

Abstract
Schizophrenia is a heritable complex phenotype associated with a background risk involving multiple common genetic variants of small effect and a multitude of environmental exposures. Early twin and family studies using proxy‐genetic liability measures suggest gene‐environment interaction in the etiology of schizophrenia spectrum disorders, but the molecular evidence is scarce. Here, by analyzing the main and joint associations of polygenic risk score for schizophrenia (PRS‐SCZ) and environmental exposures in 1,699 patients with a diagnosis of schizophrenia spectrum disorders and 1,542 unrelated controls with no lifetime history of a diagnosis of those disorders, we provide further evidence for gene‐environment interaction in schizophrenia. Evidence was found for additive interaction of molecular genetic risk state for schizophrenia (binary mode of PRS‐SCZ above 75% of the control distribution) with the presence of lifetime regular cannabis use and exposure to early‐life adversities (sexual abuse, emotional abuse, emotional neglect, and bullying), but not with the presence of hearing impairment, season of birth (winter birth), and exposure to physical abuse or physical neglect in childhood. The sensitivity analyses replacing the a priori PRS‐SCZ at 75% with alternative cut‐points (50% and 25%) confirmed the additive interaction. Our results suggest that the etiopathogenesis of schizophrenia involves genetic underpinnings that act by making individuals more sensitive to the effects of some environmental exposures.
Keywords: Schizophrenia, psychosis, genetics, environment, gene‐environment interaction, polygenic risk, childhood trauma, cannabis, bullying
Schizophrenia is a complex phenotype characterized by reality distortion, cognitive alteration and negative symptoms. Although the prevalence of schizophrenia spectrum disorders is relatively low – approximately 0.47% for schizophrenia (the poor outcome fraction) and 3.0% for other clinical diagnoses of psychotic disorders1 – they account for a tremendous personal, economic and societal burden, with 218 disability adjusted life years (DALYs) per 100,0002, making schizophrenia the fifth leading cause of DALYs in the age group of 15‐44 years. These figures indicate that there is an urgent need for breakthroughs in prevention, diagnosis and management of schizophrenia and related disorders, which can be achieved by increased understanding of etiopathology.
Decades of work consistently yielding high heritability estimates document the role of genetic background in the etiopathology of these disorders3, 4. In agreement with findings from early family‐based studies, recent results from the Danish nationwide registers confirm that the heritability estimates range from 73% for schizophrenia spectrum disorders to 79% for narrow schizophrenia diagnosis5.
Based on these findings from the field of quantitative genetic epidemiology, molecular genetics has emerged as arguably the most popular area of investigation in research targeting schizophrenia spectrum disorders. Easy and low‐cost access to high‐throughput techniques has increased genetic resolution. The Psychiatric Genomics Consortium6 was founded to achieve the power required to detect small effect sizes in a genome‐wide association (GWA) analysis. The Schizophrenia Working Group of the Consortium identified 108 genome‐wide significant loci7, and the number of novel genetic variants keeps growing as a function of sample size8. GWA findings, in line with the half‐century‐old polygenic theory of schizophrenia9, established that a large fraction of the genetic risk is explained by many common genetic variants with very small effects sizes.
However, the proportion of the genetic liability accounted for by single nucleotide polymorphisms (SNPs) detected in current GWA arrays represents only a fraction of the effect that was suggested by heritability estimates from twin studies. In other terms, there is a large “heritability gap” between twin and molecular genetics studies10. The most likely explanation for this gap is that part of the genetic effect documented by twin studies is contingent on environmental factors shared by individuals growing up in the same family10. The etiology of psychosis spectrum disorder is likely to involve genetic underpinnings that act by making individuals more sensitive to the effects of environmental exposures or by driving individuals to higher exposure rates11.
In parallel to the growing knowledge base in genetics, environmental research into schizophrenia has produced consistent findings over years. Observational studies have identified various exposures associated with risk of psychosis spectrum disorder at different levels of evidence, with varying magnitude of the effect size estimates. These environmental risk factors include cannabis use, childhood adversities (e.g., sexual abuse, emotional neglect), peer‐bullying, urban environment, proxies of social exclusion (e.g., ethnic minority, immigration, and hearing impairment), season of birth, and obstetric and pregnancy complications12, 13.
Although findings from empirical investigations relying on surrogates of genetic risk (i.e., familial history of schizophrenia) argue for a strong influence of environment in moderating genetic vulnerability11, operationalizing and translating these findings by using molecular candidate‐gene approaches have been challenging tasks14.
The utilization of polygenic risk score (PRS) as a single metric of molecular genetic risk has considerably increased the power to detect associations with phenotypes as well as gene‐environment interactions. Currently, the PRS for schizophrenia (PRS‐SCZ) of a subject can be estimated by summing the log odds ratios of individual SNPs multiplied by the number of risk alleles present at the corresponding loci15. PRS‐SCZ has been shown to explain up to 7% of variation on the liability scale to schizophrenia, at least when using the latest release of the Psychiatric Genomics Consortium in patients with more chronic forms7.
We recently discussed the challenges of evaluating the role of environmental exposures in psychiatry and the need to use exposure‐wide systematic approaches to separate genuine strong signals from selective reporting16. Guided by this, we aimed to analyze the main and joint associations of environmental exposures and PRS‐SCZ in a cross‐sectional sample that was specifically collected to test for gene‐environment interactions in schizophrenia.
METHODS
Study population
This case‐control gene‐environment interaction study used data from the Work‐package 6 (WP6) of the European Network of National Networks studying Gene‐Environment Interactions in Schizophrenia (EUGEI)17 and the Genetic Risk and Outcome of Psychosis (GROUP) study within the EUGEI18. Data were collected between 2010 and 2015 in the Netherlands, Turkey, Spain and Serbia.
Patients were diagnosed with schizophrenia spectrum disorders according to the DSM‐IV‐TR (average duration of illness since age of first contact with mental health services = 9.9 years). The diagnosis was later confirmed by the Operational Criteria Checklist for Psychotic and Affective Illness19 in the EUGEI WP6, and the Schedules for Clinical Assessment in Neuropsychiatry20 or the Comprehensive Assessment of Symptoms and History21 in the GROUP. Unrelated controls with no lifetime psychotic disorder were recruited from the same population as the cases. Exclusion criteria for all participants were a diagnosis of psychotic disorder due to another medical condition, a history of head injury with loss of consciousness, and an intelligence quotient <70.
A total of 1,866 patients and 1,583 healthy participants with genotype data available were included. As the predictive power of PRS‐SCZ has not been established in people of non‐white ethnic origin22, the present analyses were restricted to participants of Caucasian white ethnic origin. The final sample included 1,699 patients and 1,542 unrelated controls.
