

Abstract
Cardiotoxicity has historically been a major cause of drug removal from the pharmaceutical market. Several chemotherapeutic compounds have been noted for their propensities to induce dangerous cardiac-specific side effects such as arrhythmias or cardiomyocyte apoptosis. However, improved preclinical screening methodologies have enabled cardiotoxic compounds to be identified earlier in the drug development pipeline. Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) can be used to screen for drug-induced alterations in cardiac cellular contractility, electrophysiology, and viability. We previously established a novel ‘cardiac safety index’ (CSI) as a metric that can evaluate potential cardiotoxic drugs via high-throughput screening of hiPSC-CMs. This metric quantitatively examines drug-induced alterations in CM function, using several in vitro readouts, and normalizes the resulting toxicity values to the in vivo maximum drug blood plasma concentration seen in preclinical or clinical pharmacokinetic models. In this ~1-month-long protocol, we describe how to differentiate hiPSCs into hiPSC-CMs and subsequently implement contractility and cytotoxicity assays that can evaluate drug-induced cardiotoxicity in hiPSC-CMs. We also describe how to carry out the calculations needed to generate the CSI metric from these quantitative toxicity measurements.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary.
Introduction
Off-target cardiotoxicity has historically been a substantial cause of drug withdrawal from the pharmaceutical market. Compounds have passed through clinical trials only to cause potentially life-threatening cardiac side effects such as arrhythmias, reduced ventricular ejection fraction, myocardial infarction, or heart failure when administered to patients1–3. For example, chemotherapeutic agents, including some anthracyclines and tyrosine kinase inhibitors (TKIs), have been labeled with US Food and Drug Administration (FDA) black box warnings for severe cardiotoxicity associated with prolongation of the cardiac QT electrophysiological interval or CM apoptosis4. Other non-cancer drugs, such as the antibiotic erythromycin and various tricyclic antidepressants, are also known to cause QT prolongation and potentially fatal torsades de pointes5. The cardiotoxicity of approved compounds has diminished recently, in part because of improved preclinical screening methods6–8. However, there remains a high demand for screening methodologies that can accurately identify cardiotoxic compounds in a preclinical setting, as demonstrated by the recent establishment of the multisite Comprehensive In Vitro Proarrhythmia Assay (CiPA) initiative, which aims to establish a next-generation, mechanism-based standard for evaluating the arrhythmogenicity of drugs in development9. In this protocol, we describe how to implement a method we have recently described10 that enables rapid, cost-effective, preclinical screening of potential cardiotoxic drugs.
Methods for preclinical modeling of drug cardiotoxicity
Traditional methods for preclinical evaluation of drug cardiotoxicity have used animal models, which tend to be expensive and low throughput, and to exhibit species-specific differences in cardiac physiology11. Alternative approaches use heterologous expression of cardiac ion channels in noncardiac cells. However, inhibition of specific ionic currents alone is not predictive of cardiotoxicity. Drug toxicity evaluation based on the human ether-à-go-go-related gene (hERG) channel often leads to a high rate of false-positive cardiotoxic compounds and therefore, unnecessary drug attrition at the preclinical stage. Consequently, the CiPA initiative has a focus on methods that identify arrhythmogenic drugs, as opposed to those causing QT prolongation, to improve upon prior models that have largely used alterations in the hERG potassium ion channel to identify cardiotoxic drugs.
An additional drawback to the use of noncardiac cells is that they lack CM-specific structural components such as sarcomeres and ion channel β-subunits12,13 that affect ion channel gating. Other cells, such as healthy, primary human CMs, could be used for preclinical evaluation of drug cardiotoxicity, but access to these cells is extremely limited, and their long-term maintenance in culture is a major technical challenge14. In addition, because adult human CMs are mitotically inactive, they cannot be expanded in vitro to obtain enough cells for drug screening. The most predictive models for drug cardiotoxicity must recapitulate the complex spatial distribution of the physiologically distinct myocytes of the intact adult human heart. However, intact human heart preparations are inherently too costly, difficult to maintain, and, hence, too low throughput to be implemented early in the drug development pipeline. For these reasons, there remains substantial interest in developing alternative human cardiac cell types for preclinical drug testing that are cost-effective and predictive and can hence be implemented early in the drug discovery process.
The establishment of methodologies to convert hiPSCs to CMs (hiPSC-CMs) has enabled human CMs to be mass-produced in vitro for cardiovascular disease modeling and drug screening15. These hiPSC-CMs functionally express most of the ion channels and sarcomeric proteins found in adult human CMs and can spontaneously contract. They can be produced in ~2 weeks, using chemically defined cardiac differentiation protocols, and can be genetically customized, using modern genomeediting technologies16–20. In addition, hiPSC-CMs have been able to recapitulate, at the cellular level, phenotypes for a variety of cardiovascular diseases, including viral myocarditis21, Brugada syndrome22, long-QT syndrome23, dilated cardiomyopathy24,25, hypertrophic cardiomyopathy26, chemotherapy-induced cardiomyopathy10,27, and congenital heart disease28. Because hiPSC-CMs can be cultured as simple 2D or 3D structures, diseases with a cell-autonomous phenotype such as the channelopathies, can be modeled in vitro. hiPSC-CMs are also responsive to ionotropic drugs such as norepinephrine, and their beating rates can be controlled via electrical stimulation29. Because hiPSC-CMs can be rapidly purified and replated for downstream applications, research groups in academia and industry have started utilizing hiPSC-CMs, as a complementary platform to nonhuman and noncardiac model systems, for high-content imaging and drug screening assays30,31. An additional aim of the CiPA initiative is to utilize hiPSC-CMs as a novel human cardiac cellular platform for validation of drug toxicity. Recent results from the CiPA initiative have confirmed that, if utilized appropriately, the hiPSC-CM platform can serve as a reliable alternative to existing hERG assays for evaluating arrhythmogenic compounds and can sensitively detect the action potential repolarization effects associated with ion channel-blocking drugs32–34.
Development of our protocol
In part to complement the methods of the CiPA initiative, we recently established a high-throughput screening platform using hiPSC-CMs to evaluate cardiotoxicities associated with a panel of FDA-approved kinase inhibitors10. We utilized assays evaluating drug-induced alterations in hiPSC-CM contractility or edge displacement during beating, along with hiPSC-CM cytotoxicity assessments, to establish a novel CSI correlating in vitro toxicity measurements to the clinical experience of patients. By normalizing the cardiotoxic drug doses in the cell-based assays to the maximum clinical drug concentrations experienced by patients, our CSI metric could accurately rank drugs by their toxicity, with cardiotoxicity black box-labeled drugs receiving the lowest CSI scores. Thus, we believe that this high-throughput hiPSC-CM-based metric could be implemented upstream of more conventional, low-throughput cardiotoxic assays, such as ex vivo heart preparations, to rapidly and accurately evaluate the potential cardiotoxicity of preclinical drug candidates. In addition, we believe that our work complements the efforts of the electrophysiology-focused CiPA initiative by incorporating assessments of drug-induced alterations in hiPSC-CM contractility and cytotoxicity, parameters that are critical to evaluating compounds such as chemotherapeutics, which can cause direct CM damage.
This protocol details how hiPSC-CM-based assays can be used to calculate the CSI (Fig. 1). The CSI could be used to preclinically evaluate the cardiotoxicity of pharmaceutical compounds under active development using hiPSC-CMs. This protocol provides step-by-step instructions for research groups aiming to use the CSI to conduct cardiotoxicity assessment for drugs in active development, or to provide an in vitro toxicity evaluation for a cadre of existing drugs. This method is best utilized when the maximum clinical blood plasma concentration (Cmax) values for the drugs of interest have been determined, either via in vivo preclinical animal data or from human clinical trial results. For testing new compounds in a preclinical setting where Cmax values from clinical trials are obviously not available, we recommend using preclinical animal Cmax data whenever possible. In this preclinical scenario, some approximation or prediction of the potential downstream clinical dose will be highly informative for normalizing cytotoxicity and contractility values elicited while screening new drug candidates on hiPSC-CMs. Because existing methods for drug cardiotoxicity assessment do not incorporate cytotoxicity and contractility analysis into a single, easy-to-use metric, we believe that this protocol and the CSI presented will provide a next-generation method for cardiotoxicity evaluation.
Fig. 1|. Workflow for generating the cardiac safety index for drugs of interest using high-throughput contractility and cytotoxicity analysis of hiPSC-CMs.
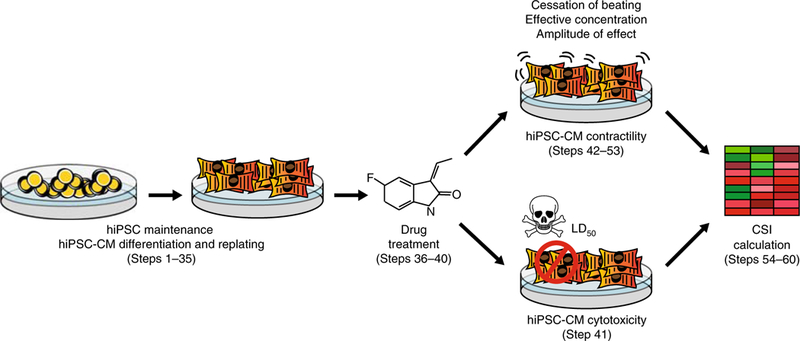
First, hiPSCs are differentiated into hiPSC-CMs. Drugs of interest are then added to hiPSC-CMs to assess drug-induced alterations in cellular contractility and cell viability. From these experiments, toxicity metrics are obtained that correspond to drug concentrations at which hiPSC-CM contractility or viability is substantially altered (e.g., cessation of beating, effective concentration, amplitude of effect, and LD50). These toxicity values are corrected by a Cmax value representing the maximum drug blood plasma concentration experienced by patients, as previously reported in FDA clinical trial literature or derived from preclinical pharmacokinetic studies in animal models. When combined, these metrics ultimately yield the CSI, which measures the cardiotoxicity of a drug based on in vitro assessment using hiPSC-CMs.
Experimental design
The Procedure begins with a description of how to maintain hiPSCs and differentiate hiPSCs to hiPSC-CMs (Steps 1–35). Alternatively, hiPSC-CMs can be obtained from a commercial or academic source, in which case the Procedure should be started at Step 36. Steps 36–40 describe how to treat cells with drugs of interest. Steps 41 and 42–53 describe how to carry out cytotoxicity and contractility assays, respectively, including how to calculate the parameters that can be obtained from these data and are needed for calculation of the CSI for each drug. Step 54 and onward describe how to calculate the CSI.
Derivation of hiPSCs
The hiPSCs needed for downstream assays can be made using established reprogramming protocols, such as that which uses a Sendai virus vector expressing the hiPSC reprogramming factors OCT4, SOX2, KLF4, and MYC to reprogram peripheral blood mononuclear cells (PBMCs) to pluripotent stem cells35. As an alternative to in-house reprogramming, low-passage hiPSCs can also be obtained from commercial vendors or from academic entities such as the Stanford University Cardiovascular Institute Biobank (http://med.stanford.edu/scvibiobank.html). Expanding and freezing newly obtained hiPSC lines is encouraged to mitigate the possible impact of later complications such as cell culture contamination. The hiPSCs must be growing optimally before use in differentiation protocols to produce hiPSC-CMs. They should exhibit a tightly packed colony structure, as well as protein markers characteristic of pluripotent stem cells, such as NANOG and TRA-1–81 (Fig. 2). Once hiPSC lines are obtained, they can be passaged nearly indefinitely.
Fig. 2|. Procedure for chemically defined differentiation of hiPSCs into hiPSC-CMs and downstream replating.
