

Abstract
Recent genome editing techniques, including CRISPR mutagenesis screens, offer unparalleled opportunities to study the regulatory non-coding genomic regions, enhancers, promoters, and functional non-coding RNAs. Heterozygous point mutations in FOXF1 and genomic deletion copy-number variants at chromosomal region 16q24.1 involving FOXF1 or its regulatory region mapping ~ 300 kb upstream to FOXF1 and leaving it intact have been identified in the vast majority of patients with a lethal neonatal lung disease, alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV). Homozygous Foxf1−/− mice have been shown to die by embryonic day 8.5 because of defects in the development of extraembryonic and lateral mesoderm-derived tissues, whereas heterozygous Foxf1+/− mice exhibit features resembling ACDMPV. We have previously defined a human lung-specific enhancer region encoding two long non-coding RNAs, LINC01081 and LINC01082, expressed in the lungs. To investigate the biological significance of lncRNAs in the FOXF1 enhancer region, we have generated a CRISPR/Cas9-mediated ~ 2.3 kb deletion involving the entire lncRNA-encoding gene Gm26878, located in the mouse region syntenic with the human FOXF1 upstream enhancer. Very recently, this mouse genomic region has been shown to function as a Foxf1 enhancer. Our results indicate that homozygous loss of Gm26878 is neonatal lethal with low penetrance. No changes in Foxf1 expression were observed, suggesting that the regulation of FOXF1 expression differs between mouse and human.
Keywords: lncRNA Gm26878, Foxf1, CRISPR/Cas9 genome editing, long-range gene regulation, alveolar capillary dysplasia with misalignment of pulmonary veins
Introduction
Transcription profiling using expression arrays and next generation RNAseq in both humans and model organisms has revealed that most of the genome is transcribed as non-coding RNAs (Carninci et al. 2005; Birney et al. 2007; Kapranov et al. 2007; Djebali et al. 2012). Long non-coding RNAs (lncRNAs) are arbitrary defined as RNAs larger than 200 bp in size; many of them are capped, spliced, polyadenylated, and synthesized from RNA polII promoters (Derrien et al. 2012). However, lncRNAs have very limited or no protein-coding potential. LncRNAs show relatively low evolutionary conservation of primary structure, contrasting with a frequent conservation of their function (Pang et al. 2006; Ulitsky et al. 2011). LncRNAs are essential regulators of cellular processes, functioning as decoys, scaffolds, or guides for protein complexes involved in regulation of gene expression, dosage compensation, genomic imprinting, nuclear organization and nuclear-cytoplasmic trafficking (Rinn and Chang 2012; Mercer and Mattick 2013). Importantly, association and expression profiling studies in humans have shown a correlation between mutations, misregulation of the expression, and copy-number variation (CNV) of lncRNAs with developmental disorders (Visel et al. 2010; Cabili et al. 2011; Wapinski and Chang 2011; Brunner et al. 2012; Szafranski et al. 2013).
We have previously reported deletion CNVs of a distant enhancer region for FOXF1 at 16q24.1 in newborns with alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV) (Stankiewicz et al. 2009; Szafranski et al. 2011; Szafranski et al. 2016a). ACDMPV is characterized by reduced number of pulmonary capillaries, muscular thickening in small pulmonary arterioles, and abnormally situated pulmonary veins running alongside pulmonary arterioles that cause respiratory distress soon after birth, usually accompanied by pulmonary hypertension (Langston 1991; Sen et al. 2004; Bishop et al. 2011). The identified FOXF1 enhancer region encodes several lncRNAs, including LINC01081 and LINC01082, expressed in fetal lungs (Szafranski et al. 2013). Based on the overlapping pathogenic CNV deletions mapping upstream to FOXF1 as well as in vitro knockdown studies, we previously hypothesized that LINC01081 and LINC01082 regulate FOXF1 expression by contributing to the chromatin looping that juxtaposes the enhancer and the FOXF1 promoter (Szafranski et al. 2013; Szafranski et al. 2014). Moreover, 32 of 33 CNV deletions in the FOXF1 locus arose de novo on the maternal chromosome 16, indicating that FOXF1 locus is epigenetically imprinted, and likely involves parent-of-origin dependent expression of some of lncRNAs. Supporting this notion, a putative promoter of LINC01081 overlaps a large CpG island that could be differentially methylated (Szafranski et al. 2016b). Another lncRNA, FENDRR that maps 1.7 kb upstream of FOXF1 in the opposite orientation and likely utilizes the same bi-directional promoter as FOXF1, interacts with chromatin-modifying polycomb repressive complex (PRC) 2 to regulate gene expression. Interestingly, homozygous loss of Fendrr, leaving Foxf1 intact, led to lethal defects of lungs and heart in mouse neonates (Sauvageau et al. 2013; Lai et al. 2015). Homozygous Foxf1−/− mice die by embryonic day 8.5 because of defects in the development of extraembryonic and lateral mesoderm-derived tissues (Mahlapuu et al. 2001), whereas heterozygous Foxf1+/− mice exhibit features resembling ACDMPV (Kalinichenko et al. 2001).