The projects were approved by the medical ethics committees of all participating sites and conducted in accordance with the Declaration of Helsinki. All respondents provided written informed consent. Participants below the age of 18 signed an assent; parent(s) also signed an informed consent.
To achieve high quality and homogeneity in clinical, experimental and environmental assessments, standardized instruments were administered by psychiatrists, psychologists or trained research assistants who completed mandatory on‐site training sessions and online training modules, including interactive interview videos and self‐assessment tools17, 18. Both on‐site and online training sessions were repeated annually to maintain high inter‐rater reliability throughout the study enrollment period.
Environmental exposures
Within the limits of data availability, we sought to examine all the environmental exposures that have previously been associated with schizophrenia spectrum disorders.
Childhood adversity was assessed using the Childhood Trauma Questionnaire Short Form (CTQ)23. This consists of 28 items, rated on a 5‐point Likert scale, measuring five domains of maltreatment (emotional and physical neglect; emotional, physical and sexual abuse). The psychometric characteristics of the translated versions (Spanish, Turkish, Dutch and Serbian) of the CTQ have been comprehensively studied24, 25, 26. To dichotomize each childhood adversity domain (0=“absent” and 1=“present”), consistent with previous work in the EUGEI27, we used the following cut‐off scores for each domain: ≥⃒9 for emotional abuse; ≥⃒8 for physical abuse; ≥⃒6 for sexual abuse; ≥⃒10 for emotional neglect; and ≥⃒8 for physical neglect.
Cannabis use was assessed by a modified version of the Cannabis Experiences Questionnaire28 in the EUGEI WP6 (0=“none”; 1=“only once or twice”; 2=“a few times a year”; 3=“a few times a month”; 4=“once or more a week”; 5=“everyday”), and by the L section of the Composite International Diagnostic Interview (CIDI)29 in the GROUP (0=“none”; 1=“less than weekly”; 2=“weekly”; 3=“daily”). Consistent with previous work30, 31, 32, a binary regular cannabis use variable was constructed by using the cut‐off value of once or more per week during the lifetime period of most frequent use.
In accordance with previous studies investigating the association between season of birth and schizophrenia in the Northern hemisphere sites33, the high‐risk birth period was defined based on the winter solstice (December‐March), and a binary winter‐birth exposure was constructed.
Hearing impairment was defined based on self‐reported hearing impairment in the last 12 months (0=“absent” and 1=“present”).
The history of bullying by peers (emotional, psychological or physical violence) before 17 years of age was assessed using the short version of the Retrospective Bullying Questionnaire (RBQ)34, 35, that measures the severity of the bullying experience: 0=“none”; 1=“some (no physical injuries)”; 2=“moderate (minor injuries or transient emotional reactions)”; 3=“marked (severe and frequent physical or psychological harm)”. Exposure to childhood bullying was dichotomized using ≥⃒1 as the cut‐off point (0=“absent” and ≥⃒1=“present”).
Genetic data processing
Samples of all individuals were genotyped at Cardiff University Institute of Psychological Medicine and Clinical Neurology, using custom Illumina HumanCoreExome‐24 BeadChip genotyping arrays containing probes for 570038 genetic variants (Illumina, San Diego, CA). Genotype data were called using the GenomeStudio package and transferred into PLINK format for further analysis.
Quality control was conducted in PLINK v1.0736 or with custom Perl scripts. Variants with call rate <98% were excluded from the dataset. Hardy‐Weinberg equilibrium p‐value was calculated separately in Turkish, Northern European and Southern European samples. Variants with Hardy‐Weinberg equilibrium p‐value <1e‐6 in any of these three regions were excluded from the dataset. After quality control, 559505 variants remained.
Samples with call rate <98% were excluded from the dataset. A linkage disequilibrium (LD) pruned set of variants was calculated using the ‐‐indep‐pairwise command in PLINK (maximum r2=0.25, window size=500 SNPs, window step size=50 SNPs) and used for further analyses. Homozygosity F values were calculated using the ‐‐het command in PLINK, and outlier samples (F<–0.11 or F>0.15) were excluded. The genotypic sex of samples was calculated from X chromosome data using the ‐‐check‐sex command in PLINK, and samples with different genotypic sex to their database sex were excluded.
Identity‐by‐descent values were calculated for the sample in PLINK. Samples with one or more siblings among the genotyped samples according to the database but no identified genotypic siblings (defined as PI‐HAT >0.35 and <0.65) were excluded. After these were removed from consideration, samples with two or more siblings in the database that were not supported by the genotypic data were also excluded.
After visually observing clustering of errors by genotyping chip, we decided to exclude chips with a high proportion of errors. All samples on chips with five or more sample exclusions due to heterozygosity or call rate (out of 12 possible samples) were excluded. All samples on chips with four or more sample exclusions due to sex or relative checks were also excluded, unless their identity was corroborated by concordance between database and genotype relatedness data with a sample on another chip.
Principal components were calculated in PLINK using LD pruned variants after combining the dataset with the Thousand Genomes reference. Due to the inherently multi‐population nature of the dataset and the variety of possible analyses, no exclusions were made to the whole dataset based on this analysis. Population effects were corrected for separately in individual analyses.
After quality control, genotypes were imputed on the Michigan Imputation Server using the Haplotype Reference Consortium reference panel (version 1.1) and the programs Eagle for haplotype phasing and Minimac3 for imputation37, 38. After imputation, variants with an imputation r2>0.6, minor allele frequency (MAF) >0.1% and call rate >99% were retained (8277535 variants). Best‐guess genotypes were generated from genotype probabilities using PLINK.
PRS‐SCZ was constructed using summary statistics from the Psychiatric Genomics Consortium genome‐wide association study, excluding samples present in the GROUP data7. Clumping was performed in imputed best‐guess genotypes for each dataset using PLINK (maximum r2=0.2, window size=500kb, minimum MAF=10%, minimum imputation information (INFO) score=0.7), and variants within regions of long‐range LD around the genome (including the human major histocompatibility complex) were excluded39. PRS‐SCZ was then constructed from best‐guess genotypes using PLINK at ten different p‐value thresholds (1, 0.5, 0.3, 0.2, 0.1, 0.05, 0.01, 1×10–4, 1×10–6, 5×10–8). Consistent with previous research in the field40, 41, 42, 43, we used p=0.05 for our primary analysis, as this threshold explained most variation in the phenotype in the Psychiatric Genomics Consortium analysis7.