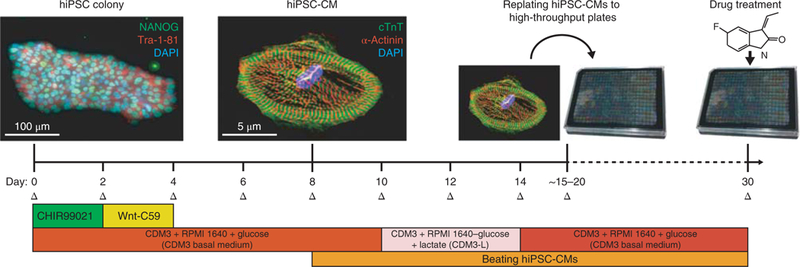
Initially, hiPSCs will exhibit a tightly packed colony morphology and will express stem cell markers such as NANOG (green) and Tra-1–81 (red). Nuclear DNA stain DAPI is also shown (blue). This differentiation approach requires small molecule-mediated modulation of the Wnt signaling pathway by GSK3β inhibitor CHIR99021 and Wnt inhibitor Wnt-C59 to drive hiPSCs toward a cardiac mesoderm and cardiomyocyte fate. Beating hiPSC-CMs are typically visible by day 8 of differentiation. After cardiomyocyte differentiation and metabolic selection are complete, hiPSC-CMs will be pure and will express cardiomyocytespecific markers such as cardiac troponin T (cTnT; green) and alpha-actinin (red). Nuclear DNA stain DAPI is also shown (blue). At this point, they can be replated into high-throughput plates for downstream drug treatment and analysis. Medium changes (Δ) should be conducted on hiPSC-CMs every 2 d. NANOG antibody (Abcam, cat. no. AB21624) and Tra-1–81 antibody (EMD Millipore, cat. no. MAB4381) were utilized at 1:200 dilutions (~5 μg/mL) for hiPSC immunofluorescence staining. Cardiac troponin T antibody (Abcam, cat. no. AB45932) and alpha-actinin antibody (Sigma, cat. no. A7811) were utilized at 1:200 dilutions (~5 μ/mL) for hiPSC-CM immunofluorescence staining. For further details on how immunofluorescence and hiPSC-CM differentiation were conducted, refer to our previous study, Sharma et al.10. The CDM3 added to RPMI1640 in this figure refers to the CDM3 supplement.
Differentiation of hiPSCs into hiPSC-CMs
The hiPSCs can be differentiated into a variety of cell types, including CMs. Recent protocols use small-molecule regulators of the Wnt signaling pathway to initiate differentiation of hiPSCs into hiPSC-CMs19. This is usually an efficient process, and we can readily obtain >95% purity hiPSC-CMs after differentiation and metabolic selection using low-glucose media30. For drug screening assays, we recommend a chemically defined differentiation approach without the use of growth factors, such as insulin, that could alter cell viability. This minimal medium further serves to reduce additional variables that may affect hiPSC-CM function. Our laboratory has recently devised a protocol allowing hiPSC-CMs to be made in a medium consisting only of ascorbic acid, human albumin, basal RPMI 1640 growth medium, and Wnt-regulating small-molecule compounds19. After an ~2-week-long differentiation protocol, hiPSC-CMs are produced (Fig. 2). These hiPSC-CMs can be further metabolically purified by removing glucose from the hiPSC-CM growth medium and supplementing it with lactate30. CMs can utilize metabolic pathways other than glycolysis, whereas most non-CMs cannot, and therefore many non-CMs die during prolonged glucose deprivation. The differentiated hiPSC-CMs will spontaneously contract and express most sarcomeric proteins and ion channels normally found in human cardiac tissue. For example, hiPSC-CMs express cardiac-specific sarcomeric markers such as cardiac troponin T (TNNT2) and alpha-actinin (ACTN2)15 (Fig. 2). We highly recommend conducting multiple rounds of differentiation and procuring additional control hiPSC-CM lines to serve as biological replicates for cell viability and contractility observations.
High-throughput imaging and quantitative viability assays using hiPSC-CMs
Once purified, hiPSC-CMs can be replated onto Matrigel or a comparable matrix in high-throughput microtiter plates, such as in a 96- or 384-well plate format, for downstream imaging and contractility assays. For optimal cell survival, we recommend replating hiPSC-CMs at high confluency (Fig. 3a). During replating, substantial manual pipetting is required to dissociate hiPSC-CMs into a single-cell format, as these cells can secrete substantial amounts of collagen during long-term culture. Collagenase treatment or filtering the dissociated cells through a straining mesh could also help separate cells into a single-cell format. After replating, we recommend not disturbing the cells for 2 d so that they can adhere to the extracellular matrix. Afterward, hiPSC-CMs can be treated with the drugs of interest (Fig. 3b). Drugs should be dissolved in water, if possible, and kept for long-term storage at –80 °C. Drugs that are insoluble in water can alternatively be dissolved in dimethyl sulfoxide (DMSO), keeping in mind that exposure to ≥0.1% (vol/vol) DMSO tends to increase toxicity in hiPSC-CMs. Note that for this protocol, DMSO was used as the vehicle negative control condition for downstream analyses, but hiPSC-CM assays that have intrinsically high well-to-well variability can also benefit from internal, well-specific baseline controls that are recorded before drug treatment.
Fig. 3|. Setup for drug-induced cytotoxicity and contractility assessment of hiPSC-CMs.
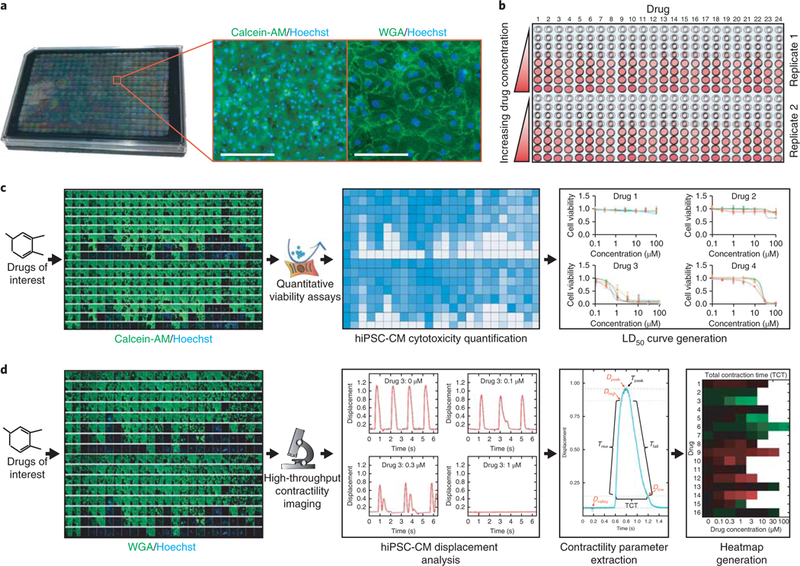
a, A typical 384-well plate containing purified hiPSC-CMs. Insets illustrate optimal hiPSC-CM plating density via a cell body stain (calcein-AM; green) and a cell boundary stain (WGA, green). For these stains, 0.5 μM calcein-AM (Thermo Fisher Scientific, cat. no. C3100MP) or 5 μg/mL WGA (Thermo Fisher Scientific, cat. no. W11261) was combined with 2 μg/mL nuclear DNA stain (blue) Hoechst 33342 (Thermo Fisher Scientific, cat. no. H3570) in CDM3 basal medium pre-warmed to 37 °C. To optimize survival, hiPSC-CMs should be plated at high confluency as shown. Scale bars, 100 μm (left); 20 μm (right). b, Diagram illustrating one format for drug treatment in a 384-well plate, which allows for up to 24 drugs to be tested in duplicate, at eight half-log concentrations from 0 to 100 μM. c, Drug treatment workflow for cytotoxicity assessment in hiPSC-CMs in a 384-well plate. After exposure to drugs, quantitative viability assays such as PrestoBlue can quantify drug-induced alterations in hiPSC-CM viability. From these data, drug dose-response curves can be generated in a statistics program such as GraphPad Prism. In these example graphs, each colored line signifies an hiPSC-CM cell line derived from a unique individual. N = 5 independent biological replicate cytotoxicity assays were conducted for each cell line and drug, with error bars representing standard error. LD50 data can be extrapolated from these dose-response curves. The calcein-AM stain (green) indicates viable cells but is not required for cytotoxicity assessment. The Hoechst stain (blue) indicates DNA in cell nuclei. d, Drug treatment workflow for contractility assessment in hiPSC-CMs in a 384-well plate. After exposure to drugs, cells are stained with WGA (green) and Hoechst nuclear DNA stain (blue) so that they can be tracked using a platform such as the Vala Sciences KIC, optimized for high-throughput contractility imaging. Next, hiPSC-CM displacement analysis can be conducted to generate graphs illustrating hiPSC-CM movement in response to drug treatment over time. From this analysis, various contractility parameters can be extracted, including time (T) parameters such as time to peak (Tpeak) and TCT. Parameters also include displacement (D) parameters such as peak displacement (Dpeak). Heatmaps can be generated for each parameter, detailing how parameters change in response to drug concentration. Red coloration indicates a decrease in contraction rate, whereas green indicates an increase in contraction rate. Another parameter obtained from contractility analysis is cessation of beating, indicating the drug concentration that causes a majority of replicate wells to cease beating. WGA, wheat germ agglutinin. Portions of this figure were adapted with permission from Sharma et al.10, American Association for the Advancement of Science.
Immunofluorescence staining can be also conducted in high-throughput microtiter plates to qualitatively assess cell viability or sarcomeric organization (Fig. 3c). CM-specific markers such as troponin or alpha-actinin can be used to examine sarcomere integrity after drug treatment. For quantitative viability measurements, cells can be treated with a variety of absorbance-, fluorescence-, or luminescence-based assays that can measure outputs such as ATP production, intracellular lactate dehydrogenase activity, troponin release, or membrane integrity36. If possible, we recommend incorporating a variety of these assays to serve as internal controls for cell viability. In addition, high-throughput imaging and viability assays can be conducted rapidly on integrated plate reading/ imaging systems that use either a 96- or 384-well plate format to generate enough replicates for robust statistics. A range of statistical analysis programs also exist that can be used to generate dose-response cell viability curves and other metrics. For calculation of the CSI, an LD50 (median lethal dose) value can be determined from cytotoxicity analyses after drug treatment on hiPSC-CMs. This value indicates the drug concentration at which there is a 50% loss in hiPSC-CM viability after drug treatment.
High-throughput contractility assessment of hiPSC-CMs
After purification, hiPSC-CMs can be replated for contractility assays, as mentioned in the previous section (Fig. 3d). Alternatively, frozen hiPSC-CMs can be purchased commercially for drug-testing purposes. However, 25% of frozen hiPSC-CMs typically do not recover after thawing, and thus we recommend using freshly differentiated hiPSC-CMs for downstream assays. For optimal cell survival, we highly recommend that cells be replated at a density to achieve 100% confluence the following day, as they will be more likely to start beating within 48 h of replating. After replating, the CM medium should be changed every 2 d until beating becomes synchronous. If cells are replated at a suboptimal density, isolated patches of hiPSC-CMs will not beat synchronously, unless paced by electrical stimulation. We recommend using either a multichannel pipettor or an automated fluid handler to treat the hiPSC-CMs with a dose-response quantity of test compound before high-throughput imaging. For contractility assessment, we have previously used an IC200 Kinetic Imaging Cytometer (KIC) in combination with wheat germ agglutinin (WGA) Alexa Fluor 488–conjugated dye and Hoechst nuclear stain to mark and track individual CMs10. CM displacements can be calculated for each well in a multi-well plate from high-frame-rate videos of contracting CMs loaded with the fluorescent dye solution and treated with varying doses of the relevant test compound.
Quantification of hiPSC-CM motion for contractility analysis
After image acquisition with a high-content microscope such as the KIC, deformation vector maps can be derived by evaluating an hiPSC-CM contraction image series with a particle image velocimetry (PIV)37. PIV determines deformation maps from two images in a time-lapse sequence by dividing each image into smaller interrogation windows and maximizing the cross-correlation between the windows in the deformed and undeformed (reference) images. The size of the interrogation window should be chosen to balance resolution, which decreases with window size; measurement floor, which decreases with window size; and signal-to-noise ratio, which increases with window size. In our method, spatial resolution is improved by multigrid PIV (multiple PIV passes using progressively smaller interrogation windows, in which each pass uses the result from the previous pass to re-center the interrogation windows)37. This procedure increases the overlap between deformed and reference interrogation windows and allows for use of smaller interrogation windows without compromising the signal-to-noise ratio, which increases the spatial resolution. In addition, it is recommended to ensure that the measured deformation field is compatible with global mechanical equilibrium. This can be achieved by setting to zero the average deformation a posteriori38 or, preferably, by enforcing the condition of global equilibrium in the maximization of window cross-correlation39. For reference, in our previous works, we have used windows of 64 × 64 pixels with a 32-pixel overlap between windows10,40,41. Image correlation signal-to-noise ratio is dependent on the quality and magnification of the microscopy images and could vary among experiments. Thus, it is recommended to set the size of the interrogation windows by applying a known synthetic distortion to the reference image(s) and running PIV on the synthetically deformed image. Comparing the prescribed deformation with the one recovered by PIV provides the spatial distribution of the PIV error as a function of window size. This quality control step can be easily automated and can result in useful information in regard to discarding of spatial regions of an image sequence that have poor quality due to poor optical focus, for example.
