

Abstract
Background:
Asymmetric dimethylarginine (ADMA) is an endogenous inhibitor of nitric oxide synthases that limits nitric oxide bioavailability. Dimethylarginine dimethylaminohydrolase-1 (DDAH1) exerts a critical role for ADMA degradation and plays an important role in NO signaling. In the heart, DDAH1 is observed in endothelial cells and in the sarcolemma of cardiomyocytes. While NO signaling is important for cardiac adaptation to stress, DDAH1 impact on cardiomyocyte homeostasis is not clear.
Methods:
Here we used the MerCreMer-LoxP model to specifically disrupt cardiomyocyte DDAH1 expression in adult mice to determine the physiological impact of cardiomyocyte DDAH1 under basal conditions and during hypertrophic stress imposed by transverse aortic constriction (TAC).
Results:
Under control conditions, Cardiomyocyte specific DDAH1 knockout (cDDAH KO) had no detectable effect on plasma ADMA and left ventricular (LV) hypertrophy or function in adult or aging mice. In response to TAC, DDAH1 levels were elevated 2.5 fold in WT mice, which exhibited no change in LV or plasma ADMA content and moderate LV hypertrophy and LV dysfunction. In contrast, cDDAH1 KO mice exposed to TAC showed no increase in LV DDAH1 expression, slightly increased LV tissue ADMA levels, no increase in plasma ADMA, but significantly exacerbated LV hypertrophy, fibrosis, nitrotyrosine production, and LV dysfunction.
Conclusions:
These findings indicate cardiomyocyte DDAH1 activity is dispensable for cardiac function under basal conditions, but plays an important role in attenuating cardiac hypertrophy and ventricular remodeling under stress conditions, possibly through locally confined regulation of subcellular ADMA and NO signaling.
Keywords: ADMA, nitric oxide, ventricular hypertrophy
Introduction
Congestive heart failure (CHF) is a major cause of morbidity and mortality in developed countries and is a major threat to human health worldwide. The common risk factors or causes of CHF include prolonged high blood pressure (chronic hypertension), myocardial infarction (heart attack) due to coronary artery disease, cardiac valve disease, idiopathic cardiomyopathy, myocarditis from inflammation, and congenital heart diseases etc.
Endogenous nitric oxide synthase (NOS) inhibitors Asymmetric dimethylarginine (ADMA) and N-monomethy L-arginine (L-NMMA) compete with L-arginine for NOS binding to attenuate nitric oxide (NO) production [6]. As ADMA is more abundant than L-NMMA, most studies have focused on the physiological or pathological effects of ADMA in various biological or clinical conditions. Increased plasma ADMA is a strong and independent predictor of all-cause mortality in the community, and the strongest predictor of mortality in patients with coronary artery disease or myocardial infarction. Importantly, ADMA levels are strongly associated with CHF [12, 25, 37], and the major risk factors of CHF such as hypertension, coronary disease, cardiomyopathy, and diabetes[2, 24, 25, 31]. In addition, experimental studies have shown that infusion of ADMA results in reduced endothelium-dependent coronary vasodilation in heart failure dogs [8], decreased cardiac output in normal human subjects [1], and hypertension in mice [17, 23].
Dimethylarginine dimethylaminohydrolase-1 (DDAH1) is the critical enzyme for ADMA and L-NMMA degradation, and DDAH1 does not degrade symmetrical dimethylarginine (SDMA). DDAH1 dysfunction by genetic DDAH1 gene deletion or pharmacological DDAH1 inhibition causes ADMA accumulation and decreased NO signaling [23]. In the heart, DDAH1 is distributed in both vascular endothelial cells and cardiomyocytes [7]. While previous studies have demonstrated that cardiomyocyte eNOS expression exerts protection against pressure overload induced ventricular remodeling through maintaining compartmentalized NO signaling in cardiomyocytes, the role of cardiomyocyte DDAH1 in regulating cardiac hypertrophy and heart failure development is unknown. Here we studied the effects of cardiomyocyte specific DDAH1 KO (cDDAH1 KO) on LV hypertrophy and dysfunction in mice under control conditions and after transverse aortic constriction (TAC). We also studied the effect of cDDAH1 KO on LV hypertrophy and function in mice during the aging process. Our data indicates an important role for cardiomyocyte DDAH1 in protecting the heart against TAC-induced ventricular hypertrophy and dysfunction.
Materials and Methods
An extended Materials and Methods section can be found in the online-only Data Supplement.
Animals and Experimental Protocol:
The experimental studies in mice were approved by the Institutional Animal Care and Use Committee at the University of Minnesota.
Generation of inducible cardiomyocyte specific DDAH1 KO mouse strain.
Adult DDAH1flox/flox mice and α-MHCMerCreMer mice [33] were used for generating DDAH1flox/flox/α-MHCMerCreMer and DDAH1flox/flox (Figure 1A). DDAH1flox/flox/α-MHCMerCreMer and DDAH1flox/flox were given 4-hydroxytamoxifen (Sigma) at 20mg/kg per day for 12 intra-peritoneal injections as we previously described [13].
Figure 1. cDDAH1 KO exacerbated TAC-induced cardiac hypertrophy and increase of lung weight in mice.
