

Abstract
Human polyomaviruses show relatively little genetic polymorphism between isolates, indicating that these viruses are genetically stable between hosts. However, it has become increasingly clear that intra-host molecular evolution is a feature of some polyomavirus (PyV) infections in humans. Mutations inducing premature stop codons in the early region of the integrated Merkel cell PyV genome lead to the expression of a truncated form of the large tumour (LT) antigen that is critical for the transformation of Merkel cell carcinoma (MCC) cells. Non-coding control region (NCCR) rearrangements and point mutations in virion protein (VP) 1 have been described in both JCPyV and BKPyV infections. In the context of JCPyV infection, molecular evolution at both these loci allows the virus to replicate effectively in the central nervous system, thereby leading to the development of progressive multifocal leukoencephalopathy (PML). In BKPyV infection, NCCR rearrangements have been linked to higher rates of virus replication in the kidney, and are proposed to play a direct causal role in the development of PyV-associated nephropathy. In all three of these infections, therefore, intra-host viral evolution appears to be an essential component of the disease process.
This article is part of the theme issue ‘Silent cancer agents: multi-disciplinary modelling of human DNA oncoviruses’.
Keywords: polyomavirus, virulence, viral evolution
1. Introduction
Polyomaviruses are small circular double-stranded DNA viruses that infect a variety of vertebrate hosts [1]. Among the 13 polyomaviruses so far classified as human polyomaviruses (HPyV) by the International Committee on Taxonomy of Viruses, 11 have been found to routinely infect humans, and six are associated with diseases. The three most extensively studied HPyV are BKPyV (HPyV1), which causes polyomavirus nephropathy (PVAN) in kidney transplant (KTx) recipients and haemorrhagic cystitis (HC) in haematopoietic stem cell transplant (HSCT) recipients [2,3], JCPyV (HPyV2), the aetiological agent of progressive multifocal leukoencephalopathy (PML) [4] and MCPyV (HPyV5), which is responsible for the most frequent form of Merkel cell carcinoma (MCC) [5]. Even in these three cases, however, the vast majority of infections are completely asymptomatic, as seroprevalence for all three of these viruses is greater than 50% in adult populations worldwide [6]. Host immunosuppression is the critical cofactor in BKPyV and JCPyV pathology, with pharmacological immunosuppression to prevent graft rejection essential for BKPyV reactivation in KTx recipients, while PML is observed in HIV/AIDS patients or under immunosuppressive and immunomodulatory medication. Similarly, immunosuppressed states including AIDS [7], solid organ transplantation [8] and haematological malignancies are associated with increased incidence of MCC [9].
The overall pattern of asymptomatic infection, with rare cases of disease mainly related to host immunosuppression, is consistent with the view that HPyV have coevolved with their hosts over long periods of time, resulting in loss of virulence. Nevertheless, even within the at-risk immunosuppressed population, BKPyV and JCPyV reactivation only leads to overt pathology in a minority of cases. That is, even when the host population is homogeneous, there is considerable heterogeneity in the outcome of virus reactivation, ranging from entirely asymptomatic replication with virus secretion in the urine, to organ dysfunction (PVAN), or even lethal (PML) pathologies. The objective of the present review is to evaluate to what extent within-host molecular evolution contributes to polyomavirus pathology in humans and how this relates to the host immune landscape.
2. T antigens in HPyV infection: focus on MCPyV
Numerous studies have provided evidence that MCPyV is the causal agent of MCC, a rare but aggressive skin cancer that develops in the elderly. The transforming potential of MCPyV is owing to the expression of genes located within the ‘early region’ of the viral genome, the T antigens [10,11]. The two main spliced products of the early region are the large T (LT) and the small T (ST) antigens whose known functions are the coordination of the viral infectious cycle, cell cycle progression and the malignant transformation of the host cell [12]. LT is a multifunctional protein composed of several domains including, from the N-terminal to the C-terminal region, a DNAJ domain, a retinoblastoma protein (pRb)-binding domain, an origin-binding domain (OBD) indispensable for viral DNA replication and the helicase/ATPase domain that activates viral and cellular transcription.
Using a subtractive transcriptomic approach, Feng and colleagues demonstrated that MCPyV is clonally integrated in 80% of MCC [5]. Integration impairs expression of late proteins and the late-encoded MCV-mir-M1 [13,14]. Loss of MCV-mir-M1 allows early region transcripts to accumulate, and therefore leads to upregulated expression of LT and ST after integration. Comparison of MCPyV genome sequences showed that truncation of the LT protein is a hallmark of MCPyV-infected transformed Merkel cells [15]. In this report, LT from nine MCC were sequenced and analysed. Mutant LT antigens retained the ability to perturb cell growth through Rb antagonism but were unable to stimulate MCPyV genome replication, because of the loss of essential C-terminal LT domains. The authors concluded that mutations were selected in MCC to avoid DNA replication of integrated virus that could be deleterious to tumour cell survival by the induction of innate responses, including the DNA damage and type I interferon responses [15]. Subsequent studies focusing on ST and LT sequencing in MCC confirmed that most of the MCPyV variability in these genes—in particular, mutations inducing stop codons or insertions/deletions—was concentrated after the pRb-binding domain
The random integration of MCPyV is considered as a prerequisite to malignant transformation in MCC. In that situation, late protein expression is hampered while early gene products like LT, while heavily mutated, can still elicit host cell transformation by forcing cell cycle progression and upregulating anti-apoptotic genes [16]. MCPyV integration/transformation appears most likely as a consequence of a fine balance allowing the host cell to escape lysis and integrated MCPyV genomes to escape clearance by the immune response. Interestingly, three original publications by Kenan et al. and Müller et al. [17–19] have shown that some LT-positive urogenital carcinomas in transplant patients were linked to the presence of genetically altered BKPyV genomes, either integrated or as episomes in cancer cells. Thus evidence is accumulating implicating aberrant LT expression as a key tumour inducer for at least two HPyV infections.