To be able to compare our estimates from the current sample with the previously reported estimates of the proportion of variance explained by PRS‐SCZ, a logistic regression model was applied to test the association of PRS‐SCZ with case‐control status (adjusted for ancestry using the first ten principal components), and Nagelkerke's R2 was calculated. PRS‐SCZ discriminated cases from controls (odds ratio, OR=1.30; 95% CI: 1.25‐1.34; p<0.001; Nagelkerke's R2=0.15), after also controlling for age, sex and country (OR=1.30; 95% CI: 1.26‐1.35; p<0.001; Nagelkerke's R2=0.20).
PRS‐SCZ was dichotomized using the quartile cut‐off points based on the control distribution of PRS‐SCZ within each country (to account for differences in PRS‐SCZ between countries that may arise due to ethnic variation). The highest quartile (PRS‐SCZ > 75% of the controls) was considered the binary genetic risk state for schizophrenia (hereafter: PRS‐SCZ75).
Statistical analyses
All analyses were carried out using the STATA version 15.044. Random intercept multilevel logistic regression models, taking into account clustering of participants within countries, were applied to test the univariate associations of exposures and PRS‐SCZ75 with case status. For each exposure, gene‐environment correlation was tested using multilevel logistic regression models in the control sample. To test gene‐environment interaction, additive models were chosen over multiplicative models prior to data collection (EUGEI consortium meeting, December 14, 2013), because they provide superior representation of biological synergy45 and inform public health decisions within the sufficient cause framework46, 47.
To test the joint effects of environmental exposures and genetic score, we entered the four states occasioned by the combination of each exposure and binary PRS‐SCZ risk state (PRS‐SCZ75) as independent variables (three dummy variables with no‐risk state as the reference category), and case status as the dependent variable, in multilevel logistic regression models.
We tested for departure from additivity using the interaction contrast ratio, also called the relative excess risk due to interaction (RERI). The RERI is considered the standard measure for interaction on the additive scale in case‐control studies48. The RERI was estimated as (ORexposure&PRS‐SCZ75 − ORexposure − ORPRS‐SCZ75 + 1)49. A RERI greater than zero was defined as a positive deviation from additivity, and considered significant when the 95% CI did not contain zero. Using the ORs derived from each model, the RERIs for each model were calculated using the delta method.
As a sensitivity measure, the alternative bootstrap percentile method50 (N=1,000 bootstrap replications) was applied to estimate the bootstrapped 95% CI for the RERI. All models were controlled for a priori covariates (age and sex), while models including PRS‐SCZ75 were additionally adjusted for ancestry, using the first ten principal components accommodating to the general recommendations. Following the extension to the STrengthening the Reporting of OBservational studies in Epidemiology (STROBE) reporting guidelines48, the interaction analyses were reported using a single reference category including the separate and joint effects of PRS‐SCZ75 and each exposure in strata of exposure and PRS‐SCZ75.
The analyses were also conducted on imputed data, given missing observations in environmental exposure assessments. Under the assumption of missing at random, the multiple imputation chained equation model51 with 20 imputations restricted to in‐range values was applied (relative efficiency ranging between 97% to 99%). Imputed data were similar to observed values in the original dataset. All analyses were run on multiply imputed data, and estimates were pooled using Rubin's rules52.
To test the robustness of our findings, sensitivity analyses of binary genetic risk thresholds were conducted using the PRS‐SCZ cut points at 50% and 25% of the controls. The nominal significance threshold was set at p=0.05.
RESULTS
Data concerning age, sex and environmental exposures in cases and controls are reported in Table 1.
Table 1.
Demographic variables and environmental exposures in cases and controls
| Total | Controls | Cases | Missing rates | |
|---|---|---|---|---|
| Age (years, mean±SD) | 32.4±9.8 | 33.4±10.6 | 31.5±9.0 | |
| Sex | ||||
| Male | 1,951 (60.2%) | 762 (49.4%) | 1,189 (70.0%) | |
| Female | 1,290 (39.8%) | 780 (50.6%) | 510 (30.0%) | |
| Cannabis use | ||||
| No | 2,390 (78.6%) | 1,366 (91.2%) | 1,024 (66.5%) | 202 (6.2%) |
| Yes | 649 (21.4%) | 132 (8.8%) | 517 (33.5%) | |
| Bullying | ||||
| No | 1,947 (72.3%) | 1,101 (83.7%) | 846 (61.4%) | 547 (16.9%) |
| Yes | 747 (27.7%) | 215 (16.3%) | 532 (38.6%) | |
| Emotional abuse | ||||
| No | 2,019 (73.0%) | 1,230 (84.8%) | 789 (60.0%) | 475 (14.7%) |
| Yes | 747 (27.0%) | 221 (15.2%) | 526 (40.0%) | |
| Physical abuse | ||||
| No | 2,477 (88.7%) | 1,362 (93.0%) | 1,115 (84.0%) | 450 (13.9%) |
| Yes | 314 (11.3%) | 102 (7.0%) | 212 (16.0%) | |
| Sexual abuse | ||||
| No | 2,269 (81.5%) | 1,309 (90.1%) | 960 (72.1%) | 456 (14.1%) |
| Yes | 516 (18.5%) | 144 (9.9%) | 372 (27.9%) | |
| Emotional neglect | ||||
| No | 1,254 (45.3%) | 789 (54.3%) | 465 (35.4%) | 473 (14.6%) |
| Yes | 1,514 (54.7%) | 664 (45.7%) | 850 (64.6%) | |
| Physical neglect | ||||
| No | 1,804 (64.8%) | 1039 (71.3%) | 765 (57.7%) | 457 (14.1%) |
| Yes | 980 (35.2%) | 419 (28.7%) | 561 (42.3%) | |
| Winter birth | ||||
| No | 1,989 (63.2%) | 951 (63.0%) | 1,038 (63.4%) | 94 (2.9%) |
| Yes | 1,158 (36.8%) | 559 (37.0%) | 599 (36.6%) | |
| Hearing impairment | ||||
| No | 2,869 (92.5%) | 1,437 (95.6%) | 1,432 (89.7%) | 141 (4.4%) |
| Yes | 231 (7.5%) | 66 (4.4%) | 165 (10.3%) |
All exposures except winter birth were associated with case status, also after adjusting for age and sex. Table 2 presents the unadjusted and adjusted ORs for PRS‐SCZ75 and each of the exposures associated with case status.
Table 2.
