

Abstract
Birth defects of the heart and face are common and most have no known genetic cause, suggesting a role for environmental factors. Maternal fever during the first trimester is an environmental risk factor linked to these defects. Neural crest cells are precursor populations essential to the development of both at-risk tissues. Here, we report that two heat-activated transient receptor potential ion channels, TRPV1 and TRPV4, were present in neural crest cells during critical windows of heart and face development. TRPV1 antagonists protected against the development of hyperthermia-induced defects in chick and zebrafish embryos. Conversely, treatment with chemical agonists of TRPV1 or TRPV4 replicated hyperthermia-induced birth defects in both model systems. To test whether transient TRPV channel permeability in neural crest cells was sufficient to induce these defects, we engineered iron-binding modifications to TRPV1 and TRPV4 that enabled remote and noninvasive activation of these channels in specific cellular locations and at specific developmental times in chick embryos with radiofrequency electromagnetic fields. Transient stimulation of radiofrequency-controlled TRP channels in neural crest cells replicated fever-associated defects in developing chick embryos. Our data provide a previously undescribed mechanism for congenital defects whereby hyperthermia activates ion channels that negatively affect fetal development.
Introduction
Birth defects of the heart and face are extremely common. Congenital heart defects affect about 1% of live births in the United States and often require surgical correction within the first year of life(1). Craniofacial clefts involving the lip and or the palate affect over 4,000 infants per year and typically require corrective surgery(2). Genetic associations have been identified in only 15% of heart or 30% of craniofacial defects leaving the majority of cases without a known etiology(3, 4). Environmental factors likely contribute. First trimester maternal fever is an environmental factor linked to both craniofacial and heart defects(5). Highly associated heart defects include right- and left-sided obstructive lesions (pulmonary atresia or pulmonary or aortic stenosis) and conotruncal defects (double outlet right ventricle (DORV), tetralogy of Fallot) (6–8). Hyperthermia-associated craniofacial defects include midface hypoplasia and cleft lip and or palate(5, 9). Specific modes of infections, such as respiratory compared to urinary or pelvic infections, confer differential susceptibilities leading some to speculate that infection itself is the critical event(8, 10). However, maternal hyperthermia following hot tub exposure is sufficient to confer risk of birth defects suggesting that hyperthermia itself is the principal teratogen(11). Nonetheless, the teratogenic mechanism(s) of maternal fever are unknown.
Craniofacial clefts, abnormal arch artery anatomy, aorticopulmonary septation and conotruncal heart defects can be linked through neural crest cell dysfunction(12, 13). Neural crest cells are a pluripotent migratory cell population that arises from the dorsal neural tube. Cranial and cardiac neural crest cells migrate to the head and pharyngeal arches where they contribute to the developing face and heart. The cranial neural crest cells differentiate into the bone and cartilage that form facial features including the jaw and palate(14). The cardiac neural crest cells are required for septation of the aorta and pulmonary trunk(12). In addition, they differentiate into the smooth muscle cells of the aortic arch arteries, the distal aorta and pulmonary trunk, and are required for aortic arch patterning(15). Further, neural crest cells influence early cardiac function and the development of the secondary heart field, a cardiogenic cell population in the pharynx required for proper alignment of the outflow vessels with respect to the ventricles(16–18). Therefore, fever-induced changes to neural crest cell function could provide a unifying mechanism to account for the types of birth defects observed following fever.
During our investigation, we discovered that two temperature-activated transient receptor potential (TRP) ion channels, TRPV1 and TRPV4, are present in cardiac and cranial neural crest cells during critical windows of heart and facial development. Several TRP channels are the principle components for temperature sensation within sensory neurons(19–21), although other TRP channel functions include modulating Ca2+ during cytoskeleton rearrangements, promoting cell polarity critical for cell migration, and regulation of cellular metabolism, suggesting highly diverse functions outside of the nervous system(22–24). We hypothesized that transient changes in the activity of temperature-activated TRP channels in neural crest cells confers susceptibility to fever-associated craniofacial and congenital heart defects. Using pharmacologic approaches to antagonize TRP activity, we protected developing chick embryos from hyperthermia-mediated birth defects. In addition, we utilized a TRP channel agonist to replicate associated birth defects in normothermic conditions. Finally, we developed a technology to remotely control transient permeability of temperature-activated TRP channels with cellular resolution using radiofrequency (RF) waves. Transient activation of these TRP channels in neural crest cells was sufficient to replicate febrile-associated birth defects.
Results
Hyperthermia induced neural crest cell-related birth defects.
We investigated the developmental effects of hyperthermia on neural crest cell-derived craniofacial and cardiovascular structures in chick embryos. The mechanisms of neural crest cell development are largely conserved between species and the chick is a well-established model to study human cardiovascular and craniofacial defects. To target critical stages in neural crest cell development, we heated Hamburger Hamilton (HH) stage 11–15 embryos to 40–41°C for 1 hour then re-incubated them at 37°C and analyzed for craniofacial and heart defects at HH36. We performed alcian blue and alizarin red stains to analyze the craniofacial features in experimental animals (Fig. 1A). Individual upper beak lengths were measured and normalized to femur lengths to account for small variations in staging (Fig. 1B). Our analysis revealed that hyperthermia exposure resulted in significant craniofacial defects including a reduction in upper beak lengths compared to normothermic control chicks with severe beak defects observed in 16% of hyperthermia exposed animals (Fig. 1C and fig. S1, A and B). Cardiovascular defects were increased in hyperthermia-exposed embryos. These defects included a significant incidence of conotruncal defects including double outlet right ventricle (DORV, Fig. 1D,E) and aortic arch patterning defects (fig. S2, A to F). All of these defects are directly linked to neural crest cell dysfunction(13). Due to the high association of outflow tract obstruction defects with maternal fevers in humans we used stereology to analyze the luminal cross-sectional areas of the aorta and pulmonary trunk using the Cavalieri probe(25). We identified a 72% and 55% reduction in luminal cross-sectional areas of the aorta and pulmonary trunk, respectively (Fig. 1F,G). The obstructive lesions were supravalvular and coincident with neural crest-derived smooth muscle vessel regions. These data demonstrate that experimentally controlled hyperthermia in chick models can produce neural crest-associated birth defects reminiscent of those observed in human infants following maternal fever.
Fig. 1. Hyperthermia-induced congenital defects in neural crest-dependent tissues.

