

Abstract
Background
Isocitrate dehydrogenase (IDH) mutant gliomas are a distinct subtype, reflected in the World Health Organization (WHO) 2016 revised diagnostic criteria. To inform IDH-targeting trial design, we sought to characterize outcomes exclusively within IDH mutant gliomas.
Methods
We retrospectively analyzed 275 IDH mutant glioma patients treated at our institution. Progression was determined using low-grade glioma criteria from Response Assessment in Neuro-Oncology. We calculated survival statistics with the Kaplan–Meier method, and survival proportions were correlated with molecular, histologic, and clinical factors.
Results
During a median follow-up of 6.4 years, 44 deaths (7.6%) and 149 first progression (PFS1) events (54.1%) were observed. Median PFS1 was 5.7 years (95% CI: 4.7–6.4) and OS was 18.7 years (95% CI: 12.2 y—not reached). Consistent with prior studies, we observed an association of grade, molecular diagnosis, and treatment with PFS1. Following the first progressive episode, 79 second progression events occurred during a median follow-up period of 4.1 years. Median PFS following an initial progressive event (PFS2) was accelerated at 3.1 years (95% CI: 2.1–4.1). PFS2 was a surrogate prognostic marker, identifying patients with poorer overall survival.
Conclusion
We report outcomes in a large cohort of IDH mutant glioma, providing a well-characterized historical control population for future clinical trial design. Notably, the interval between first and second recurrence (PFS2, 3.0 y) is shorter than time from diagnosis to first recurrence (PFS1, 5.7 y), evidence that these tumors clinically degenerate from an indolent course to an accelerated malignant phase. Thus, PFS2 represents a relevant outcome for trials investigating drug efficacy at recurrence.
Keywords: endpoint, glioma, IDH mutation, IDH1, IDH2, progression-free survival
Key Points.
The interval between first and second episodes of progression in IDH mutant glioma is a median of 3 years.
This interval (PFS2) is notably shorter than the initial progression-free interval.
PFS2 is a relevant clinical trial endpoint for investigating drug efficacy at recurrence.
Importance of the Study.
Clinical trials focused on IDH mutant glioma-targeting drugs remain challenging because long progression-free and overall survival intervals requiring lengthy follow-up periods are needed before efficacy can be determined. We performed a retrospective analysis of population of patients with WHO grade II and grade III IDH mutant gliomas and provide a characterization of clinical outcomes that serves as a representative historical cohort for future trial design. Importantly, we report the median interval between first and second progressive episode, PFS2, is 3 years, which could serve as a feasible trial endpoint for investigating drug efficacy in the recurrent setting. Further we note that time between progressive episodes shortens during the course of IDH mutant glioma disease, offering clinical evidence that these tumors degenerate from an indolent phase to a more accelerated progressive phase.
Recurrent somatic mutations in the isocitrate dehydrogenase 1 (IDH1) gene in cancer were first noted in a 2008 report of a genome-wide mutational analysis of glioblastoma (GBM).1 Further studies demonstrated that up to 80% of World Health Organization (WHO) grade II and III gliomas and virtually all secondary GBMs exhibit mutations in IDH1 and the related isoform IDH2.2–4 The presence of this alteration is associated with younger age at diagnosis and significantly longer overall survival (OS) compared with patients with IDH wild-type tumors.1–3 In addition, the presence of an IDH mutation is tightly correlated with a hypermethylation pattern at distinctive loci known as the G-CIMP (glioma cytosine-phosphate-guanine island methylator phenotype).5 IDH mutant gliomas can be further subclassified into astrocytic and oligodendroglial tumors by presence or absence of additional molecular alterations.6 Astrocytic tumors are defined by mutations in TP53 and ATRX,6–8 while oligodendroglial tumors exhibit loss of the chromosome arms 1p and 19q as well as recurrent mutations in the telomerase reverse transcriptase (TERT) gene promoter.6,8–10 Overall, work from the last decade has highlighted the distinct nature of IDH mutant gliomas, emphasizing the molecular etiology by which these tumors develop as a biologically separate entity from IDH wild-type gliomas.11 These findings have resulted in a significant change to the diagnostic categorization of adult diffuse gliomas, as reflected in the updated 2016 WHO classification of central nervous system tumors.6
As molecularly distinct tumors, IDH mutant gliomas offer an exceptional opportunity for specific therapeutic targeting. Accordingly, small-molecule inhibitors of the mutant IDH enzyme and additional classes of drugs targeting pathways uniquely altered in IDH mutant glioma are moving quickly from preclinical studies to therapeutic trials. A major practical requirement in the clinical evaluation of drug efficacy in these cohorts is the need for comparison to an appropriate control population. However, existing data regarding outcomes and prognostic factors used for historical comparison in therapeutic trials have been derived from composite cohorts consisting of a mixture of IDH mutant and IDH wild-type tumors.12–14 To adequately design a clinical trial specifically focused on patients with IDH mutant gliomas, it is necessary to establish baseline natural history and outcome characteristics solely within a population of IDH mutant glioma patients.