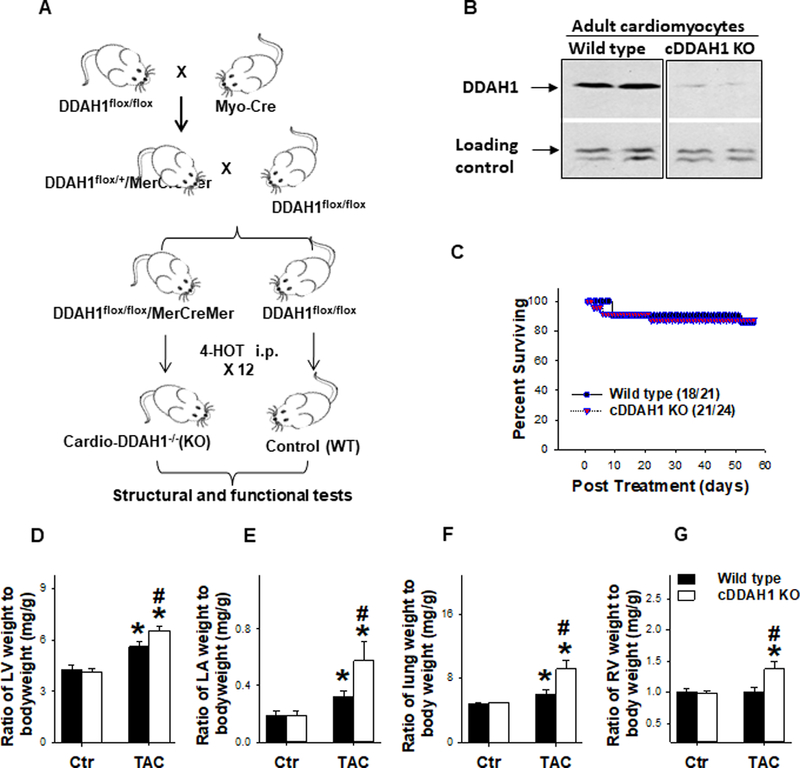
Diagram shows the approach for generation of DDAH1flox/flox-Cre mice (A); Western blot shows reduced DDAH1 expression in isolated adult cardiomyocytes (B); cDDAH1 KO did not affect chronic TAC induced mortality (C); Summarized left ventricular weight (D), left atrial weight (E), lung weight (F), and right ventricular weight (G) in WT and cDDAH1 KO mice (B-F). *p<0.05 compared to before TAC; #p<0.05 compared to the corresponding wild type group.
Measurement of ADMA, symmetrical dimethylarginine, and L-ariginine Content:
Tissue and plasma ADMA, SDMA and L-arginine were measured using a high-throughput liquid chromatographic-tandem mass spectrometric method as previously described [17, 32].
Measurements of LV hemodynamics:
At the end of the study protocol, mice were anesthetized with 2.5% isoflurane. Mice were intubated with a 20-gauge Teflon tube attached to a MiniVent type 845 mouse ventilator (Hugo Sachs Elektronik) (ventilator settings: breathing frequency, 80 breaths per minute; pressures, 9/0 cm H2O; inspiratory/expiratory ratio, 1:1). LV hemodynamics were determined using a 1.2-F pressure catheter (Scisense Inc, Ontario Canada) as previously described.
Histological staining, and evaluation of LV fibrosis:
For histological staining, tissues were sectioned to 5μm. LV sections were stained with Sirius Red (Sigma) for detection of fibrosis and FITC conjugated wheat germ agglutinin (Invitrogen) to evaluate myocyte size. The percent LV fibrosis was determined using method as previously described [33]. For mean myocyte size, the cross-sectional areas of at least 120 cells per sample and at least 5 samples per group were averaged.
Data and Statistical Analysis:
All values were expressed as mean ± standard error. Data from two groups were compared using an unpaired t-test. Two-way ANOVA was used to test for differences between KO and wild type animals under control conditions and after TAC. Post hoc pairwise comparisons were made using the Fisher least significant difference test. Statistical significance was defined as p<0.05.
Results
DDAH1 interacts with eNOS.
While studies have demonstrated that DDAH1 plays a critical role in degrading endogenous NOS inhibitor ADMA, an independent risk factor associated with CHF, hypertension, and coronary heart diseases, the role and mechanism of DDAH1 in regulating myocardial function is largely unknown. DDAH1 is abundantly expressed in areas of vascular endothelial cells, and moderately expressed on the sarcolemma and the intercalated disks on cardiomyocytes (Supplemental Figure 1A). We also observed that DDAH1 predominantly localizes to the sarcolemma of neonatal cardiomyocytes (Supplemental Figure 1B). Since the expression pattern of DDAH1 and eNOS is similar in cardiac tissue (Supplemental Figure 1, 2) [8], we hypotheisized that DDAH1 may localize within the same compartment as eNOS or interact with eNOS to maintain compartmentalized NO signaling in cardiomyocytes. We consequently determined DDAH1 and eNOS interaction by immunoprecipitation of DDAH1 followed by western blot for eNOS in cardiac tissues from pig, mouse and rats. As shown in Figure 1C, eNOS co-immunoprecipitated with DDAH1 (Supplemental Figure 1C). Immunopreciptation of eNOS followed by Western blot for DDAH1 further confirmed the association of eNOS and DDAH1 in cardiac tissue (Supplemental Figure 1D).
Generation of inducible cardiomyocyte specific DDAH1 (cDDAH1) KO mice.