Custom software can be written to implement a PIV algorithm that includes the features described above. Alternatively, there are several well-documented open-source PIV algorithms that can be downloaded from repositories (e.g., OpenPIV42). To quantify deformation, a two-step method can be used, in which the first step is the automated selection of a reference frame that represents hiPSC-CM diastolic relaxation41. This step consists of performing PIV analysis in each frame, utilizing the prior frame as a point of reference, which generates the instantaneous contraction/relaxation velocity vector map for contracting hiPSC-CMs. This is followed by computing the spatial average value of the velocity magnitude distribution, which provides an overall measurement of the movements in the hiPSC-CMs relative to the previous frame at a specific moment in time. The resulting temporal signal displays two periodic peaks, corresponding to a single cardiac contraction and relaxation cycle. Between consecutive contraction cycles, hiPSC-CMs are mostly stationary. These inactive periods are automatically identified, allowing the user to choose one or multiple reference frames. Selecting multiple frames can potentially decrease measurement noise by enabling ensemble averaging in the PIV analysis, but it also increases the computational cost of the analysis37. Next, the PIV analysis is conducted again with the selected frame(s) of reference to determine the deformation fields. Finally, the divergence of the deformation fields is computed from the PIV results, which quantifies relative changes in the area of the hiPSC-CMs during contraction and relaxation. The contractility versus time signals are obtained from the spatial average of the absolute value of the divergence distribution of each frame. Multiple kinetic parameters can be obtained from these signals for quantitative and statistical comparisons of hiPSC-CM contraction. To enhance the robustness of parameter retrieval, all peaks associated with a single contractility signal can be combined into an average peak by conducting time-dependent conditional averaging. One useful by-product of conditional averaging is a parameter for the average time between contractility peaks (Tpeak). The remaining parameters can be measured from the single-cycle contractility profile attained after conditional averaging.
The hiPSC-CM safety coefficients needed to calculate the CSI can be computed from the raw data obtained during the contractility analysis. These include parameters such as cessation of beating (CoB), defined as the drug concentration at which >50% of technical replicate wells containing hiPSC-CMs stop contracting. In our previous study, the sampling duration utilized by the KIC 200 to determine whether cells have ceased beating was 6.5 s10. Effective concentration (EC) is the drug concentration that elicits a statistically significant difference in hiPSC-CM contractility (P < 0.05) in comparison to vehicle negative control measurement. The amplitude of effect (AE) quantifies the magnitude of hiPSC-CM contractility differences at the EC in comparison to the vehicle negative control condition. The AE evaluates these contractility differences by examining changes in contractility parameters (e.g., total contraction time (TCT), contraction rate, and relaxation rate) obtained from the contractility analysis. Average EC is a weighted average of the different ECs, weighted by each respective AE. This average EC value takes into consideration all possible ECs and includes a bias toward ECs corresponding to metrics that most dramatically change normal hiPSC-CM contractility. These various hiPSC-CM contractility metrics can be used to calculate the CSI.
Calculation of a CSI from hiPSC-CM cytotoxicity and contractility metrics
After conducting hiPSC-CM cytotoxicity and contractility analysis by treating hiPSC-CMs with drugs of interest, the CSI associated with a drug can be calculated by averaging the EC, LD50, and CoB metrics for that drug. A normalized CSI (CSIN) for a specific drug can also be calculated as a metric (between 0 and 1) to compare the relative cardiotoxicity of drugs of interest in a dataset. For calculating the CSIN, we highly recommend including both positive and negative control drugs in the analysis. Examples of drugs that could serve as adequate positive controls for hiPSC-CM cytotoxicity and contractility include the TKI sorafenib, which has been shown to be highly cytotoxic to hiPSC-CMs, and the TKI nilotinib, which induces contractility abnormalities in hiPSC-CMs, as demonstrated in our previous study10. Both TKIs have been known to cause arrhythmias and heart failure in cancer patients after administration of chemotherapy. Notably, nilotinib exhibited an acute action potential duration–prolonging effect in hiPSC-CMs, as demonstrated by our previous study, and is similarly associated with an FDA black box warning for QT prolongation. Drugs that could serve as negative controls for hiPSC-CM cytotoxicity and contractility include the solvent DMSO and the TKI axitinib, which we have previously shown does not substantially alter hiPSC-CM cytotoxicity or contractility.
For calculating the CSI, the CoB, EC, and AE contractility parameters and LD50 values for drug-induced cytotoxicity are needed (Fig. 4). However, another critical metric needed to calculate the CSI is the Cmax experienced in vivo either by animal models in preclinical pharmacokinetic studies or by patients receiving the drug being tested in clinical trials. If the drug has been approved by the US FDA, the Cmax can be obtained from FDA literature associated with the approval of that drug. In this protocol, we use total Cmax values, although it should be noted that free Cmax values can also be used. Many drugs are bound to plasma proteins, with some bound >99% (e.g., warfarin), yet are efficacious because determinations of free versus total Cmax are typically static measures that do not consider the dynamic equilibriums between binding to different plasma proteins versus on- and off-targets43. Whether free or total, the Cmax value will provide information as to whether the drug concentrations observed to be cytotoxic or altering contractility in hiPSC-CM-based assays are analogous to the drug doses experienced in vivo. Thus, to calculate the CSI, the CoB, EC, and LD50 values for a drug of interest are first divided by the Cmax for the drug (if available) to give CoB, EC, and LD50 values corrected for the maximum drug concentration detected in the patient (Fig. 4a). In addition, the corrected EC is divided by the AE of that drug to provide an updated, corrected EC. To obtain a relative cardiotoxicity metric for a large dataset of drugs, the CoB, EC, and LD50 metrics for a specific drug can be normalized by the highest values for each metric in the dataset to give normalized CoB, normalized EC, and normalized LD50 viability metrics ranging between 0 and 1 (Fig. 4b). A normalized CSI can be obtained by averaging normalized CoB, EC, and LD50 metrics; it will have a value between 0 and 1, for which a value near 0 indicates a toxic drug, and a value near 1 indicates a safe drug.
Fig. 4|. Equations used to obtain the CSI.
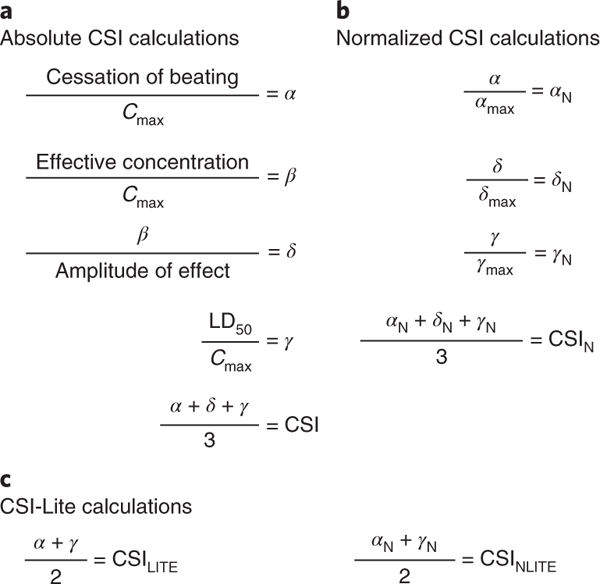
Assume that each metric corresponds to the values associated with a single drug being tested for cytotoxicity and contractility alterations on hiPSC-CMs. a, Metrics α (corrected cessation of beating), β (corrected effective concentration), δ (updated corrected effective concentration), and γ (corrected LD50) are obtained by normalizing values from contractility (cessation of beating, effective concentration, and amplitude of effect) and cytotoxicity (LD50) assays by the maximum blood plasma concentration (Cmax) for each drug being tested on hiPSC-CMs. Cmax values for each drug of interest can be found in FDA clinical trial literature or can be derived from preclinical pharmacokinetic studies in animal models. The absolute α, δ, and γ values are averaged to provide an absolute CSI associated with a specific drug. b, The CSIN metric can be used to evaluate the relative cardiotoxicity of a drug in comparison to other drugs in a large dataset. To derive the CSIN, normalized values for α, δ, and γ (αN, δN, and γN) are obtained by dividing the absoluteα;, δ, and γ values associated with a drug by the maximum α, δ, and γ values in a dataset. CSIN is determined by averaging αN, δN, and γN. c, If it is not possible to obtain δ or δN due to lack of access to a high-throughput contractility analysis platform such as the KIC, a simplified, ‘lite’ version of the final CSI can be obtained by averaging α and γ (CSILITE) or αN and γN (CSINLITE).
Our approach is very flexible. For instance, a simplified version of the CSI (CSI-Lite) can be constructed using CoB as the only contractility metric (Figs. 4c and 5). Importantly, the CSI and CSI-Lite values associated with the TKIs from our previous hiPSC-CM cardiotoxicity study were remarkably similar, illustrating the consistency of the indices (Table 1)10. Note that although it may be convenient to propose use of CSI-Lite for cases in which motion-based contractility studies are not possible, values of CSI versus CSI-Lite may not be comparable in some cases, as one is replacing a contractility-based component of the assessment with an automaticity-based parameter. In studies describing electrophysiological effects of drugs, the CoB value is sometimes simply stated as a limiting concentration for which no data are available or is used as a measure of toxicity. As native human ventricular myocytes do not display automaticity, whether this effect on hiPSC-CMs in vitro is a relevant toxicity is an open question. A limitation to assays using hiPSC-CMs is that the cells are immature, with known structural, functional, genetic, and electrophysiological consequences44. We expect that in the future, it will be possible to improve predictiveness by using more mature hiPSC-CMs and incorporating additional metrics into the CSI.
Fig. 5|. Mathematical example of CSI calculations for cardiotoxic tyrosine kinase inhibitor sorafenib.
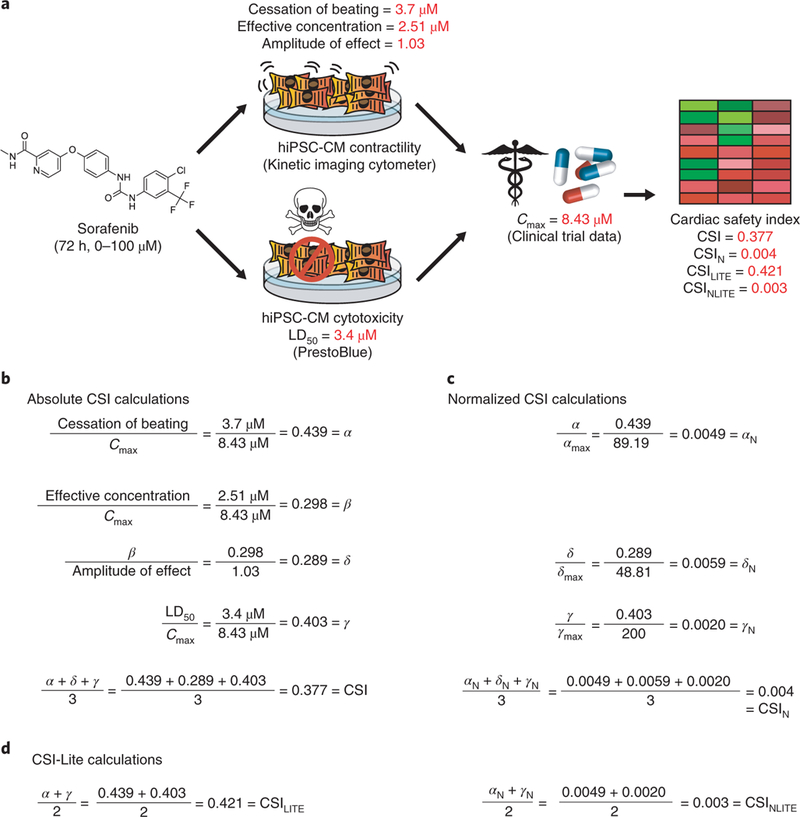
a, Workflow detailing the drug cardiotoxicity evaluation process for sorafenib, one of the most cardiotoxic tyrosine kinase inhibitors reported in our previous study (Sharma et al.10). In this study, sorafenib was added to purified hiPSC-CMs for 72 h at a concentration ranging from 0 to 100 μM. Values for cessation of beating, effective concentration, and amplitude of effect were obtained by evaluating hiPSC-CM contractility after sorafenib treatment using an IC-200 KIC from Vala Sciences. Likewise, the LD50 cytotoxicity value was obtained from drug-treated hiPSC-CMs using the colorimetric PrestoBlue cell viability assay. These metrics were then normalized to the clinical Cmax for sorafenib, reflecting the maximum blood plasma concentration experienced by patients receiving this drug. This Cmax value was obtained from published sorafenib clinical trial literature (Moore et al.10). These values were combined to create variations of the cardiac safety index. b, Calculations for absolute CSI for sorafenib, as detailed in Fig. 4a. c, Calculations for CSIN for sorafenib, as detailed in Fig. 4b. Note that values for αmax, δmax, and γmax are the maximum α, 𝛿, and γ values in the 23-drug dataset in Sharma et al.10.d, Calculations for the simplified CSILITE and CSINLITE for sorafenib, as detailed in Fig. 4c.