To this end, we conducted a retrospective analysis of a large population of IDH mutant glioma patients to examine progression-free survival (PFS) and OS characteristics to uncover appropriate metrics for the establishment of relevant clinical trial endpoints. Importantly, we discovered evidence for the clinical degeneration of these tumors from an indolent initial course, as measured from initial diagnosis to the time of first progression, into an accelerated malignant phase, as measured from first progression to second progression. In a well-characterized cohort, we quantify this second PFS interval, which, at 3 years, can provide a feasible clinical trial endpoint in IDH mutant glioma populations.
Methods
Patient Population
This study was approved by the Massachusetts General Hospital (MGH) institutional review board. We retrospectively identified a cohort of adult patients in whom WHO grade II or grade III glioma was diagnosed between 1991 and 2017 using the neuro-oncology department brain tumor database at MGH. From this, we extracted 275 patients whose tumors exhibited an IDH mutation, as determined by immunohistochemistry (IHC) using an anti-human IDH1 R132H antibody (Dianova)15 (N = 141), PCR amplification and Sanger sequencing (N = 39), or SNaPshot genotyping16 (N = 206). IDH mutation was detected by multiple methods in 105 patients, most commonly by IHC and SNaPshot genotyping (N = 87). The presence of the IDH1 mutation was detected in 65 patients by IHC alone. The disease was initially diagnosed in 32 of these patients who had undergone a surgical procedure prior to 2011, when IHC for IDH1 mutation became routine; in these cases, the diagnosis of an IDH1 mutant glioma was made when the patient underwent a repeat resection after 2011 or following IHC using the IDH mutant antibody performed on a previously obtained tumor specimen.
Clinical Data Collection
Clinical and neuroimaging data of all patients were obtained from the electronic medical record. Clinical data included age at initial diagnosis, sex, date of first and any subsequent surgeries, dates of additional treatments such as radiation therapy or chemotherapy, and dates of clinical or radiologic progression. Cranial MR imaging was used to determine dates of radiologic progression. The extent of resection was determined by analysis of pre- and postoperatively acquired cranial MRI scans using T2-weighted and fluid-attenuated inversion recovery (FLAIR) images. A surgery resulted in a gross total resection per Shaw criteria,17 as evaluated by the neuroradiologist, treating neuro-oncologist, or consensus from MGH tumor board discussion and by an independent observer (J.J.M.). For survival analysis, the last follow-up date and vital status of the patients were determined.
Histopathologic and Molecular Analysis
All cases were reviewed by a neuropathologist at MGH. We collected tumor genetic information from the medical record to allow for classification by molecular subtype, based on the status of chromosome 1p and 19q as determined by fluorescence in situ hybridization, TP53-mutation, and mutation of alpha thalassemia/mental retardation syndrome X-linked (ATRX), as determined by IHC and/or hotspot sequencing.18 A codeletion of 1p and 19q molecularly defined an oligodendroglial tumor, while absence of 1p/19q codeletion or mutation in TP53 defined astrocytic tumors. Using this histopathologic classification and molecular information, we reclassified all tumors according to the 2016 update to the WHO classification of tumors of the central nervous system.6
Survival Analysis
OS was defined as the interval from date of initial diagnosis (date of first surgery) until date of death, censored at the last follow-up visit. PFS1 was defined as the interval between the date of initial diagnosis (date of first surgery) and either disease progression or death, again censored at last follow-up visit. The second PFS interval (PFS2) was calculated from date of first progression to either second disease progression or death, censored at last follow-up visit. OS following first progression (OS2) was calculated from date of first progression to death, censored at last follow-up visit. We used the Response Assessment in Neuro-Oncology (RANO) criteria for low-grade glioma (LGG) to define disease progression, which incorporates both clinical status/deterioration and radiologic changes on MRI.19 For the small minority of tumors that were contrast enhancing, progression was defined also using RANO criteria for LGG, in which a new contrast-enhancing lesion or an increase in size of existing contrast-enhancing lesion by 25% is considered progression.19 Patient vital status and date of last follow-up were last updated on May 15, 2018.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software and R for Statistical Computing (version 3.5.0). Survival probabilities (OS and PFS) were estimated using the Kaplan–Meier method. Survival distributions were compared using the log-rank test. Multivariate analysis examining factors associated with second PFS was carried out using a Cox proportional hazards model and included age, sex, histological grade at initial surgery, and molecular diagnosis. A 2-sided P-value of less than 0.05 was considered statistically significant.
Results
Clinical Characteristics
The median age of the entire cohort (N = 275) was 38.0 years (range 19–86). As has been previously noted,20 the age of diagnosis was younger for patients with astrocytic tumors compared with oligodendroglial tumors (35 y vs 45 y). There were slightly more male subjects (149, 54.2%) than female subjects (126, 45.8%) in our study population. Histopathologic review of tumor specimens at initial diagnosis revealed 134 (48.7%) WHO grade II and 141 (51.3%) WHO grade III gliomas. The majority of tumors were astrocytic (180, 65.4%), based on the absence of 1p/19q codeletion (Table 1). Of the grade II tumors, 74 (55%) were astrocytic, while 60 (45%) were oligodendroglial. In the grade III group, 106 (75%) were astrocytic, while 35 (25%) were oligodendroglial (Supplementary Table 1). As a result, the astrocytic tumors were more commonly designated as grade III (106, 59%) compared with grade II (74, 41%), although this was not a statistically significant difference. Conversely, more oligodendroglial tumors were grade II (60, 63%) than grade III (35, 37%) (Supplementary Table 1). Three patients had tumors with IDH2 mutations, and the remaining with IDH1 mutations. Similar to what has been reported previously,2,21,22 the IDH1 R132H mutation was most frequently seen (n = 236, 86%) (Supplementary Table 2). Overall, our cohort is representative of newly diagnosed IDH mutant glioma patients.
