

Abstract
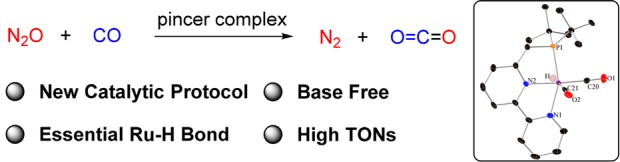
Both CO and N2O are important, environmentally harmful industrial gases. The reaction of CO and N2O to produce CO2 and N2 has stimulated much research interest aimed at degradation of these two gases in a single step. Herein, we report an efficient CO oxidation by N2O catalyzed by a (PNN)Ru–H pincer complex under mild conditions, even with no added base. The reaction is proposed to proceed through a sequence of O-atom transfer (OAT) from N2O to the Ru–H bond to form a Ru–OH intermediate, followed by intramolecular OH attack on an adjacent CO ligand, forming CO2 and N2. Thus, the Ru–H bond of the catalyst plays a central role in facilitating the OAT from N2O to CO, providing an efficient and novel protocol for CO oxidation.
Because of the widespread use of nitrogenous chemicals in agriculture and industrial processes, N2O has become the most abundant stratospheric ozone depletion substance and concomitantly one of the most potent greenhouse gases (N2O is ca. 300 times more potent than CO2) due to atmospheric concentration increases.1 To address this environmental issue, efficient destruction/conversion of N2O, including its degradation or reduction to molecular dinitrogen, is of utmost importance and has drawn much attention.2 The reaction of CO and N2O to produce CO2 and N2, which is quite exothermic and strongly driven by formation of dinitrogen, is considered an attractive reductive process due to the destruction of two environmentally harmful industrial gases in one step.3−6 Although heterogeneously catalyzed reactions have been reported,4 reactions homogeneously catalyzed by metal complexes are rare and desirable,6 since they may proceed selectively under mild conditions and may provide fundamental mechanistic information.
Experiments aimed at reduction of N2O by CO involving metal complexes were reported.5 In early stoichiometric work by Bottomley,5a a CO ligand of Cp2Ti(CO)21 was converted to free CO2 under excess N2O in 15% yield, and the process was suggested to involve reaction of Cp2Ti(CO)21 with N2O, affording [(Cp2Ti)4(CO3)2] 2, which upon further reaction with N2O produces free CO2 and [Cp2TiO] 3. A concerted transient species M1 (Scheme 1) was proposed, in which the O atom interacts with Ti and CO ligand directly. Sita reported stoichiometric photolytic CO oxidation with N2O in the presence of Cp*M[η2-N(iPr)C(Me)N(iPr)](CO)2 (M = Mo or W) 4.5b−5d The reaction was initiated by oxidation of 4 by N2O forming Cp*M(O)[η2-N(iPr)C(Me)N(iPr)] 5, releasing N2 and CO. O-atom transfer (OAT) in the presence of CO afforded CO2. While catalysis was mentioned, no catalytic data were reported. Homogeneously catalyzed reaction of CO with N2O was reported by Cheng, using metal carbonyl anions.6 In this system N2O was proposed to undergo nucleophilic attack by the anionic metal center of [Rh(CO)4]−, [Fe2(CO)8]2–, and [Ru4(CO)13]2– (M3, Scheme 1) followed by intramolecular OAT to form CO2. Although catalytic, a large excess of base was required, and low turnover numbers (TONs) were obtained (maximum ca. 50). Thus, the development of mild and efficient homogeneously catalyzed N2O reduction by CO is challenging.
Scheme 1. Homogeneous Reactions of CO and N2O.
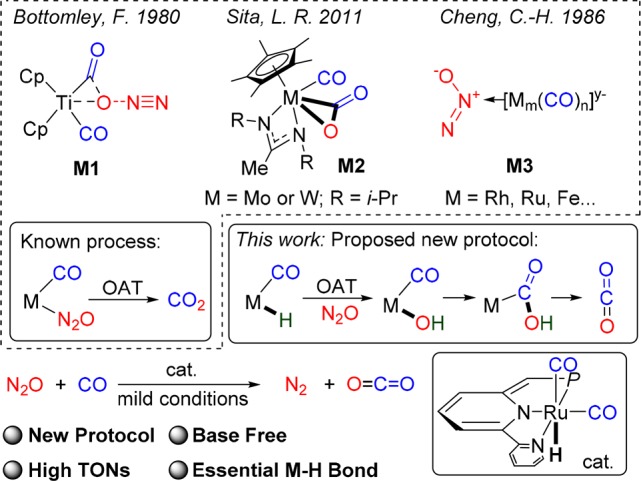
As mentioned above, direct interaction between a CO ligand and N2O was the proposed pathway for CO oxidation by N2O to form CO2 and N2.5,6 In 2017, we developed the homogeneously catalyzed hydrogenation of N2O in high TONs, in which selective mono O-transfer from N2O into a (PNP)Ru–H bond of the pincer catalyst was the key step in the catalysis.7 The efficient O-transfer into Ru–H was studied computationally,9 including specifically the (PNP)Ru–H pincer system,9b,9c and it was concluded to proceed via nucleophilic attack of the hydride ligand on the terminal nitrogen of N2O, followed by a concerted N2 liberation.9,12 This result encouraged us to explore a catalytic CO oxidation by N2O initiated by O-transfer into a M–H bond.8,9 We propose that a H–M–CO complex may undergo O-transfer from N2O into the M–H bond to generate a HO–M–CO species, followed by intramolecular interaction between M–OH and CO ligand to form a M–COOH intermediate (Scheme 1).10 CO2 release in the presence of CO regenerates the H–M–CO complex, thus completing the catalytic cycle. To enable such a homogeneously catalyzed CO oxidation, highly selective OAT from N2O into M–H bond is needed, without decomposing the complex. Herein, we report the development of mild CO oxidation by N2O catalyzed by a (PNN)Ru–H complex based on such a mechanism. The reaction proceeds smoothly, yielding high TON under mild conditions, even with no added base.