Here, we show that CRISPR/Cas9-mediated knockout of the sole lncRNA gene, Gm26878, present in the mouse enhancer region syntenic with the human FOXF1 upstream enhancer is partially neonatal lethal, but does not affect expression of Foxf1. Our data indicate involvement of Gm26878 in mouse embryonic development independently or downstream of Foxf1, and point to differences in regulation of FOXF1 expression between mouse and human.
Materials and Methods
Animal care
All mouse experiments were carried out under the approval of the Institutional Animal Care and Use Committee (IACUC) at Baylor College of Medicine (BCM). Mice were housed in the specific pathogen-free Transgenic Mouse Facility under the care of the Center for Comparative Medicine (CCM), which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
DNA and RNA isolation and DNA sequencing
DNA was isolated from mouse tails following standard proteinase K/phenol-chloroform extraction-based protocol (Sambrook et al. 1989). RNA was isolated from lung tissue using miRNeasy Mini Kit (Qiagen, Hilden, Germany). PCR products were directly sequenced by the Sanger method. Reference sequences were downloaded from the UCSC Genome Browser (NCBI build 37/hg19 or GRCm38/mm10, http://genome.ucsc.edu). Sequences were assembled using Sequencher v4.8 (GeneCodes, Ann Arbor, MI).
Generation of knockout mouse
Single guide RNA (sgRNA) sequences were selected to flank the genomic region surrounding lncRNA gene Gm26878 (upstream sgRNA chr8:120880082-120880104, 5’-TGGCCTCTGGGTCGGGACCC; downstream sgRNA chr8:120882476-120882498, 5’-TTATGGACTCCGGACTAGAA; GRCm38/mm10) using the CRISPR Design Tool (Ran et al. 2013). DNA templates for in vitro transcription of sgRNAs were produced using overlapping oligonucleotides (in a high-fidelity PCR reaction (Bassett et al. 2013) and sgRNA was transcribed using the MEGA shortscript T7 kit (ThermoFisher, Waltham, MA). Cas9 mRNA was purchased from ThermoFisher. The BCM Genetically Engineered Mouse (GEM) Core microinjected Cas9 mRNA (100 ng/µl) and sgRNA (10 ng/µl) into the cytoplasm of 100 pronuclear stage C57Bl/6J embryos. Cytoplasmic injections were performed using a microinjection needle (1 mm outer and 0.75 mm inner) with a tip diameter of 0.25-0.5 μm, an Eppendorf Femto Jet 4i to set pressure and time to control injection volume (0.5-1 pl per embryo). Injections were performed under a 200-400× magnification with Hoffman modulation contrast for visualizations.
A single PCR reaction using three primers (P1: 5’-CAGGTTCTCTGCTGCCCTAC; P2: 5’-TATCTCCAGGCCTTGGACTC; P3: 5’-CAGACACAGGGCTCTCCTTG) identified the wild-type allele (209 bp product) and the interval deletion allele (~ 540 bp product) in putative founders and resulting offspring. Deletion of Gm26878 in founder mice was verified by Sanger sequencing of the PCR-amplified deletion allele junction fragment. One founder mouse and one deletion allele were used to establish the knockout line.
Off-target mutagenesis screening
Potential sgRNA off-target sites were identified with the Wellcome Trust Sanger Institute (WTSI) Genome Editing website (http://www.sanger.ac.uk/htgt/wge/) (Hodgkins et al. 2015). High Resolution Melting (HRM) analysis (MeltDoctor HRM, ThermoFisher) on a Quantstudio 7 (ThermoFisher) was used to screen for off-target mutagenesis events at all off-target sites with ≤3 mismatches and a conventional protospacer adjacent motif (PAM) for Streptococcus pyogenes (5’-NGG) for each guide in G2 animals and C57BL/6J controls. Locations of the off-target sites analyzed for each guide and the primers used for HRM analysis are listed in Table S1.