(A) Alcian blue and alizarin red stained craniofacial features of HH36 heads for control and hyperthermia-exposed chicks. Upper beak measurement extended from the quadratojugal (white arrowhead) to the tip of the upper beak (black arrowhead). (B) Upper beak length was normalized to femur lengths for control (black) and hyperthermia-exposed (red) chicks. (C) Graph of upper beak length to femur length ratios for control and hyperthermia chicks. (D) Whole mount and histological sections of HH36 hearts from control (normothermia) and hyperthermia-exposed embryos. The white and black arrows in the whole mount images highlight the alignment of the aorta (Ao) and pulmonary trunk (P) in control heart compared to the hyperthermia-exposed heart. Histological sections of the whole mount hearts at the level of the ventricular septum and the outflow vessel semilunar valves. Dashed lines indicate the plane of outflow tract septation. Sections through the hyperthermia-exposed DORV heart with a perimembranous VSD (*) and a rightward shift of the aorta in relation to the pulmonary trunk (dashed line). Gross and histological analysis of heart anatomy was performed in 60 normothermia hearts and 49 hyperthermia-exposed hearts. Tricuspid valve (TV), mitral valve (MV). (E) Percentage of conotruncal defects in control (0%, n=60 hearts) and hyperthermia groups (12%, n=6/49 hearts). (F) Representative histological sections through the aorta and pulmonary trunk used to compare the luminal areas (arrows) of normothermic and hyperthermia-exposed hearts distal to the valves at the level of the left coronary artery (white arrowhead). Bar = 200μm. (G) Average Cavalieri probe estimates of the luminal cross-sectional areas of the aorta and pulmonary trunk immediately distal to the respective semilunar valves. Significance was determined using unpaired t-test(C,G) or Fishers exact test (E). *P<0.0001. The number of biological replicates is indicated by n in the graphs in (C, E, and G).
TRPV1 channel inhibition mitigated fever-associated birth defects.
To identify potential teratogenic mechanisms of hyperthermia, we first examined the expression profile of mRNAs encoding temperature-activated vanilloid TRP channels (TRPV1–4) in neural folds containing neural crest cells in chick embryos at HH9 using RT-PCR. As a control, mRNA transcripts for all four TRPV channels were detected in whole embryo RNA from HH14 and HH22–24 embryos. However, only transcripts for TRPV1, TRPV2, and TRPV4 were detected in dissected neural folds enriched for pre-migratory neural crest cells at HH9 (fig. S3A). TRPV2 is activated by temperatures >52°C thus making this channel less likely to be impacted by temperatures experienced during fever(20). However, TRPV1 can mediate heat-evoked Ca2+ currents >40°C and TRPV4 >34°C making these likely candidates for fever-induced activation(19, 26). To verify expression of TRPV1 and TRPV4 in migrating neural crest cells, neural tubes were electroporated with a plasmid encoding green fluorescent protein (GFP) at HH9, explanted and cultured overnight to allow for GFP+ neural crest cells to migrate from the neural tube (fig. S3B). RT-PCR analysis on FACS sorted GFP+ neural crest cells (fig. S3C,D) confirmed that both TRPV1 and TRPV4 transcripts were present in this population (fig. S3E).
The neural crest-related defects observed after hyperthermia exposures between HH11–15 suggested a critical developmental window of susceptibility. To determine if cranial and cardiac neural crest cells expressed TRPV1 and or TRPV4 during these developmental stages, we combined in situ hybridization for TRPV4 or TRPV1 mRNAs with immunohistochemistry for the neural crest cell marker HNK1. TRPV1 transcripts were detected in HNK1+ premigratory and migrating HNK1+ cranial and cardiac neural crest cells between HH11 and HH13 (fig. S4, A to J). Similarly, TRPV4 transcripts were also detected at these stages in HNK1+ cranial and cardiac neural crest cells (fig. S5, A to F. Thus both TRPV1 and TRPV4 transcripts are detected in neural crest cells during a critical developmental window of susceptibility to hyperthermia-induced defects. These TRPV1+ and TRPV4+ neural crest cells populate pharyngeal arches 1 and 2 and are the primary contributions to the bone and cartilage of craniofacial structures. Neural crest cells populating pharyngeal arches 3, 4, and 6 form the outflow septum, the smooth muscle of the great arteries, and influence aortic arch patterning. These neural crest cells are also critical to secondary heart field development and influence conotruncal alignment(18).
Next, we confirmed the responsiveness of chick TRPV 1 and TRPV4 with channel specific ligands. Treatment of chick TRPV4-expressing CHO cells with the TRPV4 agonist, GSK1016790A (GSK101), increased Ca2+ permeability as assessed by the genetically encoded Ca2+ indicator GCaMP6 (fig. S6A). Identifying an agonist for chick TRPV 1 was more challenging because it lacks the capsaicin binding site and is therefore insensitive to capsaicin and related agonists(27). However, the cysteine knot (ICK) peptide spider toxin from P. cambridgei, vanilloid toxin 3 (VaTx3), interacts with the outer pore of TRPV1 and is predicted to activate chick TRPV1(28). VaTx3 produced comparable changes in Ca2+ permeability in HEK293T cells transiently expressing either the murine or chick TRPV1 channels (fig. S6B). The TRPV1 specific antagonist SB366791 inhibited VaTx3-dependent chick TRPV1 activity (fig. S6C) suggesting the VaTx3-mediated response was TRPV1-specific.
To determine if TRPV transcripts detected in neural crest cells translated into functional channels, we electroporated chick neural tubes in ovo at HH9–10 with GCaMP6 and explanted them to culture plates to allow GCaMP6+ neural crest cells to migrate from the neural tubes (Fig. 2A). Explanted neural crest cells were exposed to the TRPV4 agonist GSK101, which produced a robust increase in Ca2+ permeability that was blocked by pretreatment with the TRPV4 antagonist RN1734 (Fig. 2B)(29). Explanted neural crest cells exposed to VaTx3 showed increased Ca2+ responses, demonstrating functional TRPV1 channels in neural crest cells (Fig. 2C). To confirm that neural crest cell-specific TRPV1 and TRPV4 channel activity extended beyond avian species, we analyzed primary mammalian neural crest cells for changes in intracellular Ca2+ following exposure to TRPV1 and TRPV4 ligands. Exposing mouse neural crest cell explants to increasing doses of GSK101 produced corresponding increases in Ca2+ responses (Fig. 2D), which were blocked by the TRPV4 inhibitor RN1734 (Fig. 2E). Capsaicin application induced Ca2+ permeability in mouse neural crest cells, an effect that was blocked with the TRPV1 inhibitor SBB366791 (Fig. 2F). These data demonstrate that TRPV 1 and TRPV4 are also present in neural crest cells during mammalian development. Exposure of primary chick neural crest cells to 40°C resulted in increased Ca2+ permeability and this response was reduced by pretreatment with the TRPV4 inhibitor RN1734 (Fig. 2G-I). Thus, neural crest cells responded to hyperthermia in a manner that was partially mediated through TRPV channels.