Initially, CO oxidation by N2O was examined by treating a premixed solution of 0.01 mmol of the (PNP)Ru complex 6 (P = P(iPr)2,11a) and 1 equiv of t-BuOK in 4 mL of toluene under 1 atm of CO and 2 atm of N2O in a 90 mL Fisher-Porter tube (1 atm of gas in 90 mL corresponds to ca. 3.7 mmol at 20 °C). After heating at 100 °C for 22 h, 0.63 mmol of CO2 (63 TON) was detected by GC in the gas phase. The reaction occurred smoothly, even using catalytic base. In the absence of 6, no CO2 was formed. Moreover, replacing 6 by RuHCl(CO)(PPh3)3 did not lead to CO2 formation. Other pincer Ru complexes were then screened (Table 1). The complex (PNP)Ru(CO)Cl27 (X-ray characterized12) did not lead to any CO2, indicating that the Ru–H bond is crucial for the transformation. With the bulkier complex 8 (P = P(tBu)2),11b only a trace of CO2 was formed. The PNN complex 9(8c,11c) was less active than the PNNH complex 10,11d which yielded 62 TON of CO2. The 2,2-bipyridine-based pincer complexes 12 (P = PPh2)11e and 14 (P = P(tBu)2)11f were less efficient and afforded 24 and 22 TON of CO2, respectively, while use of 13 (P = P(iPr)2)11g resulted in 83 TON of CO2. Interestingly, the catalytic activity of complexes 13 and 14 in THF was higher than in toluene. In THF at 70 °C, 130 and 197 TON were obtained in the gas phase, respectively, and an additional 40 TON (when using 14) was detected in solution. Without base, significantly lower catalytic activity was observed, forming only 30 TON of CO2.
Table 1. Catalyst Screening for CO Oxidation by N2Oa–d.
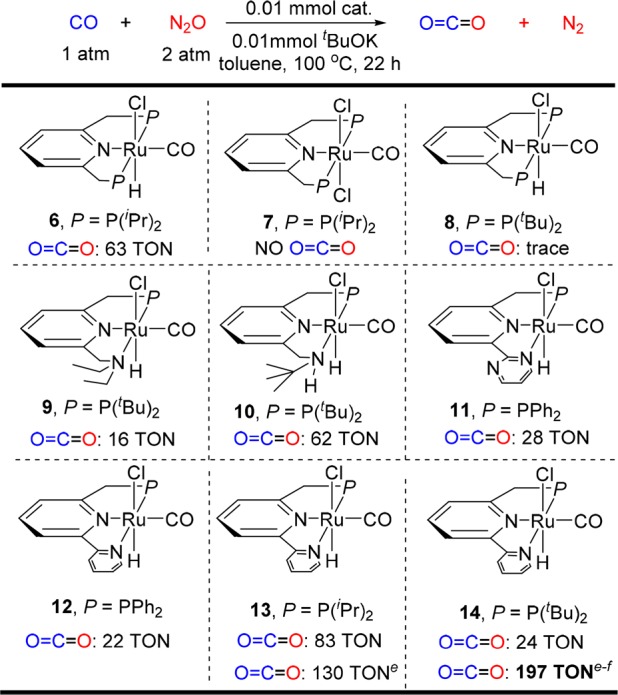
All reactions were conducted in a 90 mL Fisher-Porter tube using the catalyst (0.01 mmol), t-BuOK (0.01 mmol), 3.7 mmol of CO, and 7.4 mmol of N2O in 4 mL toluene.
The catalyst and t-BuOK were premixed in the solvent for 20 min and used directly for the reactions.
The TONs are based on the generated CO2 as measured by GC of the gas phase calibrated by a standard curve.
The reaction was conducted in THF at 70 °C.