Intergenic sequences affected by off-target mutagenesis were analyzed for functional elements using the University of California Santa Cruz (UCSC) Genome Browser (build NCBI37/mm9) and the available mouse Encyclopedia of DNA Elements (ENCODE) (Mouse ENCODE Consortium 2012) and TargetScan miRNA Regulatory Site (Agarwal et al. 2015) data tracks for transcription factor binding sites, histone modifications, and DNase hypersensitivity sites, and miRNA binding sites. Moreover, mutagenized sequences were analyzed for predicted transcription factor binding sites using Match 1.0 Public (http://gene-regulation.com/pub/programs.html) and the TRANSFAC (Fu and Weng 2005; Matys et al. 2006) positional weight matrices (vertebrates matrices with settings to minimize false positives).
Quantitative RT-PCR analysis of Foxf1 expression
RNA samples from control and Gm26878 lungs of day 18.5 mouse embryos were reverse-transcribed to cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Foxf1 transcript levels were normalized to Gapdh. Foxf1 and Gapdh TaqMan primer/probe (exon junction-spanning) sets were obtained from Applied Biosystems. qPCR was repeated three times for each of four to eight biological replicates using TaqMan Universal PCR Master Mix (Applied Biosystems). qPCR conditions included 40 cycles of 95˚C for 15 sec and 60˚C for 1 min. For relative quantification of the Foxf1 transcript, the comparative CT method was used. Wild-type 1-day pup lung was designated as a calibrator sample.
Statistical analysis
Statistical analysis of gene expression differences between genotype groups was done using a one-way analysis of variance (ANOVA). Chi-square goodness-of-fit tests were used to test for statistical deviation from expected Mendelian segregation ratios for on target and off-target mutant alleles in live or dead G2 offspring.
Results
Generation and genetic analysis of Gm26878 lncRNA knockout mice
To experimentally assess the involvement of Gm26878 in regulation of Foxf1 expression in mice, we deleted the entire Gm26878 gene using in vivo CRISPR/Cas9 genome editing (Figure 1A-B). Two sgRNAs designed to target Cas9 to sequences flanking Gm26878 were injected into pro-nuclear stage embryos with Cas9 mRNA. Nonhomologous end joining (NHEJ)-mediated repair of the two double-strand breaks (DSBs) created by sgRNA-targeted Cas9 were expected to delete the entire genomic interval containing Gm26878. In total, 40 live-born mice were obtained from 100 injected and transferred embryos. PCR genotyping results showed that 13 of the 40 animals (33%) were putative founders harboring a Gm26878 genomic deletion (Figure 1C). As expected, due to the imprecise nature of NHEJ and the insertion or deletion of bases at the DSB repair junction site, putative deletion allele PCR products of different sizes were detected in each founder. With the exception of founder M8, only one putative deletion allele PCR product was observed in the founders, suggesting low levels of mosaicism.
Figure 1. Gm26878 deletion analysis.