Main effects of environmental and genetic risk on case‐control status
| Unadjusted main effects | Adjusted main effects a | |||
|---|---|---|---|---|
| Odds ratio (95% CI) | p | Odds ratio (95% CI) | p | |
| Cannabis use | 4.85 (3.89‐6.05) | <0.001 | 3.96 (3.16‐4.97) | <0.001 |
| Bullying | 3.01 (2.48‐3.65) | <0.001 | 3.06 (2.50‐3.74) | <0.001 |
| Emotional abuse | 3.51 (2.93‐4.22) | <0.001 | 3.77 (3.12‐4.56) | <0.001 |
| Physical abuse | 2.70 (2.10‐3.48) | <0.001 | 2.83 (2.18‐3.67) | <0.001 |
| Sexual abuse | 3.66 (2.96‐4.53) | <0.001 | 4.11 (3.30‐5.13) | <0.001 |
| Emotional neglect | 2.52 (2.14‐2.96) | <0.001 | 2.65 (2.24‐3.13) | <0.001 |
| Physical neglect | 2.32 (1.96‐2.75) | <0.001 | 2.33 (1.96‐2.78) | <0.001 |
| Winter birth | 1.06 (0.92‐1.23) | 0.423 | 1.05 (0.91‐1.23) | 0.495 |
| Hearing impairment | 2.46 (1.82‐3.31) | <0.001 | 2.67 (1.96‐3.62) | <0.001 |
| PRS‐SCZ75 b | 2.91 (2.48‐3.40) | <0.001 | 2.85 (2.43‐3.35) | <0.001 |
PRS‐SCZ75 – polygenic risk score for schizophrenia (75% cut‐point)
aadjusted for sex and age, badjusted for ten principal components
Except for physical abuse, there was no evidence for gene‐environment correlation, as PRS‐SCZ75 was not associated strongly or significantly with exposures in the control group (Table 3). Physical abuse was associated with PRS‐SCZ75 (adjusted OR=1.84; 95% CI: 1.19‐2.84; p=0.006).
Table 3.
Gene‐environment correlation between PRS‐SCZ75 and environmental exposures
| Unadjusted effects | Adjusted effects a | |||
|---|---|---|---|---|
| Odds ratio (95% CI) | p | Odds ratio (95% CI) | p | |
| Cannabis use | 0.98 (0.61‐1.59) | 0.949 | 0.93 (0.57‐1.52) | 0.771 |
| Bullying | 1.27 (0.86‐1.86) | 0.228 | 1.28 (0.87‐1.89) | 0.210 |
| Emotional abuse | 1.13 (0.80‐1.58) | 0.493 | 1.13 (0.81‐1.59) | 0.476 |
| Physical abuse | 1.82 (1.18‐2.81) | 0.007 | 1.84 (1.19‐2.84) | 0.006 |
| Sexual abuse | 0.79 (0.51‐1.22) | 0.287 | 0.79 (0.51‐1.23) | 0.292 |
| Emotional neglect | 1.18 (0.91‐1.52) | 0.212 | 1.16 (0.90‐1.50) | 0.258 |
| Physical neglect | 1.18 (0.89‐1.56) | 0.246 | 1.19 (0.90‐1.58) | 0.219 |
| Winter birth | 1.13 (0.88‐1.45) | 0.338 | 1.13 (0.88‐1.45) | 0.332 |
| Hearing impairment | 1.13 (0.63‐2.02) | 0.693 | 1.18 (0.65‐2.13) | 0.592 |
PRS‐SCZ75 – polygenic risk score for schizophrenia (75% cut‐point)
aadjusted for sex, age and ten principal components
Table 4 reports the interactive effects of PRS‐SCZ75 and the exposures on the case status. There was evidence for additive interaction between PRS‐SCZ75 and regular cannabis use (RERI=5.60; 95% CI: 0.88‐10.33; p=0.020), childhood bullying (RERI=2.76; 95% CI: 0.29‐5.23; p=0.028), emotional abuse (RERI=5.52; 95% CI: 2.29‐8.75; p<0.001), sexual abuse (RERI=7.61; 95% CI: 2.05‐13.17; p=0.007), and emotional neglect (RERI=2.46; 95% CI: 0.98‐3.94; p=0.001), respectively. Figure 1 visualizes the significant interaction effects on an additive scale. No evidence was found for significant additive interaction effects between PRS‐SCZ75 and physical abuse, physical neglect, hearing impairment, and winter birth.
Table 4.
Interaction of environmental exposures and PRS‐SCZ75 on case‐control status
| PRS‐SCZ 75=0 | PRS‐SCZ 75=1 | RERI (95% CI) | |||
|---|---|---|---|---|---|
| N cases/controls | Odds ratio (95% CI) | N cases/controls | Odds ratio (95% CI) | ||
| Cannabis use = 0 | 556/1042 | 1.0 | 468/324 |
2.84 (2.36‐3.40) p<0.001 |
5.60 (0.88‐10.33) p=0.020 |
| Cannabis use = 1 | 296/102 |
4.10 (3.13‐5.36) p<0.001 |
221/30 |
11.54 (7.60‐17.51) p<0.001 |
|
| Bullying = 0 | 454/842 | 1.0 | 392/259 |
2.84 (2.31‐3.47) p<0.001 |
2.76 (0.29‐5.23) p=0.028 |
| Bullying = 1 | 296/163 |
2.97 (2.34‐3.76) p<0.001 |
236/52 |
7.56 (5.41‐10.56) p<0.001 |
|
| Emotional abuse = 0 | 464/939 | 1.0 | 325/291 |
2.39 (1.95‐2.94) p<0.001 |
5.52 (2.29‐8.75) p<0.001 |
| Emotional abuse = 1 | 273/166 |
3.26 (2.58‐4.12) p<0.001 |
253/55 |
10.17 (7.33‐14.10) p<0.001 |
|
| Physical abuse = 0 | 632/1049 | 1.0 | 483/313 |
2.71 (2.25‐3.26) p<0.001 |
1.64 (–1.07 to 4.34) p=0.235 |
| Physical abuse = 1 | 107/65 |
2.97 (2.11‐4.17) p<0.001 |
105/37 |
6.31 (4.19‐9.52) p<0.001 |
|
| Sexual abuse = 0 | 536/993 | 1.0 | 424/316 |
2.68 (2.21‐3.25) p<0.001 |
7.61 (2.05‐13.17) p=0.007 |
| Sexual abuse = 1 | 208/114 |
3.89 (2.99‐5.08) p<0.001 |
164/30 |
13.19 (8.60‐20.22) p<0.001 |
|
| Emotional neglect = 0 | 273/610 | 1.0 | 192/179 |
2.64 (2.03‐3.44) p<0.001 |
2.46 (0.98‐3.94) p=0.001 |
| Emotional neglect = 1 | 464/495 |
2.58 (2.10‐3.17) p<0.001 |
386/169 |
6.69 (5.20‐8.59) p<0.001 |
|
| Physical neglect = 0 | 438/804 | 1.0 | 327/235 |
2.81 (2.26‐3.50) p<0.001 |
1.51 (0.00‐3.03) p=0.051 |
| Physical neglect =1 | 308/306 |
2.42 (1.95 to 3.01) p<0.001 |
253/113 |
5.75 (4.36‐7.58) p<0.001 |
|
| Winter birth = 0 | 562/733 | 1.0 | 476/218 |
3.11 (2.53‐3.82) p<0.001 |
–0.55 (–1.36 to 0.27) p=0.186 |
| Winter birth = 1 | 333/414 |
1.16 (0.96 to 1.41) p=0.123 |
266/145 |
2.72 (2.14‐3.48) p<0.001 |
|
| Hearing impairment = 0 | 767/1098 | 1.0 | 665/339 |
2.97 (2.51‐3.52) p<0.001 |
1.04 (–2.65 to 4.74) p=0.579 |
| Hearing impairment = 1 | 107/50 |
3.11 (2.16 to 4.48) p<0.001 |
58/16 |
6.13 (3.43‐10.95) p<0.001 |
|
PRS‐SCZ75 – polygenic risk score for schizophrenia (75% cut‐point), RERI – relative excess risk due to interaction
Data adjusted for sex, age and ten principal components
Figure 1.