The amount of CO2 dissolved in THF was determined as ca. 0.4 mmol (40 TON).13
To gain mechanistic information on this catalytic transformation, individual stoichiometric reactions that may be involved in the catalytic cycle using complex 14 as precatalyst were explored. Reaction of 14 with t-BuOK is known to occur smoothly to afford the dearomatized complex 15.11g Alarmingly, reaction of 15 with excess N2O resulted in complete decomposition of the complex. On the other hand, reaction of 15 with excess CO afforded the dearomatized dicarbonyl complex 16, in which CO coordination stabilizes the dearomatized complex (Scheme 2). Importantly, a competitive experiment of 15 under both N2O and CO (1:1) resulted in the formation of 16 as the major product. Thus, the much faster reaction of 15 with CO prevents the decomposition of 15 caused by over-oxidation by N2O and enables the catalytic cycle.
Scheme 2. Reactions of Complex 15 and Formation of 16.

Crystals of complex 16 were obtained by recrystallization from pentane at −35 °C. The X-ray structure of 16 (Figure 1) reveals an octahedral geometry, the hydride ligand being located trans to a CO ligand. The dearomatized structure of 16 is clearly indicated by the bond length C(10)–C(11) (1.390(3) Å) being much shorter than a C–C single bond (the corresponding bond length in complex 17 is 1.508 Å), indicating a significant double bond character.
Figure 1.
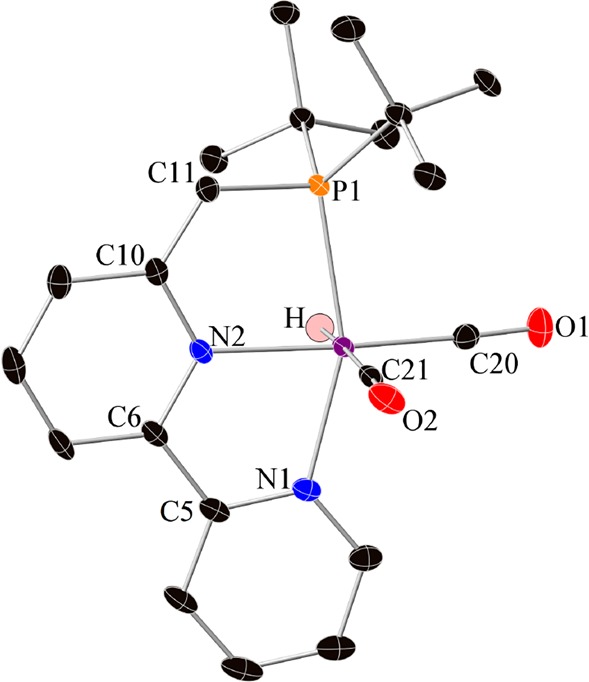
Crystal structure of complex 16. Atoms are presented as thermal ellipsoids at 50% probability level. Hydrogen atoms, except for Ru–H, are not shown. For selected bond lengths and angles, see SI.
While the corresponding dearomatized PNP pincer complexes of 6 and 8 are stable,11a,11b the PNN complex 16 is converted slowly, even at room temperature, to the aromatic complex 17 via C–H activation at the pyridine ring (eq 1).15,16 The Ru–H of 17 appears in 1H NMR in THF at −5.3 ppm (d, 2JPH = 17.6 Hz) and 31P{1H} NMR shows a singlet at 88.4 ppm.12 This transformation is much faster in toluene, providing a possible explanation for the better catalysis in THF than in toluene. Nevertheless, in real-time 31P NMR analysis of the catalytic reaction of CO and N2O using complex 14 in THF, both complexes 16 and 17 were observed. Interestingly, 17 (0.01 mmol) under 1 bar of CO and 2 bar of N2O also catalyzed the reaction, but with much lower efficiency, affording only 54 TON of CO2 in the gas phase. Thus, although 16 is the more active catalyst (see below), participation of 17 in the catalysis cannot be excluded.