Fig. 2. Temperature-activated TRPV channels in avian and mammalian neural crest cells.
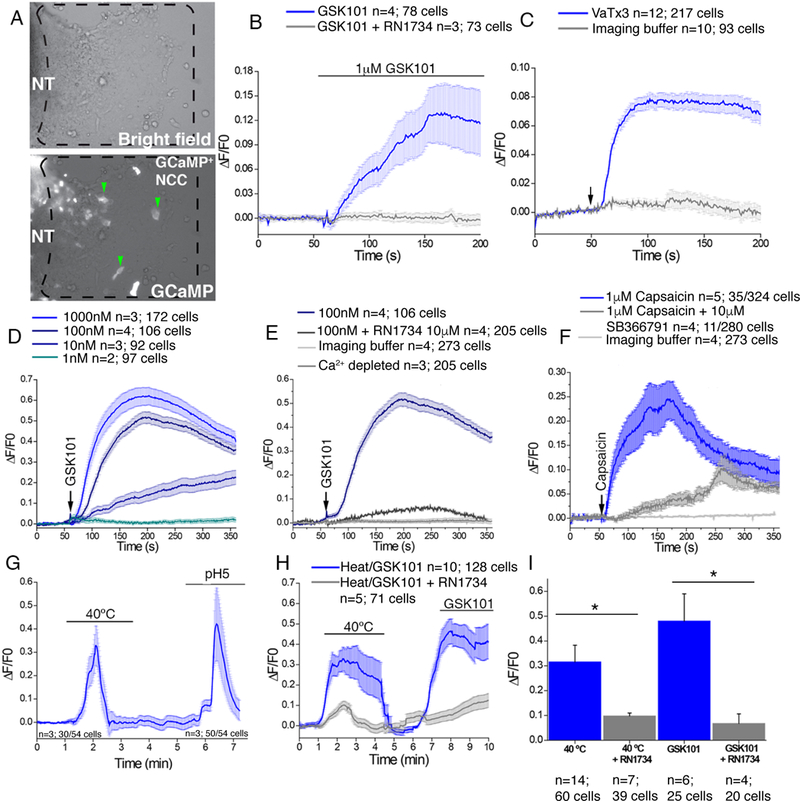
(A) Images of chick primary explanted neural tubes (NT) expressing GCaMP6 after 24 hours in culture. Dashed box region shows migrating neural crest cells used in subsequent analyses (green arrowheads). (B) Change in GCaMP6 fluorescence in chick neural crest cells following exposure to 1μM GSK101 (blue) or 1μM GSK101 combined with 10μM RN1734 (gray line). (C) GCaMP6 fluorescence in chick neural crest cells following exposure to 1μM VaTx3 (arrow, blue line) or buffer alone (gray line). (D) Change in Fluo4 fluorescence in mouse primary explanted neural crest cells from E8.5 embryos in response to the indicated doses of GSK101. (E) Fluo4 fluorescence in mouse primary explanted neural crest cells from E8.5 embryos in response to GSK101 (arrow, blue line) or to GSK101 and 10μM RN1734 (gray line). (F) Changes in Fluo4 fluorescence in mouse neural crest cells following exposure to 1μM capsaicin (arrow, blue line) or to capsaicin and SB366791 (gray line). (G) Representative GCaMP6 fluorescence in chick primary neural crest cells following exposure to 40°C imaging buffer followed by imaging buffer pH 5. (H) Representative GCaMP6 fluorescence in chick primary neural crest cells following exposure to 40°C imaging buffer or GSK101(blue) or in the presence of RN1734 inhibitor (gray line). (I) Averages of more than 4 separate experiments as in (H) analyzing the number of cells/condition as indicated. Significance was determined using unpaired t-test. *P<0.05. The number of biological replicates is indicated by n and/or the number of cells analyzed is indicated in the graphs in (B), (C), (D), (E), (F), (H), and (I).
We next hypothesized that hyperthermia-induced defects in neural crest-dependent structures in our chick model could be rescued by inhibition of TRPV channels. However, treatment of HH11 embryos with the TRPV4 inhibitor RN1734 under normothermic conditions produced a significant decrease in beak length and a 20% incidence of conotruncal heart defects (fig. S7, A to I). These defects suggested that endogenous TRPV4 activity was necessary for normal craniofacial and heart development, thus precluding us from targeting TRPV4 activity to rescue hyperthermia-associated birth defects. In contrast, treatment of HH11 embryos with the TRPV1 inhibitor SB366791 treatment at normal temperatures did not produce detectable heart or facial phenotypes. Pretreatment of embryos with SB366791 before hyperthermia prevented hyperthermia-induced craniofacial defects (Fig. 3A,B) and significantly reduced supravalvular stenosis (Fig. 3C-E). The number of conotruncal defects were reduced by ~50%, although this trend did not reach statistical significance (Fig. S8, A to G). These data link TRPV channel activation with neural crest-related congenital defects in our chick hyperthermia model.
Fig. 3. TRPV1 inhibition rescues hyperthermia-induced congenital defects.
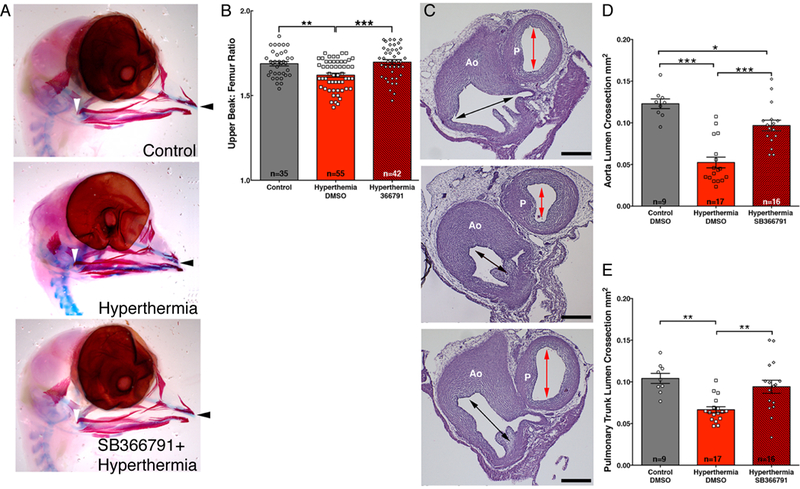
(A) Alcian blue and alizarin red stains of HH36 heads from hyperthermia-exposed and hyperthermia-exposed chicks following pretreatment with the TRPV1 inhibitor SB366791. Upper beak measurement extended from the quadratojugal (white arrowhead) to the tip of the upper beak (black arrowhead). (B) Comparison of upper beak length to femur length ratios in control, hyperthermia, or hyperthermia + SB366791 chicks. (C) Histological sections through the Aorta (Ao) and pulmonary trunk (P) comparing the luminal areas (arrows) of a control (top), hyperthermia (middle), and hyperthermia-exposed hearts with SB366791 pretreatment (lower) distal to the semilunar valves at the level of the left coronary artery (white arrowhead). Bar = 200μm. (D-E) Cavalieri probe estimates of the luminal cross-sectional area of the aorta (D) or pulmonary trunk (E) immediately above the aortic valve. Significance was determined by one-way ANOVA followed by Bonferroni’s multiple comparisons test. *P<0.05, **P<0.004, ***P<0.0001. The number of biological replicates is indicated by n in the graphs in (B), (D), and (E).
