

Abstract
Concentration of [18F]fluoride has been mentioned in literature, however, reports have lacked details about system designs, operation, and performance. Here, we describe in detail a compact, fast, fully-automated concentration system based on a micro-sized strong anion exchange cartridge. The concentration of radionuclides enables scaled-up microfluidic synthesis. Our system can also be used to provide highly concentrated [18F]fluoride with minimal water content. We demonstrate how the concentrator can produce varying concentrations of [18F]fluoride for the macroscale synthesis of N-boc-5-[18F]fluoroindole without an azeotropic drying process, while enabling high starting radioactivity. By appropriate choice of solid-phase resin, flow conditions, and eluent solution, we believe this approach can be extended beyond [18F]fluoride to other radionuclides.
Keywords: Radionuclide concentration, Positron emission tomography (PET), radiopharmaceutical preparation, microfluidic radiofluorination, azeotropic drying free radiofluorination, anion exchange cartridge, micro-cartridge
3. Introduction
Positron-emission tomography (PET) provides an increasing range of in vivo assays of specific biological receptors or biochemical processes, providing critical information for patient management in areas such as oncology (Gambhir, 2002; Kitson et al., 2009; Kubota, 2001), neurodegenerative disorders (e.g. dementia, Alzheimer’s disease, Parkinson’s disease, and epilepsy) (Kitson, 2014; Newberg and Alavi, 2005; Ravina et al., 2005; Virdee et al., 2012), cardiovascular disease (Guludec et al., 2008; Kitson et al., 2009; Schindler et al., 2010), as well as inflammation or infectious diseases (Chryssikos et al., 2008; Glaudemans et al., 2013; Stumpe et al., 2000; Tarkin et al., 2014).
There has recently been considerable interest in the development of microscale technologies for the synthesis of the short-lived radiolabeled tracers that must be injected prior to a PET scan (Rensch et al., 2013). Some microscale approaches can reduce required radiation shielding and reagent costs, compared to macroscale approaches, and could make it practical and affordable to produce small batches of diverse PET tracers on demand. Although there has been significant progress in “flow-through” type microreactors for microscale radiosynthesis, including demonstration of a wide range of tracers (Pascali et al., 2013) and clinical use of tracers produced by this approach (Liang et al., 2014), there are considerable advantages of using microscale “batch” reactors (Keng and van Dam, 2015). For example, reaction volume can be significantly smaller (e.g. ≤50 µL), reducing reagent related expenses. In addition, for microfluidic chips that do not require large supporting pumps and valves, the overall system size can be much more compact, and potentially the system can be shielded and operated on a benchtop without the need for a conventional radiochemistry hot cell. An added advantage of low reaction volume is an improvement in molar activity of the synthesized PET tracer compared to macroscale methods (Javed et al., 2014; Sergeev et al., 2018); high molar activity is critical for imaging of scarce biological targets (e.g. neuroreceptors).
However, a challenge is that the radionuclide is typically produced/supplied in a volume much greater than these microreactors. For example, [18F]fluoride is typically produced in amounts of 10s of GBq or higher as a dilute solution in ~1–5 mL of [18O]H2O. At a typical concentration of about 37 GBq/mL [1.0 Ci/mL], it would be necessary to load 40–80 µL of the radionuclide solution at the start of synthesis to produce a single patient dose (~370–740 MBq [10–20 mCi] (Hoover et al., 2016)), assuming an overall decay-corrected yield of ~50% and synthesis time of 1 half-life. To produce enough tracer for multiple human doses, or to produce enough for long-distance transport to the imaging site, especially when working with low-yield reactions, would require even more activity (and volume) to be loaded. To ensure sufficient activity can be loaded into the microreactor, the radionuclide therefore needs to be concentrated before use. If a sufficient degree of concentration can be achieved, the entire batch of radionuclide could be used for the microscale synthesis, rather than the highly undesirable situation of using e.g. 10 µL and discarding the remaining 99% of a 1 mL target bombardment.
Concentration of [18F]fluoride can also be useful to reduce water content in conventional macroscale radiosynthesizers. For nucleophilic fluorination reactions, the presence of water severely reduces the reactivity of [18F]fluoride (Lemaire et al., 2010; Seo et al., 2011), and thus water must be removed prior to fluorination, typically through a multi-step azeotropic drying process. Concentration of [18F]fluoride to a few microliters can substantially reduce water content and facilitate a reactive [18F]fluoride without the need for azeotropic drying. For example, concentration down to 5 µL would result in <0.5% (v/v) water content if added to a reactor containing 1 mL of precursor solution in organic solvent. The base content can also be reduced. This approach has been used to increase the yield and simplify the preparation of human doses of [18F]5-Fluorouracil ([18F]5-FU) (Hoover et al., 2016). This method can be considered an alternative to other approaches that have previously been reported for reducing water and/or base content for macroscale reactions. For example, Lemaire et al. (2010) demonstrated nearly quantitative elution of [18F]fluoride from a conventional QMA cartridge with a solution containing organic base in MeCN with either a small amount of alcohol or water (0.2–2.5%). The eluate was directly used in fluorination reactions and resulted in high yields without the need for azeotropic drying. Iwata et al. (2017, 2018) demonstrated efficient elution of [18F]fluoride from a conventional QMA cartridge with anhydrous MeOH containing a minimal amount of phase transfer catalyst and base. The amounts could be even further reduced by the flowing through a downstream cation exchange cartridge. This reduction of phase transfer catalyst and base combined with removing the need for azeotropic drying allowed for improved fluorination yields across various model PET tracers. Finally, Richarz et al. (2014) presented a method wherein water, base, and phase transfer catalyst could be eliminated altogether by eluting the[18F]fluoride directly from a conventional QMA cartridge using a charged precursor in anhydrous MeOH. The MeOH could then be evaporated and replaced with a more suitable solvent (e.g. DMSO, DMF) for efficient fluorination.
Due to the importance of concentrating [18F]fluoride (Figure 1), several groups, including ours, have reported methods to perform the concentration step. In one approach, water was removed by evaporation: a 200 µL open droplet of [18F]fluoride in [18O]H2O on an electrowetting-on-dieletric (EWOD) microfluidic chip was concentrated down to 5 µL in 10 min by heating the substrate (Chen et al., 2014). Once the droplet volume had been reduced, electrodes were activated to draw the droplet under the cover plate onto the EWOD transport pathways. Radioactivity loss during the concentration process, qualitatively visualized through Cerenkov imaging, proved to be negligible. This straightforward approach is suitable for modest starting volumes, but may require impractically large chip real estate, or take too much time for sequential 200 µL evaporations to handle volumes in the 1–5 mL range that are expected from most cyclotrons. Another approach is to use miniature anion exchange cartridges to trap [18F]fluoride from the large volume of [18O]H2O and to then recover this [18F]fluoride in smaller volumes ranging from 5 – 500 µL (De Leonardis et al., 2011; Elizarov et al., 2010; Lebedev et al., 2012; Salvador et al., 2017). Elizarov et al. demonstrated concentration of 32.4 GBq [876 mCi] of [18F]fluoride with a starting volume of 2 mL to a final volume of 5 μL within a custom-built micro cartridge with 2 µL bed volume filled with AG-1-X8 QMA resin (Bio-Rad Laboratories, Inc., USA) (Elizarov et al., 2010). During the concentration process, 99.5% of the starting [18F]fluoride was trapped, and 92.7% of the trapped activity was recovered during elution of the [18F]fluoride off of the cartridge (n=1). Flow rates used during concentration were 2 mL/min allowing for complete concentration in ~3 min. Lebedev et al. demonstrated concentration of [18F]fluoride (up to 110GBq [3 Ci]) in 2 mL of [18O]H2O to a final volume of 45 μL within a commercial micro-cartridge (OptiLynx, Optimize Technologies, Inc., USA; 5 µL bed volume) packed with the same resin (Lebedev et al., 2012). Trapping and release of various [18F]fluoride amounts resulted in ~95% recovery of the starting amount. Concentration was performed in ~3 min. De Leonardis et al. loaded anion exchange resin (Chromabond PS-HCO3, ABX, Germany) into a microfluidic channel and demonstrated concentration of 5–7 GBq [140–190 mCi] samples of [18F]fluoride in 4 mL [18O]H2O (De Leonardis et al., 2011). Trapping efficiency was >90% and ≥95% of the trapped activity could be eluted in 250 µL with a total processing time of 6 min. Ismail et al. demonstrated the use of a functionalized porous polymer monolith (polystyrene imidazolium chloride) instead of packed resin beads (Ismail et al., 2014). Trapping of [18F]fluoride solutions (1.5–7.4 MBq [40–200 μCi]) at a flow rate up to 250 µL/min had an efficiency of 97 ± 4% (n=39) and it was shown theoretically that higher activities could be trapped by extending the length of the monolith. Of various eluents tested, CaCO3 performed the best, recovering 94 ± 6% (n=2) of the activity in a volume of 100 μL. Recently, Salvador et al. demonstrated concentration of [18F]fluoride within a manually-operated custom built PDMS microfluidic system with 10–15 mg embedded QMA resin (from Sep-Pak Accell Plus cartridge, Waters, Inc., USA) (Salvador et al., 2017). For activities of 19 GBq [0.5 Ci], trapping of 2 mL of [18F]fluoride in [18O]H2O was achieved with 98% efficiency; trapped activity could then be eluted into a volume of 20 μL with >87% recovery. Trapping was performed with a flow rate of 180 μL/min.