 |
1 |
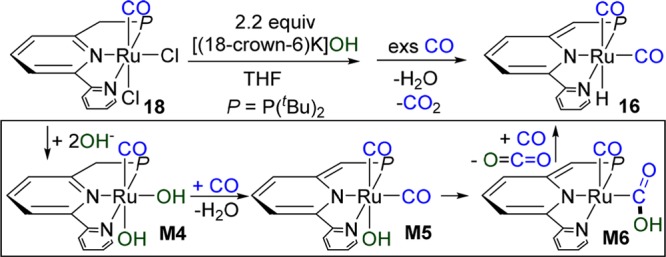
The reaction of complexes 16 or 17 with N2O (1 equiv or excess) in the absence of CO resulted in decomposition and the Ru–OH complex was not observed. Aiming at generation of (PNN)Ru(OH)X proposed in Scheme 1, the aromatized pincer complex (PNN)RuCl2(CO) 18 was prepared and crystallographically characterized.12 Complex 18 was subjected to a reaction with 2.2 equiv of [(18-crown-6)K]OH in THF at room temperature (Scheme 3). Interestingly, while the proposed ruthenium dihydroxo complex M4 and dearomatized ruthenium hydroxo complex M5 were not detected, treatment of the reaction mixture with CO resulted in formation of the dearomatized hydride complex 16, and CO2 (detected by GC), suggesting that the reaction proceeds through halide substitution by [(18-crown-6)K]OH followed by water elimination via metal–ligand cooperation (MLC)16 forming in the presence of CO the complex M5, which is unstable and leads to 16 by releasing CO2 under CO. The relatively fast intramolecular reaction between the OH group and CO results in CO2 formation, likely via a RuCOOH intermediate M6,10 and suggests that the catalysis is enabled by O-insertion from N2O into the Ru–H bond.7−9 These results suggest that the turnover limiting step in the process is the oxygen-atom-transfer step.
Scheme 3. Reaction of Complex 18 with [(18-C-6)K]OH.
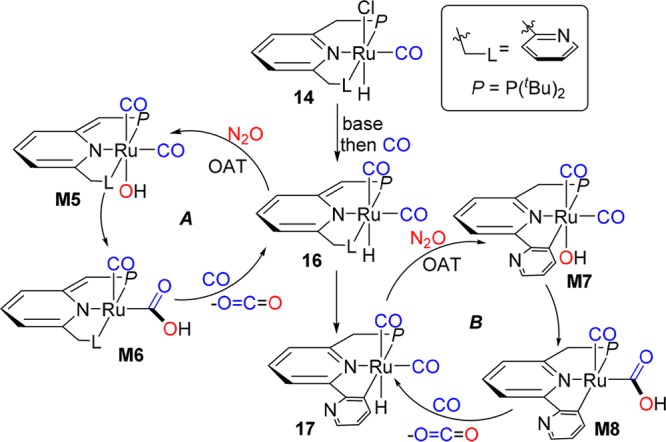
On the basis of these observations and reported DFT studies,9 a plausible mechanism for this reaction is proposed (Scheme 4). First, the premixed solution of precatalyst 14 and base generates the dearomatized ruthenium complex, which reacts with CO immediately, affording the dicarbonyl complex 16.11,12 Efficient OAT from N2O into Ru–H, which is likely initiated by nucleophilic attack of the hydride ligand on N2O,9 results in formation of a hydroxo intermediate M5,7−9 which might undergo intramolecular nucleophilic attack by hydroxide on the adjacent CO to give a RuCOOH intermediate M6,10 followed by β-H elimination to form CO2 and regeneration of 16 under CO.10 In addition, a less efficient mechanism involving complex 17, obtained during catalysis via MLC16 and C–H activation,15 might also take place to some extent, by undergoing OAT (M7) and CO2 formation (M8) (catalytic cycle B). In both processes, the Ru–H bond plays a key role in assisting OAT to CO. Throughout this catalytic cycle the formal metal oxidation state may not change, providing a novel protocol for CO oxidation by N2O.
Scheme 4. Possible Mechanism.
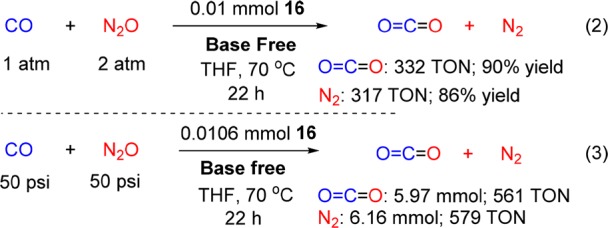
This plausible mechanism encouraged us to explore the catalytic reaction using complex 16 as catalyst with no added base. Remarkably, full conversion of CO was achieved by using 0.01 mmol of complex 16 under mild, base-free conditions (eq 2). CO2 was produced in 90% yield (2.72 mmol in the gas phase and 0.6 mmol in solution, for a total of 332 TON) and 86% yield of dinitrogen (3.17 mmol, 317 TON) was determined by GC.
 |
2 |
In addition, the highest TONs were achieved by using 0.0106 mmol of 16 as catalyst in the presence of excess of CO (50 psi) and N2O (50 psi). After heating for 22h, 6.16 mmol of dinitrogen (579 TON) together with 5.97 mmol of CO2 (4.97 mmol in gas phase and ca. 1.0 mmol in solution; 561 TON) were produced (eq 3).
In summary, a new homogeneously catalyzed reaction of CO and N2O to produce CO2 and N2 was developed. High efficiency and high TON were achieved using the ruthenium complex 14 as the precatalyst or the corresponding dearomatized complex 16 as the actual catalyst. The reaction catalyzed by 16 proceeds smoothly under base-free conditions, providing an efficient method for degradation of both CO and N2O in a single step. The Ru–H bond is necessary for the catalysis. The catalytic cycle is proposed to involve selective O-atom transfer into Ru–H of 16, intramolecular CO insertion into the resulting Ru–OH, and subsequent CO2 liberation, regenerating the catalyst 16 in the presence of CO. Quite remarkably, while N2O alone decomposes the catalyst, this is prevented by the presence of the more reactive CO.
