yield with high stereoselectivities (>20:1 dr, 94% ee). Aldehydes are known to form enamines with primary amines, and this reactivity has been used by List to achieve a DKR by reductive amination using aniline A2.[9b] We found that A2 promoted the hydroacylation with excellent reactivity (96%) but gave only 4:1 dr. However, aliphatic primary amines provided higher diastereocontrol with increased steric bulk: n-butylamine (A3) (65%, 10:1 dr), cyclohexylamine (A4) (50%, 16:1 dr), and 1-adamantylamine (A5) (73%, 14:1 dr). By using a lower loading of A5 (10 mol%) and switching the catalyst counter-ion to SbF6, 2a was obtained in high yield and stereocontrol (94%, >20:1 dr, >99% ee). The absolute configuration of 2a was determined to be (2R,4R) by X-ray crystallography.[12] To demonstrate the scalability of this DKR, we cyclized 1a on a gram-scale and obtained 2a in high yield and stereocontrol (89%, >20:1 dr, >99% ee).
Table 1.
Ligand and Amine Evaluation with α-Alkyl Aldehyde 1a[a]
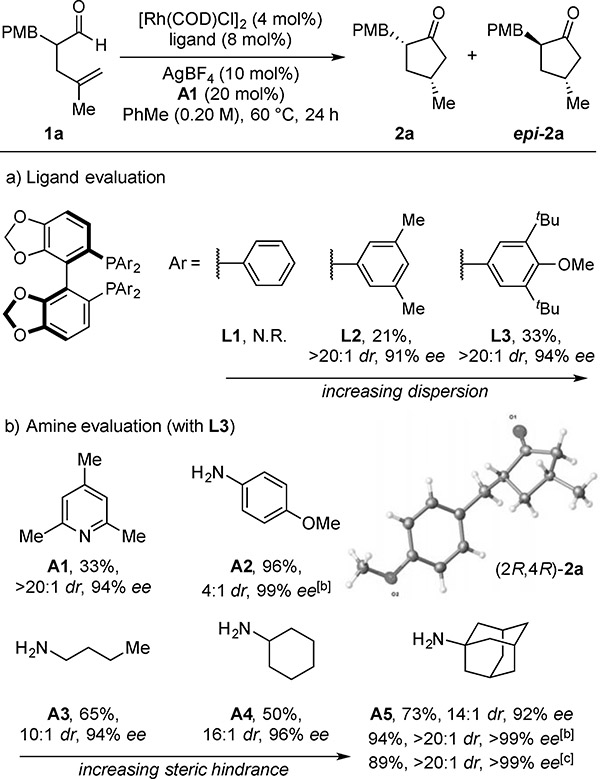 |
With 0.050 mmol of 1a.
Yields and diastereoselectivities were determined by 1H NMR analysis of the unpurified reaction mixture using triphenylmethane as an internal standard. Enantioselectivities (ee’s) were determined by chiral SFC analysis. [b] With 0.10 mmol of 1a. Reaction performed at 50 °C using AgSbF6 (10 mol%) and 10 mol% of the amine. [c] Isolated yield of 2a (4.6 mmol scale) using 2 mol% [Rh(COD)Cl]2, 4 mol% L3, 10 mol% AgSbF6, and 10 mol% A5 for 48 h. PMB = p-methoxybenzyl. COD = 1,5-cyclooctadiene.
We next examined the cyclization of various α-alkyl aldehydes (Table 2). These branched aldehydes undergo DKR with moderate to high reactivity (2b–2k, 52–94%), diastereocontrol (11–>20:1 dr), and enantiocontrol (82–>99% ee). This hydroacylation is chemoselective for the terminal olefin as styrenyl olefins (1g) and internal alkynes (1h), remain intact to afford cyclopentanones 2g and 2h (11–>20:1 dr, 94–95% ee). Placing bulkier alkyl substituents on the olefin led to diminished reactivity and diastereoselectivity (2j, 21%, 1:1 dr, 88% ee). However, high reactivity and stereoselectivities were restored by using JoSPOphos (L4) as the chiral ligand (89%, >20:1 dr, >99% ee). We also prepared a monosubstituted cyclic ketone (2k, 52%, 82% ee). A substrate containing an internal olefin (1l) failed to cyclize.