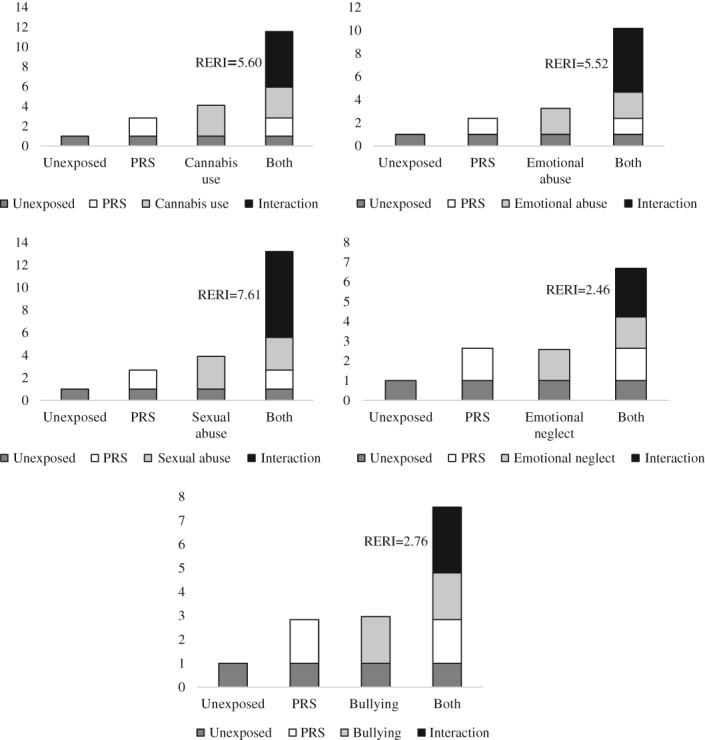
Additive effects of cannabis use, emotional abuse, sexual abuse, emotional neglect and bullying on the association between the polygenic risk score for schizophrenia, 75% cut‐point (PRS) and case‐control status, adjusted for sex, age and ten principal components; RERI – relative excess risk due to interaction
Analyses using the alternative bootstrap percentile method for estimating additive interactions yielded similar results (data not shown). The sensitivity analyses replacing the a priori set PRS‐SCZ75 as the genetic risk in the models with the alternative cut‐points of PRS‐SCZ (50% and 25%) confirmed that additive interaction was evident for regular cannabis use, childhood bullying, emotional abuse, sexual abuse, and emotional neglect across all PRS‐SCZ cut‐points (data not shown). The results from the analyses performed in the imputed data were similar (Table 5).
Table 5.
Additive interaction effects of PRS‐SCZ75 and the environmental exposures on case‐control status in the imputed data
| Main effects a | Interaction b | |||
|---|---|---|---|---|
| Odds ratio (95% CI) | p | RERI (95% CI) | p | |
| Cannabis use | 3.94 (3.15‐4.93) | <0.001 | 5.18 (0.62‐9.74) | 0.026 |
| Bullying | 2.88 (2.36‐3.51) | <0.001 | 2.88 (0.63‐5.13) | 0.012 |
| Emotional abuse | 3.49 (2.88‐4.24) | <0.001 | 5.11 (2.10‐8.13) | 0.001 |
| Physical abuse | 2.65 (2.06‐3.40) | <0.001 | 1.40 (–1.10 to 3.90) | 0.272 |
| Sexual abuse | 3.74 (3.00‐4.66) | <0.001 | 6.84 (1.77‐11.92) | 0.008 |
| Emotional neglect | 2.51 (2.14‐2.95) | <0.001 | 2.37 (0.90‐3.84) | 0.002 |
| Physical neglect | 2.14 (1.79‐2.57) | <0.001 | 1.42 (–0.05 to 2.88) | 0.058 |
| Winter birth | 1.06 (0.91‐1.23) | 0.485 | –0.53 (–1.36 to 0.30) | 0.209 |
| Hearing impairment | 2.68 (1.97‐3.66) | <0.001 | 1.24 (–2.51 to 5.00) | 0.516 |
PRS‐SCZ75 – polygenic risk score for schizophrenia (75% cut‐point), RERI – relative excess risk due to interaction
aadjusted for sex and age, badjusted for sex, age and ten principal components
DISCUSSION
In this study examining the main and joint associations of environmental exposures and genetic liability with schizophrenia spectrum disorder, evidence emerged for a positive additive interaction of genetic liability with regular cannabis use and childhood adversity domains (sexual abuse, emotional abuse, emotional neglect, and childhood bullying).
To the best of our knowledge, our study is the first to report that the sensitivity to adverse life events during childhood and exposure to cannabis is moderated by genetic risk state for schizophrenia (PRS‐SCZ75). Put simply, the positive additive interaction between genetic liability and environmental exposure indicates synergy between gene and environment; that is, the combined influence of genetic liability and environmental exposure is larger than the sum of individual effects of each.
In line with previous findings, PRS‐SCZ75 discriminated cases from controls and all environmental exposures (except for winter birth) were associated with case status. However, no evidence for an additive interaction with PRS‐SCZ75 was observed for physical abuse, physical neglect, hearing impairment, or winter birth.
The proportion of variance explained by PRS‐SCZ in our sample was comparable to previously reported estimates53 and the most recent findings from the Psychiatric Genomics Consortium7. In this dataset, we strictly conformed to previous definitions of environmental exposures to improve reproducibility and allow comparability. In agreement with previous reports, our univariate analysis demonstrated that the exposures we tested were associated with case status to varying degrees, that were similar to meta‐analytical estimates12, 13.