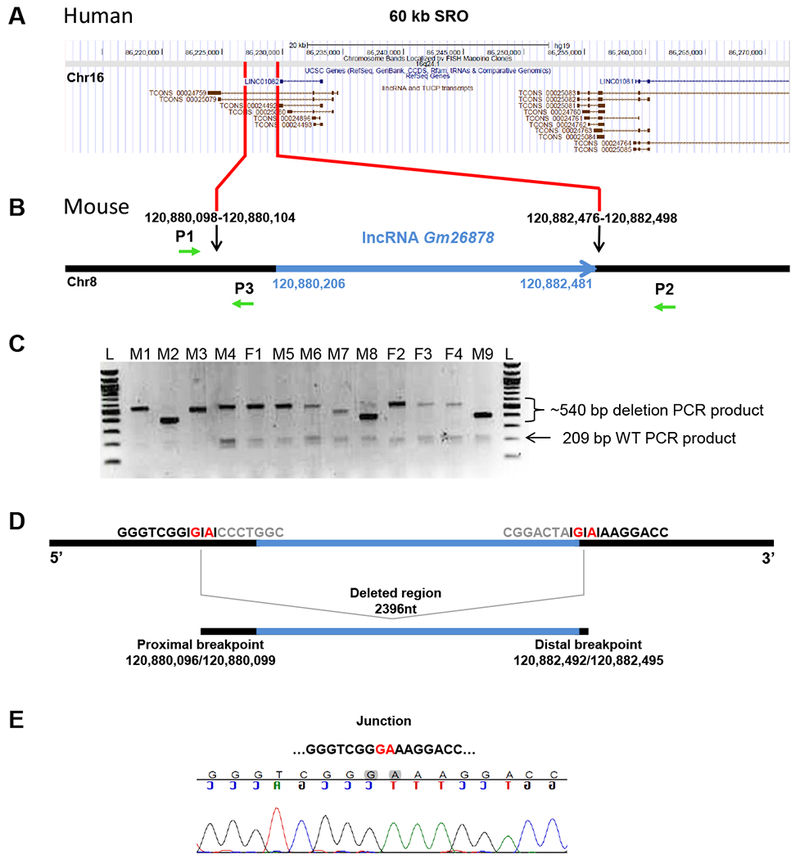
(A) RefSeq annotation (GRCh37/hg19) of the FOXF1 upstream enhancer region SRO (Smallest deletion Region of Overlap delineating the enhancer) of human chromosome 16 corresponding to a fragment of mouse chromosome 8 harboring Gm26878 lncRNA locus. (B) Schematic representation of Gm26878 lncRNA locus (blue) and neighboring sequences (black). Black arrows indicate sgRNA target sites (chr8:120,880,082-120,880,104 and chr8:120,882,476-120,882,498; GRCm38/mm10); green arrows indicate P1, P2, and P3 primers used for genotyping and sequencing (P1 and P3 amplify the wild-type allele; P1 and P2 amplify interval deletion alleles). (C) Agarose gel electrophoresis of products amplified using three PCR primers P1, P2, and P3 on gDNA extracted from 13 founder mice from co-injection of Cas9 and sgRNAs; M = male, F = female, L = ladder, 209 bp = wild-type PCR product, ~540 bp = approximate size of interval deletion PCR products. (D) Deleted region harboring Gm26878 lncRNA with proximal (chr8:120,880,096/120,880,099; GRCm38/mm10) and distal (chr8:120,882,492/120,882,495; GRCm38/mm10) breakpoints. The exact position of breakpoints is unknown due to 2 bp microhomology (marked in red). (E) Chromatogram showing DNA sequences of the nonhomologous end joining (NHEJ) repair junction.
Founders M1 and M3 were backcrossed to C57BL/6J females to produce G1 progeny and Sanger sequencing was used to confirm transmission of a Gm26878 deletion allele. Curiously, Sanger sequencing of the putative deletion allele in G1 progeny from founder M3 identified an allele consisting of the deletion interval reinserted as an inversion (Figure S1). Sanger sequencing of the predicted deletion allele confirmed the presence of a single 2,396 bp interval deletion spanning lncRNA Gm26878 in G1 progeny generated from founder M1 (Figure 1D,E).
Gm26878 gene dosage affects newborn viability but not Foxf1 expression
To test whether Gm26878 deficiency has phenotypic outcomes, G1 mice derived from founder M1 were intercrossed to produce experimental heterozygous (Gm26878+/−) and homozygous (Gm26878−/−) knockout and control wild-type (Gm26878+/+) G2 progeny. Genotyping of 113 G2 pups revealed that the Gm26878 deletion allele was transmitted to G2 offspring in the expected Mendelian frequency of 1 wild-type: 2 heterozygotes: 1 homozygote) (Table 1). However, amongst the G2 pups born, only 95 of 113 remained alive within the first few days of birth, including 27 Gm26878+/+, 52 Gm26878+/−, and 16 Gm26878−/− animals. Genotyping of the 18 pups that died revealed that only two were wild-type for Gm26878 with the remaining dead pups either homozygous (N=9) or heterozygous (N=7) for the Gm26878 deletion. Chi-square analysis of genotypes amongst the dead pups indicated a statistical deviation from the expected 1:2:1 ratio if neonatal lethality was random, with a higher than expected number of dead Gm26878−/− animals (Table 2). Using RT-qPCR, we did not observe any statistically significant difference in Foxf1 expression in the lungs of Gm26878−/− and Gm26878+/− day 18.5 mouse embryos when compared to wild-type controls (Figure 2).