Since DDAH1 binds to eNOS, and since eNOS expressed in cardiomyocyes is important in protection against TAC or infarction induced cardiac remodeling and dysfunction[5, 21, 29], we postulated that DDAH1 distributed in cardiomyocytes may protect the heart against pressure overload induced cardiomyopathy by preserving compartmentalized NO signaling in cardiomyocytes. Consequently, an inducible cardiomyocyte specific DDAH1 KO strain was generated using a strategy illustrated in Figure 1A. Briefly, adult DDAH1flox/flox mice and α-MHCMerCreMer mice [33] were used for generating DDAH1flox/flox/α-MHCMerCreMer and DDAH1flox/flox. DDAH1flox/flox/α-MHC MerCreMer mice have normal cardiac structure and function during unstressed conditions as compared with DDAH1flox/flox mice. DDAH1flox/flox/α-MHCMerCreMer and DDAH1flox/flox mice were given 4-hydroxytamoxifen at 20mg/kg per day for 12 injections. To confirm the efficacy of cardiac specific DDAH1 gene deletion, adult cardiomyocytes were isolated from cardio-DDAH1 KO mice and control wild type mice ~6 weeks after the last injection of 4-hydroxytamoxifen. Western blot demonstrated that DDAH1 expression was reduced over 89± 7% in cardiomyocytes isolated from cDDAH1 KO mice (p<0.05 as compared with wild type group) (Figure 1B).
cDDAH1 KO had no detectable effect on LV structure and function in mice under control conditions.
cDDAH1 KO over 3 months had no detectable effect on LV weight, left atrial (LA) weight, lung weight, right ventricular (RV) weight, right atrial (RA) weight, and their ratios to body weight in both male mice (Figure 1D–G, Table S1). Echocardiography demonstrated that inducible cardiac specific DDAH1 KO had no detectable effect on LV ejection fraction ( 78.0 ± 2.03 % in wild type versus 76.9 ± 1.14% in cDDAH1 KO mice), LV fractional shortening( 40.2 ± 1.83% in wild type versus 38.9 ± 1.05% in cDDAH1 KO mice), LV end systolic diameter ( 2.36 ± 0.14 mm in wild type versus 2.43 ± 0.10 mm in cDDAH1 KO mice), and LV end diastolic diameter (3.91 ± 0.13 mm in wild type versus 3.97 ± 0.11 mm in cDDAH1 KO mice) in male mice as compared with the wild type littermates under control conditions (Figure 2, Table S1). These data indicate that cDDAH1 KO has no significant impact on LV structure and function under unstressed conditions.
Figure 2. cDDAH1 KO exacerbated TAC-induced LV dysfunction in mice.

LV ejection fraction, LV dimensions, LV wall thickness and heart rates were assessed using echocardiography in wild type and cDDAH1 KO mice under control conditions and after 8 weeks TAC in mice. Echocardiography shows the changes in LV end systolic diameter (A), LV end diastolic diameter (B), LV ejection fraction (C), LV fractional shortening (D), and heart rates (E) of wild type and cDDAH1 KO mice *p<0.05 compared to before TAC; #p<0.05 compared KO mice to wild type mice.
Cardiomyocyte specific DDAH1 KO exacerbated TAC-induced cardiac hypertrophy.
eNOS deficiency does not cause LV dysfunction or cardiac hypertrophy under basal conditions, but does exacerbate cardiac hypertrophy and dysfunction in response to aortic banding[21]. Therefore, we further studied the effect of cDDAH1 KO on LV hypertrophy and dysfunction in male mice exposed to pressure overload produced by 8 weeks TAC. Chronic TAC causes LV hypertrophy and dysfunction. LV dysfunction causes an increase in LV diastolic pressure that leads to LA hypertrophy, lung remodeling and ultimately right ventricular hypertrophy or dysfunction[7, 26]. Therefore, TAC-induced CHF can be objectively evaluated by the progressive development of LA hypertrophy, increase of lung weight, and RV hypertrophy [7]. TAC-induced mortality rate was similar in wild type and cDDAH1 KO mice (Figure 1C). Interestingly, cDDAH1 KO significantly exacerbated TAC-induced LV hypertrophy, LA hypertrophy, lung congestion and RV hypertrophy, as indicated by significant more increases of the ratio of LV weight to bodyweight (5.62 ± 0.27 mg/g in wild type versus 6.56 ± 0.29 mg/g in cDDAH1 KO mice, p<0.05), the ratio of LA weight to bodyweight (0.32 ± 0.04 mg/g in wild type versus 0.58 ± 0.13 mg/g in cDDAH1 KO mice, p<0.05), and the ratio of lung weight to bodyweight (6.05 ± 0.49 mg/g in wild type versus 9.16 ± 1.13 mg/g in cDDAH1 KO mice) (Figure 1D–F, Table S1). TAC for 8 weeks also caused a greater increase of the ratio of RV weight to bodyweight (1.00 ± 0.74 mg/g in wild type versus 1.37 ± 0.12 mg/g in cDDAH1 KO mice, p<0.05) in cDDAH1 KO mice as compared with wild type mice (Figure 1G, Table S1). The ratios of LA weight, lung weight and RV weight to tibial length were also significantly increased in in cDDAH1 KO mice as compared with wild type mice (Supplemental Figure 3). It should be mentioned that our pilot study in a small number of female mice also showed that cDDAH1 KO tended to exacerbate TAC-induced LV hypertrophy and dysfunction (data not shown).
cDDAH1 KO exacerbated TAC-induced LV dysfunction, cardiomyocyte hypertrophy, and LV fibrosis in mice:
Echocardiography showed that TAC caused significant declines in LV ejection fraction and LV fractional shortening in both cDDAH1 KO and wild type mice as compared with corresponding control groups, while cDDAH1 KO mice showed significantly greater reduction of LV ejection fraction ( 55.8 ± 3.25% in wild type versus 45.0 ±2.00% in cDDAH1 KO mice, p<0.05) and LV fractional shortening (24.6 ± 2.04% in wild type versus 18.3 ± 0.99% in cDDAH1 KO mice, p<0.05) in comparing with wild type control littermates (Figure 2, Table S2). TAC caused significant increase of LV end systolic diameter and end LV diastolic diameter in both wild type and cDDAH1 KO mice, but cDDAH1 KO mice exhibited significantly greater LV end systolic diameter (3.52 ± 0.18 mm in wild type versus 3.93 ± 0.14 mm in cDDAH1 KO mice, p<0.05) (Figure 2A, Table S2). cDDAH1 KO did not exacerbate TAC-induced increase of LV end diastolic diameter (4.65 ± 0.15 mm in wild type versus 4.80 ± 0.14 mm in cDDAH1 KO mice) (Figure 2B, Table S2).