Pharmacologic activation of TRPV4 replicated heat-induced birth defects in multiple animal models.
As a complementary approach, we exposed developing chick embryos at HH11–15 to the small molecule TRPV4 agonist GSK101 under normothermic conditions to determine if pharmacologic activation of TRPV4 in ovo induced neural crest-related defects. Conotruncal defects were detected in 20% of GSK101-exposed embryos at HH36 (Fig. 4A,B and fig. S9, A to D). GSK101-treatment also replicated the severe supravalvular stenosis as indicated by a 50% and 44% reduction in luminal cross-sectional areas of the aorta and pulmonary trunk, respectively (Fig. 4C-E). As in the hyperthermia-exposed chicks, GSK101-treated embryos had significantly shorter beak-to-femur ratios compared to controls (Fig. 4F-H). Together, these data provide evidence that activation of TRPV4 channel permeability during critical windows of susceptibility can produce cardiac and craniofacial lesions that resemble those associated with maternal fevers in humans.
Fig. 4. Ligand activation of TRPV4 replicates hyperthermia induced birth defects.
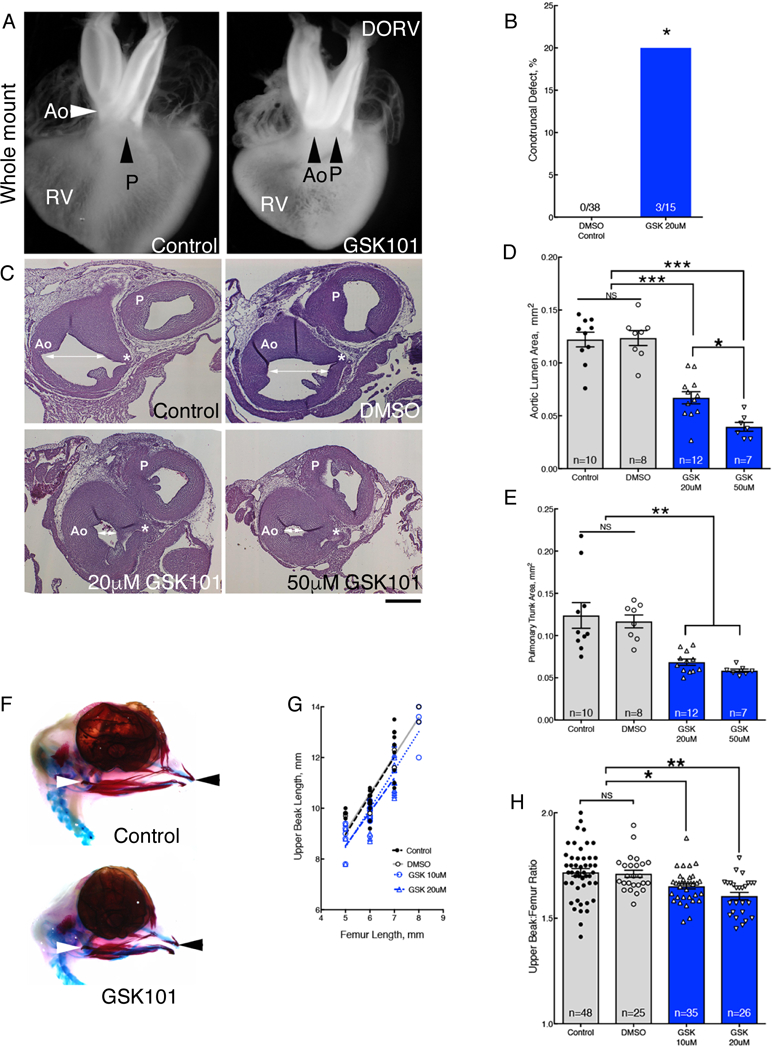
(A) Whole mounts of hearts from control and GSK101 treated-embryos. GSK-treated heart showed DORV orientation of the aorta (Ao) and pulmonary trunk (P). See fig. S9 for histological sections. Whole mount and histological analysis of heart anatomy was performed in 38 hearts from DMSO-treated embryos and 15 GSK101-treated embryos.(B) Percentage of hearts with histologically confirmed conotruncal defects in GSK101 (GSK)-treated embryos compared to DMSO controls. (C) Panel of histological sections through the aorta (Ao) and pulmonary trunk (P) comparing the luminal areas (white arrows) at the level of the left coronary artery (*) in control, DMSO, and 20μM or 50μM GSK101 treatment. Bar = 200μm. (D,E) Treatment with GSK101 reduced aortic luminal areas (D, average coefficient of error (CE)=0.03) and pulmonary trunk luminal areas (E, average CE=0.03). (F) Alcian blue and alizarin red stains of DMSO control (top) and 20μM GSK101 treated embryos (bottom) at HH36. (G) Normalization of upper beak length to femur lengths. (H) Graph of upper beak to femur ratios in untreated control, DMSO-treated control and 2 GSK101-treatment groups. Significance was determined using Fisher exact test (B) or one-way ANOVA followed by Bonferroni’s multiple comparisons test (D, E, H). *P<0.03, **P<0.004, ***P<0.0001. The number of biological replicates is indicated by n in the graphs in (D), (E), and (H).