Table 1.
Gm26878 genotype distribution in offspring from Gm26878+/− intercrosses.
| Genotype | No. observed | No. expected* | Test score (χ2, P value)† |
|---|---|---|---|
| Gm26878+/+ | 29 | 28.25 | 0.57, NS |
| Gm26878+/− | 59 | 56.5 | |
| Gm26878−/− | 25 | 28.25 |
Assumes a 1:2:1 genotype segregation ratio amongst offspring (N=113).
NS, not significant
Table 2.
Gm26878 genotype distribution in dead offspring from Gm26878+/− intercrosses.
| Genotype | No. observed | No. expected* | Test score (χ2, P value)† |
|---|---|---|---|
| Gm26878+/+ | 2 | 4.5 | 6.33, <0.05 |
| Gm26878+/− | 7 | 9 | |
| Gm26878−/− | 9 | 4.5 |
Assumes neonatal death is random and a 1:2:1 segregation ratio amongst the dead offspring (N=18).
NS, not significant
Figure 2.
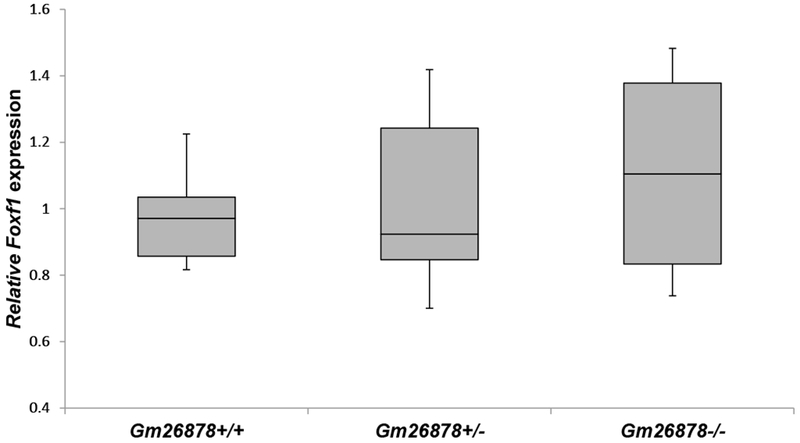
Comparative RT qPCR analysis of the Foxf1 mRNA levels in lung tissues of the P18.5 G2 Gm26878+/+, Gm26878+/−, and Gm26878−/− mice. Means of three technical replicas for each biological replica are ploted (ANOVA test comparing Gm26878−/− mice lung with wild-type mutant lungs and Gm26878+/− mice lungs: P=0.443 and P=0.704, respectively).
Off-targets analysis
To test whether either of the sgRNAs mediated an off-target mutagenesis event in founder M1 and whether that off-target mutagenesis event was transmitted to resulting progeny, high resolution melting analysis (HRM) was used to screen off-target sites with ≤3 mismatches for each sgRNA in G2 animals (N= 4-32) (Table S1). HRM analysis of an off-target site on chromosome 4 (143096216-143096238; GRCm38/mm10), with 3 sequence mismatches to the sgRNA that targeted sequence upstream of the Gm26878 transcriptional start site, identified a single mutagenesis event segregating in the G2 population (Figure 3A). Sanger sequencing confirmed a 13 bp deletion at the off-target site (Figure 3B,C). G2 animals heterozygous and homozygous for the off-target mutation were identified, indicating that G1 parents were heterozygous for the mutation. Importantly, analysis of 17 live G2 offspring from three litters produced by one mating pair of Gm26878+/− G1 animals also heterozygous for the off-target mutation revealed that the off-target mutation was segregating at the expected 1:2:1 Mendelian ratio (Table 3). Importantly, of the live Gm26878−/− G2 offspring analyzed, two were found to be homozygous for the off-target mutation. Similarly, of the 10 live Gm26878+/− G2 offspring analyzed, six were found to be heterozygous and two homozygous for the off-target mutation. Together these results indicate that the animals homozygous for both on and off-target mutations are viable and that the off-target mutation is not contributing to neonatal lethality.
Figure 3. Off-target mutagenesis on chromosome 4.