3. Non-coding control region in JCPyV and BKPyV infection
One of the most remarkable features of the HPyV is the occurrence of rearrangements of the enhancer element of the non-coding control region (NCCR). The NCCR is a key regulatory region of about 400 bp, harbouring sequences required for replication and for transcription (early and late promoters and cis-regulating elements). It is defined by a region between the ATG start codon for LT and the start of the agnoprotein open reading frame and contains several confirmed and putative binding sites for cellular transcription factors (reviewed in [20]).
The early side of the NCCR is highly conserved between different isolates of the same PyV species. It contains the origin of viral DNA replication (ORI), centred by four GAGGC pentamers that bind the OBD of the viral LT antigen. It also contains binding sites for nuclear factor kappa B (NF-kB) that play a role in the regulation of transcription of both BKPyV and JCPyV genomes [21]. Between the early and late sides of the NCCR are tandem repeat elements that function as enhancers for early and late transcription and interact with a number of cellular transcription factors.
On the basis of the NCCR sequence, archetype (ww) and rearranged (rr) NCCR have been described. The NCCR of archetype strains can be arbitrarily divided into sequence blocks (OPQRS for BKPyV, ABCDEF for JCPyV) [22,23]. The rrNCCR are characterized by mutations, partial duplications and/or deletions compared to the archetype sequence (figure 1). These rearrangements were rapidly described for BKPyV and JCPyV in cell culture as compared to sequences directly obtained from urine [24]. Archetype strains are considered as the transmissible form of the virus in the population. They are shed in the urine of immunosuppressed people, during pregnancy, but also in healthy individuals, owing to periodic, subclinical reactivation from the kidney [25]. Rearranged variants have mostly been described in the context of immunosuppressive or immunomodulatory therapy (organ transplantation, monoclonal antibody therapy) or in AIDS patients. These rearranged forms also emerge quickly in permissive cells in vitro [24], indicating that they outcompete archetype strains when replication occurs without the constraints of an effective host cellular immune response.
Figure 1.

Structure of archetype and rearranged NCCR in JCPyV and BKPyV. The archetype structures are arbitrarily divided into blocks (O-P-Q-R-S for BKPyV, A-B-C-D-E-F for JCPyV). Rearranged NCCR are characterized by duplications and deletions. As examples, the rrNCCR of the laboratory isolates BKPyV Dunlop and JCPyV Mad-1 are presented.
In BKPyV, typical rrNCCR harbour an unmodified O143-P68 segment, bearing the viral ORI and the early transcription start site, when there are complete or partial duplications of the P68 block, and deletions in Q39-R63-S63. No common sequence for rrNCCR could be identified, however, as the length, position and combination of duplications/deletions events vary extensively between isolates. In a study from Gosert et al., rrNCCR were associated with prolonged viremia and PVAN during BKPyV infection in kidney transplanted patients. Inter-compartment analysis between plasma and urine revealed a higher frequency of rrNCCR sequences in plasma than in urine, and patients displaying rrNCCR in plasma were also characterized by higher viral loads [26]. Similar to BKPyV, viral JCPyV DNA harbouring an archetype form (ABCDEF) has been detected in urine and also in tonsil tissue [23,27]. Using next generation sequencing (NGS), quasi-species analysis of JCPyV DNA from the urine of immunocompetent donors revealed the existence of mixtures of strains with point mutations or short deletions but no rearrangements [28]. A hallmark of JCPyV DNA from PML patients' brain or cerebrospinal fluid is the presence of rrNCCR, likely derived from the archetype strain by successive deletion and/or duplication events, as illustrated by inter-compartment NCCR analysis [23]. The emergence of rearranged variants may occur during JCPyV dissemination in blood, as they are also detected in plasma [29]. However, NCCR rearrangement may not be a prerequisite for pathology, as some PML patients display a major viral population with archetype NCCR in cerebrospinal fluid (CSF) [28,30].
In vitro, BKPyV rrNCCR strains are characterized by much more efficient propagation than wwNCCR strains, which grow slowly in primary human urothelial and renal epithelial cells unless infected cells overexpress large T antigen [31]. The mechanisms underlying these differences are not completely understood: using a bidirectional reporter vector, Bethge et al. showed that rearrangements affect the balance between early and late promoter activities [32]. The same author later focused on the transcription factor Sp1. Mutations disrupting an Sp1 binding site close to the early region promoter decreased both early and late gene expression, whereas mutations eliminating the Sp1 site near the late promoter increased early gene transcription [33]. In HSCT recipients, a point mutation in the Sp1 site on the late side was detected in urine of 7/16 patients with HC, but not in patients without HC [34]. Perturbing the ratio of early to late region transcripts also impacts the function of the viral miRNA, which is complementary to LT mRNA and is encoded on the late region transcript. The increased transcription of the early region from rearranged NCCR overwhelms the control of LT expression exerted by the viral miRNA, and this plays an important role in the enhanced replication observed in rearranged isolates [35].
PML-related JCPyV rearranged strains are characterized by increased early gene transcription and viral replication [29]. But once again, the functional impact of rearrangements may vary according to the pattern of rearrangement and the method used to measure its impact. Recently, L'Honneur et al., using dual reporter minicircles, showed that a deletion in the F block of JCPyV NCCR was associated with an increase in the late promoter but no change in the early promoter activities [36].