Table 2.
Hydroacylation Scope with α-Alkyl Aldehydes[a ]
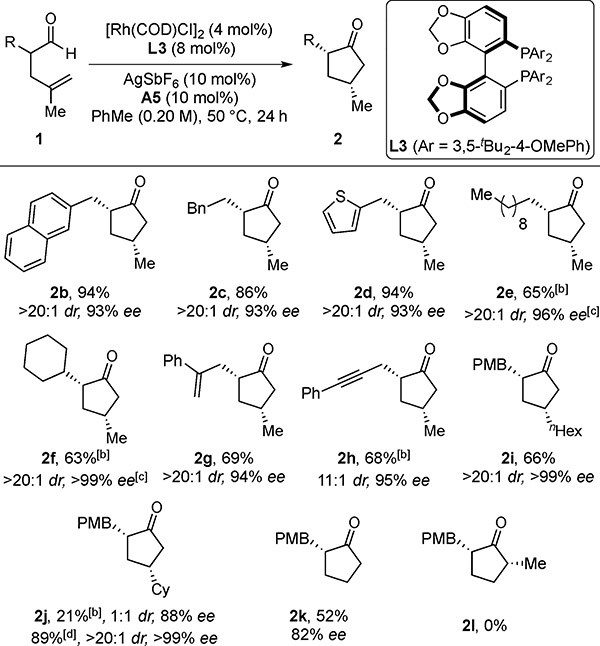 |
With 0.10 mmol of 1.
Isolated yields are given. Diastereoselectivities were determined by 1H NMR analysis of the unpurified reaction mixture. Enantioselectivities (ee’s) were determined by chiral SFC analysis. [b] Reaction performed at 60 °C. [c] SFC analysis performed with the tertiary alcohol after treatment with PhMgBr. [d] Reaction performed with L4 at 60 °C.
Aldehydes with styrenyl olefins (e.g. 3a) were slow to react with ligand L3 (Table 3). To overcome this limitation, we used amine A5 and ligand L4. This combination enabled the resolution of chiral aldehydes bearing a range of styrenyl olefins with excellent stereocontrol (4a–4h, >20:1 dr, 74–>99% ee). The absolute configuration of 4a is analogous to that of 2a, as determined by X-ray crystallography.[12a]
Table 3.
Hydroacylation Scope with Styrenyl Olefins[a]
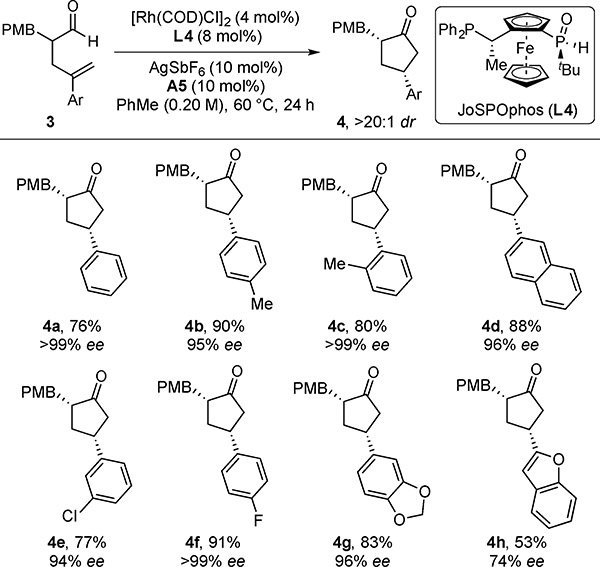 |
With 0.10 mmol of 3.
With 0.10 mmol of 3. Isolated yields are given. Diastereoselectivities were determined by 1H NMR analysis of the unpurified reaction mixture. Enantioselectivities (ee’s) were determined by chiral SFC analysis. (RP)-1-[(S)-tert-Butylphosphinoyl]-2-[(S)-1-(diphenylphosphino)ethyl]ferrocene (L4) is abbreviated as JoSPOphos.