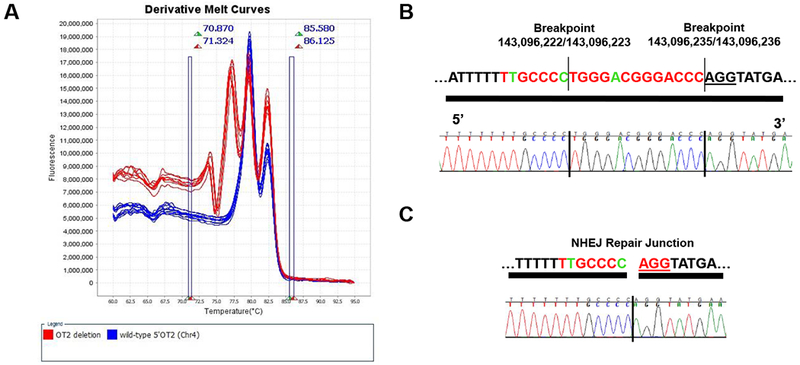
(A) High-resolution melting analysis identified a putative mutagenesis event at a predicted off-target site (chr4:143,096,216-143,096,238; GRCm38/mm10) for the guide RNA targeting sequences upstream of the Gm26878 transcriptional start site. (B) Sequence of the off-target site on chromosome 4. Bases that match the on target sgRNA target site on chromosome 8 are shown in red, mismatches are shown in green, and the PAM sequence is underlined. Flanking sequences are in black. Breakpoints (chr4; GRCm38/mm10) of a 13 base deletion allele segregating in wild-type and heterozygous and homozygous Gm26878 G2 progeny are indicated with horizontal lines. (C) Chromatogram showing DNA sequences of the nonhomologous end joining (NHEJ) repair junction at the chromosome 4 off-target mutagenesis site.
Table 3.
Chromosome 4 off target site genotype distribution in live offspring from intercrosses between G1 animals heterozygous for the on and off target site.
| Genotype | No. observed | No. expected* | Test score (χ2, P value)† |
|---|---|---|---|
| Wild-type | 4 | 4.25 | 0.06, NS |
| Heterozygous | 9 | 8.5 | |
| Homozygous | 4 | 4.25 |
Assumes a 1:2:1 segregation ratio of the off target site amongst live offspring (N=17 total; N=4 Gm26878+/+, N=10 Gm26878+/−, N=3 Gm26878−/−).
NS, not significant
The deletion on chromosome 4 is in intergenic sequence approximately 11 kb from the transcription stop site of the closest annotated gene (Prdm2; chr4:143107391-143212709; GRCm38/mm10). Analysis of ENCODE and TargetScan data accessed through the UCSC genome browser showed no annotated transcription factor binding sites, histone binding sites, DNase hypersensitivity sites, or miRNA binding sites corresponding to the 13 bp deletion (chr4:142,686,119-142,686,141; NCBI37/mm9) (data not shown). Furthermore, using the TRANSFAC database, analysis of a 60 bp window of sequence including and surrounding the deletion interval (chr4:143,096,198-143,096,257; GRCm38/mm10) failed to identify a putative transcription factor binding site (data not shown). These analyses provide further evidence that the off-target event was not causing the increased postnatal lethality of Gm26878−/− G2 animals.
Discussion
Accumulating evidence suggests that expression of lncRNAs is integral to function of many if not every enhancer (Mattick 2010; Lopes et al. 2016; Wright et al. 2016). Using CRISPR/Cas9 technology, we have generated knockout mice lacking the entire lncRNA gene Gm26878 encoded in the region corresponding to the FOXF1 upstream enhancer region in humans. This mouse genomic region was recently shown to function as a Foxf1 enhancer (Seo et al. 2016). Gm26878 is located within the 1st intron of similarly oriented gene Gm20388 encoding Galnt2 (acetyl-galacto-aminotransferase), absent in the syntenic region in humans. The human FOXF1 upstream enhancer region encodes two groups of lncRNAs: LINC01082 and RP11-805I24.3 (TCONS 00024759 and its isoforms), and LINC01081 and RP11-514D23.3 (TCONS 00025083 and its isoforms). The genomic position of Gm26878 corresponds to that of human RP11-805I24.3 although, in contrast to RP11-805I24.3, it is oppositely oriented compared to Foxf1. The analysis of the survival of Gm26878 knockout mice revealed a significant increase lethality amongst homozygous mice, in most cases occurring within the first day after birth. Lethality observed in wild-type and heterozygous mice was apparently related to genetic background of mice used to generate Gm26878 knockout. This survival pattern resembles that of heterologous Foxf1 mice. However, the lungs of Gm26878 mice were not hypoplastic or hemorrhaging as mouse lungs heterozygous for Foxf1. In agreement with the relatively normal appearance of Gm26878 lungs, Foxf1 expression in Gm26878 pups was found unaffected. Hence, we conclude that lncRNA Gm26878 does not likely regulate Foxf1 expression. The regulation of the expression of mouse Gm26878 or human lncRNAs encoded in the FOXF1 upstream enhancer region remain to be determined. Evidence from human suggests partial parent-of-origin mode of FOXF1 expression that involves epigenetically regulated LINC01081, LINC01082 and possibly other lncRNAs. Nevertheless, the evidence for epigenetic control of any of the FOXF1 lncRNAs or mouse Gm26878 is still lacking. Moreover, even if the lack of Foxf1 expression changes in response to the loss of Gm26878 would be caused by deletion of the epigenetically inactivated allele, it would be seen only in heterozygotic but not in homozygotic mice. However, we cannot exclude the effect of a modifier gene on Foxf1 expression that was masking the effect of the depletion of Gm26878, or the presence of redundant lncRNAs that substituted for the loss of Gm26878.