Acknowledgments
This research was supported by the European Research Council (ERC AdG 692775). D.M. holds the Israel Matz Professorial Chair of Organic Chemistry. R.Z. thanks the Faculty of Chemistry for being awarded a Dean’s Fellowship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b03927.
The authors declare no competing financial interest.
Supplementary Material
References
- a Prather M. Time Scales in Atmospheric Chemistry: Coupled Perturbations to N2O, NOy, and O3. Science 1998, 279, 1339–1341. 10.1126/science.279.5355.1339. [DOI] [PubMed] [Google Scholar]; b Ravishankara A. R.; Daniel J. S.; Portmann R. W. Nitrous Oxide (N2O): The Dominant Ozone-Depleting Substance Emitted in the 21st Century. Science 2009, 326, 123–125. 10.1126/science.1176985. [DOI] [PubMed] [Google Scholar]; Highlight:Dameris M. Depletion of the Ozone Layer in the 21st Century. Angew. Chem., Int. Ed. 2010, 49, 489–491. 10.1002/anie.200906334. [DOI] [PubMed] [Google Scholar]; c U.S. Greenhouse Gas Inventory Report: 1990–2014, U.S. EPA; https://www.epa.gov/ghgemissions/us-greenhouse-gas-inventory-report-1990-2014; d Hansen J.; Sato M. Greenhouse gas growth rates. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16109–16114. 10.1073/pnas.0406982101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews on N2O chemistry, see:; a Tolman W. B. Binding and Activation of N2O at Transition-Metal Centers: Recent Mechanistic Insights. Angew. Chem., Int. Ed. 2010, 49, 1018–1024. 10.1002/anie.200905364. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Konsolakis M. Decomposition over Non-Noble-Metal Oxide Catalysts: Catalytic Performance, Mechanistic Considerations, and Surface Chemistry Aspects. ACS Catal. 2015, 5, 6397–6421. 10.1021/acscatal.5b01605. [DOI] [Google Scholar]; c Severin K. Synthetic Chemistry with Nitrous Oxide. Chem. Soc. Rev. 2015, 44, 6375–6386. 10.1039/C5CS00339C. [DOI] [PubMed] [Google Scholar]; d Parmon V. N.; Panov G. I.; Uriarte A.; Noskov A. S. Nitrous oxide in oxidation chemistry and catalysis: application and production. Catal. Today 2005, 100, 115–131. 10.1016/j.cattod.2004.12.012. [DOI] [Google Scholar]; e Pauleta S. R.; Dell’Acqua S.; Moura I. Nitrous oxide reductase. Coord. Chem. Rev. 2013, 257, 332–349. 10.1016/j.ccr.2012.05.026. [DOI] [Google Scholar]; f Leont’ev A. V.; Fomicheva O. A.; Proskurnina M. V.; Zefirov N. S. Modern chemistry of nitrous oxide. Russ. Chem. Rev. 2001, 70, 91–104. 10.1070/RC2001v070n02ABEH000631. [DOI] [Google Scholar]; g Lee D.-H.; Mondal B.; Karlin K. D.. Nitrogen Monoxide and Nitrous Oxide Binding and Reduction. In Activation of Small Molecules; Tolman W. B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2006; pp 43–79. [Google Scholar]
- For a review on CO oxidation by N2O via single-atom catalysis, see:Schwarz H. Ménage-à-trois: Single-atom Catalysis, Mass Spectrometry, and Computational Chemistry. Catal. Sci. Technol. 2017, 7, 4302–4314. 10.1039/C6CY02658C. [DOI] [Google Scholar]
- For selected heterogeneous CO oxidation by N2O, see:; a McCabe R. W.; Wong C. Steady-State Kinetics of the CO-N2O Reaction over an Alumina-Supported Rhodium Catalyst. J. Catal. 1990, 121, 422–431. 10.1016/0021-9517(90)90250-N. [DOI] [Google Scholar]; b Belton D. N.; Schmieg S. J. Kinetics of CO Oxidation by N2O over Rh(111). J. Catal. 1992, 138, 70–78. 10.1016/0021-9517(92)90007-5. [DOI] [Google Scholar]; c Sadhankar R. R.; Ye J.; Lynch D. T. N2O Reducation by CO over an Alumina-Supported Pt Catalyst: Steady-State Multiplicity. J. Catal. 1994, 146, 511–522. 