By taking advantage of direct molecular measures of genetic risk, we provided further support for the putative role of gene‐environment interaction in schizophrenia spectrum disorder that was observed in previous studies applying indirect genetic liability estimates derived from family‐based (e.g., twin, relative) samples54. Our findings were corroborated by the results obtained from regression models using different genetic liability thresholds (PRS‐SCZ cut‐offs at 50% and 25%) and analyses ran in imputed data.
The RERIs and 95% CIs for emotional and sexual abuse were above 2, thereby suggesting a “mechanistic” interaction49, i.e., that there are individuals who would develop schizophrenia only when both genetic liability and environmental exposure (emotional or sexual abuse) are present, but would not develop schizophrenia when either genetic liability or environmental exposure is present alone.
PRS‐based approaches have recently gained traction in detecting gene‐environment interaction. Previously, studies investigated the possible interaction between some genetic polymorphisms possibly linked to the putative biological mechanisms underlying psychosis and cannabis use or childhood adversity. Although SNPs (in various genes) for genetic moderation (e.g., AKT1, COMT, BDNF) were identified, these findings were inconsistent across samples55 and became secondary once the genome‐wide approach took over the scene.
To date, a limited number of studies tested gene‐environment interaction across the psychosis spectrum using PRS‐SCZ. A pilot study of 80 patients with first‐episode psychotic disorders and 110 controls investigating whether PRS‐SCZ moderates the association between childhood adversities and psychosis, although yielding main effects of both PRS‐SCZ and childhood adversities, was considerably underpowered to detect gene‐environment interaction56. A recent study demonstrated that intra‐uterine environment moderates the association between PRS‐SCZ and schizophrenia, and further revealed in the pathway analysis that genes involved in cellular stress response were the main drivers of the gene‐environment interaction57. In our recent study of a general population twin cohort, we found evidence for positive interaction effects between PRS‐SCZ and exposure to childhood adversities to pleiotropically influence momentary emotional regulation and psychosis proneness58. Further, a multimodal study combining genetics and imaging techniques reported that the association between PRS‐SCZ and cortical maturation in young male adults is moderated by early‐life exposure to cannabis59. Taken together, while the area of gene‐environment research is progressing rapidly toward a more replicable path informed by the use of GWA data, conclusive evidence has yet to emerge.
There are various ways in which our findings can move forward gene‐environment interaction research in the GWA era. First, they are useful in providing direction for future pre‐registered confirmatory studies. Second, they may open up promising research lines for further exploration of gene‐environment interactions in the biological context, such as using biologically‐informative pathway scores instead of an aggregate genetic risk score for disease phenotype. These studies may help us investigate both hypotheses for biologically plausible pathways impacted by distinct exposures (e.g., hypoxia‐ischemia pathway x obstetric complications and childhood adversities x hypothalamic‐pituitary‐adrenal axis)60, 61, and putative common final pathways, such as the broad inflammatory pathway which may be influenced by many exposures cumulatively62.
However, there are important caveats: pathway scores may be less powerful than the overall polygenic scores for phenotypes, and there are almost endless options for selecting and constructing “putative” pathways. Therefore, gene‐sets for pathways should be a priori defined and frozen at a central repository to avoid data‐dredging. Further, study protocols for hypothesis‐driven selective exposure and pathway analyses (e.g., regular cannabis use and endocannabinoid pathway) should ideally be either registered or, if this is not possible, agnostic data analyses should be followed through.
In our study, data were collected through extensive interviews by trained psychiatrists, psychologists and research assistants to specifically test the role of gene‐environment interaction in schizophrenia. Further, our culturally and geographically diverse sample provided us with the advantage of observing variations in environmental exposures, which increases the power to detect interaction effects63.
However, some limitations should be acknowledged. First, the cross‐sectional design informs only on temporal association and not causality. Nevertheless, cross‐sectional analyses arguably remain an essential first step for identifying risk factors and pave the way for future longitudinal studies to investigate gene‐environment interaction in evolutionary trajectories. Second, given the sample size and explorative nature of the study, we focused on main and interaction associations of previously established environmental factors and PRS‐SCZ. However, the reality is much more complex than current statistical models can accommodate, involving dynamic interactions, causal and non‐causal associations within the exposome (e.g., dense correlation matrix of environmental factors influenced by the timing, duration, severity and extent of repeated exposures over time)16, 64; the genome (e.g., epistasis, redundancy and pleiotropy)65; and the phenome (multidimensional syndromal diversity)66. Third, instead of the commonly‐exercised selective reporting of one exposure at a time, we embraced a quasi‐systematic approach to provide an overall picture of the gene‐environment interactions findings from this dataset. However, we could not test some other known exposures (e.g., obstetric and pregnancy complications).
In conclusion, by using a molecular genetic risk measure, we have provided further evidence for the role of gene‐environment interaction in schizophrenia. Our findings warrant further validation in pre‐registered confirmatory research.
ACKNOWLEDGEMENTS
The EUGEI project was supported by the grant agreement HEALTH‐F2‐2010‐241909 from the European Community's Seventh Framework Programme. The authors are grateful to the patients and their families for participating in the project. They also thank all research personnel involved in the GROUP project, in particular J. van Baaren, E. Veermans, G. Driessen, T. Driesen, E. van't Hag and J. de Nijs. All the DNA samples from Turkey were provided by the Ankara University Brain Research Center Biobank, that was supported by Ankara University Scientific Research Projects Coordination Unit (project no. 10A6055003, 2010). B.P.F. Rutten was funded by a VIDI award (no. 91718336) from the Netherlands Scientific Organization. S. Guloksuz, L.‐K. Pries, B.P.F. Rutten and J. van Os contributed equally to this work.
APPENDIX 1.
GROUP investigators in EUGEI included: Behrooz Z. Alizadeh, Therese van Amelsvoort, Nico J. van Beveren, Richard Bruggeman, Wiepke Cahn, Lieuwe de Haan, Philippe Delespaul, Jurjen J. Luykx, Inez Myin‐Germeys, Ruud van Winkel and Jim van Os.