NCCR variants have also been described for the novel HPyV in clinical samples from immunosuppressed patients [37,38] and HPyV reporter vectors based on these rrNCCR sequences were shown to have increased early gene expression [39]. Regarding MCPyV, NCCR genotypes have been linked to geographical origin, but in patients with MCC, rrNCCR was not found in tumour tissue or normal skin specimens [40].
Recombination events occurring during viral replication have been proposed to explain the emergence of NCCR rearrangements [41], but more data are required to examine this mechanism and longitudinal NGS analyses in infected patients will help to further investigate the emergence of these rearrangements. Available data are consistent with the view that NCCR rearrangements confer increased replicative capacities to HPyV but that this gain of fitness is not compatible with long-term persistence, since higher levels of LT expression render infected cells more susceptible to lysis by cytotoxic T cells [42]. This explains why rearranged NCCR are only observed in vivo in immunosuppressed patients.
4. VP1 evolution and pathology in JCPyV and BKPyV infection
In the context of JCPyV infection, the idea that PML could be associated with specific VP1 mutations was first suggested in a 1995 paper [43] then confirmed ten years later in a Japanese cohort [44,45]. Non-synonymous VP1 mutations modifying the BC and HI loops were found in 13 of 16 sequences from the brain or CSF of PML patients, but were absent from 13 VP1 sequences derived from the urine of immunosuppressed KTx recipients.
A more systematic approach analysing VP1 sequences from 55 PML patients compared to 235 sequences from urine samples of healthy individuals reinforced the relationship between VP1 mutations and PML. This publication reported that VP1 mutations from PML patients, specifically at amino acids L55, K60 (BC-loop), S265, S267 and S269 (HI loop), emerged by positive selection independently of JCPyV genotype. Each PML-derived VP1 sequence carried only one of these five mutations, which was explained by significant epistatic interactions at these sites. That is, the presence of a mutation at position 55 reduced the likelihood of incorporation of a second mutation at position 269, and a similar interaction was found between mutations at amino acids 60 and 269. The five amino acids subject to positive selection were in close proximity to the predicted receptor binding pocket in the JCPyV VP1 pentamer, suggesting that they altered virus tropism.
The functional impact of JCPyV VP1 mutations was assessed by incorporating PML-associated mutations into either virus-like particles (VLPs) [46], VP1 pentamers [47], molecular clones of the JCPyV genome to produce infectious virus [47] and pseudotype particles produced by co-transfecting plasmids coding for JCPyV VP1, VP2 and VP3 proteins [47,48]. All of these approaches demonstrated that PML-associated mutations disrupt the sialic acid binding pocket of the VP1 pentamer and specifically abrogate binding of the JCPyV capsid to its receptor, the lactoseries tetrasaccharide c (LSTc), which terminates with an α2-6-linked sialic acid residue [49].
More recently Geoghegan et al. [48] used pseudotype JCPyV particles carrying wild-type, L55F and S269F mutant VP1 to investigate the infectious entry characteristics of PML-associated mutations. They found that entry in a gliosarcoma cell line transfected with Large T Ag (SF-539 cell line) mediated by wild-type VP1 was dependent on the presence of sialylated glycans, while entry mediated by L55F and S269F mutant VP1 was sialic acid-independent. Furthermore, they found that although the L55F and S269F mutations abrogated the infectivity of pseudotypes in an ovarian cancer cell line transfected with Large T Ag (NCI/ADR-RES cell line), infectivity was restored by supplementing these cells with the asialo-GM1 ganglioside [48]. Expression of this ganglioside in vivo is restricted to oligodendrocytes and myelin-producing cells, so asialo-GM1 is therefore an attractive candidate receptor for PML-associated JCPyV in the CNS.
The interaction between the virus and the host humoral response appears to be another important factor in JCPyV VP1 evolution, since PML-associated mutations were found to confer neutralization escape against the patient's cognate serum [50].
In the context of BKPyV infection, it has also been suggested that VP1 mutations are associated with pathology in the form of PVAN after KTx. This was first proposed by Randhawa and colleagues, in an analysis of the VP1 typing region in DNA extracted from kidney biopsies of PVAN patients [51]. This report showed first, that VP1 diversity in patients with PVAN was greater than that observed in the urine of healthy donors or asymptomatic kidney transplant recipients, and second, that intrapatient evolution can occur, because VP1 mutations accumulated over time in the three patients for whom sequential biopsies were available. A follow-up study from the same group [52] found that VP1 sequences in patients with PVAN showed higher sequence diversity in the BC-loop (AA 61-83) compared to sequences from patients with BKPyV viruria without viremia, and the dN-dS ratio in that region of the protein indicated positive selection in PVAN patients. However, an analysis of VP1 sequences in 45 KTx recipients from another group reached the conclusion that VP1 mutations in BKPyV had no impact on virus replication in vivo [53]. Similarly, VP1 mutations in the BC-loop were found both in 6 out of 8 patients with PVAN and 6 out of 7 KTx recipients characterized by high viruria without PVAN [54], indicating that the accumulation of mutations in VP1 may be a consequence of high viral load rather than specifically related to PVAN.
Recently, results from the Buck lab indicated that VP1 mutations in patients with PVAN do indeed modify viral tropism and lead to neutralization escape [55]. In this study, VP1 mutations D62N, E73Q, E73K, D77H and D77N were observed in two PVAN patients with genotype IV infection, and the functional impact of these mutations was analysed using pseudotype viruses. Pseudotype particles carrying these mutations gained the ability to agglutinate sheep red blood cells, implying that receptor use was modified. With respect to neutralization escape, patient 1 variants D62N, E73K and D77H all resulted in 70- to 90-fold greater resistance to neutralization by cognate serum compared to the wild-type VP1, whereas patient 2 variants D62N, E73Q and D77N resulted in a modest 8-fold resistance to neutralization. Interestingly, viremia became undetectable in patient 1 three months after PVAN diagnosis, without the development of a neutralizing response specific for the D62N, E73K and D77H variants. It is therefore not clear how the resistance of VP1 variants to neutralization in vitro relates to control of virus replication in vivo.