Table 1|.
Cardiac safety index metrics gathered from cytotoxicity and contractility analyses of tyrosine kinase inhibitor-treated hiPSC-CMs
| Compound | Cessation of beating (μM) |
Effective concentration (μM) |
Amplitude of effect |
LD50 (μM) |
Cmax (μM) |
Normalized CSI (CSIN) |
Normalized CSI-Lite (CSINLITE) |
Clinically reported cardiotoxicity |
|---|---|---|---|---|---|---|---|---|
| Vemurafenib | 33 | 11.00 | 0.34 | 32.10 | 126.04 | 0.003 | 0.002 | QT |
| Sorafenib | 3.7 | 2.51 | 1.03 | 3.40 | 8.43 | 0.004 | 0.003 | QT, LV, HF, MI, Hy |
| Doxorubicin | 3.7 | 1.20 | 0.60 | 0.78 | 2.93 | 0.010 | 0.008 | HF, LVa |
| Regorafenib | 11 | 3.70 | 0.84 | 7.10 | 8.08 | 0.010 | 0.010 | MI, Hyb |
| Vandetanib | 33 | 5.68 | 2.47 | 20.60 | 4.26 | 0.041 | 0.056 |
QT,
TdP,
SCD, HF, Hya |
| Crizotinib | 11 | 1.91 | 0.59 | 8.60 | 1.24 | 0.063 | 0.067 | QT, Brady |
| Nilotinib | 100 | 8.31 | 2.65 | 29.00 | 4.27 | 0.104 | 0.148 | QT, LV, Vasa |
| Imatinib | 100 | 33.00 | 1.59 | 78.20 | 5.11 | 0.126 | 0.148 | LV |
| Lapatinib | 33 | 11.00 | 0.40 | 100.76 | 2.30 | 0.209 | 0.190 | LV, QTb |
| Sunitinib | 3.7 | 0.81 | 1.33 | 12.70 | 0.18 | 0.218 | 0.292 | HF, LV, MI, QT, Hyb |
| Bosutinib | 33 | 4.73 | 1.92 | 12.39 | 0.51 | 0.315 | 0.423 | PE |
| Gefitinib | 33 | 3.11 | 1.24 | 26.30 | 0.45 | 0.409 | 0.557 | None |
| Afatinib | 3.7 | 1.65 | 1.11 | 12.30 | 0.10 | 0.444 | 0.515 | None |
| Dabrafenib | 100 | 36.75 | 0.71 | 100.68 | 4.16 | 0.459 | 0.561 | LV |
| Ponatinib | 3.7 | 3.70 | 0.54 | 4.30 | 0.14 | 0.483 | 0.225 | vas, HF, LV,Hya |
| Ibrutinib | 33 | 10.01 | 1.54 | 11.90 | 0.37 | 0.507 | 0.580 | Afib |
| Dasatinib | 3.7 | 1.20 | 0.31 | 42.00 | 0.21 | 0.524 | 0.599 | QT, PE, Hy |
| Erlotinib | NA | 63.38 | 0.51 | 87.60 | 3.11 | 0.653 | 0.570 | MI (rare) |
| Pazopanib | NA | 73.86 | 1.19 | NA | 103.08 | 0.671 | 1.000 | QT, LV (rare)b |
| Cabozantinib | NA | 91.14 | 1.37 | NA | 4.43 | 0.769 | 1.000 | Noneb |
| Trametinib | 100 | 33.00 | 2.37 | 66.80 | 0.02 | 1.000 | 1.000 | LV |
| Axitinib | NA | 71.79 | 0.44 | NA | 0.07 | 1.000 | 1.000 | HF (rare), Hy |
| DMSO | NA | 100.00 | 0.58 | NA | NA | 1.000 | 1.000 | None |
Green shading indicates values associated with lower cardiotoxicity. Red shading indicates values associated with higher cardiotoxicity. Cessation of beating is the concentration at which >50% of triplicate wells ceased beating. Effective concentration is the concentration at which a significant (P < 0.05) alteration in all listed contractility parameters was detected (see Fig. S7 and Materials and Methods in Sharma et al.10 for details). Amplitude of effect is the degree to which all listed contractility parameters were altered at the effective concentration (see Materials and Methods in Sharma et al.10 for details). LD50 values from viability assays represent the TKI concentration at which 50% loss in viability is observed in viability assays. Cmax represents the maximum TKI blood plasma concentration reported in the FDA literature. The normalized cardiac safety index (CSIN) presented here is a value from 0 to 1 that normalizes contractility and viability parameters to patient Cmax and combines these parameters to provide a relative metric for TKI cardiotoxicity. The normalized cardiacsafety index-LITE (CSINLITE) presented here is a modified version of the CSI that does not incorporate effective concentration or amplitude of effect and is optimized for researchers without access to advanced contractility analysis platforms. Clinically reported cardiotoxicities are alterations in patient cardiac function (see Table S1 in Sharma et al.10).
Afib, atrial fibrillation; Brady, bradycardia; HF, heart failure; Hy, hypertension; LV, center ventricular ejection fraction decrease; MI, myocardial infarction; PE, pericardial effusion; QT, QT interval prolongation; SCD, sudden cardiac death; TdP, torsades de pointes; Vas, vascular abnormalities. Adapted with permission from Sharma et al.10, American Association for the Advancement of Science.
(Bold type) Cardiovascular toxicity-associated boxed warning.
Non-cardiovascular toxicity-associated boxed warning.
Biological materials
Wild-type, low-passage hiPSCs adapted for feeder cell–free growth on Matrigel
 CAUTION Ethical approval, patient consent, and relevant national and institutional regulations apply when deriving hiPSCs for use in this protocol.
CAUTION Ethical approval, patient consent, and relevant national and institutional regulations apply when deriving hiPSCs for use in this protocol.  CRITICAL HiPSC lines used for differentiation into hiPSC-CMs can be either made in-house or obtained from a commercial source (e.g., from the Stanford University Cardiovascular Institute Biobank, http://med.stanford.edu/scvibiobank.html). Refer to prior protocols for more information on hiPSC derivation from blood using Sendai virus reprogramming protocols45. These hiPSCs should be obtained at low passage, ideally below passage 20. Cell lines should be regularly checked for mycoplasma contamination and genotypic authenticity.
CRITICAL HiPSC lines used for differentiation into hiPSC-CMs can be either made in-house or obtained from a commercial source (e.g., from the Stanford University Cardiovascular Institute Biobank, http://med.stanford.edu/scvibiobank.html). Refer to prior protocols for more information on hiPSC derivation from blood using Sendai virus reprogramming protocols45. These hiPSCs should be obtained at low passage, ideally below passage 20. Cell lines should be regularly checked for mycoplasma contamination and genotypic authenticity.
Reagents
Matrigel (human embryonic stem cell (hESC)-qualified; BD Sciences, cat. no. 354277)
PBS (Life Technologies, cat. no. 20012050)
DMEM/F12 medium (Thermo Fisher Scientific, cat. no. 11320033)
RPMI 1640 medium (Thermo Fisher Scientific, cat. no. 11835055)
Glucose-free RPMI 1640 medium (Thermo Fisher Scientific, cat. no. 11879–020)
L-lactic acid (Wako Chemicals, cat. no. 129–02666)
E8 medium (Thermo Fisher Scientific, cat. no. A1517001)
L-ascorbic acid 2-phosphate (Wako Chemicals, cat. no. 321–44823)
Sterile water for injection (WFI) (Corning, cat. no. 25–055-CV)
Tyrode’s solution (Sigma, cat. no. T2397)
Recombinant human albumin (ScienCell, cat. no. OsrHSA-100)
CHIR99021 (Thermo Fisher Scientific, cat. no. 508306)
Wnt-C59 (Biorbyt, cat. no. orb181132)
Rho kinase (ROCK) inhibitor (Tocris, cat. no. 1254)
0.5 M EDTA (Thermo Fisher Scientific, cat. no. 15575020)
DMSO (Sigma-Aldrich, cat. no. D-2650)
TrypLE Express (Thermo Fisher Scientific, cat. no. 12605010)
FBS (Thermo Fisher Scientific, cat. no. 10438026)
PrestoBlue cell viability reagent (Thermo Fisher Scientific, cat. no. A13261)
Wheat germ agglutinin–Alexa Fluor 488 conjugate (Thermo Fisher Scientific, cat. no. W11261)
Calcein-AM (Thermo Fisher Scientific, cat. no. C3100MP)
Trypan blue dye (Thermo Fisher, cat. no. 15250061)
Equipment
Standard cell culture laminar flow hood (e.g., Labconco, Logic+)
Cell culture centrifuge with optional plate adapter (e.g., Thermo Fisher, Heraeus Multifuge X3)
Standard 37 °C, 5% CO2 cell culture incubator (e.g., Thermo Fisher, Heracell 150i)
Tissue culture microscope (e.g., Leica Microsystems, DMi 1)
Multichannel Pipetman (e.g., Thermo Fisher, Finnpipette Novus)
Multichannel aspirator (e.g., Fisher Scientific, Corning Costar)
Hemocytometer or automated cell counter for cell counting (e.g., Thermo Fisher, Countess II)
CoolCell LX freezing container (Sigma-Aldrich, cat. no. BCS-405)
–80 °C Ultra-low-temperature freezer (e.g., Thermo Fisher, model no. Forma 88000)
Liquid nitrogen storage (e.g., Thermo Fisher, CryoPlus model)
High-throughput plate imaging/reading system (e.g., BioTek, model no. Cytation 5)
CRITICAL A high-throughput imaging system for plate imaging and reading of hiPSC-CMs with and without drug treatment is a critical requirement for obtaining viability measurements. We recommend a plate reader/imager system such as the BioTek Cytation 5, which can hold 384-well plates optimized for high-throughput analysis.Epifluorescence microscope (e.g., Zeiss, model no. LSM 510Meta confocal microscope)
High-throughput contractility assessment platform (e.g., Vala Sciences, model no. IC200 KIC)
CRITICAL A high-throughput system should ideally be used for assessing changes in hiPSC-CM contractility in response to drug treatment. For example, the IC200 KIC is a high-content screening system with the capability of obtaining high-speed time-series images and is optimal for contractility analysis. It is also possible to use an alternative commercially available screening platform to measure contractility, such as the CelOptiq (Clyde Biosciences) or xCELLigence (ACEA Biosciences).