Figure 1.
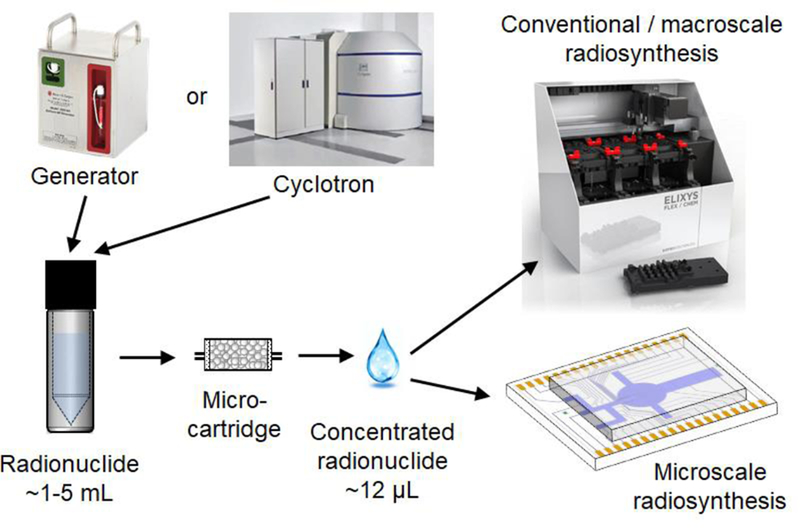
Concept image illustrating the role of radionuclide concentration. A batch of radionuclide produced either by a generator or a cyclotron is rapidly concentrated from 1–5mL to ~12 µL and can be used in either microscale or macroscale synthesis.
Concentration has also been performed using an electrochemical cell instead of anion exchange resin. Saiki et al. reported a microfluidic cell with 16 µL internal volume that could trap [18F]fluoride from 1–2 mL of [18O]H2O at up to 700 µL/min flow rate by applying a 10V potential, and then release the activity into a smaller volume of an eluent solution with an overall efficiency (deposition and release) of ~60% (Saiki et al., 2010). The release process used a total volume of 275 µL of eluent solution, but a radiation detector showed that the majority of activity was released into the first ~60 µL and concentration could thus be achieved using a switching valve (Wong et al., 2012).
In this paper, we focus on the micro-cartridge approach due to its fast operation, high efficiency, commercial availability of components, and ability to realize very small output volumes by minimizing the bed volume of the cartridge. We have developed a standalone, fully-automated system for rapid concentration of [18F]fluoride into microliter-scale volumes for a variety of applications. Though a previous version of this system has been used to avoid azeotropic drying in the preparation of clinical doses of [18F]5-FU (Hoover et al., 2016), this is the first report of the detailed design, operation, and performance of this system. As an additional demonstration we also report new data on the use of the concentrator to reduce water content (while potentially increasing starting activity) to perform Ni-mediated radiosynthesis of a model compound (N-boc-5-[18F]fluoroindole) without azeotropic drying.
4. Methods
4.1. Reagents
Anhydrous acetonitrile (MeCN), potassium bicarbonate (KHCO3), tetrabutylammonium bromide (TBAB), 1,4,7,10,13,16-hexaoxacyclooctadecane (18-crown-6), ethyl acetate (C4H8O2), hexane (C6H14), and monobasic potassium phosphate (KH2PO4) were purchased from Sigma-Aldrich (Milwaukee, WI USA). Deionized water was obtained from a Milli-Q water purification system (EMD Millipore Corporation, Berlin, Germany). Saline (0.9% w/v) was purchased from Hospira (Lake Forest, IL, USA). 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (Kryptofix 222; K222) was purchased from ABX (Radeberg, Germany). Sodium chloride (NaCl) was purchased from Fisher Scientific (Pittsburgh, PA, USA). [18F]fluoride in [18O]H2O was produced in a cyclotron (RDS-112, Siemens, Knoxville, TN, USA) through (p,n) reaction of [18O]H2O (98% isotopic purity, 18–98-050, Rotem Medical, Israel) at 11 MeV using a 1 mL internal volume tantalum target with Havar foil. Unless otherwise noted, all materials were used as received. A variety of different preconditioning solutions (based on KHCO3, NaCl, or KH2PO4 in water) and eluent solutions (based on K2CO3/K222, TBAB, NaCl, or K3PO4/18-crown-6 in water/MeCN) were prepared for different experiments (see Tables 1, 2, 4, and 5). The nickel aryl precursor complex and the hypervalent iodine oxidant used for the synthesis of N-boc-5-[18F]fluoroindole were synthesized following the methods of Lee et al. (Lee et al., 2012).
Table 1:
Efficiencies of [18F]fluoride trapping after preconditioning with various solutions measured in the intermediate vial system. The cartridge was preconditioned with 0.6 mL of preconditioning solution, rinsed twice with 0.8 mL DI water, rinsed with 1.0 mL MeCN, and air dried for 40 s.
| Preconditioning Solution | Trapping efficiency (%) |
|---|---|
| 1 M KHCO3 | 99 ± 1 (n=13) |
| 1 M NaCl | 94 ± 8 (n=10) |
| 1 M KH2PO4 | 96 ± 4 (n=16) |
Table 2:
Efficiencies of [18F]fluoride elution using different eluent solutions measured in the intermediate vial system. Elutions were performed with two plugs of eluent solution (12.4 µL total volume).
| Eluent (in DI water) | Elution efficiency based on trapped activity (%) |
|---|---|
| 0.01 M K2CO3 and 0.05 M K222 | 89 ± 7 (n=8) |
| 0.08 M TBAB | 92 ± 8 (n=2) |
| 0.15 M NaCl (Saline) | 96 ± 5(n=10) |
| 0.01 M K3PO4 and 0.07 M 18-Crown-6 | 88 (n=1) |
| 0.18 M K3PO4 | 90 (n=1) |
| 1.19 M K3PO4 | 100 (n=1) |
Table 4:
Recovery of [18F]fluoride (with respect to trapped activity) with the intermediate vial system as a function of eluent with varying compositions of MeCN. Recovery values are average of n repeats ± standard deviation.
| Solvent composition (% MeCN in DI water v/v) | K3PO4 (mM) | 18-crown-6 (mM) | Recovery (% of trapped activity, decay-corrected) |
|---|---|---|---|
| 50 | 24 | 152 | 96 (n=1) |
| 80 | 24 | 136 | 84 ± 6 (n= 6) |
| 90 | 24 | 136 | 66 (n=1) |
| 93 | 24 | 152 | 43 (n=1) |
Table 5:
Reaction conditions and radiochemical yield (RCY; decay-corrected) for synthesis of N-boc-5-[18F]fluoroindole using concentrated [18F]fluoride. In each experiment, the concentrated [18F]fluoride was recovered with two plugs (total 12.4 µL) of eluent solution (24 mM K3PO4 + 136 mM 18-crown-6 in 1:4 v/v H2O:MeCN). Drying solution 1 is 38 mM 18-crown-6 in MeCN (500 µL). Drying solution 2 is 38 mM 18-crown-6 + 10 mM K3PO4 in 1:400 H2O:MeCN (500 µL).
| Starting activity (mCi) | Ni-indole complex amount (mg) | Oxidant amount (mg) | Drying solution | H2O content in reaction (calculated) (%v/v) | H2O content (Karl-Fischer) (%v/v) | RCY (%) |
|---|---|---|---|---|---|---|
| 3.28 | 1.12 | 1.31 | 1 | 0.48 | 0.32±0.02 (n=2) | 49 |
| 1.06 | 1.02 | 1.30 | 1 | 0.48 | 52 | |
| Average ± SD | 51 ± 2 | |||||
| 1.49 | 1.31 | 1.55 | 2 | 0.73 | 0.57±0.01 (n=2) | 53 |
| 1.17 | 1.2 | 1.4 | 2 | 0.73 | 47 | |
| Average ± SD | 50 ± 4 | |||||
4.2. Miniature anion exchange cartridge
Strong anion exchange (SAX) resin (AG-MP1; 200–400 mesh size) was sourced from Bio-Rad (Hercules, CA, USA). Quaternary ammonium (QMA)-based resin was chosen for its lower affinity to the fluoride ion compared to other anions (e.g. Cl−, HCO3− ) allowing for easier elution of fluoride with these other anions. Resin was packed by Optimize Technologies (Oregon, OR, USA) into OPTI-LYNX micro trap cartridges (11–04755-ES, Optimize Technologies). The cartridges have a bed volume of ~4 µL and hold ~2 mg of resin. The cartridge was placed in an OPTI-LYNX micro holder (Optimize Technologies) to facilitate connection to fluidic paths via standard fittings and tubing. This resin/cartridge combination was selected to due to the small bed volume, commercial availability, compatibility with standard fittings, and previous reports that high amounts of activity (110 GBq [3 Ci]) could be efficiently trapped (Lebedev et al., 2012).