Contributor Information
Genetic Risk and Outcome of Psychosis (GROUP) investigators:
Behrooz Z. Alizadeh, Therese van Amelsvoort, Nico J. van Beveren, Richard Bruggeman, Wiepke Cahn, Lieuwe de Haan, Philippe Delespaul, Jurjen J. Luykx, Inez Myin‐Germeys, Ruud van Winkel, and Jim van Os
REFERENCES
- 1. Perälä J, Suvisaari J, Saarni SI et al. Lifetime prevalence of psychotic and bipolar I disorders in a general population. Arch Gen Psychiatry 2007;64:19‐28. [DOI] [PubMed] [Google Scholar]
- 2. Murray CJ, Vos T, Lozano R et al. Disability‐adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990‐2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2197‐223. [DOI] [PubMed] [Google Scholar]
- 3. McGuffin P, Farmer AE, Gottesman II et al. Twin concordance for operationally defined schizophrenia: confirmation of familiality and heritability. Arch Gen Psychiatry 1984;41:541‐5. [DOI] [PubMed] [Google Scholar]
- 4. Cardno AG, Marshall EJ, Coid B et al. Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch Gen Psychiatry 1999;56:162‐8. [DOI] [PubMed] [Google Scholar]
- 5. Hilker R, Helenius D, Fagerlund B et al. Heritability of schizophrenia and schizophrenia spectrum based on the nationwide Danish twin register. Biol Psychiatry 2018;83:492‐8. [DOI] [PubMed] [Google Scholar]
- 6. Sullivan PF, Agrawal A, Bulik CM et al. Psychiatric genomics: an update and an agenda. Am J Psychiatry 2017;175:15‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schizophrenia Working Group of the Psychiatric Genomics Consortium . Biological insights from 108 schizophrenia‐associated genetic loci. Nature 2014;511:421‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pardiñas AF, Holmans P, Pocklington AJ et al. Common schizophrenia alleles are enriched in mutation‐intolerant genes and in regions under strong background selection. Nat Genet 2018;50:381‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gottesman II, Shields J. A polygenic theory of schizophrenia. Int J Ment Health 1972;1:107‐15. [Google Scholar]
- 10. Uher R, Zwicker A. Etiology in psychiatry: embracing the reality of poly‐gene‐environmental causation of mental illness. World Psychiatry 2017;16:121‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Os J, Kenis G, Rutten BP. The environment and schizophrenia. Nature 2010;468:203. [DOI] [PubMed] [Google Scholar]
- 12. Radua J, Ramella‐Cravaro V, Ioannidis JP et al. What causes psychosis? An umbrella review of risk and protective factors. World Psychiatry 2018;17:49‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Belbasis L, Köhler C, Stefanis N et al. Risk factors and peripheral biomarkers for schizophrenia spectrum disorders: an umbrella review of meta‐analyses. Acta Psychiatr Scand 2018;137:88‐97. [DOI] [PubMed] [Google Scholar]
- 14. Duncan LE, Keller MC. A critical review of the first 10 years of candidate gene‐by‐environment interaction research in psychiatry. Am J Psychiatry 2011;168:1041‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wray NR, Lee SH, Mehta D et al. Research review: polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry 2014;55:1068‐87. [DOI] [PubMed] [Google Scholar]
- 16. Guloksuz S, Rutten BP, Pries L‐K et al. The complexities of evaluating the exposome in psychiatry: a data‐driven illustration of challenges and some propositions for amendments. Schizophr Bull 2018;44:1175‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. EUGEI, van Os J, Rutten BP et al. Identifying gene‐environment interactions in schizophrenia: contemporary challenges for integrated, large‐scale investigations. Schizophr Bull 2014;40:729‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Korver N, Quee PJ, Boos HB et al. Genetic Risk and Outcome of Psychosis (GROUP), a multi site longitudinal cohort study focused on gene‐environment interaction: objectives, sample characteristics, recruitment and assessment methods. Int J Methods Psychiatr Res 2012;21:205‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McGuffin P, Farmer A, Harvey I. A polydiagnostic application of operational criteria in studies of psychotic illness: development and reliability of the OPCRIT system. Arch Gen Psychiatry 1991;48:764‐70. [DOI] [PubMed] [Google Scholar]
- 20. Wing JK, Babor T, Brugha T et al. SCAN: Schedules for Clinical Assessment in Neuropsychiatry. Arch Gen Psychiatry 1990;47:589‐93. [DOI] [PubMed] [Google Scholar]
- 21. Andreasen NC, Flaum M, Arndt S. The Comprehensive Assessment of Symptoms and History (CASH): an instrument for assessing diagnosis and psychopathology. Arch Gen Psychiatry 1992;49:615‐23. [DOI] [PubMed] [Google Scholar]
- 22. Vassos E, Di Forti M, Coleman J et al. An examination of polygenic score risk prediction in individuals with first‐episode psychosis. Biol Psychiatry 2017;81:470‐7. [DOI] [PubMed] [Google Scholar]
- 23. Bernstein DP, Stein JA, Newcomb MD et al. Development and validation of a brief screening version of the Childhood Trauma Questionnaire. Child Abuse Negl 2003;27:169‐90. [DOI] [PubMed] [Google Scholar]
- 24. Şar V, Akyüz G, Kundakçı T et al. Childhood trauma, dissociation, and psychiatric comorbidity in patients with conversion disorder. Am J Psychiatry 2004;161:2271‐6. [DOI] [PubMed] [Google Scholar]
- 25. Hernandez A, Gallardo‐Pujol D, Pereda N et al. Initial validation of the Spanish Childhood Trauma Questionnaire‐Short Form: factor structure, reliability and association with parenting. J Interpers Viol 2013;28:1498‐518. [DOI] [PubMed] [Google Scholar]
- 26. Thombs BD, Bernstein DP, Lobbestael J et al. A validation study of the Dutch Childhood Trauma Questionnaire‐Short Form: factor structure, reliability, and known‐groups validity. Child Abuse Negl 2009;33:518‐23. [DOI] [PubMed] [Google Scholar]
- 27. Kraan TC, Velthorst E, Themmen M et al. Child maltreatment and clinical outcome in individuals at ultra‐high risk for psychosis in the EU‐GEI high risk study. Schizophr Bull 2017;44:584‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barkus EJ, Stirling J, Hopkins RS et al. Cannabis‐induced psychosis‐like experiences are associated with high schizotypy. Psychopathology 2006;39:175‐8. [DOI] [PubMed] [Google Scholar]
- 29. Robins LN, Wing J, Wittchen HU et al. The Composite International Diagnostic Interview: an epidemiologic instrument suitable for use in conjunction with different diagnostic systems and in different cultures. Arch Gen Psychiatry 1988;45:1069‐77. [DOI] [PubMed] [Google Scholar]