Histological examination demonstrated that cDDAH1 KO had no impact on LV cardiomyocyte size or LV fibrosis under control conditions (Figure 3A, B). TAC caused a significant increase of LV fibrosis, perivascular fibrosis and cardiomyocyte hypertrophy in both wild type and cDDAH1 KO mice (data not shown). The TAC-induced increases in LV fibrosis and cardiomyocyte cross-sectional area were significantly greater in cDDAH1 KO hearts (Figure 3C,D).
Figure 3. Inducible cDDAH1 KO exacerbated TAC-induced cardiomyocyte hypertrophy and ventricular fibrosis.

cDDAH1 KO did not affect LV fibrosis under control conditions, but exacerbated TAC-induced LV fibrosis (A,B); cDDAH1 KO significantly exacerbated TAC-induced increase of cardiomyocte size (C,D) in male mice. *p<0.05 compared to before TAC; #p<0.05 compared KO mice to wild type mice.
cDDAH1 KO exacerbated TAC-induced increases of ANP and 3’-nitrotyrosine in mice:
Western blots were performed to examine expression of overall LV DDAH1, eNOS, as well as atrial natriuretic peptide (ANP) and 3’-nitrotyrosine, two biochemical markers often associated with LV hypertrophy and dysfunction. Interestingly, cDDAH1 KO caused no detectable change of the overall LV DDAH1 expression under control conditions, suggesting a potential compensatory increase of endothelial DDAH1 expression in this strain. TAC caused a 2.6-fold increase of LV DDAH1 in wild type mice, and the TAC-induced DDAH1 induction was abolished in cDDAH1 KO mice. cDDAH1 KO also caused 36% compensatory increase of LV eNOS content under control condition which was maintained after TAC (Figure 4A, C, p<0.05). Under control conditions, cDDAH1 KO did not significantly affect LV ANP and 3’-nitrotyrosine levels (Figure 4A–E), but these stress markers were significantly elevated in cDDAH1 KO hearts exposed to TAC as compared to WT hearts(Figure 4A,D,E). eNOS expression was equivalent in WT and cDDAH KO mice exposed to TAC(Figure 4B).
Figure 4. cDDAH1 KO exacerbated TAC-induced changes in myocardial ANP and 3’-nitrotyrosine expression.

Tissue was collected from WT and cDDAH1 KO mice 8 weeks after TAC or control conditions, and lysates were examined by western blot for expression of DDAH1 (A,B), eNOS (A,C), atrial natrurietic peptide (ANP) (A,D), and 3’nitrotyrosine (A,D). alpha-actine was used as a loading control. * indicates p<0.05 comparing TAC to control. # indicates p<0.05 comparing WT to cDDAH1 KO.
cDDAH1 KO did not affect plasma ADMA content but caused a significant increase of LV ADMA content after TAC:
We found cDDAH1 KO had no detectable effect on plasma ADMA content under control conditions and after 8 weeks TAC (Figure 5A). While cDDAH1 KO had no detectable effect on plasma SDMA under control conditions as anticipated, to our surprise, plasma SDMA content was significantly increased in both wild type and cDDAH1 KO mice after TAC (Figure 5B). TAC had no effect on LV tissue ADMA content, but significantly increased LV ADMA content in cDDAH1 KO mice (Figure 5C). TAC caused a significant increase of LV SDMA content in cDDAH1 KO mice (Figure 5D). TAC did not affect LV L-arginine content in wild type and cDDAH1 KO mice (Figure 5E).
Figure 5. Inducible cDDAH1 KO did not affect systemic ADMA content but increased LV ADMA content after TAC.

cDDAH1 KO did not affect plasma ADMA (A), and plasma SDMA (B) under control conditions or after TAC. TAC caused significant increase of LV ADMA content (C), LV SDMA (D), and LV L-arginine (E) content in cDDAH1 KO mice. * indicates p<0.05 comparing TAC to control. # indicates p<0.05 comparing WT to cDDAH1 KO.
cDDAH1 KO had no effect on LV hypertrophy and function in middle aged mice under control conditions.
To determine whether cDDAH KO also exerts protection against age-related LV hypertrophy and dysfunction, we measured cardiac hypertrophy and function in both middle aged cDDAH1 KO and wild type littermate. The results demonstrated that cDDAH1 KO had no detectable effect on the ratio of LV weight to bodyweight (3.72 ± 0.18 mg/g in wild type versus 3.77 ± 0.18 mg/g in cDDAH1 KO mice), the ratio of LA weight to bodyweight (0.12 ± 0.02 mg/g in wild type versus 0.14 ± 0.02 mg/g in cDDAH1 KO mice), the ratio of RV weight to bodyweight (0.90 ± 0.03 mg/g in wild type versus 0.95 ± 0.03 mg/g in cDDAH1 KO mice) in male mice at age 12–14 months (Figure 6A–E, Table S3). Similar results were also observed in female mice at age of 13–14 months (Table S4). cDDAH1 KO also had no detectable effects on lung weight, liver weight, kidney weight, and spleen weight, as well as their ratios to bodyweight in both male and female mice (Table S3,S4).
Figure 6. Inducible cDDAH1 KO did not affect LV structure and function in middle age mice.