Overall, the available data show that molecular evolution of VP1 plays an essential role in the physiopathology of PML by (i) allowing the virus to escape from the host humoral response and (ii) modifying virus tropism such that cells in the CNS can be infected. The situation is analogous to that previously described in mouse polyomavirus (MuPyV) infection, in which a single amino acid substitution confers a highly pathogenic phenotype [56,57]. Furthermore, the V296A mutation in MuPyV VP1 that is responsible for enhanced pathogenicity modifies an amino acid side-chain that occupies the same position on the capsid surface as the S269F mutation in JCPyV VP1. In both cases, a single mutation modifies virus binding to glycan receptors and converts the virus to a more pathogenic form. Despite such a low genetic barrier PML remains a rare manifestation of JCPyV infection, presumably because the emergence of neurotropic strains requires a combination of host immunosuppression and transport across the blood–brain barrier of viruses carrying the initially rare VP1 variant.
In contrast, the contribution of VP1 mutations to pathology in the context of BKPyV infection has not been so clearly established. Unlike the case of PML-associated VP1 mutations, which are found only in the CNS and plasma [46], mutations in BKPyV VP1 in PVAN patients are also found in virus excreted in the urine. Therefore, BKPyV VP1 mutations do not entail the loss of virus replication in the reno-urinary epithelium. This can be explained by examining the position of BKPyV VP1 mutations from PVAN patients on the viral capsid (figure 2). In contrast to L55F and S269F in JCPyV and V296A mutation in MuPyV, mutations in BKPyV VP1 do not appear to disrupt the sialic acid binding pocket on the BKPyV capsid. Indeed, functional analysis of pseudotypes carrying the D62N, E73Q/K, and D77N/H mutations showed that virus entry mediated by mutant VP1 remained dependent on sialic acid [55]. It therefore seems likely that BKPyV VP1 mutations result in relatively minor changes to virus tropism—the virus may switch to a different entry receptor on the same host cell type, but this does not alter its tissue tropism.
Figure 2.
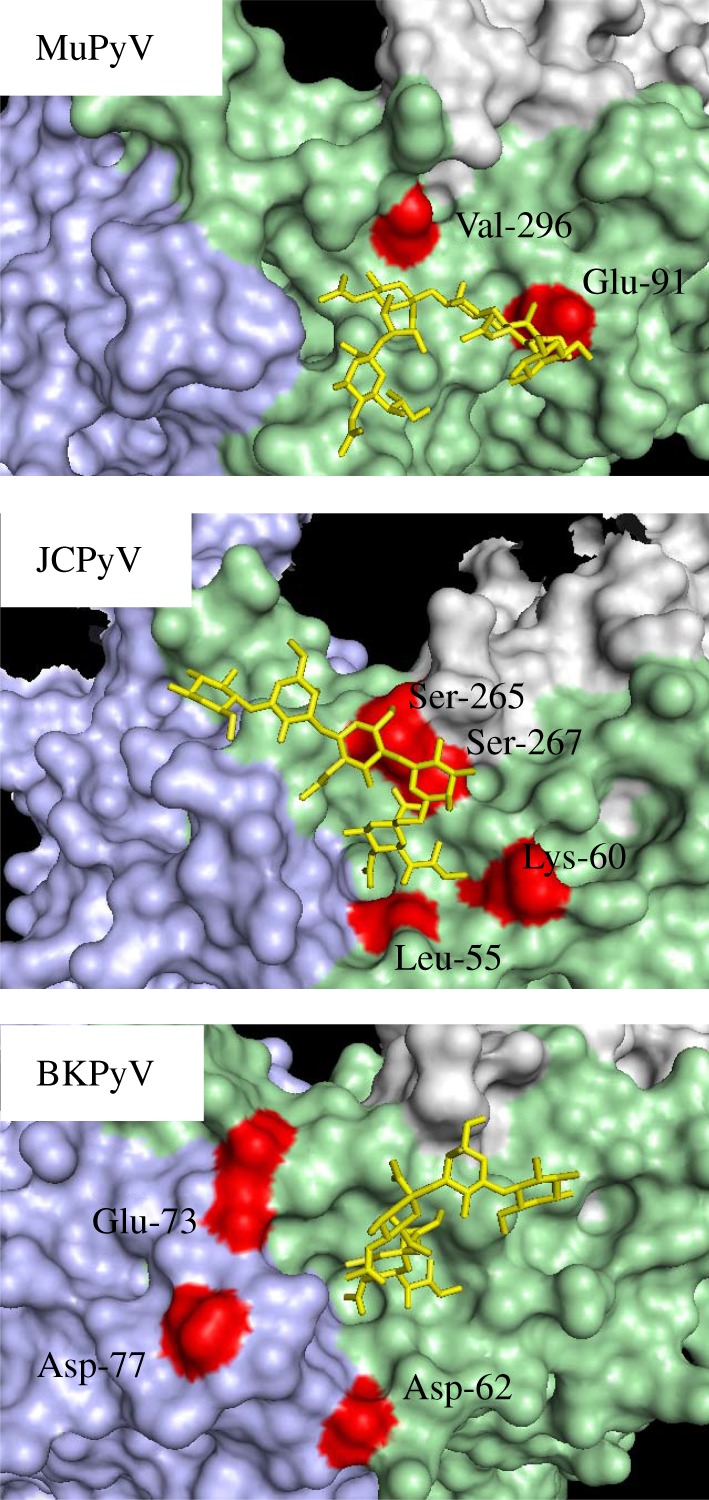
Position of VP1 mutations on MuPyV, JCPyV and BKPyV VP1 pentamers. Adjacent VP1 monomers are shown in mauve, green and grey. Glycan receptor fragments complexed with VP1 pentamers are shown in yellow and VP1 mutations detailed in the text are shown in red. For BKPyV the VP1 pentamer structure was obtained for genotype I VP1, whereas the functional impact of mutations has only been determined for genotype IV VP1. Labels refer to amino acids in genotype IV VP1, while the residues highlighted in red are the amino acids at the corresponding position in the primary structure of genotype I VP1. It is assumed that the relevant amino acids occupy equivalent positions on the surface of genotype I and genotype IV VP1 pentamers. The figure was prepared using Pymol with the following structures—1VPS (MuPyV), 4X13 (JCPyV), and 4MJ0 (BKPyV).