Table 1.
Clinical characteristics of patients with IDH mutant gliomas included in analysis
| Number of patients | 275 |
| Median age at diagnosis (range) | 38 (19–86) |
| Male:female ratio | 1.17 |
| Histologic grade (%) | |
| WHO grade II | 134 (48.7) |
| WHO grade III | 141 (51.3) |
| Molecular diagnosis (%) | |
| Astrocytic | 180 (65.4) |
| Oligodendroglial | 95 (34.6) |
| Extent of resection (%) | |
| Gross total resection | 142 (51.6) |
| Subtotal resection | 100 (36.4) |
| Biopsy | 33 (12.0) |
| Adjuvant treatment (%) | |
| RT only | 44 (16.0) |
| RT with chemotherapy | 108 (39.3) |
| Chemotherapy only | 31 (11.3) |
| None | 92 (33.4) |
| Median follow-up | |
| Non-progressors | 6.4 y |
| Survivors | 5.5 y |
Survival Characteristics of the Population
The estimated median OS of the entire cohort was 18.7 years (95% CI: 12.2 y—not reached), based on 44 deaths and a median follow-up time in survivors of 5.5 years (Supplementary Figure 1A). This survival is similar in range to what has been reported in other retrospective studies focused specifically on grade II and grade III IDH mutant gliomas.10,22–25
Disease progression occurred in 147 patients (53.5%), with progression events defined using RANO criteria for LGG.19 These criteria consist of radiographic evidence of a 25% increase in the T2/FLAIR lesion, development of new lesions or new contrast enhancement within existing tumor margins, or clinical progression (seizure, focal deficit, or neurologic decline). If a patient with MRI changes was referred for surgical resection and pathology showed reactive changes or treatment effects, this patient was considered a non-progressor. The median PFS1 for patients was 5.7 years (95% CI: 4.7–6.4), exceeded by a median follow-up time of 6.4 years in non-progressive survivors (Supplementary Figure 1B).
Interestingly, we did not observe a significant association between histopathologic grade and PFS1 within this cohort of IDH mutant gliomas (Fig. 1A), with a median PFS of grade II tumors of 4.7 years (95% CI: 4.0–6.1) compared with 6.7 years (95% CI: 5.3–8.9) for grade III tumors (P = 0.086). However, when comparing OS between IDH mutant grade II and grade III tumors, an association did emerge, with patients with grade II tumors living significantly longer, undefined versus 14.4 years (P = 0.015) (Fig. 1B).
Fig. 1.

Kaplan–Meier plots showing (A) PFS and (B) OS stratified by grade, with grade II tumors in blue and grade III tumors in red. (C) PFS and (D) OS stratified by molecular status, with astrocytic tumors in blue and oligodendroglial tumors in red.
Regarding the influence of molecular diagnosis on survival characteristics, we did not observe a difference in PFS1 between astrocytic and oligodendroglial tumors (P = 0.93) (Fig. 1C; Supplementary Table 3), with the median PFS1 in both groups estimated to be 5.7 years. When compared with prior studies, it is important to note that significant differences in PFS between astrocytic and oligodendroglial tumors have been described in studies like Radiation Therapy Oncology Group (RTOG) 9802, RTOG 9402, European Organisation for Research and Treatment of Cancer (EORTC) 26951, and Lassman et al,26–29 which draw comparisons using either histologic definitions or only partial molecular characterization. In these situations, the populations being compared consist of both mixed IDH mutant and wild-type tumors that are likely not balanced between oligo or 1p/19q codeleted cohorts and astrocytic or 1p/19q intact cohorts. Indeed, in a large molecularly defined cohort comparison,30 a scant difference in PFS was detectable between astrocytic and oligodendroglial tumors.
Nonetheless, similar to prior studies, the OS in our cohort of molecularly defined oligodendroglial tumors was significantly longer than that of astrocytic tumors (median OS undefined for oligo vs 14.4 years for astro, P = 0.025) (Fig. 1D). Interestingly, receipt of radiation treatment, either alone or combined with chemotherapy, was independently associated with PFS1 in a multivariate analysis including grade and molecular group (discussed in greater detail below) (Supplementary Table 4). Overall, the survival characteristics of our cohort are consistent with what has been previously reported in the literature, particularly that observed in a number of randomized clinical trials assessing the efficacy of chemotherapy and radiation therapy26,31 and a comprehensive retrospective analysis.10
Influence of Treatment on Survival
At the time of diagnosis, patients within this cohort most commonly underwent a gross total resection (52%) compared with a subtotal resection (36%) or a biopsy (12%) (Table 1). The study population received a heterogeneous group of treatments. Following initial surgery, 92 (33%) patients received no adjuvant therapy within 6 months, while the remaining patients received adjuvant treatment with radiation therapy (RT) alone (44, 13%), combination of chemotherapy and radiation (108, 39%), or chemotherapy alone (31, 11%) during this timeframe (Table 1). Adjuvant chemotherapy consisted of temozolomide (92, 85%); the combination procarbazine, lomustine, and vincristine (14, 13%); BCNU in one case; and combination temozolomide plus bevacizumab in one case.