Specificity of the low penetrance, neonatal lethality phenotype to Gm26878 deficiency was verified by screening for mutagenesis at off-target sites with ≤ 3 mismatches to the on target for each sgRNA. We did observe mutagenesis at an off-target site with 3 mismatches, highlighting the importance of performing quality control assays to verify observed phenotypes in CRISPR/Cas9-generated alleles. Importantly, the off-target mutation was randomly segregating from the on target Gm26878 deletion, was observed in wild-type, heterozygous knockout, and homozygous knockout G2 littermates, and was present in viable offspring of all genotypes. Moreover, the closest annotated gene to the off-target mutation, Prdm2, was previously reported to cause a wide spectrum of tumors but not neonatal lethality when knocked out in mice (Steele-Perkins et al. 2001). Thus, it is unlikely that the off-target event was causing the postnatal lethality that occurred at a higher incidence in Gm26878−/− mice.
We have previously shown that siRNA-mediated decrease of LINC01081 level results in a decrease of the expression of FOXF1 (Szafranski et al. 2014), and that homozygous deletions, including LINC01082 and RP11-805I24.3, result in weaker but still lethal ACDMPV phenotype, suggesting involvement also of LINC01082 or RP11-805I24.3 lncRNAs in the regulation of FOXF1. Thus, the data presented here indicate that the regulation of FOXF1 expression differs between human and mouse.
However, this conclusion does not seem to apply to all lncRNA genes encoded in Foxf1 region because lncRNA Fendrr, located immediately upstream of Foxf1, is involved in lung development both in mouse and human (Sauvageau et al. 2013; Lai et al. 2015). Future expression profiling of the Gm26878 mice will likely shed more light on the nature of developmental process regulated by this lncRNA.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (RO1HL101975) to P.S., and National Organization for Rare Disorders (NORD grants 2012 and 2014) to P.Sz. Resources accessed through cores were supported by a National Institutes of Health-National Cancer Institute grant (CA125123) to the Dan L. Duncan Cancer Center.