5. Conclusion
Intra-host evolution of human polyomaviruses occurs and is clearly responsible for pathology in MCPyV and JCPyV infections (figure 3) and although the contribution of NCCR and VP1 evolution and pathology is less clear in the case of BKPyV infection, the available data indicate that it is likely to play an important role.
Figure 3.

Summary of intra-patient evolution leading to pathology in MCPyV, JCPyV and BKPyV infection. DDR, DNA damage response.
In MCPyV infection, virus integration, followed by the selection of cellular clones that express a truncated LT protein, results in oncogenic transformation of the host cell. This process is similar to that observed during induction of cervical cancer by the human papillomaviruses (HPV), HPV16 and HPV18. Integrated HPV genomes are frequently detected in malignant tissue, accompanied by disruption of late gene expression, with conservation of the early genes, E6 and E7 that are essential for cancer cell transformation. Interestingly, the E2 gene is frequently disrupted by HPV integration, and this is associated with increased expression of E6 and E7 [58]. Therefore, chronic upregulated expression of early gene products essential for tumour cell transformation, combined with loss of other early gene functions, appears to be a common theme linking the carcinogenic process driven by oncogenic HPV and HPyV.
In JCPyV and BKPyV infection, in-host virus evolution appears to be a response to the unique immune environment of the infected host. In multiple sclerosis patients treated with Natalizumab, JCPyV reactivation presents the virus with a host in which cellular immunity is blocked specifically in the CNS, while systemic immunity, notably the humoral response, remains intact. The presence of a robust humoral response therefore exerts selection pressure on VP1, while the absence of cellular immunity in the CNS gives a selective advantage to rrNCCR variants. The immune landscape is similar in KTx recipients with BKPyV reactivation. Even though cellular immune responses are abrogated by the immunosuppressive drugs given to prevent graft rejection, these patients mount a strong antiviral humoral response, hence the same pattern of VP1 point mutations and NCCR rearrangements is observed.
These processes are observed within individual patients on a time scale of months to years, and this raises two important questions—first, what causes this high intra-patient mutation rate, and second, how can we reconcile relatively rapid intra-patient evolution with the very low sequence diversity of these viruses observed at the population level?
With respect to high intra-patient mutation rates, two lines of evidence have incriminated the host cytosine deaminase, APOBEC3B, as the cause of VP1 mutations in BKPyV infection. First, its expression is induced by BKPyV infection [59], and second, the pattern of VP1 mutations observed in vivo by deep sequencing displays an APOBEC3B signature [59]. Interestingly, the two main PML-associated VP1 mutations, L55F and S269F, can be induced by cytosine deamination (TTG=>TTC, and TCT=>TTT, respectively) so APOBEC3B activation could presumably also be involved in the emergence of pathogenic JCPyV variants. APOBEC3B activation may also be implicated in the development of reno-urinary cancers in solid organ transplant recipients secondary to BKPyV reactivation [60]. In particular, urothelial bladder carcinoma genomes carry a strong APOBEC3B signature [61]. Overall, the in-host evolution of BKPyV, and possibly JCPyV, VP1 appears to be the result of a fascinating interplay between the innate antiviral response, leading to the induction of APOBEC3B, which creates the mutant viruses that are then selected by the host humoral response [47]. In contrast, the molecular mechanism of NCCR rearrangements and the cause of LT mutations in integrated MCPyV genomes that lead to MCC have not been identified.
With regard to the second question, it is clear that the mutations involved in JCPyV and MCPyV pathology are evolutionary dead ends for these viruses. In MCPyV, both integration into the host chromosome and the incorporation of premature stop codons in LT are incompatible with productive virus replication. Thus, the LT mutations that contribute to MCC never leave the transformed cell. Similarly, transmission of neurotropic strains of JCPyV has never been observed, as they are found only in the CNS and blood of PML patients, but not in the urine [50]. Therefore, the neurotropic JCPyV variants that arise as a result of selection pressure in the immunosuppressed host never leave that particular individual. In the case of BKPyV, the situation is not so clear, since VP1 mutant viruses are excreted in the urine. However, these mutations only arise in KTx recipients, so it is possible that VP1 mutant viruses are indeed transmitted, but comprise such a small proportion of circulating viruses that they are not detected in the general population. Furthermore, the reduction in infectivity reported for BKPyV VP1 mutations [43] may lead to them being outcompeted by revertants in a new host.
It therefore seems likely that intra-host molecular evolution driven by the unique selection pressures encountered in immunosuppressed hosts results in the emergence of pathogenic polyomaviruses that are, fortunately, either incapable of transmission to, or at a competitive disadvantage in new hosts.