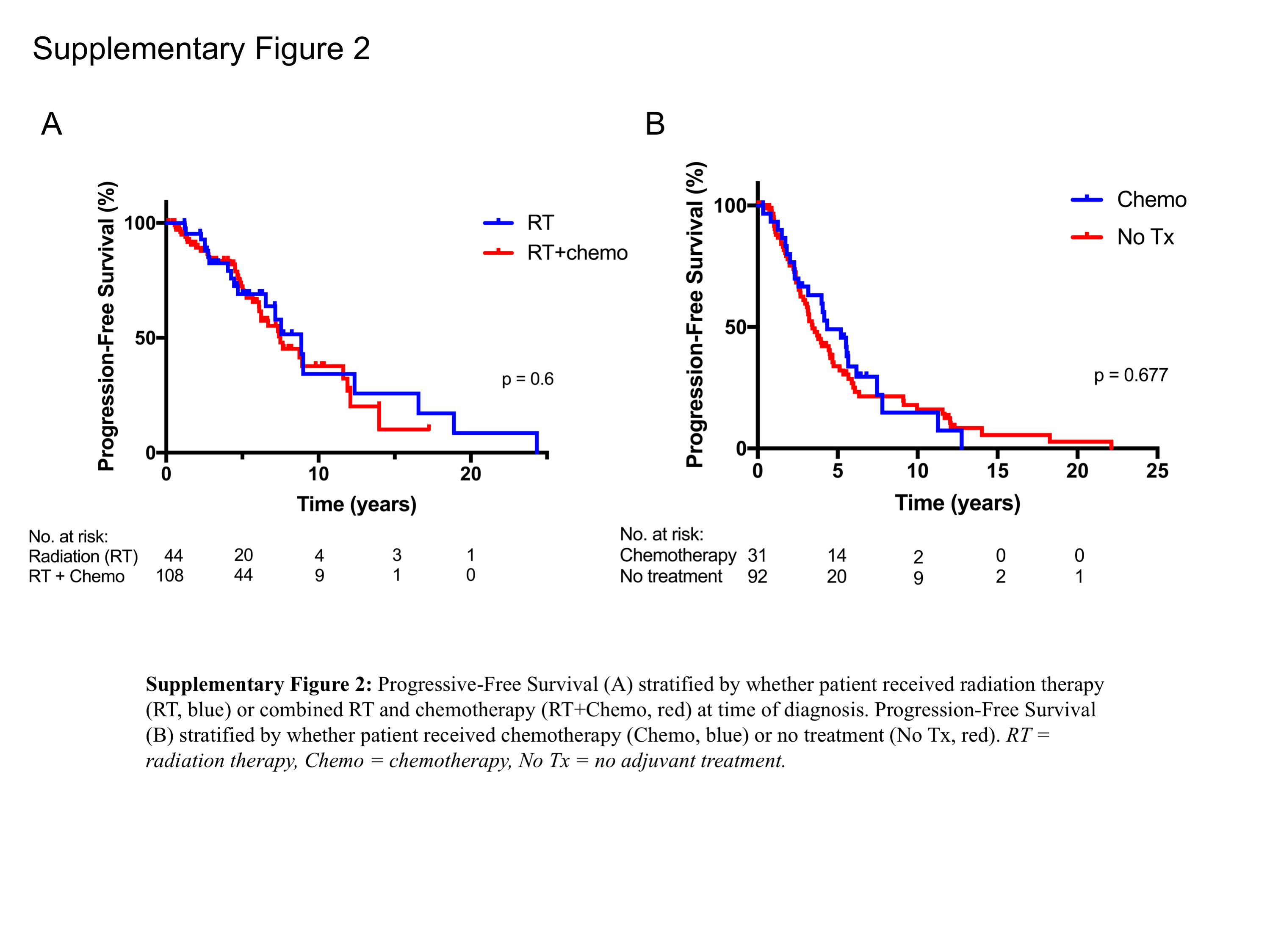
Consistent with the results of the EORTC 22845 study in histologically defined LGGs,32 we observed a significantly longer PFS1 in those who received any type of adjuvant RT compared with those who did not undergo RT (median PFS 7.6 y versus 4.0 y, P < 0.0001) (Fig. 2A). Radiation alone and chemoradiation were associated with improved PFS1 versus chemotherapy alone or observation, with median PFS1 of 8.7 years and 7.5 years for RT and chemoradiation, respectively, compared with 4.3 years for chemotherapy only and 3.4 years for observation (Fig. 2B). Otherwise, there were no detectable differences in PFS1 when comparing patients who received concurrent chemotherapy with those who received radiation only (P = 0.6; Supplementary Figure 2A), nor was there a discernable PFS1 benefit derived in association with receiving chemotherapy alone compared with observation (P = 0.68; Supplementary Figure 2B).
Fig. 2.

(A) PFS stratified by whether patient received RT at time of diagnosis. (B) PFS stratified by type of adjuvant therapy received. No Tx = no adjuvant treatment.