Summarized heart weight, left ventricular weight, left atrial weight, lung weight, and right ventricular weight (A-E) in male cDDAH1 KO mice and wild type littermates at age over 12 months old. LV pressure, aortic pressure, LV contractility, LV ejection fraction, LV dimensions and wall thickness were unaffected in cDDAH1 KO mice at age over 12 months (F-O).
Echocardiography demonstrated that cDDAH1 KO had no detectable effect on LV ejection fraction (78.4 ± 1.65 % in wild type versus 79.9 ±1.56% in cDDAH1 KO mice), LV fractional shortening (40.2 ± 1.55 % in wild type versus 41.6 ± 1.61% in cDDAH1 KO mice), LV end diastolic diameter, and LV end systolic diameter in these middle aged male mice (Figure 6, Table S6). LV hemodynamics showed that cardio-DDAH1 KO caused no changes of mean aortic pressure, LV dP/dtmax and LV dP/dtmin in these middle aged mice (Figure 6,Table S7). cDDAH1 KO also had no detectable effect on LV ejection fraction, LV fractional shortening, LV end diastolic diameter, LV end systolic diameter in female mice (Figure 6, Table S6).
Discussion
The major finding of this study is that inducible cardiomyocyte specific DDAH1 gene deletion in mice had no observable effects on cardiac structure or function under control conditions, but dramatically exacerbated pressure overload-induced LV hypertrophy and dysfunction. Furthermore, the disruption of cardiomyocyte DDAH KO did not increase plasma ADMA levels or blood pressure, and only modestly increased LV tissue ADMA levels, suggesting DDAH1 exerts actions specifically within cardiomyocytes which facilitate adaptation to hemodynamic overload.
ADMA levels are strongly associated with CHF and common risk factors for CHF. ADMA occurs in hypertension[34], coronary disease, and cardiomyopathy [3, 15]. Elevated plasma ADMA levels are also associated with an increased risk for developing myocardial infarction or cardiac death [4]. Using global DDAH1 KO mice, previous studies indicate that moderate, chronic increase of ADMA alone does not independently induce LV dysfunction in mature adult mice under unstressed control conditions. However, chronic accumulation of ADMA in global DDAH1 KO causes reduced vascular NO production, moderate hypertension, and diminished acetylcholine-induced vasodilatation [16, 23]. ADMA accumulation in global DDAH1 KO mice or endothelial specific DDAH1 KO mice also results in reduced angiogenesis and impaired vascular injury repair capacity [11, 39, 40]. Conversely, over-expression of DDAH1 results in decreases of plasma and tissue ADMA levels, which was associated decrease of systemic blood pressure [10], increase of insulin sensitivity [35], increase of angiogenesis [19], and reduced high fat diet induced atherosclerosis [38]. Since blood pressure, angiogenesis, atherosclerosis, coronary artery disease all contribute CHF development and progression, DDAH1 is expected to modulate heart failure development at least partially through modulating systemic ADMA metabolism and the overall cardiovascular NO bioavailability[25]. Our finding that cardiomyocyte specific DDAH1 KO still exacerbates cardiac hypertrophy and LV dysfunction in absence of changes in plasma ADMA and only mildly increased LV tissue ADMA suggests that cardiomyocyte DDAH1 exerts systemic ADMA independent protective effects or that DDAH1 protects against deleterious effects of cardiomyocyte derived intracellular ADMA.
The main source of ADMA and SDMA is degradation of proteins containing methylated arginines. Cardiac hypertrophy involves intracellular and extracellular remodeling processes that promote degradation and rebuilding of structural proteins. Our finding that plasma ADMA levels did not increase in response to TAC and that cDDAH1 KO only caused a moderate increase of LV ADMA in mice after TAC may be due to the fact that DDAH1 is strongly expressed in myocardial endothelial cells. ADMA produced by hypertrophying cardiomyocytes could be rapidly degraded by surrounding myocardial endothelial cells or rapidly taken into the circulation and degraded by DDAH1 in other tissues. The increased LV ADMA levels observed in cDDAH KO mice may represent the specific change in cardiomyocyte intracellular ADMA, before it is degraded outside of cardiomyocytes or removed into the circulation. Unfortunately, the measurement of intracellular ADMA in cardiomyocytes in LV tissue is not possible now.
Unlike ADMA, SDMA is not degraded by DDAH1[17] and is mainly removed through renal excretion. While plasma ADMA levels did not change in response to TAC, plasma and tissue SDMA levels were elevated in both WT and cDDAH KO mice. Thus, it is likely that increased cardiomyocyte protein degradation (or arginine methylation) induced by hypertrophic remodeling elevates production of both ADMA and SDMA levels, but ADMA is rapidly degraded by DDAH1. The further increase of SDMA observed in cDDAH KO mice may represent the greater hypertrophy and remodeling observed in these hearts. While many studies have focused on ADMA as a cardiovascular risk factor, several recent studies have demonstrated that increased SDMA is also a risk factor for mortality and cardiovascular events, as well as a marker of renal failure [28, 30]. However, the direct impact of SDMA accumulation on cardiac hypertrophy and dysfunction can’t be addressed in the present study.
The mechanism by which cardiomyocyte DDAH1 disruption exacerbates hypertrophy and LV dysfunction is not clear, but likely involves altered NO signaling. In the heart, eNOS is not only expressed in vascular endothelial cells, but is also expressed on the sarcolemma of cardiomyocytes [14, 18, 20]. The cellular distribution of eNOS in cardiomyocytes is important in protection against pressure overload or infarction induced LV remodeling and dysfunction[5, 21, 29]. We find that the cellular distribution of DDAH1 in the heart is similar to that of eNOS[8] and more importantly, that DDAH1 directly interacts with eNOS in cardiac tissues. This close association may have evolved to protect eNOS from inhibition by ADMA and facilitate compartmentalized NO production at the plasma membrane of endothelial cells and cardiomyocytes.