Data accessibility
This article has no additional data.
Authors' contributions
C.B.-B.—literature analysis and drafting of section on NCCR variation. F.H.—literature analysis and drafting of section on LT variation. D.M.—literature analysis and drafting of section on VP1 variation. All co-authors approve the final submitted manuscript.
Competing interests
D.M., F.H. and C.B.-B. have an ongoing scientific collaboration with guest editor I. Bravo.
Funding
Université de Nantes (D.M., C.B.-B.), INSERM (F.H.), CHU Nantes (C.B.-B.).
References
- 1.Buck CB, et al. 2016. The ancient evolutionary history of polyomaviruses. PLoS Pathog. 12, e1005574 ( 10.1371/journal.ppat.1005574) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen S, Harmon W, Krensky AM, Edelson PJ, Padgett BL, Grinnell BW, Rubino MJ, Walker DL. 1983. Tubulo-interstitial nephritis associated with polyomavirus (BK type) infection. N. Engl. J. Med. 308, 1192–1196. ( 10.1056/NEJM198305193082004) [DOI] [PubMed] [Google Scholar]
- 3.Arthur RR, Shah KV, Baust SJ, Santos GW, Saral R. 1986. Association of BK viruria with hemorrhagic cystitis in recipients of bone marrow transplants. N. Engl. J. Med. 315, 230–234. ( 10.1056/NEJM198607243150405) [DOI] [PubMed] [Google Scholar]
- 4.Padgett BL, Walker DL, ZuRhein GM, Hodach AE, Chou SM. 1976. JC Papovavirus in progressive multifocal leukoencephalopathy. J. Infect. Dis. 133, 686–690. ( 10.1093/infdis/133.6.686) [DOI] [PubMed] [Google Scholar]
- 5.Feng H, Shuda M, Chang Y, Moore PS. 2008. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096–1100. ( 10.1126/science.1152586) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamminga SS, van der Meijden Els PZ, Feltkamp, Mariet MCW, Zaaijer HHL. 2018. Seroprevalence of fourteen human polyomaviruses determined in blood donors. PLoS ONE 13, e0206273 ( 10.1371/journal.pone.020627) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. 2002. Merkel cell carcinoma and HIV infection. Lancet 359, 497–498. ( 10.1016/S0140-6736(02)07668-7) [DOI] [PubMed] [Google Scholar]
- 8.Clarke CA, et al. 2015. Risk of Merkel cell carcinoma after solid organ transplantation. J. Natl Cancer Inst. 107 ( 10.1093/jnci/dju382) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rotondo JC, et al. 2017. Merkel cell carcinomas arising in autoimmune disease affected patients treated with biologic drugs, including anti-TNF. Clin. Cancer Res. 23, 3929–3934. ( 10.1158/1078-0432.CCR-16-2899) [DOI] [PubMed] [Google Scholar]
- 10.Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, Moore PS, Becker JC. 2010. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 84, 7064–7072. ( 10.1128/JVI.02400-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arora R, et al. 2012. Survivin is a therapeutic target in Merkel cell carcinoma. Sci. Transl. Med. 4, 133ra56 ( 10.1126/scitranslmed.3003713) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.An P, Sáenz Robles MT, Pipas JM. 2012. Large T antigens of polyomaviruses: amazing molecular machines. Annu. Rev. Microbiol. 66, 213–236. ( 10.1146/annurev-micro-092611-150154) [DOI] [PubMed] [Google Scholar]
- 13.Seo GJ, Chen CJ, Sullivan CS. 2009. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 383, 183–187. ( 10.1016/j.virol.2008.11.001) [DOI] [PubMed] [Google Scholar]
- 14.Martel-Jantin C, et al. 2012. Genetic variability and integration of Merkel cell polyomavirus in Merkel cell carcinoma. Virology 426, 134–142. ( 10.1016/j.virol.2012.01.018) [DOI] [PubMed] [Google Scholar]
- 15.Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, Chang Y. 2008. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl Acad. Sci. USA 105, 16 272–16 277. ( 10.1073/pnas.0806526105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gjoerup O, Chang Y. 2010. Update on human polyomaviruses and cancer. Adv. Cancer Res. 106, 1–51. ( 10.1016/S0065-230X(10)06001-X) [DOI] [PubMed] [Google Scholar]
- 17.Kenan DJ, Mieczkowski PA, Latulippe E, Côté I, Singh HK, Nickeleit V. 2017. BK polyomavirus genomic integration and large T antigen expression: evolving paradigms in human oncogenesis. Am. J. Transplant. 17, 1674–1680. ( 10.1111/ajt.14191) [DOI] [PubMed] [Google Scholar]
- 18.Kenan DJ, Mieczkowski PA, Burger-Calderon R, Singh HK, Nickeleit V. 2015. The oncogenic potential of BK-polyomavirus is linked to viral integration into the human genome. J. Pathol. 237, 379–389. ( 10.1002/path.4584) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Müller DC, et al. 2018. Donor-derived, metastatic urothelial cancer after kidney transplantation associated with a potentially oncogenic BK polyomavirus. J. Pathol. 244, 265–270. ( 10.1002/path.5012) [DOI] [PubMed] [Google Scholar]
- 20.White MK, Safak M, Khalili K. 2009. Regulation of gene expression in primate polyomaviruses. J. Virol. 83, 10 846–10 856. ( 10.1128/JVI.00542-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wollebo HS, Melis S, Khalili K, Safak M, White MK. 2012. Cooperative roles of NF-κB and NFAT4 in polyomavirus JC regulation at the KB control element. Virology 432, 146–154. ( 10.1016/j.virol.2012.06.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moens U, Johansen T, Johnsen JI, Seternes OM, Traavik T. 1995. Noncoding control region of naturally occurring BK virus variants: sequence comparison and functional analysis. Virus Genes 10, 261–275. ( 10.1007/BF01701816) [DOI] [PubMed] [Google Scholar]