For optimal performance, the manufacturer recommends sonicating the cartridge in 50:50 v/v MeCN/DI water for 5 min to hydrate and redistribute the internal resin after the cartridges are stored dry. This procedure was performed whenever cartridges could not be immediately removed from the system and stored in 50:50 MeCN/DI water.
4.3. System design
4.3.1. Overview
The overall concentration system has several components. One portion controls the flows for trapping the [18F]fluoride onto the micro-cartridge and later releasing it. Another part generates the low volumes of eluent solution for the release step. Finally, the third part is responsible for controlling which reagents are passing through the cartridge. Each of these components is described in detail below.
Reagents were driven either by inert gas pressure or vacuum. The inert gas was provided from an electronic pressure regulator (ITV0010–2BL, SMC Corporation, Japan) connected to a nitrogen source. Vacuum was supplied from an electronic vacuum regulator (ITV2090, SMC) connected to a central “house” vacuum (1.9 L/min, −90 kPa). A small vacuum pump could be used instead. Unless otherwise specified, liquid pathways were implemented with 0.02’’ ID, 1/16” OD ETFE tubing (1516L, IDEX).
4.3.2. Trapping and elution
At the core of the system, the micro-cartridge is connected to a low dead volume HPLC injection valve (“cartridge valve”, Titan HT 715–000, IDEX Health & Science) via 0.01” ID 1/16” OD PEEK tubing (Figure 2). In one state, the valve allows large volumes (up to several mL) of solution (i.e., preconditioning solution, [18F]fluoride/[18O]H2O, or wash solutions) from the reagent selection system (described in Section 4.3.4) to flow through the cartridge, and then to one of two waste outputs (“cartridge waste” or “[18O]H2O recovery”), selected by a downstream 3-way valve (D; “cartridge waste valve”; LVM105R, SMC Corporation, Japan). A liquid sensor (#1, OCB350L062Z, OPTEK Technologies, Carrollton, TX, USA) was positioned at this output to enable automatic determination of when a reagent had entirely passed through the cartridge.
Figure 2.

Fluidic and pneumatic diagram for micro-cartridge and associated valve. Configuration for (A) preconditioning the micro-cartridge, (B) trapping of [18F]fluoride on the cartridge, and (C) elution of [18F]fluoride from the cartridge.
After trapping [18F]fluoride and drying the micro-cartridge, the cartridge valve is switched to its other state, allowing a small volume of eluent solution to efficiently flow through the cartridge to an output where the concentrated radionuclide is collected for downstream use (e.g. in a microfluidic radiosynthesis chip or macroscale reaction vessel).
4.3.3. Metering eluent solution
To measure the desired small volumes of the eluent solution, another identical rotary injection valve (“eluent metering valve”) was used in conjunction with a 6.2 µL “loop”, consisting of a 12.25 cm length of 0.01” ID 1/16” OD PEEK tubing (1531, IDEX). Note that this loop size was the minimum length of tubing that could reliably be connected into the eluent metering valve. The fluidic connections are shown in Figure 3.
Figure 3.

Fluidic and pneumatic diagram for the eluent metering subsystem. Configuration for (A) metering of eluent solution, and (B) elution of [18F]fluoride to the micro-cartridge.
In one state of the valve, fluids (i.e. either eluent solution to fill the loop, or wash solution to clean the loop) flow through the loop and out to an “eluent waste vial” (60942A40, Kimble Chase, Vineland, NJ, USA) via a 3-way “eluent waste valve” (H; LVM105R, SMC Corporation, Japan). Reagents were driven with vacuum by connecting the headspace of the eluent waste vial to a regulated vacuum source. Additional 3-way valves (I, E, F, G; LVM105R, SMC Corporation, Japan) are used to select which input reagent flows through the loop (i.e. eluent solution or eluent washing solutions: MeCN or DI water). Details of these reagents connections are in the Supplemental Information, Section 1.1. A liquid sensor (#3; OCB350L062Z, OPTEK Technologies, Carrollton, TX, USA) was positioned at the eluent waste output of the eluent metering valve to determine when the loop had been completely filled.
In the other position of the rotary valve, the metered plug of eluent solution in the loop is driven by inert gas toward the cartridge valve via a ~3 cm segment of 0.02” ID 1/16” OD PFA HP plus tubing (1902L, IDEX). A liquid sensor (#2; OCB350L062Z, OPTEK Technologies, Carrollton, TX, USA) was positioned half way along this tubing segment to monitor the passage of the metered eluent plug toward the cartridge valve.
4.3.4. Reagent delivery
During operation, several different reagents (e.g. cartridge preconditioning solution, preconditioning wash solution (DI water), [18F]fluoride/[18O]H2O, and rinse solution (MeCN)) must be flowed through the micro-cartridge connected to the cartridge valve. Selection of the reagent is controlled via a 7-port, 6-position rotary stream selection valve (“reagent selection valve”; Titan HT 715–105, IDEX). This type of valve connects any one of its 6 inlets to a common outlet.
Two generations of prototype systems were built during this study with slightly different methods of measuring and delivering the solutions. In the “intermediate” vial system, each reagent was delivered by first filling the desired amount of the reagent from a larger reservoir into an intermediate vial using vacuum and then pushing this volume out of the vial to the inlet of the cartridge valve (and through the micro-cartridge). A calibration was performed for each reagent to determine the vacuum pressure and time needed to transfer a certain volume into the intermediate vial. Details of fluid connections and operation are discussion in the Supplemental Information, Section 1.2.1. In the direct loading system the output of the reagent selection valve is connected directly to the inlet of the cartridge valve via 1/16’’OD PEEK tubing. We chose to operate the system such that each reagent vial contained a pre-measured amount of the reagent, but it would be possible to meter reagent volumes by performing time- or pressured-based calibrations as was done for the intermediate vial system. For reagents that are needed twice during the concentration process (i.e. DI water and MeCN), we filled two vials with the same reagent. Details of fluid connections and operation are discussed in the Supplemental Information, Section 1.2.2.
4.3.5. Control system
Detailed documentation of electronic components used to control and automate the system can be found in Supplemental Information, Section 2.
4.4. Concentration process
4.4.1. Overview
To carry out the concentration process, first, all reagent vials are filled except for the radionuclide. The cartridge is then preconditioned and rinsed, and the eluent metering loop is filled with eluent solution. Next, the radionuclide is introduced into the system, and is trapped on the micro-cartridge. Finally, the radionuclide is recovered by flowing the contents of the eluent loop through the cartridge. Additional elution steps can be performed if desired. A flowchart representation of these steps is shown in Supplemental Information, Figure S5.
4.4.2. Setup
Before operation, all waste vials (cartridge waste, [18O]H2O recovery, and eluent loop waste) were emptied.
In the intermediate vial system, reagent vials were loaded with the following volumes of reagents: DI water for precondition rinsing (40 mL), MeCN (40 mL), preconditioning solution (10 mL), eluent solution (2 mL), MeCN for eluent metering valve (40 mL), DI water for eluent metering valve (40 mL).
In the direct loading system, the following volumes were used: preconditioning solution (1 mL), DI water for precondition rinsing (0.5 mL x 2 vials), eluent solution (2 mL), MeCN (0.5 mL x2 vials), MeCN for eluent metering valve (40 mL), and DI water for eluent metering valve (40 mL)
4.4.3. Preconditioning
Before trapping [18F]fluoride, the micro-cartridge must be pre-conditioned (Figure 2A). First, the cartridge valve is set into the “trap” position and the cartridge waste valve is set to cartridge waste (Figure 2A). Pre-conditioning solution (0.6 mL for intermediate vial system, 1.0 mL for direct loading system) was passed through the micro-cartridge to the waste vial. In the intermediate vial system, 3.0 mL of DI water was then loaded to rinse the intermediate vial and discarded to waste by temporarily switching the cartridge valve to the “elute” position to bypass the cartridge. Next, the cartridge was rinsed twice with DI water (0.8 mL each time for the intermediate vial system, 0.5 mL each time for the direct loading system). Finally, DI water was eliminated from the cartridge by flowing MeCN (1.0 mL for the intermediate vial system and 0.5 mL for the direct loading system) through the cartridge, followed by a flush with inert gas (40 s). Note that the volumes were slightly higher for the intermediate vial system to improve reliability by accounting for losses that could occur within the intermediate vial.
4.4.4. [18F]fluoride trapping
In typical experiments, the [18F]fluoride vial was loaded with ~0.5 mL (~ 9–1220 MBq [0.25–33 mCi]) [18F]fluoride/[18O]H2O prior to trapping. In practice, this vial could be filled via tubing directly from the cyclotron or other [18F]fluoride sources. In the case of the intermediate vial system, a vacuum pull time of 11 s was used to ensure complete transfer of the [18F]fluoride into the intermediate vial, even though calibration indicated that only 5 s was needed to transfer 0.5 mL.