We posited that if TRPV activation is a broadly relevant driver mechanism for neural crest-dependent congenital defects, then pharmacologic TRPV4 activation should reproduce these defects in another vertebrate. Because TRPV4 transcripts are detected in neural crest-derived jaw structures of zebrafish during early head development(30), we targeted TRPV4 activation in developing zebrafish larvae. While zebrafish single ventricle heart anatomy limits our ability to assess conotruncal alignment defects, it offers an excellent model of craniofacial development. First, we confirmed that GSK101 activates zebrafish TRPV4 transiently expressed in CHO cells (Fig. 5A). We next used 1.4cola1:egfp transgenic zebrafish larvae which express egfp gene under the cola1 promoter to drive expression in jaw structures(31) to monitor craniofacial development in live fish over time. We analyzed for craniofacial defects in zebrafish larvae treated at 2dpf with GSK101 or GSK1153218 (a structurally related compound which lacks TRPV4 activity, table S1). By 3dpf and 4dpf, only GSK101 exposed larvae exhibited a significant reduction in jaw length, as indicated by the decreased distance between Meckel’s and ceratohyal cartilages (Fig. 5B-D). Moreover, histological examination revealed disruption of the cellular organization in the ethmoid plate suggesting defective palatogenesis (fig. S10, A and B). Consistent with the developmental timing and observed phenotypes of our chick studies, these data demonstrate the impact of aberrant TRPV channel activation on neural crest-derived craniofacial development extends beyond an avian model.
Fig. 5. TRPV4 activation disrupts jaw extension in zebrafish larvae.
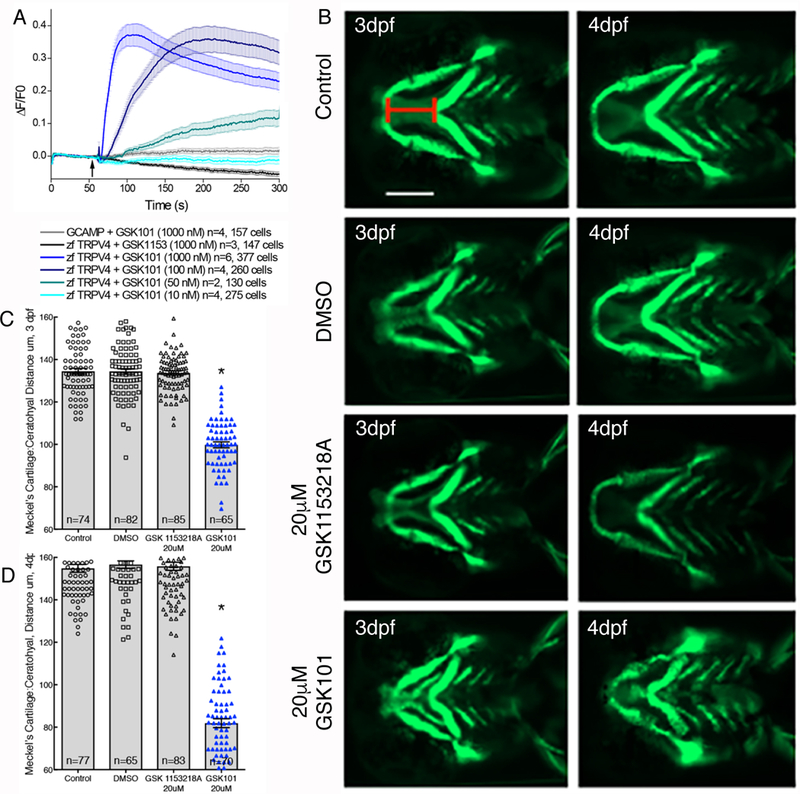
(A) The activity of GSK101 at the indicated concentrations and of GSK1153218 (black tracing), a structurally related compound that lacks activity on mammalian TRPV4 (Table S1), in cloned zebrafish TRPV4 in CHO cells using GCaMP6 to assess Ca2+ permeability. (B) Representative images of 1.4cola1:egfp transgenic zebrafish larvae treated with 20μM GSK101, vehicle control, or GSK1153218A. Replicate batches were imaged live at 3 and 4 dpf. Scale bar approximately 130μm. (C,D) Quantification of the distance between Meckel’s cartilage and the ceratohyal was measured (red line in B). Significance was determined using one-way ANOVA followed by Bonferroni’s multiple comparisons test. *P<0.0001, GSK101-treated group compared to all other groups). The number of biological replicates is indicated by n and/or the number of cells analyzed is indicated in the graphs in (A), (C), and (D).
Remote activation of TRP channels in neural crest cells phenocopied fever-associated birth defects.
To test the hypothesis that congenital defects can be caused by transient activation of TRPV channels specifically within neural crest cells, we needed temporal and cell-specific control over these channels during development. Under specific conditions, modified ferritin-bound TRPV channels can be remotely controlled in vivo using radio frequency electromagnetic fields(32–34) or static magnetic fields(35). We engineered FeRIC (Fe3+ Redistribution to Ion Channels) modifications to TRPV1 and TRPV4 channels to direct endogenous intracellular ferritin to the cytoplasmic domains of TRPV 1 or TRPV4 channels. Specifically, we fused the ferritin-binding domain 5 (D5) of Kininogen-1 (36) with the C-terminal cytoplasmic domain of either TRPV1 (TRPV1FeRIC) or TRPV4 (TRPV4FeRIC) to confer responsiveness to RF (Fig. 6A,B and fig. S11, A to C, and S12, A to C). By targeting endogenous ferritin found in cells, we eliminated the need for potentially cell-toxic exogenous iron administration or co-expression of modified ferritin(33, 37).
Fig. 6. Development of remotely controlled TRPVFeRIC channels.