- 23.Pietropaolo V, Videtta M, Fioriti D, Mischitelli M, Arancio A, Orsi N, Degener AM. 2003. Rearrangement patterns of JC virus noncoding control region from different biological samples. J. Neurovirol. 9, 603–611. ( 10.1080/13550280390246507) [DOI] [PubMed] [Google Scholar]
- 24.Rubinstein R, Schoonakker BC, Harley EH. 1991. Recurring theme of changes in the transcriptional control region of BK virus during adaptation to cell culture. J. Virol. 65, 1600–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markowitz RB, Eaton BA, Kubik MF, Latorra D, McGregor JA, Dynan WS. 1991. BK virus and JC virus shed during pregnancy have predominantly archetypal regulatory regions. J. Virol. 65, 4515–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gosert R, Rinaldo CH, Funk GA, Egli A, Ramos E, Drachenberg CB, Hirsch HH. 2008. Polyomavirus BK with rearranged noncoding control region emerge in vivo in renal transplant patients and increase viral replication and cytopathology. J. Exp. Med. 205, 841–852. ( 10.1084/jem.20072097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato A, Kitamura T, Takasaka T, Tominaga T, Ishikawa A, Zheng H-Y, Yogo Y. 2004. Detection of the archetypal regulatory region of JC virus from the tonsil tissue of patients with tonsillitis and tonsilar hypertrophy. J. Neurovirol. 10, 244–249. ( 10.1080/13550280490468663) [DOI] [PubMed] [Google Scholar]
- 28.Van Loy T, Thys K, Ryschkewitsch C, Lagatie O, Monaco MC, Major EO, Tritsmans L, Stuyver LJ.. 2015. JC virus quasispecies analysis reveals a complex viral population underlying progressive multifocal leukoencephalopathy and supports viral dissemination via the hematogenous route. J. Virol. 89, 1340–1347. ( 10.1128/JVI.02565-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gosert R, Kardas P, Major EO, Hirsch HH. 2010. Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalopathy patient samples increase virus early gene expression and replication rate. J. Virol. 84, 10 448–10 456. ( 10.1128/JVI.00614-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seppälä H, Virtanen E, Saarela M, Laine P, Paulín L, Mannonen L, Auvinen P, Auvinen E. 2017. Single-molecule sequencing revealing the presence of distinct JC polyomavirus populations in patients with progressive multifocal leukoencephalopathy. J. Infect. Dis. 215, 889–895. ( 10.1093/infdis/jiw399) [DOI] [PubMed] [Google Scholar]
- 31.Broekema NM, Imperiale MJ. 2012. Efficient propagation of archetype BK and JC polyomaviruses. Virology 422, 235–241. ( 10.1016/j.virol.2011.10.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bethge T, Hachemi HA, Manzetti J, Gosert R, Schaffner W, Hirsch HH. 2015. Sp1 sites in the noncoding control region of BK polyomavirus are key regulators of bidirectional viral early and late gene expression. J. Virol. 89, 3396–3411. ( 10.1128/JVI.03625-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bethge T, Ajuh E, Hirsch HH. 2016. Imperfect symmetry of Sp1 and core promoter sequences regulates early and late virus gene expression of the bidirectional BK polyomavirus noncoding control region. J. Virol. 90, 10 083–10 101. ( 10.1128/JVI.01008-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Priftakis P, Bogdanovic G, Kalantari M, Dalianis T. 2001. Overrepresentation of point mutations in the Sp1 site of the non-coding control region of BK virus in bone marrow transplanted patients with haemorrhagic cystitis. J. Clin. Virol. 21, 1–7. ( 10.1016/S1386-6532(00)00171-2) [DOI] [PubMed] [Google Scholar]
- 35.Broekema NM, Imperiale MJ. 2013. miRNA regulation of BK polyomavirus replication during early infection. Proc. Natl Acad. Sci. USA 110, 8200–8205. ( 10.1073/pnas.1301907110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.L'Honneur A-S, Leh H, Laurent-Tchenio F, Hazan U, Rozenberg F, Bury-Moné S. 2018. Exploring the role of NCCR variation on JC polyomavirus expression from dual reporter minicircles. PLoS ONE 13, e0199171 ( 10.1371/journal.pone.0199171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lednicky JA, Butel JS, Luetke MC, Loeb JC. 2014. Complete genomic sequence of a new human polyomavirus 9 strain with an altered noncoding control region. Virus Genes 49, 490–492. ( 10.1007/s11262-014-1119-z) [DOI] [PubMed] [Google Scholar]
- 38.Nguyen KD, et al. 2017. Human polyomavirus 6 and 7 are associated with pruritic and dyskeratotic dermatoses. J. Am. Acad. Dermatol. 76, 932–940. ( 10.1016/j.jaad.2016.11.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ajuh ET, Wu Z, Kraus E, Weissbach FH, Bethge T, Gosert R, Fischer N, Hirsch HH. 2018. Novel human polyomavirus noncoding control regions differ in bidirectional gene expression according to host cell, large T-antigen expression, and clinically occurring rearrangements. J. Virol. 92, pii: e02231-17. ( 10.1128/JVI.02231-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashida Y, Higuchi T, Matsui K, Shibata Y, Nakajima K, Sano S, Daibata M. 2018. Genetic variability of the noncoding control region of cutaneous Merkel cell polyomavirus: identification of geographically related genotypes. J. Infect. Dis. 217, 1601–1611. ( 10.1093/infdis/jiy070) [DOI] [PubMed] [Google Scholar]