With the cartridge valve in “trap” position, the [18F]fluoride solution was then driven by inert gas (20 psig) through the micro-cartridge and to the [18O]H2O recovery vial (Figure 2B). The micro-cartridge traps the fluoride ions while the [18O]H2O passes through. After trapping, a portion of MeCN (1.0 mL for the intermediate vial system and 0.5 mL for the direct loading system) is passed through the cartridge (to remove residual water) to the cartridge waste vial. Finally, the cartridge is dried by flowing inert gas at 20 psig for 40 s through the same fluid pathway.
4.4.5. [18F]Fluoride elution
For each elution step, a 6.2 µL plug of eluent solution is prepared. With the eluent metering valve in the “load” position (Figure 3A), the eluent path is primed by applying vacuum (-10 psig) to the eluent waste reservoir. Once the loop is filled (detected by an air-to-liquid transition at the eluent waste liquid sensor (#3)), valve H is closed, and the eluent metering valve is switched to the “elute” position (Figure 3B). At the same time, the cartridge valve is switched to the “elute” position (Figure 2C). Inert gas pressure is then applied to push the eluent plug toward the cartridge valve, through the micro-cartridge, and to the concentrated radionuclide outlet for downstream radiosynthesis. To ensure the small plug of eluent solution remains intact, the inert gas pressure is gradually ramped up (0.5 psi every 5 s). Once liquid sensor #2 detects that the whole eluent plug has passed (via air-to-liquid then liquid-to-air transitions), pressure is increased by 5 psig for an additional 10 s. The majority of the eluent volume is ejected from the cartridge within 4 s following the pressure ramp, but an additional 10 s was chosen to provide a safety margin as well as to recover any residual droplets formed on the concentrated radionuclide outlet tubing during ejection.
The eluent loop can be refilled to perform additional elution steps. Note that upon switching the eluent metering valve back to the load position, the eluent loading system is still full of eluent except for a gap of air in the loop. Thus the system can be re-primed by applying vacuum to the eluent waste vial until the liquid sensor (#3) detects a liquid-to-gas followed by a gas-to-liquid transition. The elution process can then be repeated exactly as above. The eluent loop can also be filled with water or MeCN to perform rinsing steps or to perform cleaning of the system (Supplementary Information, Section 4).
4.5. Characterization of trapping and elution efficiency
Characterization of trapping and elution efficiency was performed by taking radioactivity measurements with a calibrated dose calibrator (CRC-25 PET, Capintec, Inc., Ramsey, NJ) during the trapping and elution processes. For the purposes of calculations, all radioactivity measurements were decay-corrected to a common timepoint. Measurements were made of the starting activity in the [18F]fluoride (“source”) vial before trapping (A0source), activity in the source vial after trapping (Asource), activity on the cartridge after trapping (Acartridge), activity in the [18O]H2O recovery vial after trapping (Awaste), and the collected activity after elution (Acollect). Trapping efficiency (%) was computed as Acartridge / (A0source – Asource) x 100%. Elution efficiency (%) was calculated as Acollect / (Acartridge – Asource) x 100%. In early experiments, Acartridge was measured directly, however in later experiments Acartridge was measured indirectly (i.e. calculated as A0source – (Awaste + Asource)) to prevent unnecessary radiation exposure to the operator. Measurements via the two approaches were found to agree within ~0.5% of the starting activity.
4.6. Synthesis of N-boc-5-[18F]fluoroindole
Using concentrated [18F]fluoride to limit water content, we performed the synthesis of N-boc-5-[18F]fluoroindole via nickel-mediated oxidative fluorination (Figure 5A) as reported by Lee et al. (Lee et al., 2012).
Figure 5.
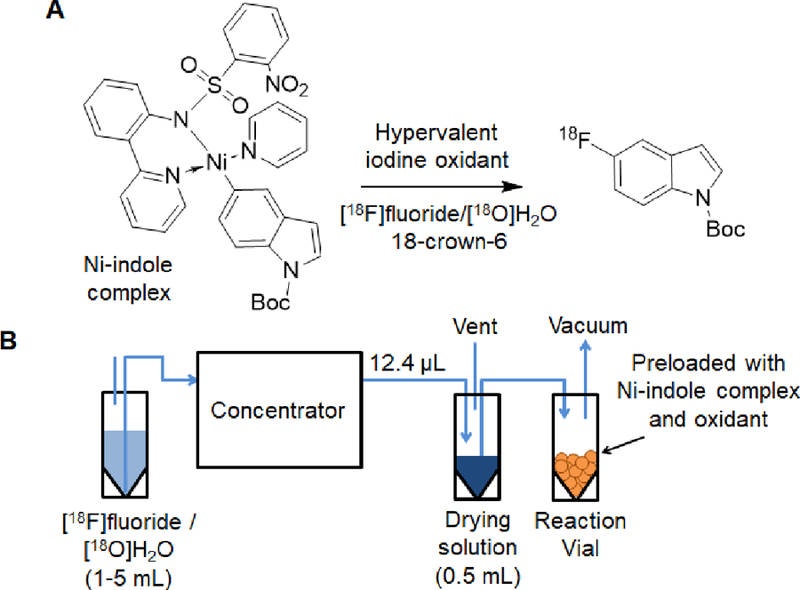
(A) Reaction scheme for synthesis of N-boc-5-[18F]fluoroindole from Ni-indole complex. (B) System configuration for production of N-boc-5-[18F]fluoroindole.
The setup of the experiment is shown in Figure 5B. Concentration of [18F]fluoride was performed using 1 M KH2PO4 for preconditioning the micro-cartridge, and 24 mM K3PO4 + 136 mM 18-crown-6 in a 20:80 v/v mixture of DI water and MeCN for elution. The eluted solution had a volume of 12.4 µL (two elution plugs). The concentrator output was connected into a 3 mL v-vial (Wheaton) containing 500 µL of a “drying” solution (salt in MeCN). The concentrated [18F]fluoride was thus “dried” by dilution, resulting in a [18F]fluoride solution with a low and well-controlled amount of water. A #23 needle in this vial provided a vent during the transfer of the concentrated [18F]fluoride. Two drying solutions were tested – one containing 38 mM 18-crown-6 in MeCN (drying solution #1), and the other containing 38 mM 18-crown-6 + 10 mM K3PO4 in 1:400 v/v H2O:MeCN (drying solution #2).
The drying/dilution vial was connected via a dip-tube to a second “reaction” vial. This vial was pre-filled with a 2:3 mixture of the Ni-indole complex (1.0 – 1.3 mg) and hypervalent iodine oxidant (1.3 – 1.6 mg) prepared under argon prior to each experiment. The dried [18F]fluoride was transferred by applying vacuum to the headspace of the second vial, and the resulting mixture was then allowed to react for 1 min. Determination of water content in the reaction mixture for synthesis of N-boc-5-[18F]fluoroindole was performed using Karl Fischer titration. Detailed description of the equipment and methods used can be found in Supplemental Information, Section 6.
The crude product was then analyzed by radio thin layer chromatography (radio-TLC) for determination of radiochemical conversion (RCC). A 1 μL droplet of the crude product was spotted on a silica TLC plate (JT4449–2, J.T. Baker, Center Valley, PA, USA) with a micropipette. The TLC plate was developed in 10% v/v ethyl acetate in hexane and then analyzed with a radio-TLC reader (MiniGITA Star, Raytest, Germany). The chromatograms contained two peaks (Supplemental Figure S8): [18F] fluoride (Rf = 0.0) and N-boc-5-[18F]fluoroindole (Rf = 0.58). The RCC of N-boc-5-[18F]fluoroindole was calculated as the area under the N-boc-5-[18F]fluoroindole peak divided by the area under both peaks.
5. Results and Discussion
5.1. Duration of concentration process
The times required to complete each of the main operations (i.e. preconditioning, trapping, and elution) were measured for the intermediate vial and direct loading systems were measured using the procedure described in the Supplemental Information, Section 7. Results for each configuration are summarized in Supplemental Tables 2 and 3, respectively. These experiments were performed with 0.5 mL of water as a mock [18F]fluoride solution.
For the intermediate vial system, the preconditioning process took 355 ± 3 s (n= 3), trapping took 134 ± 6 s (n=3), and elution took 137 ± 1 s (n=3) for two elution plugs. In total, the entire concentration process for the intermediate vial system took 625 ± 9 s (n=3). Of the preconditioning time, 119 ± 4 s (n=3) was spent rinsing the intermediate vial to eliminate residue of the preconditioning solution.
The direct loading system reduces the overall concentration time as certain steps, including rinsing of the intermediate vial and transferring from reagent reservoirs to intermediate vial, are not needed. The total time in this case was ~3 min shorter, i.e. 452 ± 4 s (n=3).
Likely, one would eventually use micro-cartridges that are pre-conditioned at the manufacturer, or one would perform the preconditioning ahead of time (i.e. before addition of [18F]fluoride solution). In such a case, the total time for concentration after adding the [18F]fluoride solution would be 271 ± 7 s (n=3) and 250 ± 7 s (n=3) for the intermediate vial and direct loading systems, respectively.