References
- Agarwal V, Bell GW, Nam JW, Bartel DP. (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AR, Tibbit C, Ponting CP, Liu JL (2013) Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep 4:220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos J, Dutta A, Guigó R, Gingeras TR, Margulies EH, et al. ; ENCODE Project Consortium; NISC Comparative Sequencing Program; Baylor College of Medicine Human Genome Sequencing Center; Washington University Genome Sequencing Center; Broad Institute; Children’s Hospital Oakland Research Institute (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NB, Stankiewicz P, Steinhorn RH (2011) Alveolar capillary dysplasia. Am J Respir Crit Care Med 184:172–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner AL, Beck AH, Edris B, Sweeney RT, Zhu SX, Li R, et al. (2012) Transcriptional profiling of long non-coding RNAs and novel transcribed regions across a diverse panel of archived human cancers. Genome Biol 13:R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, et al. FANTOM Consortium; RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group) (2005) The transcriptional landscape of the mammalian genome. Science 309:1559–1563 [DOI] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22:1775–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. (2012) Landscape of transcription in human cells. Nature 489:101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Weng Z (2005) Improvement of TRANSFAC matrices using multiple local alignment of transcription factor binding site sequences. Genome Inform 16:68–72. [PubMed] [Google Scholar]
- Hodgkins A, Farne A, Perera S, Grego T, Parry-Smith DJ, Skarnes WC, Iyer V (2015) WGE: a CRISPR database for genome engineering. Bioinformatics. 31:3078–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V et al. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V, Shen B, Zhang W, Hodgkins A, Keane T, Huang X, et al. (2015) Off-target mutations are rare in Cas9-modified mice. Nat Methods 12:479. [DOI] [PubMed] [Google Scholar]
- Kalinichenko VV, Lim L, Shin B, Costa RH (2001) Differential expression of forkhead box transcription factors following butylated hydroxytoluene lung injury. Am J Physiol Lung Cell Mol Physiol 280:L695–L704 [DOI] [PubMed] [Google Scholar]
- Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. (2007) RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316:1484–1488 [DOI] [PubMed] [Google Scholar]
- Lai KM, Gong G, Atanasio A, Rojas J, Quispe J, Posca J, et al. (2015) Diverse Phenotypes and Specific Transcription Patterns in Twenty Mouse Lines with Ablated LincRNAs. PLoS One 10:e0125522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston C (1991) Misalignment of pulmonary veins and alveolar capillary dysplasia. Pediatr Pathol 11:163–170 [DOI] [PubMed] [Google Scholar]
- Lopes R, Korkmaz G, Agami R (2016) Applying CRISPR-Cas9 tools to identify and characterize transcriptional enhancers. Nat Rev Mol Cell Biol 17:597–604 [DOI] [PubMed] [Google Scholar]
- Mahlapuu M, Enerback S, Carlsson P (2001) Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signalling, causes lung and foregut malformations. Development 128:2397–2406 [DOI] [PubMed] [Google Scholar]
- Mattick JS (2010) Linc-ing Long noncoding RNAs and enhancer function. Dev Cell 19:485–486 [DOI] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, et al. (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34(Database issue):D108–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Mattick JS (2013) Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol 20:300–307 [DOI] [PubMed] [Google Scholar]
- Mouse ENCODE Consortium, Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, et al. (2012) An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 13:418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KC, Frith MC, Mattick JS (2006) Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 22:1–5 [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch E. Maniatis T (1989) Molecular Cloning: A Laboratory Manual, 2nd ed. CSH Laboratory Press, Cold Spring Harbor [Google Scholar]
- Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, et al. (2013) Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2:e01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Thakur N, Stockton D W, Langston C, Bejjani BA (2004) Expanding the phenotype of alveolar capillary dysplasia (ACD). J Pediatr 145:646–651 [DOI] [PubMed] [Google Scholar]
- Seo H, Kim J, Park GH, Kim Y, Cho SW (2016) Long-range enhancers modulate Foxf1 transcription in blood vessels of pulmonary vascular network. Histochem Cell Biol 146:289–300 [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, et al. (2009) Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet 84:780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele-Perkins G, Fang W, Yang XH, Van Gele M, Carling T, Gu J (2001) Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev 15:2250–2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranski P, Dharmadhikari AV, Brosens E, Gurha P, Kolodziejska KE, Zhishuo O, et al. (2013) Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res 23:23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranski P, Dharmadhikari AV, Wambach JA, Towe CT, White FV, Grady RM, et al. (2014) Two deletions overlapping a distant FOXF1 enhancer unravel the role of lncRNA LINC01081 in etiology of alveolar capillary dysplasia with misalignment of pulmonary veins. Am J Med Genet A 164A:2013–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranski P, Gambin T, Dharmadhikari AV, Akdemir KC, Jhangiani SN, Schuette J, et al. (2016a) Pathogenetics of alveolar capillary dysplasia with misalignment of pulmonary veins. Hum Genet 135:569–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranski P, Herrera C, Proe LA, Coffman B, Kearney DL, Popek E, Stankiewicz P (2016b) Narrowing the FOXF1 distant enhancer region on 16q24.1 critical for ACDMPV. Clin Epigenet 8:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP (2011) Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147:1537–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, et al. (2010) Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 464:409–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O, Chang HY (2011) Long noncoding RNAs and human disease. Trends Cell Biol 21:354–361 [DOI] [PubMed] [Google Scholar]
- Wright JB, Sanjana NE (2016) CRISPR Screens to Discover Functional Noncoding Elements. Trends Genet 32:526–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R (2013) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154:1370–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.