The importance of compartmentalized cardiomyocyte NO signaling in protection against systolic overload-induced LV remodeling has been previously reported. Thus, several previous studies have demonstrated that eNOS derived NO in cardiomyocytes protects against cardiac remodeling and dysfunction under stress conditions such as myocardial infarction, and pressure overload[21, 29]. While eNOS KO was shown to exacerbate TAC-induced LV hypertrophy and dysfunction in mice [21], cardiomyocyte-restricted restoration of eNOS (over-expressing eNOS in eNOS KO mice) reversed the exacerbated TAC-induced ventricular remodeling in eNOS KO mice [5], indicating an important role of NO derived from cardiomyocytes in attenuating TAC-induced LV remodeling and function. In addition, progressive LV hypertrophy, fibrosis, and dysfunction that develops in surviving tissue after myocardial infarction, are exacerbated in eNOS KO mice as compared to wild type mice[29], while transgenic mice over-expressing eNOS are protected from myocardial infarction-induced LV remodeling, cardiac death, and dysfunction[21, 29]. In addition, previous study demonstrated that NO regulates cardiac contractility in humans [27]. Thus, DDAH1 activity is likely to promote these previously described protective effects of eNOS. In addition, while many studies indicate protective effects of eNOS in cardiac hypertrophy, some studies also indicate eNOS has deleterious effects through uncoupling and production of reactive oxygen species[36]. ADMA competes with L-arginine for NOS binding to attenuate NO production. By preventing L-arginine binding to NOS, ADMA not only reduces NO formation, ADMA promotes NOS uncoupling to produce superoxide formation. Moreover, NO promotes cGMP production by activating soluble guanylate cyclase. Increased cGMP production subsequently activates Protein Kinase G and its downstream targets to cause smooth muscle relaxation and vessel dilatation. cGMP is degraded by PDE5 and PDE9A [22, 26, 36]. Studies from us and others have demonstrated that CHF is associated with reduced coronary blood flow [8, 9], increased expression of PDE5 and PDE9A [22,26], and reduced cGMP activity [22, 26]. Thus DDAH1 activity may exert protective effects in cardiomyocytes not only by increasing NO production, preventing NOS dependent superoxide production, but also by enhancing the downstream of NO signaling through stimulating cGMP/PKG. Our finding that LV nitrotyrosine levels are dramatically increased in cDDAH KO mice is consistent with an increase in cardiomyocyte oxidative stress due to increased cardiomyocyte ADMA and uncoupled NOS.
Interestingly, both cardiomyocyte-restricted eNOS gene deletion and eNOS overexpression had no significant impact on LV structure and function in mice under unstressed condition[5, 21]. Our finding that cardiomyocytes-restricted DDAH1 gene deletion exacerbated TAC-induced LV hypertrophy and dysfunction without affecting LV structure and function in mature adult and middle aged adult mice under control conditions is conceptually consistent with the previous reports that compartmentalized NO signaling in cardiomyocytes exerts protective effects which are only noticeable under stress conditions [5, 21].
In WT mice, we observed that TAC caused 2.6-fold increase of myocardial DDAH1, but this increase was absent in cDDAH1 KO mice. This finding suggests that pressure overload induces DDAH1 expression mainly in cardiomyocytes. This would support the idea that DDAH1 upregulation is an adaptive response to cardiac hypertrophy and remodeling, presumably to remove ADMA that accumulates during the hypertrophic remodeling process. Interestingly, we also observed a small but significant increase of myocardial eNOS in cDDAH1 KO mice, which may be a compensatory response to impaired NO signaling in these mice under basal conditions.
In conclusion, our findings indicate cardiomyocyte DDAH1 activity is dispensable for cardiac function under basal conditions, but plays an important role in attenuating cardiac hypertrophy and heart failure under stress conditions, indicating that maintaining ADMA metabolism in cardiomyocytes by DDAH1 is important in protection against development of CHF.
Supplementary Material
Acknowledgements
Sources of funding
This study was supported by Grants 81370197, 81470512 and 81570355 from National Natural Science Foundation, Grants HL102597 and R01HL105406 from the National Institutes of Health, a Grant 09GRNT2260175 from the American Heart Association, and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (to Xu X).
Footnotes
Disclosures
None.