5.2. Trapping efficiency
The efficiency of trapping [18F]fluoride was assessed for several pre-conditioning solutions (KHCO3, KH2PO4, and NaCl) using the intermediate vial system. KHCO3 is commonly used in conjunction with K2CO3 / Kryptofix 2.2.2 or KHCO3 / Kryptofix 2.2.2 as an eluent. KH2PO4, in conjunction with K3PO4 / 18-crown-6 has been shown to be useful for metal-mediated fluorination reactions where certain precursors unfavorably react with the amine functionality found in Kryptofix 2.2.2 (Kamlet et al., 2013). Use of NaCl as both preconditioning solution and eluent has been demonstrated in isotopic exchange reactions (Liu et al., 2015), where it helps to simplify the purification and quality control processes since NaCl is injectable, though introduction of chloride ion can interfere with nucleophilic fluorination reactions.
Since flow rates of reagents through the cartridge determine how long the solutes have to interact with the resin within the cartridge, flow rates were set to 1 mL/min, which is slower than the report of Lebedev et al. (Lebedev et al., 2012) (in which the same cartridges were used) by a safety factor of 2.
Under all conditions tested, at least 94% trapping efficiency was observed (Table 1). Of the three preconditioning solutions tested, KHCO3 and KH2PO4 resulted in the highest trapping efficiencies of 99 ± 1% (n=13) and 96 ± 4% (n=16), respectively. Of the anions used for preconditioning, Cl− has the highest affinity for the resin, while HCO3− and H2PO4− are lower (“AG® MP-1M Anion Exchange Resins | Process Separations | Bio-Rad,” n.d.), explaining the higher displacement by [18F]fluoride (and thus higher trapping efficiency) for these latter anions.
5.3. Effect of initial volume of radionuclide solution
We anticipated that the starting volumes of radioactivity for a downstream radiosynthesis may vary (e.g. preparing multiple tracers from a single master batch of [18F]fluoride), therefore we explored the effect of the volume of [18F]fluoride solution in the source vial on trapping efficiency. We hypothesized that there are some dead volumes associated with the tubing interface into the source vial, and that losses would become more significant as starting volume was reduced, resulting in lower apparent trapping efficiencies. For example, the liquid could become distributed on the vial surface, on tubing, and inside valves before reaching the cartridge valve.
Trapping efficiencies for various starting volumes, using the direct loading system, are summarized in Figure 6. Pre-conditioning was performed with 1 M NaCl. The volume can be scaled down quite far without adverse effect on the trapping efficiency. For volumes ranging from 1.0 down to 0.125 mL, activity lost within the system (i.e. activity not in [18F]fluoride vial, trapped on cartridge, or in cartridge waste vial) is low (< 2%). For starting volumes of 0.06, 0.03, and 0.01 mL, losses increase dramatically to 5.0 ± 2.9% (n = 3), 15.0 ± 4.6% (n=3) and 44.0 ± 5.3% (n=3), respectively.
Figure 6.
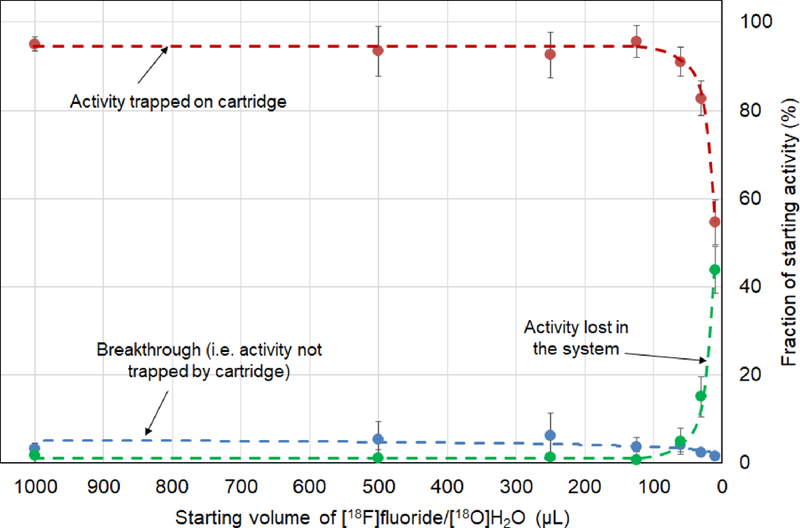
Trapping efficiency on the cartridge as a function of the starting volume of [18F]fluoride. Also shown is the “breakthrough”, i.e. the fraction of initial [18F]fluoride found in the cartridge waste as well as fraction of activity lost within the system (i.e. not in radionuclide vial, trapped in cartridge, or in cartridge waste). Data points represent an average of 3 repeats and error bars represent the standard deviation. Dashed lines are guides for the eye.
These results suggest at least 0.06 mL should be used in the source vial to ensure efficient overall operation of the concentrator. To accommodate smaller volumes, one could always dilute the [18F]fluoride source with DI water to increase the volume into this range (at the expense of requiring more time for trapping), or potentially could rinse the full fluid path with DI water (through the cartridge) after trapping with little effect on overall duration or system complexity.
5.4. Effect of number of eluent plugs
Recovery of trapped [18F]fluoride from strong anion exchange cartridges has been shown to be more efficient when eluted with multiple smaller elution plugs rather than one larger single plug (Lebedev et al., 2012). With the intermediate vial system, we explored the influence of the number of eluent plugs on elution efficiency in a set of experiments using 1 M NaCl for preconditioning and 0.15 M NaCl for elution. In Table 3, we observe that only a small fraction of the activity (21.9 ± 2.6%, n=3) is recovered with one elution rinse. The cumulative amount recovered by two rinses was 88.4 ± 1.3 % (n=3). An additional 2 rinses recovered an additional 10.1 ± 2.9% (n=3) of the initial amount, and further rinses recovered negligible amounts of additional activity. To ensure the highest concentration and lowest water content, we used 2 elution steps for most experiments.
Table 3:
Performance as a function of number of eluent and rinse plugs. Each plug is 6.2µL in volume. The eluent solution and rinse solution were 0.15 M NaCl and MeCN, respectively. Trapping data represents decay-corrected fraction of starting activity trapped on cartridge ± standard deviation (n=3). Elution data represents decay-corrected fraction of trapped activity ± standard deviation (n=3).
| Step in concentration process | Radioactivity measurement (% of trapped activity, decay-corrected) |
||
|---|---|---|---|
| Protocol 1: 6x eluent plugs | Protocol 2: 2x eluent plugs, 4x MeCN plugs | Protocol 3: 1x eluent plug, 5x MeCN plugs | |
| After trapping | 95.6 ± 3.5 | 91.8 ± 1.0 | 92.6 ± 1.3 |
| Elution #1 | Not measured | Not measured | 21.9 ± 2.6 |
| Elution #2 | Not measured | Not measured | 51.6 ± 8.1 |
| Elutions #1, #2 (combined) | 88.4 ± 1.3 | 87.6 ± 3.3 | 73.6 ± 5.6 |
| Elutions #3, #4 (combined) | 10.1 ± 2.9 | 6.1 ± 1.4 | 5.5 ± 1.9 |
| Elutions #5, #6 (combined) | 1.0 ± 0.4 | 0.3 ± 0.1 | 0.8 ± 0.2 |
| Total eluate collected | 99.5 ± 2.1 | 94.0 ± 1.9 | 79.9 ± 3.7 |
We were curious as to why the recovery was so low for a single elution plug. Since other reports had shown highly efficient recovery in 5 µL (Elizarov et al., 2010) (lower than the volume of one elution plug in our setup), we hypothesized that the majority of activity may successfully be released from the cartridge with a single elution plug but is lost between the cartridge and the system output. In order to explore this hypothesis, we trapped [18F]fluoride, then performed one rinse with eluent solution, followed by multiple rinses with MeCN and measured the activity recovered at each step. To minimize carryover of eluent solution, paths in the eluent metering subsystem, with the exception of the tubing connecting the eluent valve to the cartridge valve were rinsed with MeCN three times prior to filling the eluent loop with MeCN. It was not possible to rinse the tubing between the eluent metering valve and cartridge valve, but residual eluent solution was expected to be negligible in this region. Results are summarized in Table 3. Indeed, by following the elution rinse with just one MeCN rinse improved the recovery from 21.9 ± 2.6% (n=3), to 73.6 ± 5.6% (n=3). Though this is still less than the amount recovered with 2 eluent rinses (i.e. 88.4 ± 1.3% (n=3)), this result strongly supports the hypothesis.
Next, we compared the amount recovered using 1 eluent plug followed by 5 MeCN rinses, 2 eluent plugs followed by 4 MeCN rinses, and 6 eluent plugs and found recoveries of 79.9 ± 3.7% (n=3), 94.0 ± 1.9% (n=3), and 99.5 ± 2.1% (n=3), respectively. It can be seen in the second and third cases that additional eluent plugs help to further release residual fluoride from the cartridge, and thus that the overall efficiency is related to both release of fluoride from the resin as well as flushing this fluoride through the fluid pathway to the output tubing.
5.5. Elution efficiency
Elution efficiency was explored for several different eluent solutions using the intermediate vial system and two elution plugs (12.4 µL total volume). Results are summarized in Table 2. Recovery was found to be >88% under all conditions tested.