- 41.Johnson EM, Wortman MJ, Dagdanova AV, Lundberg PS, Daniel DC. 2013. Polyomavirus JC in the context of immunosuppression: a series of adaptive, DNA replication-driven recombination events in the development of progressive multifocal leukoencephalopathy. Clin. Dev. Immunol. 2013, 197807 ( 10.1155/2013/197807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. 2005. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435, 682–686. ( 10.1038/nature03576) [DOI] [PubMed] [Google Scholar]
- 43.Stoner GL, Ryschkewitsch CF. 1995. Capsid protein VP1 deletions in JC virus from two AIDS patients with progressive multifocal leukoencephalopathy. J. Neurovirol. 1, 189–194. ( 10.3109/13550289509113965) [DOI] [PubMed] [Google Scholar]
- 44.Zheng H-Y, et al. 2005. Characterization of the VP1 loop mutations widespread among JC polyomavirus isolates associated with progressive multifocal leukoencephalopathy. Biochem. Biophys. Res. Commun. 333, 996–1002. ( 10.1016/j.bbrc.2005.06.012) [DOI] [PubMed] [Google Scholar]
- 45.Zheng H-Y, et al. 2005. New sequence polymorphisms in the outer loops of the JC polyomavirus major capsid protein (VP1) possibly associated with progressive multifocal leukoencephalopathy. J. Gen. Virol. 86, 2035–2045. ( 10.1099/vir.0.80863-0) [DOI] [PubMed] [Google Scholar]
- 46.Gorelik L, et al. 2011. Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change its receptor specificity. J. Infect. Dis. 204, 103–114. ( 10.1093/infdis/jir198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maginnis MS, Ströh LJ, Gee GV, O'Hara BA, Derdowski A, Stehle T, Atwood WJ. 2013. Progressive multifocal leukoencephalopathy-associated mutations in the JC polyomavirus capsid disrupt lactoseries tetrasaccharide c binding. MBio 4, e00247-13 ( 10.1128/mBio.00247-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geoghegan EM, et al. 2017. Infectious entry and neutralization of pathogenic JC polyomaviruses. Cell Rep. 21, 1169–1179. ( 10.1016/j.celrep.2017.10.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neu U, Maginnis MS, Palma AS, Ströh LJ, Nelson CDS, Feizi T, Atwood WJ, Stehle T. 2010. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 8, 309–319. ( 10.1016/j.chom.2010.09.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ray U, Cinque P, Gerevini S, Longo V, Lazzarin A, Schippling S, Martin R, Buck CB, Pastrana DV. 2015. JC polyomavirus mutants escape antibody-mediated neutralization. Sci. Transl. Med. 7, 306ra151 ( 10.1126/scitranslmed.aab1720) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Randhawa PS, Khaleel-Ur-Rehman K, Swalsky PA, Vats A, Scantlebury V, Shapiro R, Finkelstein S. 2002. DNA sequencing of viral capsid protein VP-1 region in patients with BK virus interstitial nephritis. Transplantation 73, 1090–1094. ( 10.1097/00007890-200204150-00013) [DOI] [PubMed] [Google Scholar]
- 52.Luo C, Hirsch HH, Kant J, Randhawa P. 2012. VP-1 quasispecies in human infection with polyomavirus BK. J. Med. Virol. 84, 152–161. ( 10.1002/jmv.22147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krautkrämer E, Klein TM, Sommerer C, Schnitzler P, Zeier M. 2009. Mutations in the BC-loop of the BKV VP1 region do not influence viral load in renal transplant patients. J. Med. Virol. 81, 75–81. ( 10.1002/jmv.21359) [DOI] [PubMed] [Google Scholar]
- 54.Tremolada S, Delbue S, Castagnoli L, Allegrini S, Miglio U, Boldorini R, Elia F, Gordon J, Ferrante P. 2010. Mutations in the external loops of BK virus VP1 and urine viral load in renal transplant recipients. J. Cell. Physiol. 222, 195–199. ( 10.1002/jcp.21937) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peretti A, et al. 2018. Characterization of BK polyomaviruses from kidney transplant recipients suggests a role for APOBEC3 in driving in-host virus evolution. Cell Host Microbe 23, 628–635. ( 10.1016/j.chom.2018.04.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bauer PH, Bronson RT, Fung SC, Freund R, Stehle T, Harrison SC, Benjamin TL. 1995. Genetic and structural analysis of a virulence determinant in polyomavirus VP1. J. Virol. 69, 7925–7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freund R, Calderone A, Dawe CJ, Benjamin TL. 1991. Polyomavirus tumor induction in mice: effects of polymorphisms of VP1 and large T antigen. J. Virol. 65, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park TW, Fujiwara H, Wright TC. 1995. Molecular biology of cervical cancer and its precursors. Cancer 76, 1902–1913. [DOI] [PubMed] [Google Scholar]
- 59.Verhalen B, Starrett GJ, Harris RS, Jiang M. 2016. Functional upregulation of the DNA cytosine deaminase APOBEC3B by polyomaviruses. J. Virol. 90, 6379–6386. ( 10.1128/JVI.00771-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Papadimitriou JC, Randhawa P, Rinaldo CH, Drachenberg CB, Alexiev B, Hirsch HH. 2016. BK polyomavirus infection and renourinary tumorigenesis. Am. J. Transplant. 16, 398–406. ( 10.1111/ajt.13550) [DOI] [PubMed] [Google Scholar]
- 61.Burns MB, Temiz NA, Harris RS. 2013. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983. ( 10.1038/ng.2701) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.