CRITICAL A simplified version of the CSI can be calculated without the use of a high-throughput contractility assessment platform by using only the CoB and LD50 metrics for a given drug.Statistical analysis software (e.g., GraphPad Prism, https://www.graphpad.com/scientific-software/prism/)
6-, 12-, 24-, and 96-well tissue culture-treated plates (Corning)
Imaging optimized 384-well plates (Greiner Bio-One, cat. no. 781091)
15- and 50-mL conical centrifuge tubes (e.g., BD Falcon)
500-mL cell culture medium filters (Corning, cat. no. 431118)
Disposable plastic pipettes and tips (USA Scientific)
Nunc biobanking and cell culture cryogenic tubes (Fisher Scientific, cat. no. 375418)
70-μm cell strainers (Corning, cat. no. 352350)
12 × 12 mm glass coverslips (VWR, cat. no. 89015–725)
Reagent reservoir (Thermo Fisher Scientific, cat. no. 95128095)
Reagent setup
Chemically defined differentiation basal medium
To make chemically defined differentiation basal medium (CDM3 basal medium), first make aliquots of the CDM3 supplement. To make the CDM3 supplement, slowly add 10.56 g of L-ascorbic acid 2-phosphate to 500 mL of WFI water, mixing occasionally until the solution is transparent. To this solution, add 25 g of recombinant human albumin and mix until totally dissolved. Filter-sterilize the mixture and make 5-mL CDM3 supplement aliquots in 15-mL conical tubes, which can be frozen at –20 °C for 6 months. To make a stock of CDM3 basal medium for hiPSC-CM differentiation or maintenance, add one aliquot of CDM3 supplement to 500 mL of RPMI 1640 medium containing glucose. This medium is stable for up to 1 month at 4 °C. As referred to throughout this protocol, CDM3 basal medium contains glucose unless otherwise specified. See Troubleshooting section.
Chemically defined differentiation basal medium, low glucose
To make a stock of CDM3 basal medium, low glucose (CDM3-L) for hiPSC-CM purification, add one aliquot of CDM3 supplement to 500 mL of glucose-free RPMI 1640 medium. Then add 4 mM L-lactic acid. This medium is stable for up to 1 month at 4 °C19.
10 mM Y27632 rho kinase inhibitor
Prepare 10 mM stocks of Y27632 rho kinase inhibitor dissolved in water. These stocks can be kept at –80 °C for up to 6 months.
10 mM CHIR99021 GSK3β inhibitor stock
Prepare 10 mM CHIR99021 stocks in DMSO. The DMSO should ideally be obtained fresh from small aliquots, especially if used as a drug solvent. These stocks can be kept at –80 °C for up to 6 months.
10 mM Wnt-C59 Wnt inhibitor stock
Prepare 10 mM Wnt-C59 stocks in DMSO. The DMSO should ideally be obtained fresh from small aliquots, especially if used as a drug solvent. These stocks can be kept at –80 °C for up to 6 months.
Matrigel-coated plates
Thaw Matrigel extracellular matrix at 4 °C overnight or on ice. Cool all pipettes and pipette tips to 4 °C before pipetting cold Matrigel, to prevent premature solidification of the Matrigel. Gently add 125 μL of ice-cold Matrigel to 50 mL of ice-cold DMEM/F12, at a dilution of 1:400. Add ~2 mL of the Matrigel solution in DMEM/F12 to each well of a six-well dish. Make multiple Matrigel plates for downstream passaging steps. Allow the Matrigel solution to coat the bottom of each well for minimum of 1 h before using the plate. Typically, the Matrigel-coated plates can be stored in a standard cell culture incubator at 37 °C and 5% CO2 for 2 weeks. CRITICAL Before adding the hiPSCs, immediately before use, ensure that the Matrigel mixture is aspirated from the six-well plate.
10 mM stocks of drugs of interest
Dissolve drugs for testing in the appropriate solvent, ideally water or DMSO, per the manufacturer’s recommendations. These stocks can usually be kept at –80 °C for up to 6 months.
Procedure
Maintenance of hiPSCs  Timing 1 week
Timing 1 week
-
1
Resuspend 1 × 106 low-passage hiPSCs (at or below passage 20) either fresh or from thawed vials in 2 mL of E8 medium with 10 μM rho kinase inhibitor.
CRITICAL STEP Rho kinase inhibitor markedly improves survival of pluripotent stem cells during plating and passaging46. -
2
Plate resuspended hiPSCs in a single well of a Matrigel-coated six-well plate by gently pipetting them in a circular motion around the well. Rock the plate horizontally once and then vertically once to distribute the cells evenly in the well. Return the cells to the 37 °C incubator and do not disturb for 24 h.
-
3
At 24 h after plating hiPSCs, observe the cells. Some cell death will probably occur, but many hiPSCs should have stuck to the culture plate. At this point, the hiPSCs should have a spread-out morphology due to treatment with rho kinase inhibitor.
-
4
Aspirate the medium and replace it with 2 mL of regular E8 medium containing no rho kinase inhibitor. Return the cells to the 37 °C incubator.
-
5
Continue daily medium change with E8 medium until the hiPSCs become 80–100% confluent.
CRITICAL STEP Do not allow the hiPSCs to become overconfluent, as this may interfere with downstream differentiation and expression of pluripotency genes. Typically, cells should be passaged when they are in the logarithmic phase of growth. When the hiPSCs reach the optimal confluency for passaging, passage one well from a six-well plate of hiPSCs in a 1:10 ratio, as described below. This passage will maintain the cells for future differentiations. Optimal confluency for hiPSC-CM differentiation may vary based on the hiPSC line. Figure 2 and its accompanying legend give details of how immunofluorescence staining for NANOG and Tra-1–81 can be used to confirm hiPSC identity. -
6
When hiPSCs achieve optimal confluency, prepare 25 mL of E8 medium with 10 μM rho kinase inhibitor.
-
7
Aspirate the Matrigel from the wells of a new six-well plate pre-coated with Matrigel. Add 1 mL of fresh E8 medium with 10 μM rho kinase inhibitor to each well of this new six-well plate.
-
8
Begin passaging by aspirating the E8 medium from an ~80% confluent well of hiPSCs. Add 1 mL of 0.5 mM EDTA in PBS to this well of hiPSCs. Incubate the plate at 37 °C for 5 min to partially dissociate the hiPSCs.
-
9
Very gently, aspirate the 1 mL of 0.5 mM EDTA.
CRITICAL STEP The hiPSCs should not be completely dissociated from the well. When observed under a microscope after EDTA addition, the hiPSC colonies should be lightly adhered to the plate but breaking apart into individual cells. -
10
With a 1,000-μL manual Pipetman, spray off the EDTA-loosened hiPSCs with 1 mL of new E8 medium containing 10 μM rho kinase inhibitor. Move the resuspended hiPSCs to a new 15-mL conical tube.
-
11
Add 9 mL of E8 medium with 10 μM rho kinase inhibitor to the conical tube to obtain a 1:10 dilution of cells.
-
12
To each well of the newly prepared six-well plate, add 1 mL of the 1:10 dilution of hiPSCs resuspended in E8 medium containing 10 μM rho kinase inhibitor. Return the plate to the 37 °C incubator for culture.
CRITICAL STEP Evenly distribute the cells in the new six-well plate by pipetting in a circular motion around each well. Rock the plate vertically and horizontally once to help evenly distribute the cells. We recommend not touching the plate for 5 min after passaging, so that cells can begin adhering to the bottom of the well. -
13
After 24 h, replace the E8 medium with rho kinase inhibitor in every well of this plate with 2 mL of E8 medium without rho kinase inhibitor.
-
14
Change the replacement E8 medium daily for ~4 d, at which point the cells should be ready to passage again (as described in Steps 6–13) or for differentiation (described in the following steps).
 TROUBLESHOOTING
TROUBLESHOOTING
Chemically defined differentiation of hiPSCs to hiPSC-CMs Timing 2 weeks
-
15
To begin hiPSC differentiation, remove the E8 medium from a well of 80–100% confluent hiPSCs (Fig. 2). Add 2 mL of fresh CDM3 basal medium, supplemented with 6 μM CHIR99021, to each well that will be differentiated. Return the cells to 37 °C and do not disturb for 48 h.
CRITICAL STEP The optimal starting confluency (typically 85–100%) and CHIR99021 concentration (typically 6–12 μM) can vary among hiPSC lines. We recommend optimizing these conditions for each hiPSC line being differentiated. -
16
After 48 h of treatment with CHIR99021 (day 2 of differentiation), remove old CDM3 basal medium containing CHIR99021 and substitute it with 2 mL of fresh CDM3 basal medium containing 5 μM small-molecule Wnt inhibitor Wnt-C59 dissolved in DMSO. Leave differentiating hiPSCs in this medium for 48 h.
-
17
After 48 h (day 4 of differentiation), remove the CDM3 basal medium containing Wnt-C59 and replace it with fresh CDM3 basal medium without any small molecules.
-
18
Continue changing 2 mL per well of the CDM3 basal medium with fresh CDM3 basal medium without any small molecules every 48 h. Beating hiPSC-CMs should become apparent between days 8 and 10 of differentiation.
TROUBLESHOOTING -
19
At day 10 of differentiation, metabolically purify the hiPSC-CMs from the non-CMs. To do this, remove old CDM3 basal medium and replace with 2 mL of CDM3-L. Leave the hiPSC-CMs in this medium for 48 h, replacing the medium with CDM3-L every 48 h as needed (Fig. 2).
CRITICAL STEP Apoptosis of cells that are not hiPSC-CMs will begin after 24 h of treatment with CDM3-L. The hiPSC-CMs may cease contracting, and there may still be some hiPSC-CM apoptosis due to the metabolic immaturity of these cells.
TROUBLESHOOTING -
20
Stop the metabolic selection process by aspirating off the CDM3-L and add CDM3 basal medium (medium containing glucose) after purification is complete (usually after 2–4 d) or if there is a large amount of hiPSC-CM death.
CRITICAL STEP The metabolically purified hiPSC-CMs can be replated for downstream contractility and cytotoxicity analysis. -
21
Continue medium changes every 2 d with 2 mL per well of CDM3 basal medium containing glucose until the cells are 15–20 d old.
CRITICAL STEP
Figure 2 and its accompanying legend give details of how immunofluorescence staining for cardiac troponin T (cTnT) and alpha-actinin can be used to confirm that CMs have been successfully differentiated.
Replating and growing hiPSC-CMs for high-throughput cytotoxicity and contractility assays Timing 2 weeks
CRITICAL Once the hiPSC-CMs are purified, they can be replated into a 384-well plate for high-throughput cytotoxicity and contractility analysis. We recommend replating hiPSC-CMs between days 15 and 20 of differentiation for best survival. Cells older than day 15 are optimal because this is soon after CDM3-L selection, and cells younger than day 20 tend to be easier to dissociate due to less collagen secretion.
-
22
Prepare a 384-well plate to receive hiPSC-CMs for high-throughput cytotoxicity and contractility assays by adding, to each well, 25 μL of DMEM/F12 with 100 μg/mL (or alternatively a 1:200 dilution) of Matrigel. Allow the plate to coat for at least 1 h at 37 °C.
CRITICAL STEP As usual, when preparing Matrigel, ensure that all materials encountering Matrigel are precooled. -
23
Prepare hiPSC-CM passaging medium by combining 80 mL of CDM3 basal medium (with glucose) with 20 mL of FBS and 10 μM rho kinase inhibitor in a new bottle. Filter-sterilize this solution, using a vacuum filter with a 0.22-pm filter. Warm this passaging medium containing FBS and rho kinase inhibitor at 37 °C for 1 h.
CRITICAL STEP As an alternative to FBS, chemically defined or serum-free alternative supplements can be used, as FBS has the potential to alter hiPSC-CM physiology due to an abundance of growth factors and other undefined materials. Note that we recommend using FBS only during these hiPSC-CM passaging steps to aid with cell survival. After passaging, return hiPSC-CMs to CDM3 basal medium. -
24
Prepare TrypLE dissociation reagent by adding 10 mL of TrypLE to a 15-mL conical tube and warming it at 37 °C for 1 h.
-
25
After all reagents have been warmed, select enough wells of hiPSC-CMs for passaging, keeping in mind that up to 1 × 107 cells may be required to fill every well of a 384-well plate at 2.5 × 104 cells per well.
-
26
Wash each well of hiPSC-CMs to be passaged with 1 mL of PBS. Remove the PBS after the wash.