With other conditions constant, we expected that elution efficiency would depend on the anion strength and concentration of the eluent solution as well as the amount of the anion present. Indeed, affinity of Br– and Cl– anions to the cartridge are high, and elution efficiencies with the corresponding eluents (TBAB and NaCl, respectively) were very high, i.e. 92 ± 8% (n=2) and 96 ± 5% (n=10), respectively. Relative strengths of CO32− and PO43− to the cartridge were not provided by the manufacturer. In addition, we observed that increasing amount of K3PO4 in the eluent leads to increasing recovery of [18F]fluoride. For eluent containing 0.01 M, 0.18 M and 1.19 M K3PO4, the elution efficiencies were 88, 90, and 100%, respectively.
5.6. Elution with organic solvent containing eluent
We showed above that one could elute with a single eluent plug (6.2 µL water) followed by organic solvent (6.2 µL MeCN) rinse instead of two eluent plugs, as long as one is willing to tolerate the ~20% loss in recovered activity (i.e. 73.6 ± 5.6% (n=3) versus 88.4 ± 1.3 % (n=3)). To further reduce water content, e.g. to avoid the need for azeotropic drying, we explored the possibility of introducing portions of organic solvents into the eluent solution itself to further reduce water content. (Another approach would be to reduce the volume of each eluent plug, but for practical reasons, it was difficult to reduce the volume of the eluent loop.) Since it is known that decreasing water content decreases elution efficieny (Lemaire et al., 2010), we explored the impact in our system to determine the lower limit of water content. Experiments were performed with two eluent plugs (12.4 µL total) containing 0.02 M K3PO4 and 0.14–0.15 M 18-crown-6 in various mixtures of DI water and MeCN. (The same eluent is used later in a fluorination reaction.) Preconditioning solution used was 1 M KH2PO4.
Results of these experiments are shown in Table 4. Even with 50% (v/v) MeCN content, elution efficiency was high, i.e. 96% (n=1), but as MeCN content further increased, recovery diminished further. At 80% MeCN, recovery using 2 eluent plugs was 84 ± 6% (n=6), suggesting that a further ~60% reduction in water content is possible if one is willing to tolerate a ~12% loss in elution efficiency.
5.7. Synthesis of N-boc-5-[18F]fluoroindole
As a proof of concept of using the concentrator to reduce water content for radiofluorination, we explored the synthesis of a model compound, N-boc-5-[18F]fluoroindole. This reaction has previously been performed without azeotropic drying, but because the amount of water was limited to ≤1% v/v, this greatly limited the amount of starting activity of [18F]fluoride/[18O]H2O that could be used in the reaction (Lee et al., 2012).
Results are summarized in Table 5. Starting activities ranged from 41 – 122 MBq [1.1 – 3.3 mCi]. Two pairs of reactions were carried out, each pair using a different solution for “drying” the concentrated [18F]fluoride by dilution. The final water content of the reaction mixtures for drying solution 1 and drying solution 2 are estimated to be 0.48% v/v and 0.73% v/v, respectively. Interestingly, the water contents as measured via Karl Fischer titration were found to be slightly lower, i.e. 0.32 ± 0.02% v/v (n = 2) and 0.57 ± 0.01% v/v (n = 2), respectively, suggesting that transfer of the eluent solution through the cartridge may pick up some residual MeCN remaining after the cartridge is rinsed following the [18F]fluoride trapping step. Radiochemical conversions for the two pairs of experiments were found to be 51% ± 2% (n=2) and 50% ± 4% (n=2), respectively. The results were nearly identical despite the higher amount of K3PO4 and water in the second pair of experiments. Notably, the yields were comparable to those reported by Lee et al. (53 ± 7%, n = 6) using 18-crown-6 in 100% MeCN (no salts) as the drying solution (Lee et al., 2012).
This proof of concept experiment suggests that [18F]fluoride concentrated within our platform can be used to increase the activity scale of nickel-mediated oxidative fluorination reactions. The activity levels used in experiments by Lee et al. were low, i.e. 3.7 – 18.5 MBq [100–500 µCi] per reaction, due to the ≤1% v/v limit in the amount of water (2–5 µL) that could be added to the reaction volume (0.2 – 0.5 mL) (Lee et al., 2012). Even by using more concentrated [18F]fluoride (e.g. 37 GBq/mL [1.0 Ci/mL] is routinely available from cyclotrons), the maximum starting activity would have been 185 MBq [5.0 mCi], making it impractical to produce a clinically-relevant dose (~370 MBq [~10 mCi]), especially after accounting for losses during reaction (~50% conversion) and purification/formulation. Notably, by using these small volumes of [18F]fluoride out of 99.5% of the initial radioactivity would have been wasted. By using the [18F]fluoride concentrator, we were able to boost activity levels by 10x compared to the report of Lee et al., and further increase in output could be achieved by concentrating a larger amount of initial activity. In fact, we have previously demonstrated the ability to concentrate ~63 GBq [1.7 Ci] of activity down to ~12.4 µL with 94.3% (n=1) efficiency (Hoover et al., 2016). 5 µL of this solution (identical to the volume used by Lee et al.) would contain ~24 GBq [650 mCi], sufficient for a larger number of human doses, even after accounting for losses during the fluorination reaction and subsequent processing.
5.8. Comparison of operational differences between concentration system architectures
Overall, the intermediate vial and direct loading systems functioned similarly, but the direct loading system was slightly simpler and faster (since it was not necessary to perform the rinsing of the intermediate vial. It should be appreciated, however, that the reagent loading method is independent of the system setup: pre-metering could be used in conjunction with the intermediate vial system, or a time or pressure-based calibration could be used in the direct loading system, if desired.
5.9. Concentration of other radionuclides
In addition to concentrating [18F]fluoride, this system would likely be useful for concentrating other radionuclides. For example, researchers often use radiometals, such as Cu-64, Ga-68, and Zr-89, for labeling of peptides and antibodies. Ga-68 is recovered from a generator in volumes of several mL and the output of the generator decreases over time requiring larger volumes of eluent (HCl) to collect the desired amount of activity. These concentrations are not only too dilute for microscale synthesis but may also present a challenge for macroscale synthesis of clinical doses. Several groups have developed techniques to minimize the volume (e.g. using only the initial fraction), or to concentrate the Ga-68 after recovery. Gebhardt et al. described a QMA-cartridge-based method that reduces volumes from 3.5 mL to 0.2 mL in 15 min with ~67% overall recovery (Gebhardt et al., 2010). Potentially, using our setup with a micro-QMA cartridge, this final volume could be reduced to ~12.4 µL and the time reduced to ~3 min. Zr-89 is produced in a cyclotron and is typically recovered in oxalic acid after a process to separate Zr-89 from the Y-89 target material (Holland et al., 2009; Lin et al., 2016). Due to the toxicity of oxalic acid, several groups have presented a method to convert [89Zr]Zr-oxalate to [89Zr]ZrCl2 prior to chelation through the use of a QMA cartridge followed by elution with 300–500 µL of saline or HCl (Holland et al., 2009; Lin et al., 2016). Using the micro-QMA cartridge in our system offers the potential to further shrink this volume and time required. Similar to microfluidic advancements for [18F]fluoride chemistry, several groups have also turned to leveraging microfluidic technologies for labeling radiometals (Causey et al., 2009; Wright et al., 2016; Zeng et al., 2013). Zeng et al. has demonstrated that increasing starting radiometal amounts (e.g. 64Cu2+, 68Ga3+) in microfluidic radiosynthesizers results in increased radiolabeling yields (Zeng et al., 2013). The ability to concentrate radiometals, therefore, not only enables the loading of more radioactivity into an experiment but could also enable higher synthesis yields. In the near future we hope to explore concentration of these other radionuclides within our platform.
6. Conclusions
In this paper, we have developed, optimized and automated a compact microfluidic platform to concentrate radionuclides such as [18F]fluoride into microliter-scale volumes. The standalone system can easily fit into a hot cell or mini-cell along with other equipment. It can be easily integrated with various types of radiosynthesis platforms (e.g. microfluidic droplet based systems, microfluidic flow-through systems, and macroscale systems). The system has applications in microfluidic radiochemistry, enabling the delivery of high amounts of activity into small-volume microreaction devices, e.g. based on droplet radiochemistry (Keng and van Dam, 2015; Wang et al., 2017), an area of active investigation in our laboratory.
It also has valuable applications in macroscale radiochemistry, such as enabling quick “drying” of [18F]fluoride simply by the reduction of water volume followed by dilution in an anhydrous reaction solvent. An application of the latter was demonstrated: using ≤1% v/v water content in the reaction mixture. Radiochemical conversion of N-boc-5-[18F]fluoroindole was similar to that reported in literature, but with the advantage of being able to introduce orders of magnitude higher quantities of [18F]fluoride into a single reaction. For chemistries relying on such an approach to reduce water content, the concentrator will facilitate the production of clinically relevant amounts of tracers. Furthermore, low eluent volumes used in the system can enable significant reduction in eluent salts/base that are carried into the downstream reaction, potentially providing a way to improve the performance of base-sensitive reactions.