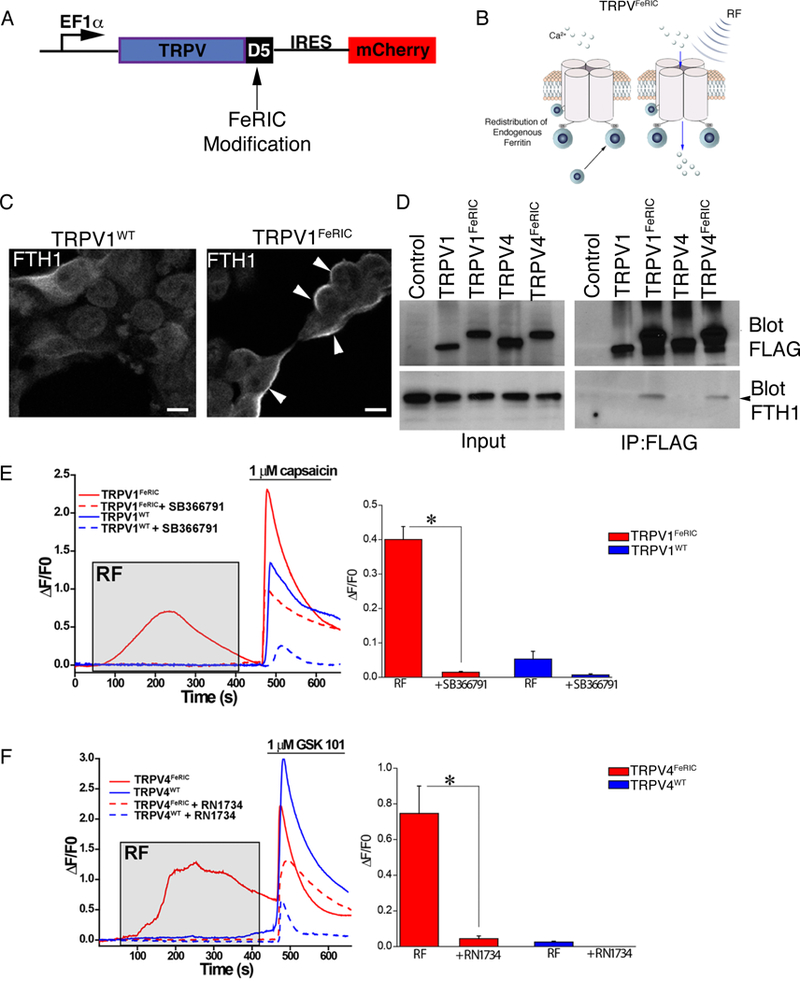
(A) TRPV channels were tagged with domain 5 of kininogenl (D5) and were cloned into the PLVX vector with an internal ribosomal entry site (IRES) for mCherry (fig. S12 and S13). (B) FeRIC channels were designed to recruit endogenous cellular ferritin to the modified TRPV channel at the cell membrane. (C) Cytoplasmic distribution of ferritin heavy chain fused with mCherry (FTH1mCherry) in HEK293T cells expressing TRPV1WT and membrane redistribution of FTH1mCherry (white arrowheads) in TRPV 1FeRIC-expressing cells. Images were representative of 2 independent experiments. (D) Representative immunoprecipitation western blot of HEK293T cells expressing FLAG tagged TRPV 1WT, TRPV1FeRIC, TRPV4WT, or TRPV4FeRIC. The blot was probed for FLAG and FTH1. 4 independent experiments were conducted using TRPV lFeRIC and 3 independent experiments were conducted using TRPV4FeRIC. (E) GCaMP6 fluorescence in TRPV 1WT-expressing (blue) or TRPV1FeRIC-expressing (red) HEK293T cells following RF (gray box) then 1μM capsaicin (bar). Bar graphs are DF/F0 averages of 4 experiments with 50–100 cells/group analyzed. (F) GCaMP6 fluorescence in TRPV4WT-expressing (blue) or TRPV4FeRIC-expressing (red) HEK293T cells following RF (gray box) then 1μM GSK101 (bar). Bar graphs are ΔF/F0 averages of 5 experiments with 106–123 cells/group analyzed. Significance was determined using unpaired t-test. *P<0.05.
To confirm FeRIC-mediated ferritin redistribution to the cell membrane, we investigated the subcellular localization of a ferritin heavy chain-mCherry fusion (FTH1mCherry) protein in TRPV1WT and TRPV1FeRIC-expressing HEK293T cells. In TRPV1WT cells, FTH1mCherry showed a cytoplasmic distribution consistent with normal FTH1 expression. However, when FTH1mCherry was co-transfected with TRPV1FeRIC, FTH1mCherry was redistributed to the cell membrane by 24 hours (Fig. 6C). In addition, both TRPV1FeRIC and TRPV4FeRIC, but not TRPV1WT, TRPV4WT or FLAG constructs co-immunoprecipitated FTH1 (Fig. 6D). Next, we tested the influence of FeRIC-mediated ferritin redistribution on the bioavailability of cellular iron metabolism by measuring changes in four well-characterized iron responsive genes. Redistribution of cellular ferritin using TRPV1FeRIC or TRPV4FeRIC did not significantly change mRNA concentrations of Fpn1 (which encodes ferroportin-1), TfR1 (which encodes transferrin receptor 1), SLC11A2 (which encodes divalent metal transporter 1), or mRNAs encoding heavy and light chains of ferritin (fig. S13, A to E), suggesting that iron bioavailability was not substantially altered.
To remotely activate FeRIC channels, we used a 175 MHz RF coil that resonates at a frequency similar to that generated by a standard 3-Tesla MRI scanner. Measurement of the current in the RF coil was 1.5A peak-to-peak, generating a mean magnetic field of 36 μT and a specific absorption rate (SAR) of 6.8 W/kg (fig. S14, A to D). We confirmed that this magnetic field did not induce bulk temperature increases that could activate FeRIC channels by measuring the temperature of the cell media immediately before and after applying RF. The temperature fluctuation was measured at < ±1°C. Because membrane-localized Fe3+ combined with RF could potentially be cytotoxic, we investigated cellular viability and membrane integrity. Treating TRPV1FeRIC–positive HEK293T cells with 10 minutes of RF had no detectable impact on membrane integrity and did not affect cell viability at 24 hours (fig. S15, A to C). We next tested the responsiveness of TRPV1FeRIC and TRPV4FeRIC to RF in HEK293T cells co-expressing GCaMP6. RF stimulation of unmodified TRPV1WT or TRPV4WT did not alter Ca2+ permeability. However, channels responded to capsaicin (TRPV1, Fig. 6E) or GSK101 (TRPV4, Fig. 6F) agonists as expected. Conversely, only TRPV 1FeRIC or TRPV4FeRIC were transiently responsive to RF stimulation and their respective ligands. In both cases, the FeRIC-dependent RF responses were abolished in the presence of channel-specific inhibitors (Fig. 6E,F). We next confirmed that remote activation of FeRIC channels required ferritin expression. Using CRISPR/Cas9, we deleted the ferritin heavy chain gene (FTH1) in HEK293T cells (HEKFTH1-KO, fig. S16A-C). Deletion of FTH1 abolished TRPV1FeRIC responses to RF but did not interfere with capsaicin responses (fig. S16D,E,G). Transfection of HEKFTH1KO cells with human FTH1 rescued RF responsiveness to TRPV1FeRIC (fig. S16D-G).
To gain mechanistic insight into how RF activates TRPV1FeRIC we generated TRPV1ΔT FeRIC which has mutations that abolish temperature activation (N628K, N652T, and Y653T) (38) without affecting chemical sensitivity (fig. S17A-C). When expressed in HEK293T cells, the FeRIC-modified triple mutant was not responsive to temperature changes as expected but retained its responsiveness to capsaicin (fig. S17D,E). In addition to the disruption of temperature-activation, these three point mutations also abolished RF-induced changes in permeability, suggesting that RF stimulation may act on TRPV1FeRIC through a temperature-specific mechanism (fig. S17F).