-
27
Add 1 mL of warm TrypLE per well of hiPSC-CMs and leave the cells at 37 °C for 5 min.
-
28
After 5 min, add 1 mL per well of warm hiPSC-CM passaging medium containing 20% (vol/vol) FBS and rho kinase inhibitor to deactivate the TrypLE. Begin manually dissociating the hiPSC-CMs from the six-well plate with a 1,000-μL Pipetman.
CRITICAL STEP Owing to extracellular matrix deposition, it may require up to 50 pipetting repetitions to ensure that the hiPSC-CMs are adequately dissociated, and more repetitions may be needed to dissociate older cells. As an alternative to FBS, chemically defined or serum-free alternative supplements can be used, as FBS has the potential to alter hiPSC-CM physiology due to an abundance of growth factors and other undefined materials. -
29
Place all dissociated hiPSC-CMs in a 15-mL conical tube, and centrifuge at 200g for 5 min at room temperature (22 °C). Discard the supernatant and suspend the hiPSC-CMs in 10 mL of fresh, warmed hiPSC-CM passaging medium containing FBS and rho kinase inhibitor.
CRITICAL STEP Rigorously pipette the hiPSC-CMs to make sure that they are as close to singlecell format as possible. Optionally, a 70-μm cell strainer can be used to further dissociate hiPSC-CMs into single cells. -
30
Use a hemocytometer or automated cell counter to determine the number of dissociated hiPSC-CMs resuspended in passaging medium.
-
31
Use a multichannel aspirator to remove the Matrigel diluted in DMEM/F12 from the 384-well plate (s) that will receive hiPSC-CMs for downstream assays.
-
32
For cytotoxicity and contractility assays, use a multichannel pipettor to add 50 μL of medium containing 2.5 × 104 hiPSC-CMs to each well of a 384-well plate.
-
33
Return the hiPSC-CMs plated in the 384-well plate to 37 °C, and do not disturb for 48 h.
TROUBLESHOOTING -
34
After 48 h have passed, replace the hiPSC-CM passaging medium with fresh CDM3 basal medium containing glucose.
-
35
Keep replacing the CDM3 basal medium every 48 h until day 30, at which point the cells are ready for drug treatment (Fig. 3a).
Drug treatment of hiPSC-CMs replated into high-throughput plates Timing 3 d
CRITICAL As an alternative to differentiation (Steps 1–35), day-30 hiPSC-CMs can be obtained from commercial or academic sources such as the Stanford University Cardiovascular Institute Biobank and thawed directly into high-throughput plates for drug treatment. To thaw hiPSC-CMs, quickly resuspend unfrozen cells in fresh, pre-warmed hiPSC-CM passaging medium (sterile-filtered CDM3 basal medium with 20% FBS, or a serum-free alternative, and 10 μM rho kinase inhibitor). Spin the cells in a 15-mL conical tube at 200g for 5 min at room temperature (22 °C), aspirate the supernatant, and then resuspend the pellet in fresh hiPSC-CM passaging medium. Count the cells with a hemocytometer, and then plate 50 μL of medium containing 2.5 × 104 hiPSC-CMs in each well of a 384-well plate.
-
36
Warm 50 mL of CDM3 basal medium at 37 °C for 1 h before beginning drug dilutions.
TROUBLESHOOTING -
37
Using a blank 96-well plate and a multichannel pipettor, prepare a serial dilution of the drugs of interest in CDM3 basal medium by diluting from 100 μM in half-log increments down to 0 μM (Fig. 3b).
CRITICAL STEP For the drugs of interest, note the EC50, which represents the concentration of a drug of interest that gives the half-maximal response. Verify that this value falls within the dose response being tested on hiPSC-CMs. Always include a negative control serial dilution for the solvent in which the drugs are dissolved (e.g., DMSO, water). The advantage of a 384-well plate is that on a single plate, up to 24 unique drugs can be tested with technical duplicates for eight halflog concentrations. The range for 0–100 μM is ideal for drugs for which the EC50 is >1 μM and <10 μM. However, in a scenario in which a drug’s EC50 value does not fall within this range, the dosing range should be changed accordingly. -
38
Aspirate all medium from the 384-well plate containing beating day-30 hiPSC-CMs. Use separate 384-well plates for cytotoxicity and contractility assessment.
-
39
Add the drugs of interest in 25 μL of CDM3 basal medium per well, serially diluted in half-log concentrations ranging from 0 to 100 μM. We recommend conducting drug treatment on hiPSC-CMs in at least three technical replicate wells to enhance reproducibility in downstream contractility and cytotoxicity analyses.
-
40
Return the hiPSC-CMs plated in the 384-well plate to 37 °C for the duration of the drug treatment period.
CRITICAL STEP The time for drug treatment can vary based on preference and the study being conducted. In our prior study examining kinase inhibitor-induced cardiotoxicity, we treated hiPSC-CMs with drugs for 72 h before conducting cytotoxicity and contractility assessment10.
Quantitative drug-induced cytotoxicity assessment of hiPSC-CMs Timing 1 d
-
41After treating hiPSC-CMs with the drugs of interest, begin a quantitative cytotoxicity assessment (Fig. 3c, option A) or a qualitative imaging-based assessment of drug-induced cytotoxicity (Fig. 3c, option B) of one drug-treated 384-well plate. Option A is preferred for most instances when quantitative toxicity measurements are required, as this enables the derivation of downstream LD50 values. Option B is primarily used as a qualitative means of confirming quantitative toxicity results from option A.
-
(A)Quantitative cytotoxicity assessment
-
(i)Add 2.5 μL of PrestoBlue reagent directly to each well of a 384-well plate containing hiPSC-CMs treated with 25 μL of drug dissolved in CDM3 basal medium.
-
(ii)Return the hiPSC-CMs treated with PrestoBlue reagent to 37 °C for 2 h.CRITICAL STEP After treatment with PrestoBlue reagent, wells with a higher number of living cells will shift in color from blue to red due to the reducing environment of viable cells.
-
(iii)After the 2-h incubation, use a high-throughput plate reader (e.g., BioTek Cytation 5) to quantify the fluorescence intensity of PrestoBlue-treated wells containing hiPSC-CMs and drugs of interest. Use an excitation/emission range of 560/590 nm when measuring fluorescence.
-
(iv)For each drug, normalize dose responses by dividing the fluorescence intensity value at all drug concentrations by the fluorescence intensity at the 0 μM (untreated) condition. This should give values ranging from 0 to 1, where 0 is complete cell loss due to drug and 1 is no loss in cell viability.
-
(v)Using a curve-fitting software such as GraphPad Prism, generate a sigmoidal LD50 dose–response curve that will be able to provide the LD50 value for a specific drug in hiPSC-CMs.CRITICAL STEP A variety of curve-fitting equations can be used to generate LD50 values. We recommend a model such as the following.
In this model, top and bottom are asymptotic plateaus in the units of the y axis, typically 1 and 0, respectively, in this scenario. LD50 is the concentration of drug that gives a response halfway between the bottom and top values. HillSlope describes the steepness of the curve. A HillSlope of –1.0 is standard, whereas a HillSlope more negative than –1 is steeper.
TROUBLESHOOTING
-
(i)
-
(B)Qualitative imaging-based assessment of drug-induced cytotoxicity
-
(i)Resuspend 0.5 μM calcein-AM live imaging dye and 2 pg/mL Hoechst nuclear stain in PBS.
-
(ii)Incubate this dye suspension with post drug-treated hiPSC-CMs for 10 min before imaging at a 377/447 excitation/emission spectrum on a fluorescence microscope.
-
(iii)After imaging, aspirate the dye suspension and replenish the cells with fresh CDM3 basal medium before proceeding to PrestoBlue-based quantitative cytotoxicity analysis (option A).
-
(i)
-
(A)
Quantitative drug-induced contractility assessment of hiPSC-CMs Timing 1 d
CRITICAL It may not possible to conduct advanced contractility analysis to determine the EC metric described below if access to a high-throughput contractility platform such as the KIC or a highresolution fluorescent microscope with an integrated camera is not available. However, a simplified contractility analysis can still be performed. To do this, observe the drug-treated hiPSC-CMs and determine the drug concentration at which a majority of drug-treated replicate wells cease beating. Designate this concentration as the CoB concentration and skip directly to the next subsection (Steps 54–60) to calculate a simplified version of the CSI.
-
42
Prepare fresh imaging solution by diluting Hoechst 33342 to 2 μg/mL and WGA Alexa Fluor 488–conjugated dye to 5 μg/mL in Tyrode’s solution.
TROUBLESHOOTING -
43
Use a multichannel aspirator to aspirate all medium from each well of a second 384-well plate containing hiPSC-CMs. Ideally, this should be a separate 384-well plate from the one used for cytotoxicity assessment.
-
44
Add 25 μL of imaging solution per well to the hiPSC-CMs and incubate them at 37 °C and 5% CO2 for 15 min.
-
45
Use a multichannel aspirator to aspirate all medium from each well of the 384-well plate and add 25 μL of Tyrode’s solution to wash the cells; then incubate the plate at 37 °C and 5% CO2 for 15 min before imaging.
-
46
Using a high-throughput imaging system such as the IC200 KIC; record a 6.5-s time series of contracting hiPSC-CMs at 100 Hz at 20× magnification per well of a 384-well plate (Fig. 3d).
TROUBLESHOOTING -
47
Calculate the deformation vector map by PIV37. From a recorded hiPSC-CM video of interest, first select a reference frame representing hiPSC-CM diastolic relaxation, or the ‘non-contracted’ state. This can be done by running PIV and using the previous frame as reference, resulting in a measurement of the instantaneous contraction/relaxation velocity vector map for hiPSC-CMs. There are several well-documented open-source PIV algorithms that can be downloaded from repositories (e.g., OpenPIV, http://www.openpiv.net/)42. We recommend an interrogation window size twice the nominal size in each direction to speed up this part of the algorithm (we used 128 × 128 pixels with a 64-pixel overlap in our previous experiments). The spatially averaged value of the magnitude of the velocity fields measures the overall motion of a sheet of hiPSC-CMs at a single moment in time. The resulting signal presents two periodic peaks corresponding to an hiPSC-CM contraction and relaxation cycle. Between consecutive contraction cycles, hiPSC-CMs are mostly at rest. These rest periods can be identified to obtain multiple reference frames.
-
48
Run the PIV routine again using the reference frame selected in Step 47 to obtain deformation fields at nominal resolution (i.e., with a smaller interrogation window) to obtain finer spatial resolution and more accurate deformation measurements. Although we previously used a nominal interrogation window size of 64 × 64 pixels with a 32-pixel overlap, this parameter can be dependent on image magnification, image quality (e.g., speckle size, spatial distribution, and brightness), time resolution, magnitude of the deformations, and so on. We recommend performing window size sensitivity analyses with prescribed synthetic deformation fields.
-
49
Use the cell deformation vector maps obtained from PIV to apply Gauss’s divergence theorem to automatically quantify cell contractility as the relative change in area of beating hiPSC-CMs. Divergence, mathematically, is defined as du/dx + dv/dy, where u is the deformation in the x direction and v is the deformation in the y direction. In other words, compute spatial derivatives that numerically (i.e., with a computer) are obtained with finite differences. Gauss’s divergence theorem states that divergence is equal to the relative change of area. Contractility signals can be obtained as the spatial average of the magnitude of relative change in area, resulting in periodic signals with one peak per cycle whose magnitude is proportional to the overall axial tension of the beating hiPSC-CMs.
CRITICAL STEP Several contractility parameters can be extracted from these signals to perform quantitative and statistical comparisons. To improve the robustness of the parameter retrieval, all the peaks present in one contractility signal can be collapsed into a most-representative peak by performing time-dependent conditional averaging. In layman terms, time-dependent conditional averaging means to align all the peaks and then obtain an ‘averaged’ peak. As a by-product of conditional averaging, the mean time between contractility peaks (Tpeak) and the contractility parameter can be obtained. Additional contractility parameters (Trise, Tfall, TCT, Dpeak, Dvalley, Dhigh, Dlow, contraction rate, and relaxation rate) can be measured from the single-cycle contractility profile obtained after conditional averaging. -
50
Determine the first cardiac safety coefficient, CoB, by calculating the drug concentration at which >50% of drug-treated, replicate hiPSC-CM wells stop contracting.