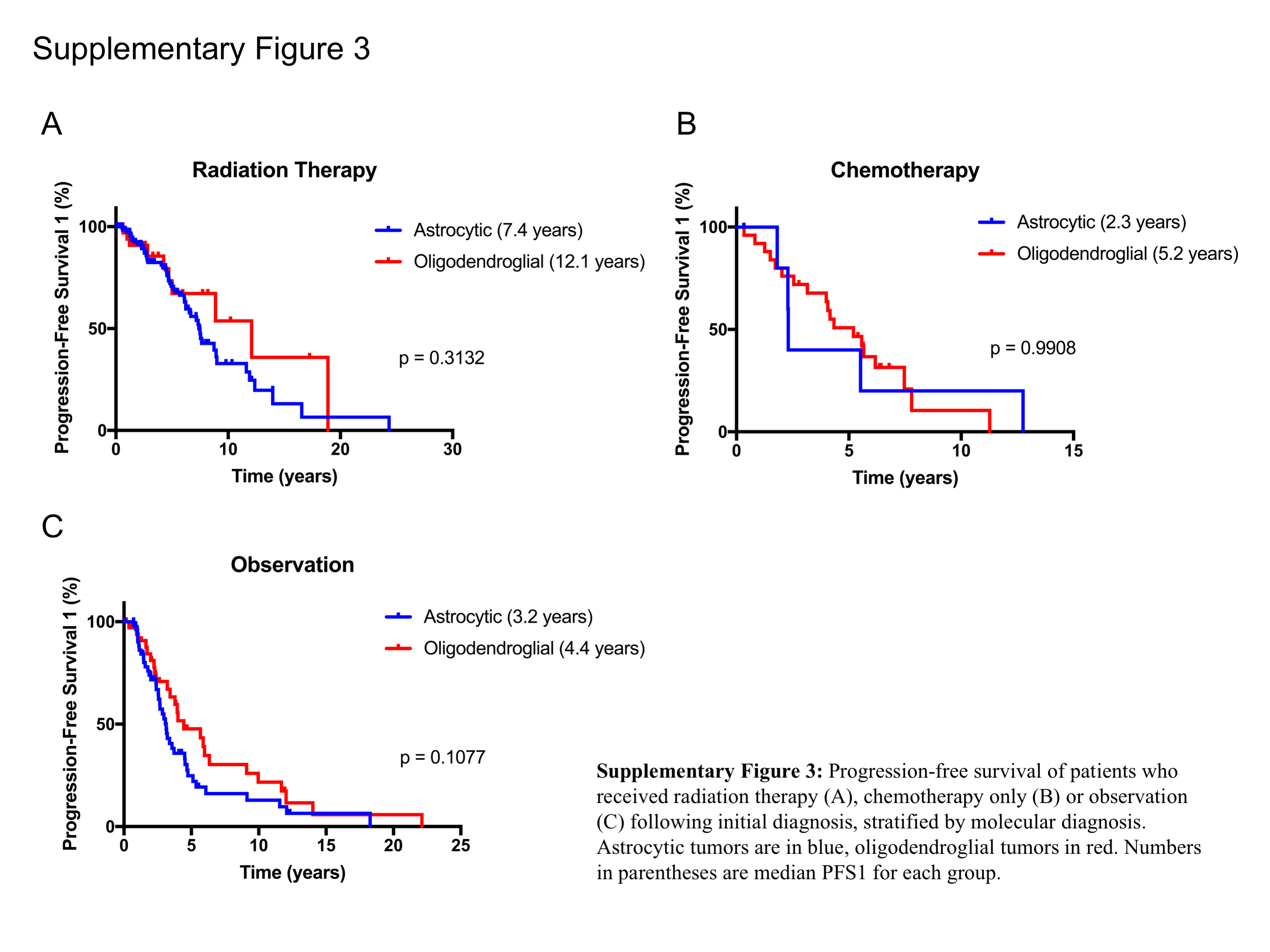
Radiation therapy has a strong impact of PFS1, therefore we were interested in investigating whether the type of treatment received differentially influenced PFS1 when the patients were stratified by different molecular diagnosis categories. Although this comparison is limited by the small number of patients per category, there was no significant difference in PFS1 between astrocytic and oligodendroglial tumors in each individual treatment category (RT, chemotherapy only, or observation; Supplementary Figure 3). Given the size of these cohort subsets (Supplementary Table 6), potential differences may be difficult to detect.
Patient Characteristics at the First Episode of Recurrence
Of the 275 IDH mutant glioma patients within the initial cohort, 147 experienced a progressive disease event during the follow-up period. The group who experienced first progression exhibited similar clinical characteristics to the initial study population (Table 2); however, in all cases, disease progression resulted in the recommendation of initiation of a salvage treatment by the treating physician. As noted above and in Fig. 1A, there was no significant difference in PFS1 between patients with grade II and grade III tumors. Accordingly, the breakdown by histopathologic grade was similar to that of the entire population, with 81/147 (55%) of tumors designated as grade II at diagnosis and 66/147 (45%) of tumors designated as grade III. Likewise, the relative proportion of astrocytic and oligodendroglial tumors in the progressive disease cohort was the same as in the initial cohort (Table 2). Although the proportion of astrocytic tumors that were grade II was larger at progression than at initial diagnosis (50.5% vs 41.1%), this difference was not significant (P = 0.16, Fisher’s exact test) (Supplementary Table 5). It is notable that 60% of patients with grade II tumors received no adjuvant treatment compared with only 7.8% of patients with grade III tumors (Supplementary Table 6), yet similar proportions of grades II and III tumors experienced first progression events. It is therefore difficult to determine if there is an association between treatment and grade as determinants of first progression in this retrospective analysis.