10.1006/jcat.1994.1089. [DOI] [Google Scholar]; d Gluhoi A. C.; Dekkers M. A. P.; Nieuwenhuys B. E. Comparative Studies of The N2O/H2, N2O/CO, H2/O2 and CO/O2 Reactions on Supported Gold Catalysts: Effect of the Addition of Various Oxides. J. Catal. 2003, 219, 197–205. 10.1016/S0021-9517(03)00185-4. [DOI] [Google Scholar]; e Zhdanov V. P.; Ma Y.; Matsushima T. Analysis of the Kinetics of N2O-CO reaction on Pd(110). Surf. Sci. 2005, 583, 36–45. 10.1016/j.susc.2005.03.020. [DOI] [Google Scholar]; f Zhdanov V. P.; Nakagoe O.; Matsushima T. Kinetics of the N2O-CO reaction on Rh(110). Surf. Sci. 2007, 601, L49–L54. 10.1016/j.susc.2007.03.019. [DOI] [Google Scholar]; g Jia A.-P.; Jiang S.-Y.; Lu J.-Q.; Luo M.-F. Study of Catalytic Activity at the CuO-CeO2 Interface for CO oxidation. J. Phys. Chem. C 2010, 114, 21605–21610. 10.1021/jp108556u. [DOI] [Google Scholar]
- For stoichiometric CO oxidation by N2O in solution, see:; a Bottomley F.; Lin I. J. B.; Mukaida M. Reactions of Dinitrogen Oxide (Nitrous Oxide) with Dicyclopentadienyltitanium Complexes Including a Reaction in Which Carbon Monoxide Is Oxidized. Carbon Monoxide-Induced N–N Bond Cleavage of Nitrous Oxide That Is Competitive with Oxygen Atom Transfer to Carbon Monoxide as Mediated by a Mo(II)/Mo(IV) Catalytic Cycle. J. Am. Chem. Soc. 1980, 102, 5238–5242. 10.1021/ja00536a020. [DOI] [Google Scholar]; b Reeds J. P.; Yonke B. L.; Zavalij P. Y.; Sita L. R. Carbon Monoxide-Induced N–N Bond Cleavage of Nitrous Oxide That Is Competitive with Oxygen Atom Transfer to Carbon Monoxide as Mediated by a Mo(II)/Mo(IV) Catalytic Cycle. J. Am. Chem. Soc. 2011, 133, 18602–18605. 10.1021/ja208669s. [DOI] [PubMed] [Google Scholar]; c Yonke B. L.; Reeds J. P.; Zavalij P. Y.; Sita L. R. Catalytic Degenerate and Nondegenerate Oxygen Atom Transfers Employing N2O and CO2 and a MII/MIV Cycle Mediated by Group 6 MIV Terminal Oxo Complexes. Angew. Chem., Int. Ed. 2011, 50, 12342–12346. 10.1002/anie.201106074. [DOI] [PubMed] [Google Scholar]; d Xie H.; Yang L.; Ye X.; Cao Z. Mechanism of Carbon Monoxide Induced N–N Bond Cleavage of Nitrous Oxide Mediated by Molybdenum Complexes: A DFT Study. Organometallics 2014, 33, 1553–1562. 10.1021/om400935f. [DOI] [Google Scholar]; A related paper:; e Horn B.; Limberg C.; Herwig C.; Feist M.; Mebs S. CO Oxidation at Nickel Centres by N2O or O2 to Yield a Novel Hexanuclear Carbonate. Chem. Commun. 2012, 48, 8243–8245. 10.1039/c2cc33846g. [DOI] [PubMed] [Google Scholar]
- For homogeneously catalyzed CO oxidation by N2O, see:; a Fang W. P.; Cheng C. H. Homogeneous Catalytic Reduction of Nitrous and Nitric Oxides to Dinitrogen by Carbon Monoxide. J. Chem. Soc., Chem. Commun. 1986, 503–504. 10.1039/c39860000503. [DOI] [Google Scholar]; b Lee J.-D.; Fang W.-P.; Li C.-S.; Cheng C.-H. Catalytic Reduction of Nitrous Oxide by Carbon Monoxide in the Presence of Rhodium Carbonyl and Hydroxide. Evidence for an Electron-transfer and an Oxygen-transfer Mechanism. J. Chem. Soc., Dalton Trans. 1991, 1923–1927. 10.1039/dt9910001923. [DOI] [Google Scholar]; c Li C.-S.; Sun K.-S.; Cheng C.-H. Catalytic Reduction of Nitrous Oxide to Dinitrogen by Carbon Monoxide using Group 8 Metal Carbonyl Anions. J. Chem. Soc., Dalton Trans. 1992, 1025–1029. 10.1039/dt9920001025. [DOI] [Google Scholar]