References
- 1.Achan V, Broadhead M, Malaki M, Whitley G, Leiper J, MacAllister R, Vallance P (2003) Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol 23:1455–1459 doi: 10.1161/01.atv.0000081742.92006.59 [DOI] [PubMed] [Google Scholar]
- 2.Anderssohn M, Rosenberg M, Schwedhelm E, Zugck C, Lutz M, Luneburg N, Frey N, Boger RH (2012) The L-Arginine-asymmetric dimethylarginine ratio is an independent predictor of mortality in dilated cardiomyopathy. J Card Fail 18:904–911 doi: 10.1016/j.cardfail.2012.10.011 [DOI] [PubMed] [Google Scholar]
- 3.Anderssohn M, Schwedhelm E, Luneburg N, Vasan RS, Boger RH (2010) Asymmetric dimethylarginine as a mediator of vascular dysfunction and a marker of cardiovascular disease and mortality: an intriguing interaction with diabetes mellitus. Diab Vasc Dis Res 7:105–118 doi: 10.1177/1479164110366053 [DOI] [PubMed] [Google Scholar]
- 4.Boger RH, Sullivan LM, Schwedhelm E, Wang TJ, Maas R, Benjamin EJ, Schulze F, Xanthakis V, Benndorf RA, Vasan RS (2009) Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation 119:1592–1600 doi: 10.1161/circulationaha.108.838268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buys ES, Raher MJ, Blake SL, Neilan TG, Graveline AR, Passeri JJ, Llano M, Perez-Sanz TM, Ichinose F, Janssens S, Zapol WM, Picard MH, Bloch KD, Scherrer-Crosbie M (2007) Cardiomyocyte-restricted restoration of nitric oxide synthase 3 attenuates left ventricular remodeling after chronic pressure overload. Am J Physiol Heart Circ Physiol 293:H620–627 doi: 10.1152/ajpheart.01236.2006 [DOI] [PubMed] [Google Scholar]
- 6.Cardounel AJ, Cui H, Samouilov A, Johnson W, Kearns P, Tsai AL, Berka V, Zweier JL (2007) Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J Biol Chem 282:879–887 doi: 10.1074/jbc.M603606200 [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Guo H, Xu D, Xu X, Wang H, Hu X, Lu Z, Kwak D, Xu Y, Gunther R, Huo Y, Weir EK (2012) Left ventricular failure produces profound lung remodeling and pulmonary hypertension in mice: heart failure causes severe lung disease. Hypertension 59:1170–1178 doi: 10.1161/HYPERTENSIONAHA.111.186072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Li Y, Zhang P, Traverse JH, Hou M, Xu X, Kimoto M, Bache RJ (2005) Dimethylarginine dimethylaminohydrolase and endothelial dysfunction in failing hearts. Am J Physiol Heart Circ Physiol 289:H2212–2219 doi: 10.1152/ajpheart.00224.2005 [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Traverse JH, Du R, Hou M, Bache RJ (2002) Nitric oxide modulates myocardial oxygen consumption in the failing heart. Circulation 106:273–279 [DOI] [PubMed] [Google Scholar]
- 10.Dayoub H, Achan V, Adimoolam S, Jacobi J, Stuehlinger MC, Wang BY, Tsao PS, Kimoto M, Vallance P, Patterson AJ, Cooke JP (2003) Dimethylarginine dimethylaminohydrolase regulates nitric oxide synthesis: genetic and physiological evidence. Circulation 108:3042–3047 doi: 10.1161/01.cir.0000101924.04515.2e [DOI] [PubMed] [Google Scholar]
- 11.Dowsett L, Piper S, Slaviero A, Dufton N, Wang Z, Boruc O, Delahaye M, Colman L, Kalk E, Tomlinson J, Birdsey G, Randi AM, Leiper J (2015) Endothelial Dimethylarginine Dimethylaminohydrolase 1 Is an Important Regulator of Angiogenesis but Does Not Regulate Vascular Reactivity or Hemodynamic Homeostasis. Circulation 131:2217–2225 doi: 10.1161/circulationaha.114.015064 [DOI] [PubMed] [Google Scholar]
- 12.Duckelmann C, Mittermayer F, Haider DG, Altenberger J, Eichinger J, Wolzt M (2007) Asymmetric dimethylarginine enhances cardiovascular risk prediction in patients with chronic heart failure. Arterioscler Thromb Vasc Biol 27:2037–2042 doi: 10.1161/atvbaha.107.147595 [DOI] [PubMed] [Google Scholar]
- 13.Fassett JT, Xu X, Kwak D, Wang H, Liu X, Hu X, Bache RJ, Chen Y (2013) Microtubule Actin Cross-linking Factor 1 regulates cardiomyocyte microtubule distribution and adaptation to hemodynamic overload. PLoS One 8:e73887 doi: 10.1371/journal.pone.0073887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T (1996) Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem 271:22810–22814 [DOI] [PubMed] [Google Scholar]
- 15.Haghikia A, Podewski E, Libhaber E, Labidi S, Fischer D, Roentgen P, Tsikas D, Jordan J, Lichtinghagen R, von Kaisenberg CS, Struman I, Bovy N, Sliwa K, Bauersachs J, Hilfiker-Kleiner D (2013) Phenotyping and outcome on contemporary management in a German cohort of patients with peripartum cardiomyopathy. Basic Res Cardiol 108:366 doi: 10.1007/s00395-013-0366-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu X, Atzler D, Xu X, Zhang P, Guo H, Lu Z, Fassett J, Schwedhelm E, Boger RH, Bache RJ, Chen Y (2011) Dimethylarginine dimethylaminohydrolase-1 is the critical enzyme for degrading the cardiovascular risk factor asymmetrical dimethylarginine. Arterioscler Thromb Vasc Biol 31:1540–1546 doi: 10.1161/ATVBAHA.110.222638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu X, Xu X, Zhu G, Atzler D, Kimoto M, Chen J, Schwedhelm E, Luneburg N, Boger RH, Zhang P, Chen Y (2009) Vascular endothelial-specific dimethylarginine dimethylaminohydrolase-1-deficient mice reveal that vascular endothelium plays an important role in removing asymmetric dimethylarginine. Circulation 120:2222–2229 doi: 10.1161/CIRCULATIONAHA.108.819912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, Groszmann RJ, Shah VH, Sessa WC (2006) Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A 103:19777–19782 doi: 10.1073/pnas.0605907103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobi J, Sydow K, von Degenfeld G, Zhang Y, Dayoub H, Wang B, Patterson AJ, Kimoto M, Blau HM, Cooke JP (2005) Overexpression of dimethylarginine dimethylaminohydrolase reduces tissue asymmetric dimethylarginine levels and enhances angiogenesis. Circulation 111:1431–1438 doi: 10.1161/01.cir.0000158487.80483.09 [DOI] [PubMed] [Google Scholar]