- 30. Van Winkel R, Van Beveren NJ, Simons C et al. AKT1 moderation of cannabis‐induced cognitive alterations in psychotic disorder. Neuropsychopharmacology 2011;36:2529‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pries L‐K, Guloksuz S, ten Have M et al. Evidence that environmental and familial risks for psychosis additively impact a multidimensional subthreshold psychosis syndrome. Schizophr Bull 2018;44:710‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Radhakrishnan R, Guloksuz S, ten Have M et al. Interaction between environmental and familial affective risk impacts psychosis admixture in states of affective dysregulation. Psychol Med (in press). [DOI] [PubMed]
- 33. Davies G, Welham J, Chant D et al. A systematic review and meta‐analysis of Northern Hemisphere season of birth studies in schizophrenia. Schizophr Bull 2003;29:587‐93. [DOI] [PubMed] [Google Scholar]
- 34. Schäfer M, Korn S, Smith PK et al. Lonely in the crowd: recollections of bullying. Br J Dev Psychol 2004;22:379‐94. [Google Scholar]
- 35. Hunter SC, Mora‐Merchan J, Ortega R. The long‐term effects of coping strategy use in victims of bullying. Spanish J Psychol 2004;7:3‐12. [DOI] [PubMed] [Google Scholar]
- 36. Purcell S, Neale B, Todd‐Brown K et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 2007;81:559‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Das S, Forer L, Schonherr S et al. Next‐generation genotype imputation service and methods. Nat Genet 2016;48:1284‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loh PR, Danecek P, Palamara PF et al. Reference‐based phasing using the Haplotype Reference Consortium panel. Nat Genet 2016;48:1443‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Price AL, Weale ME, Patterson N et al. Long‐range LD can confound genome scans in admixed populations. Am J Hum Genet 2008;83:132‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Allardyce J, Leonenko G, Hamshere M et al. Association between schizophrenia‐related polygenic liability and the occurrence and level of mood‐incongruent psychotic symptoms in bipolar disorder. JAMA Psychiatry 2018;75:28‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Escott‐Price V, Smith DJ, Kendall K et al. Polygenic risk for schizophrenia and season of birth within the UK Biobank cohort. Psychol Med (in press). [DOI] [PMC free article] [PubMed]
- 42. Sørensen HJ, Debost J‐C, Agerbo E et al. Polygenic risk scores, school achievement, and risk for schizophrenia: a Danish population‐based study. Biol Psychiatry 2018;84:684‐91. [DOI] [PubMed] [Google Scholar]
- 43. van Os J, van der Steen Y, Islam MA et al. Evidence that polygenic risk for psychotic disorder is expressed in the domain of neurodevelopment, emotion regulation and attribution of salience. Psychol Med 2017;47:2421‐37. [DOI] [PubMed] [Google Scholar]
- 44. StataCorp . STATA Statistical Software: Release 15. Texas: College Station, 2017. [Google Scholar]
- 45. Rothman KJ. The estimation of synergy or antagonism. Am J Epidemiol 1976;103:506‐11. [DOI] [PubMed] [Google Scholar]
- 46. Kendler KS, Gardner CO. Interpretation of interactions: guide for the perplexed. Br J Psychiatry 2010;197:170‐1. [DOI] [PubMed] [Google Scholar]
- 47. Rothman KJ, Greenland S, Walker AM. Concepts of interaction. Am J Epidemiol 1980;112:467‐70. [DOI] [PubMed] [Google Scholar]
- 48. Knol MJ, VanderWeele TJ. Recommendations for presenting analyses of effect modification and interaction. Int J Epidemiol 2012;41:514‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. VanderWeele TJ, Knol MJ. A tutorial on interaction. Epidemiol Methods 2014;3:33‐72. [Google Scholar]
- 50. Richardson DB, Kaufman JS. Estimation of the relative excess risk due to interaction and associated confidence bounds. Am J Epidemiol 2009;169:756‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Royston P, White IR. Multiple imputation by chained equations (MICE): implementation in Stata. J Stat Softw 2011;45:1‐20. [Google Scholar]
- 52. Rubin DB. Multiple imputation for nonresponse in surveys, Vol. 81. Chichester: Wiley, 2004. [Google Scholar]
- 53. Mistry S, Harrison JR, Smith DJ et al. The use of polygenic risk scores to identify phenotypes associated with genetic risk of schizophrenia: systematic review. Schizophr Res 2018;197:2‐8. [DOI] [PubMed] [Google Scholar]
- 54. Van Os J, Rutten BP, Poulton R. Gene‐environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull 2008;34:1066‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Modinos G, Iyegbe C, Prata D et al. Molecular genetic gene‐environment studies using candidate genes in schizophrenia: a systematic review. Schizophr Res 2013;150:356‐65. [DOI] [PubMed] [Google Scholar]
- 56. Trotta A, Iyegbe C, Di Forti M et al. Interplay between schizophrenia polygenic risk score and childhood adversity in first‐presentation psychotic disorder: a pilot study. PLoS One 2016;11:e0163319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ursini G, Punzi G, Chen Q et al. Convergence of placenta biology and genetic risk for schizophrenia. Nat Med 2018;24:792‐801. [DOI] [PubMed] [Google Scholar]
- 58. Pries LK, Klingenberg B, Menne‐Lothmann C et al. Interaction between polygenic liability for schizophrenia and childhood adversity influences daily‐life emotional dysregulation and subtle psychosis expression. Submitted for publication. [DOI] [PMC free article] [PubMed]
- 59. French L, Gray C, Leonard G et al. Early cannabis use, polygenic risk score for schizophrenia and brain maturation in adolescence. JAMA Psychiatry 2015;72:1002‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schmidt‐Kastner R, Van Os J, Esquivel G et al. An environmental analysis of genes associated with schizophrenia: hypoxia and vascular factors as interacting elements in the neurodevelopmental model. Mol Psychiatry 2012;17:1194‐205. [DOI] [PubMed] [Google Scholar]
- 61. Binder EB. Understanding gene x early adversity interactions: possibilities for insight in the biology of psychiatric disorders. Eur Arch Psychiatry Clin Neurosci 2017;267:183‐5. [DOI] [PubMed] [Google Scholar]
- 62. Radhakrishnan R, Kaser M, Guloksuz S. The link between the immune system, environment, and psychosis. Schizophr Bull 2017;43:693‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ritz BR, Chatterjee N, Garcia‐Closas M et al. Lessons learned from past gene‐environment interaction successes. Am J Epidemiol 2017;186:778‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guloksuz S, van Os J, Rutten BP. The exposome paradigm and the complexities of environmental research in psychiatry. JAMA Psychiatry 2018;75:985‐6. [DOI] [PubMed] [Google Scholar]
- 65. Pingault J‐B, O'Reilly PF, Schoeler T et al. Using genetic data to strengthen causal inference in observational research. Nat Rev Genet 2018;19:566‐80. [DOI] [PubMed] [Google Scholar]
- 66. Guloksuz S, Van Os J. The slow death of the concept of schizophrenia and the painful birth of the psychosis spectrum. Psychol Med 2018;48:229‐44. [DOI] [PubMed] [Google Scholar]