- Zeng R.; Feller M.; Ben-David Y.; Milstein D. Hydrogenation and Hydrosilylation of Nitrous Oxide Homogeneously Catalyzed by a Metal Complex. J. Am. Chem. Soc. 2017, 139, 5720–5723. 10.1021/jacs.7b02124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For “O”-atom transfer into M–H bond of N2O to form M–OH, see the following. M = Ru:; a Kaplan A. W.; Bergman R. G. Nitrous Oxide Mediated Oxygen Atom Insertion into a Ruthenium-Hydride Bond. Synthesis and Reactivity of the Monomeric Hydroxoruthenium Complex (DMPE)2Ru(H)(OH). Organometallics 1997, 16, 1106–1108. 10.1021/om960991i. [DOI] [Google Scholar]; b Kaplan A. W.; Bergman R. G. Nitrous Oxide Mediated Synthesis of Monomeric Hydroxoruthenium Complexes. Reactivity of (DMPE)2Ru(H)(OH) and the Synthesis of a Silica-Bound Ruthenium Complex. Organometallics 1998, 17, 5072–5085. 10.1021/om980295d. [DOI] [Google Scholar]; c Kohl S. W.; Weiner L.; Schwartsburd L.; Konstantinovski L.; Shimon L. J. W.; Ben-David Y.; Iron M. A.; Milstein D. Consecutive Thermal H2 and Light-Induced O2 Evolution from Water Promoted by a Metal Complex. Science 2009, 324, 74–77. 10.1126/science.1168600. [DOI] [PubMed] [Google Scholar]; M = Rh:; d Gianetti T. L.; Annen S. P.; Santiso-Quinones G.; Reiher M.; Driess M.; Grützmacher H. Nitrous Oxide as a Hydrogen Acceptor for the Dehydrogenative Coupling of Alcohols. Angew. Chem., Int. Ed. 2016, 55, 1854–1858. 10.1002/anie.201509288. [DOI] [PubMed] [Google Scholar]; M = Hf:; e Vaughan G. A.; Rupert P. B.; Hillhouse G. L. Selective O-Atom Transfer from Nitrous Oxide to Hydride and Aryl Ligands of Bis(pentamethylcyclopentadienyl)hafnium Derivatives. J. Am. Chem. Soc. 1987, 109, 5538–5539. 10.1021/ja00252a047. [DOI] [Google Scholar]; Other selected related references:; f Lee J.-H.; Pink M.; Tomaszewski J.; Fan H.; Caulton K. G. Facile Hydrogenation of N2O by an Operationally Unsaturated Osmium Polyhydride. J. Am. Chem. Soc. 2007, 129, 8706–8707. 10.1021/ja071452f. [DOI] [PubMed] [Google Scholar]; g Doyle L. E.; Piers W. E.; Borau-Garcia J. Ligand Cooperation in the Formal Hydrogenation of N2O Using a PCsp2P Iridium Pincer Complex. J. Am. Chem. Soc. 2015, 137, 2187–2190. 10.1021/ja512602m. [DOI] [PubMed] [Google Scholar]; h Co complex catalyzed O-transfer from N2O to phosphines:Gianetti T. L.; Rodriguez-Lugo R. E.; Harmer J. F.; Trincado M.; Vogt M.; Santiso-Quinones G.; Grützmacher H. Zero-Valent Amino-Olefin Cobalt Complexes as Catalysts for Oxygen Atom Transfer Reactions from Nitrous Oxide. Angew. Chem., Int. Ed. 2016, 55, 15323–15328. 10.1002/anie.201609173. [DOI] [PubMed] [Google Scholar]
- The mechanism of “O” insertion into Ru–H was studied by DFT, see:; a Yu H.; Jia G.; Lin Z. Theoretical Studies on O-Insertion Reactions of Nitrous Oxide with Ruthenium Hydride Complexes. Organometallics 2008, 27, 3825–3833. 10.1021/om8000845. [DOI] [Google Scholar]; b Luque-Urrutia J. A.; Poater A. The Fundamental Noninnocent Role of Water for the Hydrogenation of Nitrous Oxide by PNP Pincer Ru-based Catalysts. Inorg. Chem. 2017, 56, 14383–14387. 10.1021/acs.inorgchem.7b02630. [DOI] [PubMed] [Google Scholar]; c Yao L.; Li Y.; Huang L.; Guo K.; Ren G.; Wu Z.; Lei Q.; Fang W.; Xie H. A DFT Study on the Mechanisms of Hydrogenation and Hydrosilylation of Nitrous Oxide Catalyzed by a Ruthenium PNP Pincer Complex. Comput. Theor. Chem. 2018, 1128, 48–55. 10.1016/j.comptc.2018.02.010. [DOI] [Google Scholar]
- For a review on hydroxycarbonyl complexes as key intermediates, see:Sinha A.; Ghatak T.; Bera J. K. Hydroxycarbonyl Complexes as Key Intermediates in the Base-assisted Reduction of Ruthenium Carbonyls. Dalton Trans. 2010, 39, 11301–11313. 10.1039/c0dt00679c. [DOI] [PubMed] [Google Scholar]