-
51
Determine the EC (for the ith metric (ECi)). This is the concentration that demonstrates a statistically significant departure (P < 0.05) from the contractility rate at the vehicle negative control condition. Note that i = 1, ..., M; M is the number of contractility parameters (e.g., Tpeak, Trise, Tfall, TCT, Dpeak, Dvalley, Dp2v, contraction rate, and relaxation rate) obtained from the contractility analysis.
CRITICAL STEP If it is not possible to conduct high-throughput contractility analysis to obtain the EC, a simplified version of the CSI can be obtained by averaging only CoB and LD50 viability. We refer to this simplified version of the CSI as CSI-Lite and believe that it may prove useful to those without access to advanced high-throughput contractility analysis platforms. Refer to Step 59 for details. -
52
Determine the amplitude of the effect (AEi), which quantifies the magnitude of such departures from baseline (vehicle negative control) as the log2 of the ratio of EC; (or effective value (EVi)), to the value at baseline value (BVi). Again, i = 1, ..., M; M is the number of contractility parameters (e.g., Tpeak, Trise, Tfall, TCT, Dpeak, Dvalley, Dp2v, contraction rate, and relaxation rate) obtained from the contractility analysis.
Average AE is obtained by averaging all AEi values.
-
53
Determine the average EC, which is a weighted average of all ECs, using each respective AE as weight.
The reasoning behind this average EC formula is that it considers ECs associated with all contractility parameters (e.g., Tpeak, Trise, Tfall, TCT, Dpeak, Dvalley, Dp2v, contraction rate, and relaxation rate) and introduces a bias for the ECs corresponding to parameters that most prominently alter normal cell behavior.
Calculation of the cardiac safety index from cytotoxicity and contractility parameters Timing 1 d
-
54
If possible, obtain the maximum blood plasma concentration (Cmax) experienced either in preclinical pharmacokinetic studies using animal models or in clinical trials by patients receiving the drug being tested (Fig. 4a). If the drug being tested has been approved by the FDA, Cmax information can be found in the ‘clinical pharmacology and biopharmaceutics review’ document associated with the drug and published by the FDA Center for Drug Evaluation and Research. This information can be found online at https://www.fda.gov. In the data we show here, we utilize total Cmax values, although it should be noted that free Cmax values can also be used.
CRITICAL STEP The CSI is most accurately calculated by first normalizing the EC, AE, CoB, and LD50 values to the Cmax for a drug being tested on hiPSC-CMs. -
55To calculate the CSI, first divide the CoB contractility value of a drug by the Cmax for the drug to obtain a value corrected for the maximum drug concentration observed in vivo, either in preclinical models or in clinical trials. This corrected CoB is referred to as .
-
56Next, divide the EC contractility value of the drug by the Cmax for the drug to obtain a value corrected for the maximum drug concentration observed in vivo. This corrected EC is referred to as β If a high- throughput contractility analysis platform is not available to generate the EC, skip this step.
-
57Divide the corrected EC (β) by the AE of the drug to obtain an updated corrected EC (δ). If a high- throughput contractility analysis platform is not available to generate the EC, skip this step.
-
58Divide the LD50 hiPSC-CM cytotoxicity value associated with the drug by the Cmax for the drug to obtain a value corrected for the maximum drug concentration observed in vivo. This corrected LD50 value is referred to as γ.
-
59Obtain the absolute CSI for a drug of interest by averaging the a, 5, and y values for that drug. This CSI is an absolute value, specific to one drug.
.CRITICAL STEP If a high-throughput contractility analysis platform is not available to generate the EC, then a simplified CSI (CSILITE) can be calculated by averaging α and γ.
-
60In a large dataset of drugs with pre-calculated CSIs, the relative cardiotoxicity of a drug can be compared to the cardiotoxicity of other drugs in the dataset simply by comparing CSIs. To perform this comparison, derive a CSIN value for each drug in a dataset that normalizes the absolute CSI value of a drug by the maximum CSI value observed in a drug dataset. This CSIN value provides a comparative value between 0 and 1 (Fig. 4b). To derive the CSIN value for a drug, first obtain the normalized CoB (αN), normalized EC (δN), and normalized LD50 viability (γN) for that drug by comparing the absolute α, δ, and γ of the drug to the maximum α, δ, and γ values, respectively, observed in the dataset.A CSIN value is obtained by averaging αN, δN, and γN. This will give a value between 0 and 1, where a value close to 0 indicates a cardiotoxic drug and a value close to 1 indicates a safe drug. This value can be used as a relative metric to evaluate the cardiotoxicity of drugs in a dataset.CRITICAL STEP If it is not possible to conduct high-throughput contractility analysis to obtain the normalized EC (δN), a simplified version of the CSIN can be obtained by averaging only normalized CoB (αN) and normalized LD50 viability (γn). We refer to this simplified version of the CSIN as CSINLITE and believe that it may prove useful to those without access to advanced high- throughput contractility analysis platforms (Fig. 4c).
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2|. Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| Reagent setup | No hiPSCs produced during reprogramming | Low-quality reprogramming virus and reagents | Use fresh reprogramming virus and reagents; use a higher MOI or more PBMCs during reprogramming; use a hypoxia incubator during reprogramming |
| 14 | Spontaneously differentiating hiPSCs | Low-quality hiPSC lines with possible genetic irregularities | Scrape and repick hiPSC colonies with ideal morphologies; check passage number and expiration date of E8 medium |
| 18 | No hiPSC-CMs produced during differentiation | Line-to-line variability in hiPSC differentiation capacity | Try different concentrations of Wnt agonist (6–24 μM) CHIR99021; alter the starting confluency for hiPSC differentiation |
| 19 | Excessive hiPSC-CM death during glucose starvation selection process | hiPSC-CMs are somewhat immature and may not have completely shifted metabolically to utilizing lactate | End glucose starvation early for hiPSC-CMs; subject the cells to glucose starvation for a maximum of 2 d only if the cells cannot tolerate this environment |
| 33 | hiPSC-CMs do not survive well when replated into high-throughput screening plates | hiPSC-CM survival is dependent on cell confluency and extracellular matrix deposition | Use younger hiPSC-CMs for replating; increase Matrigel concentration; replate hiPSC-CMs at higher confluency; do not disturb hiPSC-CMs for 48 h after replating |
| 36 | Substantial hiPSC-CM death when thawing frozen hiPSC-CMs | Substantial cell loss is a known problem when thawing hiPSC-CMs | Use >10% FBS when thawing hiPSC-CMs; thaw at a higher confluency; use rho kinase inhibitor while thawing |
| 41A(v) | High background during fluorescence imaging for drug-induced hiPSC-CM cytotoxicity assays |
Plastic plates cause autofluorescence | Use low-autofluorescence multi-well imaging plates |
| 42 | Unable to compute the effective concentration metric for assessing hiPSC-CM contractility | Unable to gain access to a high-throughput contractility analysis platform such as the KIC | A simplified CSI (CSI-Lite) uses only the CoB concentration for assessing drug-induced alterations in hiPSC-CM contractility. Drug concentration causing CoB can be easily foun simply by looking at drug-treated wells |
| 46 | High well-to-well variability in hiPSC-CM contractility | Slightly different numbers of hiPSC-CMs plated per well or impure cell populations | Increase the number of technical replicates for contractility analysis; purify hiPSC-CMs for a longer time before replating; utilize well-specific baseline controls instead of vehicle controls |
Timing
Steps 1–14: maintenance of hiPSCs: 1 week
Steps 15–21: chemically defined differentiation of hiPSCs into hiPSC-CMs: 2 weeks
Steps 22–35: replating and growing hiPSC-CMs for high-throughput cytotoxicity and contractility assays: 2 weeks
Steps 36–40: drug treatment of hiPSC-CMs replated into high-throughput plates: 3 d
Step 41: quantitative drug-induced cytotoxicity assessment of hiPSC-CMs: 1 d
Steps 42–53: quantitative drug-induced contractility assessment of hiPSC-CMs: 1 d
Steps 54–60: calculation of the CSI from cytotoxicity and contractility parameters: 1 d
Anticipated results
Using the approaches described here, a CSI for evaluating drug cardiotoxicity can be readily obtained with data derived from high-throughput hiPSC-CM-based cytotoxicity and contractility assays. Although we do not describe it in detail here, the process for generating hiPSCs has been well established and should take ~1 month to complete45. These cells can be either produced in-house or obtained from commercial or academic sources. One advantage to creating hiPSCs in-house is patient specificity, as the hiPSCs created will have the same genotype as the patient from whom they came. This allows for possible identification of patient-specific drug responses using hiPSC-CMs, as has been previously described27. However, hiPSCs should be adequately maintained upon production. Specifically, newly obtained hiPSCs should not be allowed to grow to overconfluency while maintaining these cells, as this may lead to cell senescence or reduced pluripotency gene expression. In addition, if the hiPSCs exhibit signs of spontaneous differentiation, these non-hiPSCs should be scraped away or a new set of hiPSCs should be used. Ideally, differentiation experiments should start with low-passage hiPSCs (under passage 50), as extended hiPSC maintenance may lead to chromosomal abnormalities. To test for potential chromosomal or other abnormalities due to long-term passaging, hiPSCs should be regularly karyotyped or genetically characterized using a commercially available pluripotency single-nucleotide polymorphism (SNP) array.
After the establishment of stable hiPSC lines, these cells can be differentiated into a variety of somatic cell types, including CMs. It is anticipated that the hiPSC-CM differentiation process will take ~2 weeks (Fig. 2). The early stages of differentiation involving small-molecule modulation of Wnt signaling are typically the most critical. Within the first 4 d, there should be an observable change in hiPSC morphology, indicating that the hiPSCs are successfully responding to the small-molecule treatment. Cells should begin beating as early as 1 week after initiating differentiation. If no beating cells are observed by day 10 post differentiation, then the results from that round of differentiation are unlikely to yield hiPSC-CMs, and a new differentiation round should be started. However, poorly differentiated hiPSC-CMs could still be purified via lactate selection and glucose starvation, which can increase the percentage of viable hiPSC-CMs to be used for downstream assays. Note that the number of cells required for high-throughput assays can be quite substantial (up to 10 million per full 384-well plate), and therefore multiple six-well plates containing hiPSCs should be differentiated into hiPSC-CMs. Purified hiPSC-CMs should be ready to replate as early as day 15 post differentiation, and we highly recommend replating hiPSC-CMs as early as possible to minimize collagen secretion. Recent advances in the chemically defined differentiation approach for hiPSC-CM production have greatly simplified differentiation protocols, enabling the reproducible production of cells for drug screening assays19,47,48. Undefined factors in earlier protocols might have been responsible for variable efficiencies of differentiation and might also alter hiPSC-CM viability or contractility in response to drug treatment. Physiological limitations of the hiPSC-CMs made using current protocols include a relatively high resting potential, a low sodium channel density, and an underdeveloped sarcoplasmic reticulum and T-tubule network; circumventing these limitations, as well as correcting metabolic and mechanical properties of hiPSC-CMs, is the focus of ongoing studies41. As improved differentiation strategies emerge, they will likely increase the fidelity of the CSI protocol described herein.