In contrast to the previous aldehydes, we found that the DKR of α-aryl aldehydes 5 requires an aniline co-catalyst (Table 4). Using A5 and L3, 5a transformed into cyclopentanone 6a with high selectivity (>20:1 dr, >99% ee), albeit with low yield (24%). Switching to other biaryl ligand scaffolds produced similar results. In contrast, changing the amine to 2,6-disubstituted anilines A7 and A8 resulted in improved reactivity and diastereocontrol (78–80%, 13–14:1 dr).[13] Aniline A9, which is more sterically hindered, provided higher diastereocontrol (16:1 dr) but lower yield (68%). Using A8, we found that Garphos-derived ligand L5 promoted the formation of 6a in 87% yield with high stereoselectivities (>20:1 dr, >99% ee) (Garphos = 2,2’-bis(diphenylphosphino)-4,4’,6,6’-tetramethoxybiphenyl).
Table 4.
Ligand and Amine Evaluation with α-Aryl Aldehyde 5a[a]
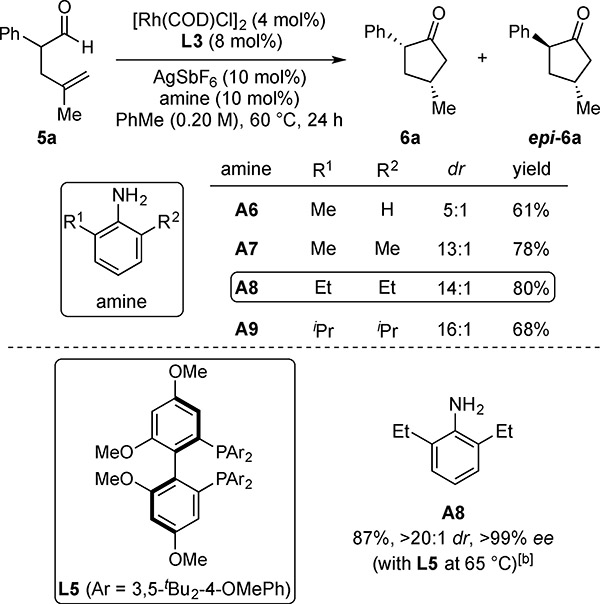 |
With 0.050 mmol 5a.
Yields and diastereoselectivities were determined by 1H NMR analysis of the unpurified reaction mixture using triphenylmethane as an internal standard. Enantioselectivities (ee’s) were determined by chiral SFC analysis. [b] Using 0.10 mmol of 5a.
We found that 6a epimerizes on silica and decomposes to form hydroxyketone 6aa and keto acid 6ab, which we isolated as the methyl ester (Scheme 1).[12a] This observation is consistent with those reported by Houminer and others that α-aryl cyclopentanones undergo oxidation via a hydroperoxide intermediate.[14] To circumvent this oxidation, we treat the reaction mixture with L-Selectride® to produce the all-syn cyclopentanol 7a with high diastereoselectivity (>20:1:1:1 dr) (Table 5). The absolute configuration of 7a was determined by X-ray crystallography after derivatization to the corresponding 3,5-dinitrobenzoic ester.[12a]
Scheme 1.

Decomposition of 6a.
Table 5.
Hydroacylation Scope with α-Aryl Aldehydes[a]
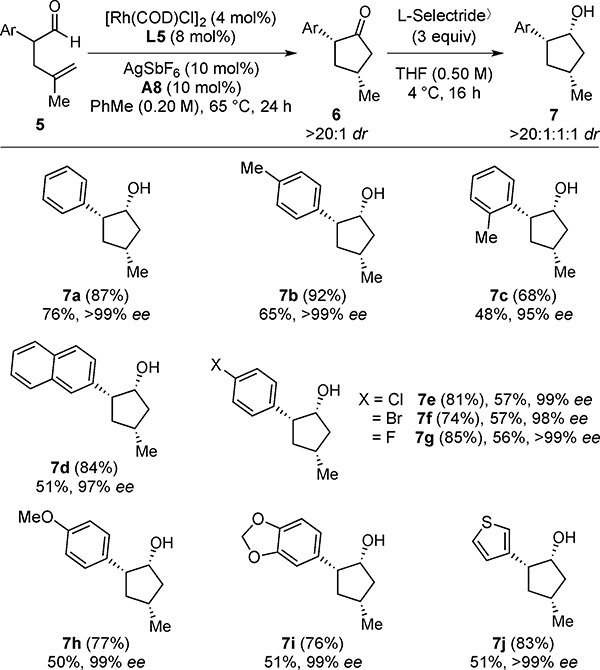 |
With 0.10 mmol of 5.