We next sought to test whether cell-specific activation of the candidate TRPV channels phenocopied fever-associated heart defects. RF applied to chick neural crest cells expressing TRPV1FeRIC increased Ca2+ permeability, which was blocked by the TRPV1 inhibitor SB366791 (Fig. 7A), thus confirming that TRPV1FeRIC was responsive to RF in these cells. For in ovo experiments, TRPV1FeRIC or TRPV1WT plasmid DNA was bilaterally electroporated into premigratory neural crest cells at HH9. Expression of TRPV 1FeRIC alone did not appreciably affect neural crest cell migration to the pharyngeal arches at HH13–14 (Fig. 7B). Twenty-four hours after electroporation, eggs were either exposed for 10 min to RF (RF10min) or not (RPneg). Of the TRPV1WT groups, RFnegTRPV1WT and RF10minTRPV1WT did not show conotruncal defects (Fig. 7C,D). In contrast, RF10minTRPV1FeRIC embryos exhibited conotruncal alignment defects in 48% of the embryos (Fig. 7C,D and movie S1). These defects included overriding aorta, DORV, and tetralogy of Fallot and were accompanied by a perimembranous VSD (fig S18, A to L). In contrast, we observed only a single heart with an overriding aorta and VSD in RPnegTRPV1FeRIC embryos (Fig. 7D). Remotely induced conotruncal defects were rescued by pretreatment with the TRPV1 inhibitor SB366791 (Fig. 7D and fig. S18, A to L). To control for generalized RF effects, unelectroporated eggs were exposed to 10 minutes of RF and all unelectroporated RF10min control eggs had structurally normal hearts (Fig. 7D).
Fig. 7. Fever-associated heart defects following remote activation of TRPV1 in neural crest cells.
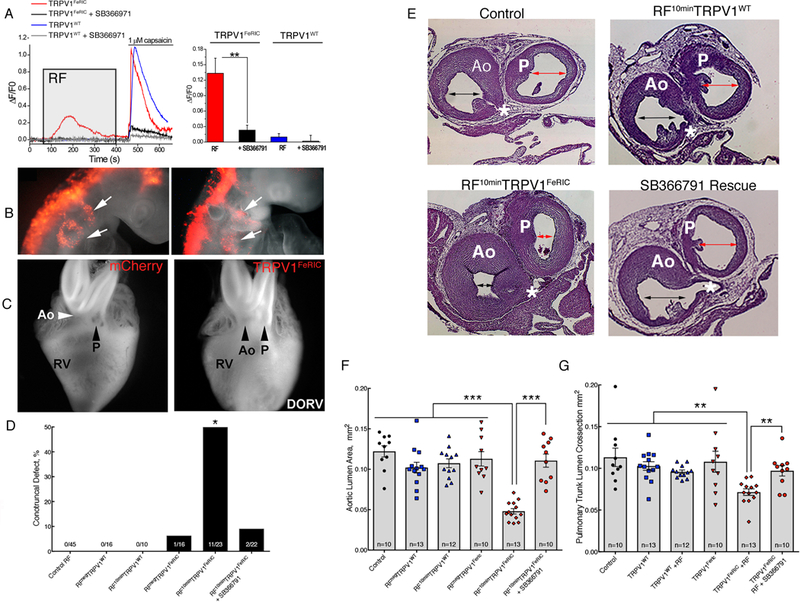
(A) GCaMP6 fluorescence in primary chick neural crest cells electroporated with TRPV1WT (blue lines) or TRPV1FeRIC (red lines) in the absence or presence of the TRPV1 inhibitor SB366971 (gray and black lines) following RF (gray box) then 1μM capsaicin (bar). Bar graph shows cumulative responses representing 3–4 separate experiments with 22–138 cells/group analyzed. (B) mCherry+ neural crest streams migrating to the pharyngeal arches (white arrows) in HH14 control or TRPV1FeRIC electroporated embryos. 6 independent experiments. (C) Whole-mounted hearts from RFNegTRPV1FeRIC embryo and RF10minTRPV1FeRIC embryo with arrows noting the position of the aorta (Ao) and pulmonary trunk (P) with respect to the right ventricle (RV). 5 independent experiments were performed. See Supplemental Movie 1 for an MRI reconstruction of a RF10minTRPV 1FeRIC-induced DORV heart defect in chick. (D) Percent of embryos with histologically confirmed conotruncal heart defects within indicated groups at HH36. (E) Representative histological sections through the aorta and pulmonary trunk at the level of the coronary artery (*) in Control RF, RF10minTRPV 1WT, RF10minTRPV1FeRIC, and RF10minTRPVFeRIC with SB366791 pretreatment. The double arrowheads highlight the luminal areas of the aorta and pulmonary trunk estimated using the Cavalieri probe. Bar = 200μm. (F) Graph of the Cavalieri probe estimates of cross sectional areas through the aorta at the level of the coronary artery in the indicated treatment groups (CE=0.03). (G) Graph of the Cavalieri probe estimates of the crosssectional areas through the pulmonary trunk distal to the semilunar valve in the indicated treatment groups (CE=0.03). Significance was determined using unpaired t-test (A), Fisher exact test (D) or one-way ANOVA followed by Bonferroni’s multiple comparisons test (F,G). *P<0.02, **P<0.005, ***P<0.0001. The number of biological replicates is indicated by n in the graphs in (F) and (G).
Next, we examined the vessels for outflow obstructive lesions and found no significant differences in cross-sectional luminal areas of the outflow tracts between TRPV 1WT embryos with or without RF stimulation or between RPnegTRPV1FeRIC embryos and controls (Fig. 7E-G). However, similar to hyperthermia and GSK101-exposed embryos, RF10minTRPV1FeRIC embryos displayed a 61% and 37% decrease in luminal cross-sectional areas in the aorta and pulmonary trunk respectively (Fig. 7E-G). Supravalvular stenosis was also rescued by pretreatment with SB366791 (Fig. 7E-G).
We also analyzed the craniofacial structures in TRPV1FeRIC-expressing embryos following remote activation in cranial neural crest cells and compared them to RPnegTRPV1FeRIC controls. Remote activation of TRPV1FeRIC resulted in a significant reduction in upper beak length compared to control or RPnegTRPV1FeRIC embryos (fig. S19, A to C). These data demonstrate that transient activation of TRPV1 in cranial neural crest cells is sufficient to phenocopy craniofacial defects associated with maternal fevers.