Reliable concentration of [18F]fluoride was performed starting with 60 – 1000 µL volumes, but even larger volumes (e.g. a full cyclotron target volume, i.e. 1–5 mL) could readily be used if a longer trapping time could be tolerated. Indeed, concentration of ~ 63 GBq [1.7 Ci] with a prototype version of the system described here has been reported (Hoover et al., 2016). The entire concentration process can be completed in 452 ± 4 s (n=3) using the direct loading system. If certain steps (e.g. preconditioning) are performed in advance, then the trap and release process only requires 250 ± 7 s (n=3).
Different preconditioning solutions were tested resulting in 94–99% trapping efficiencies, and different aqueous eluent solutions resulted in 85–99% elution efficiencies. We also explored the relationship of recovered activity and number of eluent plugs and identified that two elution plugs (12.4 µL total volume) provides an excellent tradeoff between overall efficiency and final output volume. Water content could be reduced by replacing the second eluent plug with MeCN or by diluting the eluent solution in a solvent / DI water mixture (e.g. up to 80% v/v MeCN in DI water).
This standalone automated concentrator enables fast, reliable concentration of [18F]fluoride enabling high starting activities, low water and salt content, leading to efficient fluorination of PET tracers. With the possibility of concentrating radiometals, the benefits of this system can be further extended for peptide- and antibody- based PET imaging.
Supplementary Material
Figure 4.

Fluidic and pneumatic diagram for the complete direct loading system (left), and a photograph of the direct loading system (right)
8. Acknowledgments
The authors thank Dr. Saman Sadeghi, Dr. Umesh Gangadharmath, and the staff of the UCLA Biomedical Cyclotron Facility for providing [18F]fluoride for these studies. The authors also thank Jeffery Collins for helpful discussions and for providing assistance with concentration experiments. The authors also thank the Harran Lab for allowing us to use their equipment to perform Karl Fischer titrations. This work was supported in part by the Department of Energy Office of Biological and Environmental Research (DE-SC0005056, DE-SC00001249), the National Institute on Aging (R21 AG049918), the National Institute of Mental Health (R44MH097271), and the National Cancer Institute (R21 CA212718, U54 CA151819).
Footnotes
Disclosure statement
This work was funded in part by a subcontract of an SBIR awarded to Sofie Biosciences, Inc. R.M. van Dam is a founder of, and consultant for, Sofie Biosciences, Inc.
10 References
- AG® MP-1M Anion Exchange Resins | Process Separations | Bio-Rad [WWW Document], n.d URL http://www.bio-rad.com/en-ch/product/ag-mp-1m-anion-exchange-resins (accessed 12.5.17).
- Berridge MS, Apana SM, Hersh JM, 2009. Teflon radiolysis as the major source of carrier in fluorine-18. J. Label. Compd. Radiopharm 52, 543–548. 10.1002/jlcr.1672 [DOI] [Google Scholar]
- Causey P, Stephenson K, Valliant J, 2009. A microfluidic approach to the preparation of radiometal-based probes. J. Nucl. Med 50, 148–148.19091886 [Google Scholar]
- Chen S, Javed MR, Kim H-K, Lei J, Lazari M, Shah GJ, Dam M van, Keng PY, Kim C-J, 2014. Radiolabelling diverse positron emission tomography (PET) tracers using a single digital microfluidic reactor chip. Lab. Chip 14, 902–910. 10.1039/C3LC51195B [DOI] [PubMed] [Google Scholar]
- Chryssikos T, Parvizi J, Ghanem E, Newberg A, Zhuang H, Alavi A, 2008. FDG-PET Imaging Can Diagnose Periprosthetic Infection of the Hip. Clin. Orthop 466, 1338–1342. 10.1007/s11999-008-0237-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leonardis F, Pascali G, Salvadori PA, Watts P, Pamme N, 2011. On-chip pre-concentration and complexation of [18F]fluoride ions via regenerable anion exchange particles for radiochemical synthesis of Positron Emission Tomography tracers. J. Chromatogr. A 1218, 4714–4719. 10.1016/j.chroma.2011.05.062 [DOI] [PubMed] [Google Scholar]
- Elizarov AM, van Dam RM, Shin YS, Kolb HC, Padgett HC, Stout D, Shu J, Huang J, Daridon A, Heath JR, 2010. Design and Optimization of Coin-Shaped Microreactor Chips for PET Radiopharmaceutical Synthesis. J Nucl Med 51, 282–287. 10.2967/jnumed.109.065946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambhir SS, 2002. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2, 683–693. 10.1038/nrc882 [DOI] [PubMed] [Google Scholar]
- Gebhardt P, Opfermann T, Saluz HP, 2010. Computer controlled 68Ga milking and concentration system. Appl. Radiat. Isot 68, 1057–1059. [DOI] [PubMed] [Google Scholar]
- Glaudemans AWJM, de Vries EFJ, Galli F, Dierckx RAJO, Slart RHJA, Signore A, 2013. The Use of 18F-FDG-PET/CT for Diagnosis and Treatment Monitoring of Inflammatory and Infectious Diseases. Clin. Dev. Immunol 2013, 1–14. 10.1155/2013/623036 [DOI] [PMC free article] [PubMed]
- Guludec DL, Lautamäki R, Knuuti J, Bax JJ, Bengel FM, Cardiology (ECNC), on behalf of the E.C. of N., 2008. Present and future of clinical cardiovascular PET imaging in Europe—a position statement by the European Council of Nuclear Cardiology (ECNC). Eur. J. Nucl. Med. Mol. Imaging 35, 1709–1724. 10.1007/s00259-008-0859-1 [DOI] [PubMed] [Google Scholar]
- Holland JP, Sheh Y, Lewis JS, 2009. Standardized methods for the production of high specific-activity zirconium-89. Nucl. Med. Biol 36, 729–739. 10.1016/j.nucmedbio.2009.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover AJ, Lazari M, Ren H, Narayanam MK, Murphy JM, van Dam RM, Hooker JM, Ritter T, 2016. A Transmetalation Reaction Enables the Synthesis of [18F]5-Fluorouracil from [18F]Fluoride for Human PET Imaging. Organometallics 35, 1008–1014. 10.1021/acs.organomet.6b00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail R, Irribarren J, Javed MR, Machness A, van Dam M, Keng PY, 2014. Cationic imidazolium polymer monoliths for efficient solvent exchange, activation and fluorination on a continuous flow system. RSC Adv 4, 25348–25356. 10.1039/c4ra04064c [DOI] [Google Scholar]
- Iwata R, Pascali C, Terasaki K, Ishikawa Y, Furumoto S, Yanai K, 2018. Practical microscale one‐pot radiosynthesis of 18F‐labeled probes. J. Label. Compd. Radiopharm 10.1002/jlcr.3618 [DOI] [PubMed]
- Iwata R, Pascali C, Terasaki K, Ishikawa Y, Furumoto S, Yanai K, 2017. Minimization of the amount of Kryptofix 222 - KHCO3 for applications to microscale 18F-radiolabeling. Appl. Radiat. Isot 125, 113–118. 10.1016/j.apradiso.2017.04.021 [DOI] [PubMed] [Google Scholar]
- Javed MR, Chen S, Kim H-K, Wei L, Czernin J, Kim C-J“CJ,” Dam RM van, Keng PY, 2014. Efficient Radiosynthesis of 3′-Deoxy-3′-18F-Fluorothymidine Using Electrowetting-on-Dielectric Digital Microfluidic Chip. J. Nucl. Med 55, 321–328. 10.2967/jnumed.113.121053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamlet AS, Neumann CN, Lee E, Carlin SM, Moseley CK, Stephenson N, Hooker JM, Ritter T, 2013. Application of Palladium-Mediated 18F-Fluorination to PET Radiotracer Development: Overcoming Hurdles to Translation. PLoS ONE 8, e59187 10.1371/journal.pone.0059187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keng PY, van Dam RM, 2015. Digital Microfluidics: A New Paradigm for Radiochemistry. Mol. Imaging 14, 579–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitson S, Cuccurullo V, Ciarmiello A, Salvo D, Mansi L, 2009. Clinical Applications of Positron Emission Tomography (PET) Imaging in Medicine: Oncology, Brain Diseases and Cardiology. Curr. Radiopharm 2, 224–253. 10.2174/1874471010902040224 [DOI] [Google Scholar]
- Kitson SL, 2014. Positron Emission Tomography Neuro-Imaging. Neuro 1 10.17140/NOJ-1-102 [DOI]