1H NMR yields of 6 are given in parentheses. Isolated yields over two steps are given of 7. Diastereoselectivities of each step were determined by 1H NMR analysis of the unpurified reaction mixture. Enantioselectivities (ee’s) were determined by chiral SFC analysis.
With this two-step protocol, various cyclopentanols can be prepared (7a–7j, >20:1:1:1 dr, 95–>99% ee) (Table 5). Cyclopentanols containing aryl halides (7e–7g) can be accessed with high stereoselectivities (>20:1:1:1 dr, 98–>99% ee). Electron-deficient (7g) and electron-rich arenes (7h and 7i) are tolerated. Cyclopentanols bearing heterocycles (7j) are obtained with excellent stereocontrol (>20:1:1:1 dr, >99% ee). By merging DKR with desymmetrization,[15] the α,β,γ-trisubstituted cyclopentanone 9 can be generated in 53% yield as a single stereoisomer (>20:1:1:1 dr, >99% ee) [Eq. (1)]. This example illustrates enantioselective construction of three contiguous stereocenters via a single C–H oxidation.
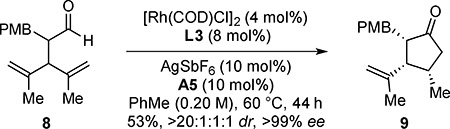 |
(1) |
We propose a mechanism involving two catalysts (Scheme 2). The primary amine catalyst condenses with aldehyde 1 to form an achiral enamine (A) that then undergoes hydrolysis. The R-enantiomer ((R)-1) undergoes oxidative addition with the Rh-catalyst to generate the Rh-acyl-hydride B. Subsequent migratory insertion makes metallacycle C, which undergoes reductive elimination to afford cyclopentanone 2. When 1a was subjected to the Rh-catalyst in the absence of amine A5, we observed hydroacylation with the same diastereo- and enantiocontrol (>20:1 dr, >99% ee), although in lower yield as expected (38%) (Scheme 3a). This experiment points to the aldehyde as being the substrate for hydroacylation, as opposed to the imine intermediate.[16] In the absence of aldehyde, 2a can be epimerized. When 2a (>20:1 dr) was subjected to the standard reaction conditions with L3 and A5, it was recovered with lower diastereoselectivity (14:1 dr). (Scheme 3b). When treated with n-butylamine (A3), 2a epimerized more rapidly (5:1 dr). Due to unfavorable steric interactions, enamine formation with the product should be more challenging with bulky amines. Moreover, the bulky amine should favor the less substituted enamine 2ab to avoid allylic strain (Scheme 3c).
Scheme 2.

Amine-catalyzed racemization and Rh-catalyzed hydroacylation
Scheme 3.

Amine-free hydroacylation and product epimerization.
An isotope labeling experiment with 1a-d showed that the deuterium label is fully incorporated at the γ-position (Scheme 4a). This result is consistent with a highly regioselective olefin insertion step. We reason that reductive elimination is the turnover-limiting step. When a 1:1 mixture of 1a and 1a-d was used for the reaction, no primary kinetic isotope effect (KIE) was observed (Scheme 4b), which suggests that oxidative addition and migratory insertion are not turnover-limiting.[17]
Scheme 4.

Isotopic labeling and KIE experiments.
By using tandem catalysis,[18] we have added a dynamic twist to hydroacylation. The empirical trends we observed for catalyst choice provides a useful guide for accessing a wide range of enantiopure cyclopentanones that are relatively unique.[19] Our study contributes to a growing class of DKR’s that feature aldehyde racemization.[9] The identification of an efficient amine-catalyst for racemization will impact future studies that feature DKR of aldehydes.
Supplementary Material
Acknowledgements
Funding provided by UC Irvine and the National Institutes of Health (R35GM127071). Z.C. and H.M.H.N. are grateful for Allergan Fellowships. We thank Kyle B. Brook and Alexander Y. Jiu for help with initial studies and characterization data. We acknowledge Dr. Curtis Moore and Dr. Arnold Rheingold (UC San Diego) for X-ray crystallographic analysis.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Zhiwei Chen, Department of Chemistry, University of California, Irvine, Irvine, California 92697 (United States).