- 20.Jagnandan D, Sessa WC, Fulton D (2005) Intracellular location regulates calcium-calmodulin-dependent activation of organelle-restricted eNOS. Am J Physiol Cell Physiol 289:C1024–1033 doi: 10.1152/ajpcell.00162.2005 [DOI] [PubMed] [Google Scholar]
- 21.Jones SP, Greer JJ, van Haperen R, Duncker DJ, de Crom R, Lefer DJ (2003) Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proc Natl Acad Sci U S A 100:4891–4896 doi: 10.1073/pnas.0837428100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA (2015) Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 519:472–476 doi: 10.1038/nature14332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O’Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P (2007) Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med 13:198–203 doi: 10.1038/nm1543 [DOI] [PubMed] [Google Scholar]
- 24.Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, Kimoto M, Tsuji H, Reaven GM, Cooke JP (2002) Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation 106:987–992 [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Hou L, Xu D, Chen A, Yang L, Zhuang Y, Xu Y, Fassett JT, Chen Y (2016) Effect of asymmetric dimethylarginine (ADMA) on heart failure development. Nitric Oxide 54:73–81 doi: 10.1016/j.niox.2016.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu Z, Xu X, Hu X, Lee S, Traverse JH, Zhu G, Fassett J, Tao Y, Zhang P, dos Remedios C, Pritzker M, Hall JL, Garry DJ, Chen Y (2010) Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation 121:1474–1483 doi: 10.1161/circulationaha.109.906818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rassaf T, Poll LW, Brouzos P, Lauer T, Totzeck M, Kleinbongard P, Gharini P, Andersen K, Schulz R, Heusch G, Modder U, Kelm M (2006) Positive effects of nitric oxide on left ventricular function in humans. Eur Heart J 27:1699–1705 doi: 10.1093/eurheartj/ehl096 [DOI] [PubMed] [Google Scholar]
- 28.Schepers E, Speer T, Bode-Boger SM, Fliser D, Kielstein JT (2014) Dimethylarginines ADMA and SDMA: the real water-soluble small toxins? Semin Nephrol 34:97–105 doi: 10.1016/j.semnephrol.2014.02.003 [DOI] [PubMed] [Google Scholar]
- 29.Scherrer-Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, Lindsey ML, Vancon AC, Huang PL, Lee RT, Zapol WM, Picard MH (2001) Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation 104:1286–1291 [DOI] [PubMed] [Google Scholar]
- 30.Schlesinger S, Sonntag SR, Lieb W, Maas R (2016) Asymmetric and Symmetric Dimethylarginine as Risk Markers for Total Mortality and Cardiovascular Outcomes: A Systematic Review and Meta-Analysis of Prospective Studies. PLoS One 11:e0165811 doi: 10.1371/journal.pone.0165811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnabel R, Blankenberg S, Lubos E, Lackner KJ, Rupprecht HJ, Espinola-Klein C, Jachmann N, Post F, Peetz D, Bickel C, Cambien F, Tiret L, Munzel T (2005) Asymmetric dimethylarginine and the risk of cardiovascular events and death in patients with coronary artery disease: results from the AtheroGene Study. Circ Res 97:e53–59 doi: 10.1161/01.res.0000181286.44222.61 [DOI] [PubMed] [Google Scholar]
- 32.Schwedhelm E, Maas R, Tan-Andresen J, Schulze F, Riederer U, Boger RH (2007) High-throughput liquid chromatographic-tandem mass spectrometric determination of arginine and dimethylated arginine derivatives in human and mouse plasma. J Chromatogr B Analyt Technol Biomed Life Sci 851:211–219 doi: 10.1016/j.jchromb.2006.11.052 [DOI] [PubMed] [Google Scholar]
- 33.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD (2001) Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89:20–25 [DOI] [PubMed] [Google Scholar]
- 34.Surdacki A, Nowicki M, Sandmann J, Tsikas D, Boeger RH, Bode-Boeger SM, Kruszelnicka-Kwiatkowska O, Kokot F, Dubiel JS, Froelich JC (1999) Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetric dimethylarginine in men with essential hypertension. J Cardiovasc Pharmacol 33:652–658 [DOI] [PubMed] [Google Scholar]
- 35.Sydow K, Mondon CE, Schrader J, Konishi H, Cooke JP (2008) Dimethylarginine dimethylaminohydrolase overexpression enhances insulin sensitivity. Arterioscler Thromb Vasc Biol 28:692–697 doi: 10.1161/atvbaha.108.162073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA (2005) Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest 115:1221–1231 doi: 10.1172/JCI21968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang WH, Tong W, Shrestha K, Wang Z, Levison BS, Delfraino B, Hu B, Troughton RW, Klein AL, Hazen SL (2008) Differential effects of arginine methylation on diastolic dysfunction and disease progression in patients with chronic systolic heart failure. Eur Heart J 29:2506–2513 doi: 10.1093/eurheartj/ehn360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weis M, Kledal TN, Lin KY, Panchal SN, Gao SZ, Valantine HA, Mocarski ES, Cooke JP (2004) Cytomegalovirus infection impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine in transplant arteriosclerosis. Circulation 109:500–505 doi: 10.1161/01.cir.0000109692.16004.af [DOI] [PubMed] [Google Scholar]
- 39.Zhang P, Hu X, Xu X, Chen Y, Bache RJ (2011) Dimethylarginine dimethylaminohydrolase 1 modulates endothelial cell growth through nitric oxide and Akt. Arterioscler Thromb Vasc Biol 31:890–897 doi: 10.1161/atvbaha.110.215640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang P, Xu X, Hu X, Wang H, Fassett J, Huo Y, Chen Y, Bache RJ (2013) DDAH1 deficiency attenuates endothelial cell cycle progression and angiogenesis. PLoS One 8:e79444 doi: 10.1371/journal.pone.0079444 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.