Table 2.
Clinical characteristics of patients with IDH mutant grade II and grade III gliomas who experienced first episode of progression
| Number of patients | 147 |
| Median age at diagnosis (range) | 36.7 (18–64) |
| Male:female ratio | 1.2:1 |
| Histologic grade (%) | |
| WHO grade II | 81 (55.1) |
| WHO grade III | 66 (44.9) |
| Molecular diagnosis (%) | |
| Astrocytic | 95 (64.6) |
| Oligodendroglial | 52 (35.4) |
| Median follow-up | |
| Non-progressors | 4.1 y |
| Survivors | 4.0 y |
Progression-Free Interval Between First and Second Episode of Recurrence
During a median follow-up of 4.1 years, a second episode of disease recurrences was noted in 79 patients. The PFS interval between first episode of disease progression following diagnosis and the second episode of recurrence (PFS2) was 3.1 years (95% CI: 2.1–3.9 y) (Fig. 3A). The estimated median OS following first episode of disease progression (OS2) was 8.3 years (95% CI: 5.8–13.1) (Fig. 3B), based on 43 events.
Fig. 3.

Kaplan–Meier plots showing (A) PFS and (B) OS in 147 patients with IDH mutant gliomas who experienced first episode of disease progression, in which time 0 indicates date of first progression. PFS in patients who experienced first episode of disease progression, stratified by (C) grade and (D) molecular status.
Interestingly, in contrast to what we found for PFS1, initial histopathologic grading and molecular diagnosis were significantly associated with the length of PFS2. Median PFS2 was 4.2 years (95% CI: 3.1–4.8 y) for patients with initial diagnoses of grade II tumors, while PFS2 was 1.7 years (95% CI: 1.5–3.2 y) for grade III tumors (P = 0.0029) (Fig. 3C). For those with oligodendroglial tumors, median PFS2 was 4.2 years (95% CI: 3.3–not defined) compared with 2.6 years (95% CI: 1.6–3.3 y) for astrocytic tumors (P = 0.016) (Fig. 3D).
Of note, in a multivariate Cox proportional hazards model that included age at diagnosis, sex, initial WHO grade, and molecular diagnosis (Table 3), both histopathologic grade (hazard ratio [HR] 1.7, 95% CI: 1.0–2.7) and molecular diagnosis (HR 0.6 for oligodendroglial status, 95% CI: 0.3–1.0) remained independently associated with a longer PFS2.
Table 3.
Multivariate analysis using Cox proportional hazards model of variables influencing PFS2
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| Variable | HR | 95% CI | P-value | HR | 95% CI | P-value |
| Age | 0.99 | 0.97–1 | 0.340 | 1.00 | 0.98–1.03 | 0.470 |
| Grade | 2.00 | 1.3–3.1 | 0.004 | 1.68 | 1.04–2.7 | 0.033 |
| Molecular diagnosis | 0.56 | 0.35–0.9 | 0.017 | 0.57 | 0.33–0.99 | 0.046 |
| Sex | 0.58 | 0.37–0.92 | 0.021 | 0.64 | 0.39–1.05 | 0.076 |
Notably, the median OS1 of patients who experienced a second progression event was 11.3 years (95% CI: 10.2–16.8 y), shorter than the median OS1 of 18.7 years of the entire cohort.
Discussion
IDH mutant gliomas represent a distinct subset of gliomas with unique clinical behavior and prognosis. While the updated 2016 WHO classification guidelines for CNS tumors reflects these molecular determinants, much of the available data for PFS and OS gleaned from clinical trials and used as historical control comparators are derived from outcomes from heterogeneous groups of patients with both IDH mutant and IDH wild-type tumors. Accordingly, survival data for historical control populations are largely based on mixed IDH mutant and wild-type populations. In concert with the updated classification guidelines, clinical trials testing novel therapeutics are now being designed to focus only on patients with IDH mutant gliomas and may include both WHO grades II and III tumors. Unfortunately, the necessary historical control data required to accurately estimate effect size, survival endpoints, and sample sizes to optimally design a trial are not available in the existing, prospective trial literature. The design of a phase II study in IDH mutant gliomas requires an appropriate comparison cohort to determine if an experimental agent is showing signs of efficacy. To this end, careful retrospective analyses of large cohorts of grades II and III IDH mutant tumors have been instrumental in capturing these survival characteristics.22,33
A recurrent theme emerging from these studies, which is well appreciated within the field, is that the median OS for patients with grade II or III IDH mutant tumors is estimated to be greater than 10 years.10,22,33,34 This is corroborated by subgroup analysis from recent prospective trials.26,27,31 Likewise, PFS estimates found in these datasets are also lengthy, with median PFS of IDH mutant groups estimated between 4–5 years.12,26 The use of these endpoints becomes difficult and at times infeasible when conducting early phase therapeutic clinical trials in IDH mutant patients owing to the extensive follow-up time required. Alternative, but clinically meaningful, endpoints are urgently needed to allow for more rapid execution of phase II trials in order to determine whether drug candidates are worth further consideration in the phase III setting.