Yusuke Aota, Department of Chemistry, University of California, Irvine, Irvine, California 92697 (United States); Department of Chemistry, Graduate School of Sciences, Kyoto University, Sakyo, Kyoto 606-8502 (Japan).
Hillary M. H. Nguyen, Department of Chemistry, University of California, Irvine, Irvine, California 92697 (United States)
Vy M. Dong, Department of Chemistry, University of California, Irvine, Irvine, California 92697 (United States)
References
- [1].a) For selected reviews on dynamic kinetic resolution, see: Bhat V, Welin ER, Guo X, Stoltz BM, Chem. Rev 2017, 117, 4528–4561; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nakano K, Kitamura M in Separation of Enantiomers: Synthetic Methods (Ed. Todd M), Wiley-VCH: Weinheim, 2014, pp. 161–216; [Google Scholar]; c) Verho O, Bäckvall J-E, J. Am. Chem. Soc 2015, 137, 3996–4009; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pàmies O, Bäckvall J-E, Chem. Rev 2003, 103, 3247–3262. [DOI] [PubMed] [Google Scholar]
- [2].a) For selected examples using hydrogenation, see: Steward KM, Gentry EC, Johnson JS, J. Am. Chem. Soc 2012, 134, 7329–7332; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang DS, Chen Q-A, Li W, Yu C-B, Zhou Y-G, Zhang X, J. Am. Chem. Soc 2010, 132, 8909–8911; [DOI] [PubMed] [Google Scholar]; c) Xie J-H, Liu S, Kong W-L, Bai W-J, Wang X-C, Wang L-X, Zhou Q-L, J. Am. Chem. Soc 2009, 131, 4222–4223. [DOI] [PubMed] [Google Scholar]; d) For selected examples using acylation, see:Piotrowski DW, Kamlet AS, Dechert-Schmitt A-MR, Yan J, Brandt TA, Xiao J, Wei L, Barrila MT, J. Am. Chem. Soc 2016, 138, 4818–4823; [DOI] [PubMed] [Google Scholar]; e) Lee SY, Murphy JM, Ukai A, Fu GC, J. Am. Chem. Soc 2012, 134, 15149–15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].For a review of C–C bond formations using DKR, see Bartlett SL, Johnson JS, Acc. Chem. Res 2017, 50, 2284–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]; Also, see Doyle AG, Jacobsen EN, Angew. Chem. Int. Ed 2007, 46, 3701–3705. [DOI] [PubMed] [Google Scholar]
- [4].Trost BM, Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- [5].a) For selected reviews, see: Murphy SK, Dong VM, Chem. Commun 2014, 50, 13645–13649; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Willis MC, Chem. Rev 2010, 110, 725–748. [DOI] [PubMed] [Google Scholar]
- [6].a) James BR, Young CG, J. Chem. Soc., Chem. Commun 1983, 1215–1216; [Google Scholar]; b) James BR, Young CG, J. Organomet. Chem 1985, 285, 321–332; [Google Scholar]; c) Tanaka K, Fu GC, J. Am. Chem. Soc 2003, 125, 8078–8079; [DOI] [PubMed] [Google Scholar]; d) González-Rodríguez C, Parsons SR, Thompson AL, Willis MC, Chem. Eur. J 2010, 16, 10950–10954. [DOI] [PubMed] [Google Scholar]
- [7].a) For selected examples of kinetic resolutions by C–H functionalization, see: Xiao K-J, Chu L, Chen G, Yu J-Q, J. Am. Chem. Soc 2016, 138, 7796–7800; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zheng J, You S-L, Angew. Chem. Int. Ed 2014, 53, 13244–13247; [DOI] [PubMed] [Google Scholar]; c) Chu L, Xiao K-J, Yu J-Q, Science 2014, 346, 451–455; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Larrow JF, Jacobsen EN, J. Am. Chem. Soc 1994, 116, 12129–12130. [Google Scholar]