Freshly produced or frozen hiPSC-CMs can be replated into a high-throughput format, although these cells should be replated at high confluency for optimal survival (Fig. 3a). We recommend up to 25,000 cells per well of a 384-well plate to ensure full confluency and best survival post passage. Cells should begin beating within 48 h after passage. For drug treatments, a 384-well plate is preferred because it allows for up to 24 drugs to be tested in duplicate on a single plate, with up to eight drug concentrations per replicate (Fig. 3b). Using the maximum number of technical and biological replicate experiments is highly recommended to obtain the most accurate data possible. It should be noted that there is known batch-to-batch and cell line-to-cell line variability in hiPSC-CM differentiation and culture, and such differences should be tested and verified on the basis of the hiPSC-CM differentiation protocol being used49. To obtain drug-induced cytotoxicity measurements of hiPSC-CMs, a variety of fluorescence-, luminescence-, or absorbance-based assays are available. These assays should allow for the rapid quantification of drug-induced cytotoxicity in hiPSC-CMs, and from these quantitative data, drug dose-response curves can be generated with statistical analysis programs (Fig. 3c). These curves should allow for the identification of LD50 values that will indicate a standard metric reflecting the general cytotoxicity of a drug on hiPSC-CMs. Likewise, drug-induced contractility measurements can also be obtained for hiPSC-CMs replated in a high-throughput format. If one has access to a high-throughput microscope, high-resolution cell displacement measurements can be made after cells are labeled with fluorescent dyes such as WGA. Modifications to the protocol that provide an automated algorithm for taking the raw hiPSC-CM displacement images, performing the PIV analysis, creating divergence peaks, phase-averaging them, and calculating contractility parameters would further improve the protocol. This contractility assessment can elicit a vast amount of data, including many displacement- and time-related parameters, finely detailing how a drug alters hiPSC-CM contractility (Fig. 3d). Contractility and cytotoxicity measurements are critical to the generation of the CSI.
The CSI can be calculated from the various values obtained. For each drug, the relevant LD50, CoB, and EC parameters should first be normalized to the Cmax value associated with that drug (Fig. 4a). This Cmax value should be readily available in FDA literature, if the drug has been approved by the US FDA. However, if the candidate drug has not yet entered clinical trials, pharmacokinetic studies in preclinical animal models can also provide a suitable Cmax value. After Cmax normalization and exclusion of substantial outliers, these updated LD50, CoB, and EC metrics can be averaged to generate an absolute CSI reflecting drug cardiotoxicity (Fig. 4a). A CSIN can also be generated to compare drugs within a large dataset (Fig. 4b). If it is not possible to obtain an EC because of the lack of a high-throughput microscope, then a CSINLITE value can be obtained by averaging the normalized CoB and LD50 values for each drug (Fig. 4c). However, the CSIN value should ideally be considered in the context of positive and negative controls for cardiotoxicity. The usefulness and accuracy of the CSIN value depend on the quality of cytotoxicity and contractility data from the high-throughput screening assays, as well as the number of other drugs being comparatively tested. Thus, we highly recommend including as many candidate compounds as possible, as well as positive and negative controls, in the CSIN calculation. An example CSI calculation is provided for sorafenib, a highly cardiotoxic TKI according to our previous study and other published literature (Fig. 5)10,50. We have also included a comparison between CSIN and CSINLITE values for sorafenib and the other TKIs tested in our previous study (Table 1)10. As better metrics emerge in the future, they can be incorporated into new iterations of the CSI to improve cardiotoxicity predictiveness. For example, additional metrics could indicate alterations in hiPSC-CM mitochondrial function, action potential, or calcium transients.
To further confirm the cardiotoxicities of drugs with low CSI values, electrophysiological assessments of drug-treated hiPSC-CMs can be conducted using the gold-standard patch-clamp technique. We did not include such assessments in calculating the CSI, in part because patch clamp remains notoriously difficult to conduct in high throughput, in contrast to the contractility and cytotoxicity assays presented here. Follow-up in vitro electrophysiological assessments can provide information about the extent to which drugs with low CSI values alter cardiac action potential. As we observed in our previous study examining kinase inhibitor–induced hiPSC-CM cardiotoxicity, results from such electrophysiological assessments align well with the results from corresponding contractility analyses10. For example, we found that the clinically cardiotoxic and low-CSI-value kinase inhibitor vandetanib elicited arrhythmias and alterations in hiPSC-CM contractility at a comparable concentration to which it caused changes in hiPSC-CM electrophysiology and action potential duration. This is probably because at the cellular level, the cardiac action potential is directly mechanistically linked to CM contractility. Combining the results from assays evaluating a variety of cardiac cellular properties, such as electrophysiology, contractility, and viability, will provide a more complete picture of how a pharmaceutical compound can alter cardiac function.
Supplementary Material
Acknowledgements
We acknowledge the Stanford High-Throughput Bioscience Center for assistance with high-throughput imaging and plate reader assays. We acknowledge support from an American Heart Association (AHA) Predoctoral Fellowship (13PRE15770000) and an NSF Graduate Research Fellowship (DGE-114747) (A.S.); Burroughs Wellcome Foundation Innovation grant 1015009, AHA grant 17MERIT3361009, and National Institutes of Health (NIH) grants R01 HL132875, R01 HL130020, R01 HL128170, R01 HL123968, and R24 HL117756 (J.C.W.); and NIH grants NIH R01 HL130840, R01 HL128072, and R01 HL132225 (M.M.).
Footnotes
Competing interests
M.M. holds equity in and is on the scientific advisory board for Vala Sciences, a company offering high-content screening services and instrumentation for measuring the electrical and contractile physiology of CMs. J.C.W. is a cofounder of and scientific advisory board member for Khoris, a company using hiPSCs to accelerate drug discovery. The remaining authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41596–018-0076–8.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data availability
Source data are provided or are available from the corresponding author upon request.
References
- 1.Chang HM, Okwuosa TM, Scarabelli T, Moudgil R & Yeh ETH Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 2. J. Am. Coll. Cardiol. 70, 2552–2565 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang HM, Moudgil R, Scarabelli T, Okwuosa TM & Yeh ETH Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 1. J. Am. Coll. Cardiol. 70, 2536–2551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Force T, Krause DS & Van Etten RA Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer 7, 332–344 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Moslehi JJ & Deininger M Tyrosine kinase inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia. J. Clin. Oncol. 33, 4210–4218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu AH Cardiotoxic drugs: clinical monitoring and decision making. Heart 94, 1503–1509 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Magdy T, Schuldt AJT, Wu JC, Bernstein D & Burridge PW Human induced pluripotent stem cell (hiPSC)-derived cells to assess drug cardiotoxicity: opportunities and problems. Annu. Rev. Pharmacol. Toxicol. 58, 83–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferri N et al. Drug attrition during pre-clinical and clinical development: understanding and managing drug-induced cardiotoxicity. Pharmacol. Ther. 138, 470–484 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Stack JP, Moslehi J, Sayed N & Wu JC Cancer therapy-induced cardiomyopathy: can human induced pluripotent stem cell modelling help prevent it? Eur. Heart J. 10.1093/eurheartj/ehx811 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colatsky T et al. The comprehensive in vitro proarrhythmia assay (CiPA) initiative—update on progress. J. Pharmacol. Toxicol. Methods 81, 15–20 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Sharma A et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 9, eaaf2584 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercola M, Colas A & Willems E Induced pluripotent stem cells in cardiovascular drug discovery. Circ. Res. 112, 534–548 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guth BD Preclinical cardiovascular risk assessment in modern drug development. Toxicol. Sci. 97,4–20 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann P & Warner B Are hERG channel inhibition and QT interval prolongation all there is in drug-induced torsadogenesis? A review of emerging trends. J. Pharmacol. Toxicol. Methods 53, 87–105 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Mitcheson JS, Hancox JC & Levi AJ Cultured adult cardiac myocytes: future applications, culture methods, morphological and electrophysiological properties. Cardiovasc. Res. 39, 280–300 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Sharma A, Wu JC & Wu SM Induced pluripotent stem cell-derived cardiomyocytes for cardiovascular disease modeling and drug screening. Stem Cell Res. Ther. 4, 150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garg P et al. Genome editing of induced pluripotent stem cells to decipher cardiac channelopathy variant. J. Am. Coll. Cardiol. 72, 62–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma N et al. Determining the pathogenicity of a genomic variant of uncertain significance using CRISPR/ Cas9 and human-induced pluripotent stem cells. Circulation 10.1161/CIRCULATI0NAHA.117.032273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burridge PW, Sharma A & Wu JC Genetic and epigenetic regulation of human cardiac reprogramming and differentiation in regenerative medicine. Annu. Rev. Genet. 49, 461–484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burridge PW et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 11, 855–860 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma A et al. CRISPR/Cas9-mediated fluorescent tagging of endogenous proteins in human pluripotent stem cells. Curr. Protoc. Hum. Genet. 96, 21.11.1–21.11.20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma A et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circ. Res. 115, 556–566 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang P et al. Patient-specific and genome-edited induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of Brugada syndrome. J. Am. Coll. Cardiol. 68, 2086–2096 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itzhaki I et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471, 225–229 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Sun N et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 4, 130ra147 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu H et al. Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised beta-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell 17, 89–100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lan F et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 12, 101–113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burridge PW et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the pre¬dilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 22, 547–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kodo K et al. iPSC-derived cardiomyocytes reveal abnormal TGF-beta signalling in left ventricular noncompaction cardiomyopathy. Nat. Cell Biol. 18, 1031–1042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Navarrete EG et al. Screening drug-induced arrhythmia [corrected] using human induced pluripotent stem cell-derived cardiomyocytes and low-impedance microelectrode arrays. Circulation 128, S3–S13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tohyama S et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12, 127–137 (2013). [DOI] [PubMed] [Google Scholar]
- 31.McKeithan WL et al. An automated platform for assessment of congenital and drug-induced arrhythmia with hiPSC-derived cardiomyocytes. Front. Physiol. 8, 766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Millard D et al. Cross-site reliability of human induced pluripotent stem-cell derived cardiomyocyte based safety assays using microelectrode arrays: results from a blinded CiPA pilot study. Toxicol. Sci. 164, 550–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ando H et al. A new paradigm for drug-induced torsadogenic risk assessment using human iPS cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 84, 111–127 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Blinova K et al. Comprehensive translational assessment of human-induced pluripotent stem cell derived cardiomyocytes for evaluating drug-induced arrhythmias. Toxicol. Sci. 155, 234–247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Churko JM et al. Transcriptomic and epigenomic differences in human induced pluripotent stem cells generated from six reprogramming methods. Nat. Biomed. Eng. 1, 826–837 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riss TL et al. in Assay Guidance Manual (eds. Sittampalam GS et al. ) https://www.ncbi.nlm.nih.gov/books/NBK53196/(2004).
- 37.Adrian RJ & Westerweel J Particle Image Velocimetry (Cambridge University Press, Cambridge, UK, 2011). [Google Scholar]
- 38.Del Alamo JC et al. Spatio-temporal analysis of eukaryotic cell motility by improved force cytometry. Proc. Natl. Acad. Sci. USA 104, 13343–13348 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serrano R, Aung A & Varghese S & del Alamo JC Three-dimensional monolayer stress cytometry. Biophys. J. 112, 271a (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerjee I et al. Cyclic stretch of embryonic cardiomyocytes increases proliferation, growth, and expression while repressing Tgf-beta signaling. J. Mol. Cell. Cardiol. 79, 133–144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Del Alamo JC et al. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim. Biophys. Acta 1863, 1717–1727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor ZJ, Gurka R, Kopp GA & Liberzon A Long-duration time-resolved PIV to study unsteady aerodynamics. IEEE Trans. Instrum. Meas. 59, 3262–3269 (2010). [Google Scholar]
- 43.Smith DA, Di L & Kerns EH The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discov. 9, 929–939 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Veerman CC et al. Immaturity of human stem-cell-derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev. 24, 1035–1052 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Churko JM, Burridge PW & Wu JC Generation of human iPSCs from human peripheral blood mononuclear cells using non-integrative Sendai virus in chemically defined conditions. Methods Mol. Biol. 1036, 81–88 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watanabe K et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25, 681–686 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Pei F et al. Chemical-defined and albumin-free generation of human atrial and ventricular myocytes from human pluripotent stem cells. Stem Cell Res. 19, 94–103 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Lian X et al. Chemically defined, albumin-free human cardiomyocyte generation. Nat. Methods 12, 595–596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huo J et al. Evaluation of batch variations in induced pluripotent stem cell-derived human cardiomyocytes from 2 major suppliers. Toxicol. Sci. 156, 25–38 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Hasinoff BB & Patel D Mechanisms of myocyte cytotoxicity induced by the multikinase inhibitor sorafenib. Cardiovasc. Toxicol. 10, 1–8 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Moore M et al. Phase I study to determine the safety and pharmacokinetics of the novel Raf kinase and VEGFR inhibitor BAY 43–9006, administered for 28 days on/7 days off in patients with advanced, refractory solid tumors. Ann. Oncol. 16, 1688–1694 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.