In our cohort of 275 patients with IDH mutant glioma, we find a median PFS1 of 4.7 years and estimated median OS of 18.7 years. These values are in line with both prospective and retrospective reports from the literature that involve both grade II and grade III tumors.10,12,22,26,33,35 Further, the baseline clinical characteristics in this cohort are highly similar to those previously described for IDH mutant patients.1,2,33 Overall, these data suggest that our cohort is representative of a typical population of grade II and grade III IDH mutant gliomas.
Despite the limitation that this is a retrospective review of patients treated at a single institution, the similarity between median PFS and OS in our cohort and in the literature suggests that this group is representative of IDH mutant grades II and III tumors at large. The work of Olar and colleagues demonstrated no clear difference in survival characteristics between patients with IDH mutant grade II and grade III tumors.22 We similarly did not uncover a significant association between initial histologic grade and PFS1 in our cohort. However, our analysis here shows that PFS2 and OS are influenced by initial grade, which we speculate may be a manifestation of differing treatment strategies between our cohort and those reported by Olar and colleagues.22,36 Along the same lines, despite finding no relationship between PFS1 and molecular diagnosis, we note a significantly longer PFS2 for patients with oligodendroglial tumors, consistent with the well-known treatment responsiveness of tumors with 1p/19q codeletion.31,37,38
An additional potential limitation in this study is the use of RANO criteria to determine progression events. Though this method is the consensus standard in the field,19 the generally slow, incremental growth of these tumors over time limits the precision with which progression is definitively defined when the lesions remain non-enhancing. Further, radiation-induced posttreatment changes can be misinterpreted for tumor growth in non-enhancing tumors. However, in the absence of other well-validated criteria for progression in this population, these criteria represent the standard utilized for trial design in the field (NCT03343197, NCT03212274).
From this group of IDH mutant grade II and grade III gliomas we estimate the median length of the interval between the first episode of progression and the second episode of progression to be 3.1 years, which we have designated PFS2. This interval is nearly half of the initial median PFS of 5.7 years. These data suggest that time to progression accelerates over time in IDH mutant glioma patients. Consistent with the PFS findings, median survival time following a first progression event of 8.3 years is much shorter than the median OS from initial diagnosis of nearly 19 years. Additionally, we find that patients who experienced an episode of second progression have a shortened OS time compared with the cohort at large. These findings emphasize the idea that PFS can serve as a strong predictor of OS, as has been demonstrated from analysis of GBM populations.39,40
Most importantly, our finding of an acceleration of time to progressive events during the course of IDH mutant disease is in keeping with the concept of malignant degeneration of IDH mutant tumors. These tumors exhibit mutations in IDH, considered truncal or primary mutations, and alterations in TP53 and ATRX or loss of 1p/19q, considered secondary events, at the time of diagnosis. Over time, however, additional, tertiary genetic events accumulate, including the acquisition of receptor tyrosine kinase amplifications, mutations in PI3K signaling pathways,41–43 and deletion of tumor suppressors like CDKN2A.44,45 Activation of these oncogenic pathways are observed in tumors obtained at progression, and they may represent targetable drivers of the more accelerated malignant phase these tumors manifest later in the natural history of the disease.
In conclusion, the magnitude of PFS2 at 3 years offers a feasible endpoint for design of therapeutic clinical trials. This measure can serve both as a historical control for single-arm trials involving recurrent IDH mutant grade II and grade III gliomas, as well as for the design of randomized phase II trials in this setting. In this patient population, PFS2 is a surrogate marker of OS, the gold standard in oncology trials, and therefore merits consideration for use in early clinical trials.
Funding
This work was supported by the National Institutes of Health (NIH K12CA090354 to J.J.M., R01CA227821 to D.P.C., P50CA165962 to D.P.C. and T.T.B.).
Conflict of interest statement.
Merck & Co (DPC), Eli Lilly (DPC), Genomicare (TTB), Pfizer (TTB), Oncoceutics (TTB), Agios, Boehringer Ingelheim and Karus Therapeutics (IAR).
Authorship statement.
Conception/design: JJM, FL, TAJ, TTB, IAR, DPC. Acquisition of data: JJM, FL, TAJ, SST, EAW, TTB, IAR, DPC. Analysis and interpretation of data: JJM, TAJ, DPC. Drafting of manuscript: JJM, FL, TAJ, SST, EAW, TTB, IAR, DPC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We appreciate the advice from Rebecca Betensky at the Harvard T. H. Chan School of Public Health regarding statistical analysis.