- Kubota K, 2001. From tumor biology to clinical PET: A review of positron emission tomography (PET) in oncology. Ann. Nucl. Med 15, 471–486. 10.1007/BF02988499 [DOI] [PubMed] [Google Scholar]
- Lebedev A, Miraghaie R, Kotta K, Ball CE, Zhang J, Buchsbaum MS, Kolb HC, Elizarov A, 2012. Batch-reactor microfluidic device: first human use of a microfluidically produced PET radiotracer. Lab. Chip 13, 136–145. 10.1039/C2LC40853H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-C, Sui G, Elizarov A, Shu CJ, Shin Y-S, Dooley AN, Huang J, Daridon A, Wyatt P, Stout D, Kolb HC, Witte ON, Satyamurthy N, Heath JR, Phelps ME, Quake SR, Tseng H-R, 2005. Multistep Synthesis of a Radiolabeled Imaging Probe Using Integrated Microfluidics. Science 310, 1793–1796. 10.1126/science.1118919 [DOI] [PubMed] [Google Scholar]
- Lee E, Hooker JM, Ritter T, 2012. Nickel-Mediated Oxidative Fluorination for PET with Aqueous [18F] Fluoride. J. Am. Chem. Soc 134, 17456–17458. 10.1021/ja3084797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire CF, Aerts JJ, Voccia S, Libert LC, Mercier F, Goblet D, Plenevaux AR, Luxen AJ, 2010. Fast Production of Highly Reactive No-Carrier-Added [18F]Fluoride for the Labeling of Radiopharmaceuticals. Angew. Chem. Int. Ed 49, 3161–3164. 10.1002/anie.200906341 [DOI] [PubMed] [Google Scholar]
- Liang SH, Yokell DL, Normandin MD, Rice PA, Jackson RN, Shoup TM, Brady TJ, El Fakhri G, Collier TL, Vasdev N, 2014. First Human Use of a Radiopharmaceutical Prepared by Continuous-Flow Microfluidic Radiofluorination: Proof of Concept with the Tau Imaging Agent [18F]T807. Mol. Imaging 13, 1–5. https://doi.org/ DOI 10.2310/7290.2014.00025 [DOI] [PubMed] [Google Scholar]
- Lin M, Mukhopadhyay U, Waligorski GJ, Balatoni JA, González-Lepera C, 2016. Semi-automated production of 89Zr-oxalate/89Zr-chloride and the potential of 89Zr-chloride in radiopharmaceutical compounding. Appl. Radiat. Isot 107, 317–322. 10.1016/j.apradiso.2015.11.016 [DOI] [PubMed] [Google Scholar]
- Liu Z, Lin K-S, Bénard F, Pourghiasian M, Kiesewetter DO, Perrin DM, Chen X, 2015. One-step 18F labeling of biomolecules using organotrifluoroborates. Nat. Protoc 10, 1423–1432. 10.1038/nprot.2015.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon BS, Kil HS, Park JH, Kim JS, Park J, Chi DY, Lee BC, Kim SE, 2011. Facile aromatic radiofluorination of [18F]flumazenil from diaryliodonium salts with evaluation of their stability and selectivity. Org. Biomol. Chem 9, 8346–8355. 10.1039/C1OB06277H [DOI] [PubMed] [Google Scholar]
- Newberg AB, Alavi A, 2005. The role of PET imaging in the management of patients with central nervous system disorders. Radiol. Clin. North Am 43, 49–65. [DOI] [PubMed] [Google Scholar]
- Pascali G, Watts P, Salvadori P, 2013. Microfluidics in radiopharmaceutical chemistry. Nucl. Med. Biol 40, 776–787. 10.1016/j.nucmedbio.2013.04.004 [DOI] [PubMed] [Google Scholar]
- Ravina B, Eidelberg D, Ahlskog JE, Albin RL, Brooks DJ, Carbon M, Dhawan V, Feigin A, Fahn S, Guttman M, Gwinn-Hardy K, McFarland H, Innis R, Katz RG, Kieburtz K, Kish SJ, Lange N, Langston JW, Marek K, Morin L, Moy C, Murphy D, Oertel WH, Oliver G, Palesch Y, Powers W, Seibyl J, Sethi KD, Shults CW, Sheehy P, Stoessl AJ, Holloway R, 2005. The role of radiotracer imaging in Parkinson disease. Neurology 64, 208–215. 10.1212/01.WNL.0000149403.14458.7F [DOI] [PubMed] [Google Scholar]
- Rensch C, Jackson A, Lindner S, Salvamoser R, Samper V, Riese S, Bartenstein P, Wängler C, Wängler B, 2013. Microfluidics: A Groundbreaking Technology for PET Tracer Production? Molecules 18, 7930–7956. 10.3390/molecules18077930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensch C, Lindner S, Salvamoser R, Leidner S, Böld C, Samper V, Taylor D, Baller M, Riese S, Bartenstein P, Wängler C, Wängler B, 2014. A solvent resistant lab-on-chip platform for radiochemistry applications. Lab. Chip 14, 2556–2564. 10.1039/C4LC00076E [DOI] [PubMed] [Google Scholar]
- Richarz R, Krapf P, Zarrad F, Urusova EA, Neumaier B, Zlatopolskiy BD, 2014. Neither azeotropic drying, nor base nor other additives: a minimalist approach to 18F-labeling. Org. Biomol. Chem 12, 8094–8099. 10.1039/C4OB01336K [DOI] [PubMed] [Google Scholar]
- Saiki H, Iwata R, Nakanishi H, Wong R, Ishikawa Y, Furumoto S, Yamahara R, Sakamoto K, Ozeki E, 2010. Electrochemical concentration of no-carrier-added [18F]fluoride from [18O]water in a disposable microfluidic cell for radiosynthesis of 18F-labeled radiopharmaceuticals. Appl. Radiat. Isot 68, 1703–1708. 10.1016/j.apradiso.2010.02.005 [DOI] [PubMed] [Google Scholar]
- Salvador B, Luque A, Fernandez-Maza L, Corral A, Orta D, Fernández I, Quero JM, 2017. Disposable PDMS Chip With Integrated [18F]Fluoride Pre-Concentration Cartridge for Radiopharmaceuticals. J. Microelectromechanical Syst 26, 1442–1448. 10.1109/JMEMS.2017.2764121 [DOI] [Google Scholar]
- Schindler TH, Schelbert HR, Quercioli A, Dilsizian V, 2010. Cardiac PET Imaging for the Detection and Monitoring of Coronary Artery Disease and Microvascular Health. JACC Cardiovasc. Imaging 3, 623–640. 10.1016/j.jcmg.2010.04.007 [DOI] [PubMed] [Google Scholar]
- Seo JW, Lee BS, Lee SJ, Oh SJ, Chi DY, 2011. Fast and Easy Drying Method for the Preparation of Activated [18F]Fluoride Using Polymer Cartridge. Bull. Korean Chem. Soc 32, 71–76. 10.5012/bkcs.2011.32.1.71 [DOI] [Google Scholar]
- Sergeev M, Lazari M, Morgia F, Collins J, Javed MR, Sergeeva O, Jones J, Phelps ME, Lee JT, Keng PY, Dam RM van, 2018. Performing radiosynthesis in microvolumes to maximize molar activity of tracers for positron emission tomography. Commun. Chem 1, 10 10.1038/s42004-018-0009-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpe KDM, Dazzi H, Schaffner A, Schulthess G.K. von, 2000. Infection imaging using whole-body FDG-PET. Eur. J. Nucl. Med 27, 822–832. 10.1007/s002590000277 [DOI] [PubMed] [Google Scholar]
- Tarkin JM, Joshi FR, Rudd JHF, 2014. PET imaging of inflammation in atherosclerosis. Nat. Rev. Cardiol 11, 443–457. 10.1038/nrcardio.2014.80 [DOI] [PubMed] [Google Scholar]
- Virdee K, Cumming P, Caprioli D, Jupp B, Rominger A, Aigbirhio FI, Fryer TD, Riss PJ, Dalley JW, 2012. Applications of positron emission tomography in animal models of neurological and neuropsychiatric disorders. Neurosci. Biobehav. Rev 36, 1188–1216. 10.1016/j.neubiorev.2012.01.009 [DOI] [PubMed] [Google Scholar]
- Wang J, Chao PH, Hanet S, Dam R.M. van, 2017. Performing multi-step chemical reactions in microliter-sized droplets by leveraging a simple passive transport mechanism. Lab. Chip 17, 4342–4355. 10.1039/C7LC01009E [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong R, Iwata R, Saiki H, Furumoto S, Ishikawa Y, Ozeki E, 2012. Reactivity of electrochemically concentrated anhydrous [18F]fluoride for microfluidic radiosynthesis of 18F-labeled compounds. Appl. Radiat. Isot 70, 193–199. 10.1016/j.apradiso.2011.09.022 [DOI] [PubMed] [Google Scholar]
- Wright BD, Whittenberg J, Desai A, DiFelice C, Kenis PJA, Lapi SE, Reichert DE, 2016. Microfluidic Preparation of a 89Zr-Labeled Trastuzumab Single-Patient Dose. J. Nucl. Med 57, 747–752. 10.2967/jnumed.115.166140 [DOI] [PubMed] [Google Scholar]
- Zeng D, Desai AV, Ranganathan D, Wheeler TD, Kenis PJA, Reichert DE, 2013. Microfluidic radiolabeling of biomolecules with PET radiometals. Nucl. Med. Biol 40, 42–51. 10.1016/j.nucmedbio.2012.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatopolskiy BD, Zischler J, Krapf P, Zarrad F, Urusova EA, Kordys E, Endepols H, Neumaier B, 2015. Copper-Mediated Aromatic Radiofluorination Revisited: Efficient Production of PET Tracers on a Preparative Scale. Chem. – Eur. J 21, 5972–5979. 10.1002/chem.201405586 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.