- [8].For a dynamic kinetic asymmetric transformation (DYKAT) of 1,3-disubstituted allenes using hydroacylation, see Osborne JD, Randell-Sly HE, Currie GS, Cowley AR, Willis MC, J. Am. Chem. Soc 2008, 130, 17232–17233. [DOI] [PubMed] [Google Scholar]
- [9].a) For selected dynamic kinetic resolutions of aldehydes, see: Xie J-H, Zhou Z-T, Kong W-L, Zhou Q-L, J. Am. Chem. Soc 2007, 129, 1868–1869; [DOI] [PubMed] [Google Scholar]; b) Hoffmann S, Nicoletti M, List B, J. Am. Chem. Soc 2006, 128, 13074–13075; [DOI] [PubMed] [Google Scholar]; c) Cheng X, Goddard R, Buth G, List B, Angew. Chem. Int. Ed 2008, 47, 5079–5081; [DOI] [PubMed] [Google Scholar]; d) Lee A, Michrowska A, Sulzer-Mosse S, List B, Angew. Chem. Int. Ed 2011, 50, 1707–1710. [DOI] [PubMed] [Google Scholar]
- [10].Jurkauskas V, Buchwald SL, J. Am. Chem. Soc 2002, 124, 2892–2893. [DOI] [PubMed] [Google Scholar]
- [11].For a review of London dispersion, see Wagner JP, Schreiner PR, Angew. Chem. Int. Ed 2015, 54, 12274–12296. [DOI] [PubMed] [Google Scholar]
- [12].a) See the Supporting Information for more details; b) CCDC 1869120 contains the supplementary crystallographic data for 2a.
- [13].Sterically encumbered anilines are also effective for racemizing α-alkyl aldehydes. Using L3 and A8, 1a cyclized to 2a with high diastereo- and enantioselectivity (86%, >20:1 dr, 96% ee).
- [14].a) Houminer Y, J. Org. Chem. 1985, 50, 786–789; [Google Scholar]; b) For selected additional reports, see:Schröder K, Join B, Amali AJ, Junge K, Ribas X, Costas M, Beller M, Angew. Chem. Int. Ed 2011, 50, 1425–1429; [DOI] [PubMed] [Google Scholar]; c) Paju A, Kanger T, Pehk T, Lopp M, Tetrahedron 2002, 58, 7321–7326. [Google Scholar]
- [15].a) For selected examples of desymmetrizations using olefin hydroacylation, see: Park J-W, Kou KGM, Kim DK, Dong VM, Chem. Sci 2015, 6, 4479–4483; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Phan DHT, Kou KGM, Dong VM J. Am. Chem. Soc 2010, 132, 16354–16355; [DOI] [PubMed] [Google Scholar]; c) Tanaka M, Imai M, Fujio M, Sakamoto E, Takahashi M, Eto-Kato Y, Wu XM, Funakoshi K, Sakai K, Suemune H, J. Org. Chem 2000, 65, 5806–5816. For an example using alkyne hydroacylation, see: [DOI] [PubMed] [Google Scholar]; d) Tanaka K, Fu GC, J. Am. Chem. Soc 2002, 124, 10296–10297. [DOI] [PubMed] [Google Scholar]
- [16].a) For selected examples of an olefin hydroacylation via an imine intermediate, see: Beletskiy EV, Sudheer C, Douglas CJ, J. Org. Chem 2012, 77, 5884–5893; [DOI] [PubMed] [Google Scholar]; b) Jun C-H, Lee D-Y, Lee H, Hong J-B, Angew. Chem. Int. Ed 2000, 39, 3070–3072; [DOI] [PubMed] [Google Scholar]; c) Jun C-H, Lee H, Hong J-B, J. Org. Chem 1997, 62, 1200–1201 [Google Scholar]
- [17].Simmons EM, Hartwig JF, Angew. Chem. Int. Ed. 2012, 51, 3066–3072. [DOI] [PubMed] [Google Scholar]
- [18].a) For selected reviews on tandem catalysis, see: Lohr TL, Marks TJ, Nat. Chem 2015, 7, 477–482; [DOI] [PubMed] [Google Scholar]; b) Wasilke J-C, Obrey SJ, Baker RT, Bazan GC, Chem. Rev 2005, 105, 1001–1020; [DOI] [PubMed] [Google Scholar]; c) Fogg DE, dos Santos EN, Coord. Chem. Rev 2004, 248, 2365–2379. [Google Scholar]
- [19].We evaluated each model aldehyde (1a, 3a, and 5a) using each of the ligand and amine combinations. See the Supporting Information for more details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.