- For catalyst synthesis, see:; a Zhang J.; Leitus G.; Ben-David Y.; Milstein D. Efficient Homogeneous Catalytic Hydrogenation of Esters to Alcohols. Angew. Chem., Int. Ed. 2006, 45, 1113–1115. 10.1002/anie.200503771. [DOI] [PubMed] [Google Scholar]; b Khaskin E.; Iron M. A.; Shimon L. J. W.; Zhang J.; Milstein D. N–H Activation of Amines and Ammonia by Ru via Metal–Ligand Cooperation. J. Am. Chem. Soc. 2010, 132, 8542–8543. 10.1021/ja103130u. [DOI] [PubMed] [Google Scholar]; c Zhang J.; Leitus G.; Ben-David Y.; Milstein D. Facile Conversion of Alcohols into Esters and Dihydrogen Catalyzed by New Ruthenium Complexes. J. Am. Chem. Soc. 2005, 127, 10840–10841. 10.1021/ja052862b. [DOI] [PubMed] [Google Scholar]; d Fogler E.; Garg H. A.; Hu P.; Leitus G.; Shimon L. J. W.; Milstein D. System with Potential Dual Modes of Metal–Ligand Cooperation: Highly Catalytically Active Pyridine-Based PNNH–Ru Pincer Complexes. Chem. - Eur. J. 2014, 20, 15727–15731. 10.1002/chem.201405295. [DOI] [PubMed] [Google Scholar]; e Srimani D.; Balaraman E.; Hu P.; Ben-David Y.; Milstein D. Formation of Tertiary Amides and Dihydrogen by Dehydrogenative Coupling of Primary Alcohols with Secondary Amines Catalyzed by Ruthenium Bipyridine-Based Pincer Complexes. Adv. Synth. Catal. 2013, 355, 2525–2530. 10.1002/adsc.201300620. [DOI] [Google Scholar]; f Balaraman E.; Gnanaprakasam B.; Shimon L. J.; Milstein D. Direct Hydrogenation of Amides to Alcohols and Amines under Mild Conditions. J. Am. Chem. Soc. 2010, 132, 16756–16758. 10.1021/ja1080019. [DOI] [PubMed] [Google Scholar]; g Milstein D.; Balaraman E.; Gunanathan C.; Gnanaprakasam B.; Zhang J. PCT Int. Appl. WO 2012052996, 2012.
- For more details, see SI.
- For a book on the solubility of CO2, see:Carbon Dioxide in Non-Aqueous solvents at Pressure Less Than 200 KPA, Solubility Data Series, Vol 50, Fogg P. G. T., Ed.; IUPAC, 1992. [Google Scholar]
- The mole fraction of CO2 in toluene under 1 atm is ca. 0.01 at 293 K.
- For reports on C–H activation of PNN ligand, see:; a Zhang L.; Huang Z. Synthesis of 1,1,1-Tris(boronates) from Vinylarenes by Co-Catalyzed Dehydrogenative Borylations–Hydroboration. J. Am. Chem. Soc. 2015, 137, 15600–15603. 10.1021/jacs.5b11366. [DOI] [PubMed] [Google Scholar]; b Barrios-Francisco R.; Balaraman E.; Diskin-Posner Y.; Leitus G.; Shimon L. J. W.; Milstein D. PNN Ruthenium Pincer Complexes Based on Phosphinated 2,2′-Dipyridinemethane and 2,2′-Oxobispyridine. Metal–Ligand Cooperation in Cyclometalation and Catalysis. Organometallics 2013, 32, 2973–2982. 10.1021/om400194w. [DOI] [Google Scholar]
- For reviews on bond activation via metal–ligand cooperation (MLC), see:; a Gunanathan C.; Milstein D. Metal–Ligand Cooperation by Aromatization–Dearomatization: A New Paradigm in Bond Activation and “Green” Catalysis. Acc. Chem. Res. 2011, 44, 588–602. 10.1021/ar2000265. [DOI] [PubMed] [Google Scholar]; b Gunanathan C.; Milstein D. Applications of Acceptorless Dehydrogenation and Related Transformations in Chemical Synthesis. Science 2013, 341, 1229712. 10.1126/science.1229712. [DOI] [PubMed] [Google Scholar]; c Gunanathan C.; Milstein D. Bond Activation and Catalysis by Ruthenium Pincer Complexes. Chem. Rev. 2014, 114, 12024–12087. 10.1021/cr5002782. [DOI] [PubMed] [Google Scholar]; d Khusnutdinova J. R.; Milstein D. Metal–Ligand Cooperation. Angew. Chem., Int. Ed. 2015, 54, 12236–12273. 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]; e Zell T.; Milstein D. Hydrogenation and Dehydrogenation Iron Pincer Catalysts Capable of Metal–Ligand Cooperation by Aromatization/Dearomatization. Acc. Chem. Res. 2015, 48, 1979–1994. 10.1021/acs.accounts.5b00027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.