References
- 1. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ichimura K, Pearson DM, Kocialkowski S, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009;11(4):341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116(6):597–602. [DOI] [PubMed] [Google Scholar]
- 5. Noushmehr H, Weisenberger DJ, Diefes K, et al. ; Cancer Genome Atlas Research Network Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Louis DN, Ohgaki H, Wiestler O, et al. WHO Classification of Tumours of the Central Nervous System, Revised. 4th ed.Lyon: International Agency for Research on Cancer; 2016. [Google Scholar]
- 7. Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3(7):709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brat DJ, Verhaak RG, Aldape KD, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eckel-Passow JE, Lachance DH, Molinaro AM, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015;372(26):2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller JJ, Shih HA, Andronesi OC, Cahill DP. Isocitrate dehydrogenase-mutant glioma: evolving clinical and therapeutic implications. Cancer. 2017;123(23):4535–4546. [DOI] [PubMed] [Google Scholar]
- 12. Baumert BG, Hegi ME, van den Bent MJ, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016;17(11):1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wahl M, Chang SM, Phillips JJ, et al. Probing the phosphatidylinositol 3-kinase/mammalian target of rapamycin pathway in gliomas: a phase 2 study of everolimus for recurrent adult low-grade gliomas. Cancer. 2017;123(23):4631–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Levin VA, Tonge PJ, Gallo JM, et al. CNS anticancer drug discovery and development conference white paper. Neuro Oncol. 2015;17(Suppl 6):vi1–vi26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009;118(5):599–601. [DOI] [PubMed] [Google Scholar]
- 16. Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shaw EG, Berkey B, Coons SW, et al. Recurrence following neurosurgeon-determined gross-total resection of adult supratentorial low-grade glioma: results of a prospective clinical trial. J Neurosurg. 2008;109(5):835–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chi AS, Batchelor TT, Dias-Santagata D, et al. Prospective, high-throughput molecular profiling of human gliomas. J Neurooncol. 2012;110(1):89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van den Bent MJ, Wefel JS, Schiff D, et al. Response assessment in neuro-oncology (a report of the RANO group): assessment of outcome in trials of diffuse low-grade gliomas. Lancet Oncol. 2011;12(6):583–593. [DOI] [PubMed] [Google Scholar]
- 20. Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3(7):709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118(4):469–474. [DOI] [PubMed] [Google Scholar]
- 22. Olar A, Wani KM, Alfaro-Munoz KD, et al. IDH mutation status and role of WHO grade and mitotic index in overall survival in grade II-III diffuse gliomas. Acta Neuropathol. 2015;129(4):585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lassman AB, Iwamoto FM, Cloughesy TF, et al. International retrospective study of over 1000 adults with anaplastic oligodendroglial tumors. Neuro Oncol. 2011;13(6):649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Panageas KS, Reiner AS, Iwamoto FM, et al. Recursive partitioning analysis of prognostic variables in newly diagnosed anaplastic oligodendroglial tumors. Neuro Oncol. 2014;16(11):1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van den Bent MJ, Chang SM. Grade II and III oligodendroglioma and astrocytoma. Neurol Clin. 2018;36(3):467–484. [DOI] [PubMed] [Google Scholar]
- 26. Buckner JC, Shaw EG, Pugh SL, et al. Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N Engl J Med. 2016;374(14):1344–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cairncross JG, Wang M, Jenkins RB, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32(8):783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344–350. [DOI] [PubMed] [Google Scholar]
- 29. Lassman AB, Iwamoto FM, Cloughesy TF, et al. International retrospective study of over 1000 adults with anaplastic oligodendroglial tumors. Neuro Oncol. 2011;13(6):649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tabouret E, Nguyen AT, Dehais C, et al. ; For POLA Network Prognostic impact of the 2016 WHO classification of diffuse gliomas in the French POLA cohort. Acta Neuropathol. 2016;132(4):625–634. [DOI] [PubMed] [Google Scholar]
- 31. Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31(3):337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van den Bent MJ, Afra D, de Witte O, et al. ; EORTC Radiotherapy and Brain Tumor Groups and the UK Medical Research Council Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet. 2005;366(9490):985–990. [DOI] [PubMed] [Google Scholar]
- 33. Reuss DE, Mamatjan Y, Schrimpf D, et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol. 2015;129(6):867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Juratli TA, Kirsch M, Robel K, et al. IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J Neurooncol. 2012;108(3):403–410. [DOI] [PubMed] [Google Scholar]
- 35. Houillier C, Wang X, Kaloshi G, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75(17):1560–1566. [DOI] [PubMed] [Google Scholar]
- 36. Wahl M, Phillips JJ, Molinaro AM, et al. Chemotherapy for adult low-grade gliomas: clinical outcomes by molecular subtype in a phase II study of adjuvant temozolomide. Neuro Oncol. 2017;19(2):242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473–1479. [DOI] [PubMed] [Google Scholar]
- 38. Jenkins RB, Blair H, Ballman KV, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66(20):9852–9861. [DOI] [PubMed] [Google Scholar]
- 39. Polley MY, Lamborn KR, Chang SM, Butowski N, Clarke JL, Prados M. Six-month progression-free survival as an alternative primary efficacy endpoint to overall survival in newly diagnosed glioblastoma patients receiving temozolomide. Neuro Oncol. 2010;12(3):274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lamborn KR, Yung WK, Chang SM, et al. ; North American Brain Tumor Consortium Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol. 2008;10(2):162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wakimoto H, Tanaka S, Curry WT, et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res. 2014;20(11):2898–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnson BE, Mazor T, Hong C, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343(6167):189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cohen A, Sato M, Aldape K, et al. DNA copy number analysis of Grade II-III and Grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol Commun. 2015;3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bai H, Harmancı AS, Erson-Omay EZ, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48(1):59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shirahata M, Ono T, Stichel D, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018;136(1):153–166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.