We analyzed neural crest cell-specific remote activation of TRPV4FeRIC in chick neural crest explants. RF induced transient Ca2+ permeability in neural crest cells expressing TRPV4FeRIC that was blocked using RN1734 (Fig. 8A). Similar to FeRIC modified TRPV1, remote activation of TRPV4FeRIC in neural crest cells produced a 57% incidence of conotruncal defects in the RF10minTRPV4FeRIC group compared to no defects in the RPnegTRPV4FeRIC group (Fig. 8B,C). Remote activation of TRPV4FeRIC also resulted in significant supravalvular stenosis of the aorta and pulmonary trunk (Fig. 8B, D, and E). Taken together, these data indicate that transient activation of neural crest cell-specific TRPV1 or TRPV4 can replicate the types of clinically important heart defects that have been linked to maternal fevers in humans.
Fig. 8. Fever-associated heart defects following remote activation of TRPV4 in neural crest cells.
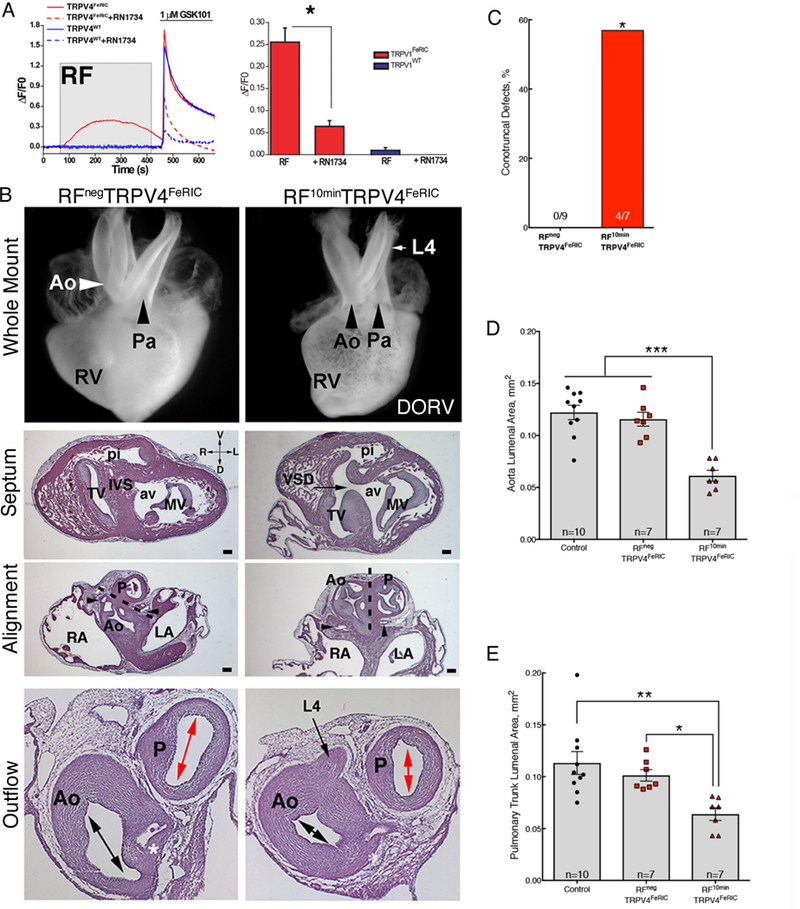
(A) GCaMP6 fluorescence in chick neural crest cells electroporated with TRPV4wt (blue lines) and TRPV4FeRIC (red lines) following RF (gray box) then GSK101 (bar). TRPV4 inhibitor RN1734 inhibits response (dashed lines). Bar graph shows cumulative responses representing 4 separate experiments with 45–49 cells/group analyzed. (B) Whole mount and histological sections of HH63 hearts from RFNegTRPV4FeRIC embryo and RF10minTRPV4FeRIC embryo with DORV and a persistent L4 arch artery. The white and black arrows in the whole mount images highlight the alignment of the aorta (Ao) and pulmonary trunk (P) in the RFNegTRPV4FeRIC and RF10minTRPV4FeRIC hearts. Histological sections of the whole mount in RPnegTRPV4FeRIC and RF10minTRPV4FeRIC hearts (above) at the level of the ventricular septum (IVS), at the level of the semilunar valves of the aorta (coronary arteries, black arrowheads) and pulmonary trunk and more distally through the smooth muscle walls of the aorta and pulmonary trunk at the level of the left coronary artery (*). The dashed line indicates the plane of outflow tract septation. Black arrows indicate a VSD and persistent L4 arch artery in the section through the RF10minTRPV4FeRIC heart. Double headed arrows indicate the luminal cross sectional areas of the aorta and pulmonary trunk measured in the Calvalieri estimates in (D-E). Bar = 200μm. 3 separate experiments were performed and a total of 9 RFNegTRPV4FeRIC and 7 RF10minTRPV4FeRIC hearts were analyzed. Tricuspid valve (TV), mitral valve (MV), aortic vestibule (av), pulmonary infundibulum (pi) right atrium, (RA), and left atrium (LA). (C) Percentage of histologically confirmed conotruncal defects in RFNegTRPV4FeRIC embryos compared to RF10minTRPV4FeRIC embryos. (D) Graph of the Cavalieri probe estimates of cross sectional areas through the aorta at the level of the coronary artery in the indicated treatment groups (CE=0.05). (E) Graph of the Cavalieri probe estimates of the cross-sectional areas through the pulmonary trunk distal to the semilunar valve in the indicated treatment groups. (CE=0.05). Significance was determined using unpaired t-test (A), Fisher exact test (C) or one-way ANOVA followed by Bonferroni’s multiple comparisons test (D,E). *P<0.02, **P<0.005, ***P<0.0001. The number of biological replicates is indicated by n in the graphs in (C, D and E).
To confirm that FeRIC-mediated defects were not caused by RF-induced bulk temperature increases in tissues, we non-invasively mapped temperature changes throughout the egg and embryo at the beginning and end of the 10 min RF exposure using MRI thermometry(39). No substantial temperature differences were observed between RF10min and RPneg groups (fig. S20, A to H). Thus, RF exposure does not significantly heat the embryo to induce congenital defects, which is also supported by the 0% incidence of cardiovascular defects in control eggs exposed to RF (Fig. 7D).
Discussion
Our study identified the presence of two temperature-activated ion channels, TRPV 1 and TRPV4, in cranial and cardiac neural crest cells during critical windows of heart and facial development. We demonstrated that experimentally controlled activation of these channels through temperature, pharmacological, or genetic approaches was sufficient to produce congenital heart and craniofacial defects similar to those associated with human fever in the first trimester. We linked TRP channel activity to hyperthermia-induced defects by showing that TRPV1 inhibition during heat exposure mitigated the majority of the congenital defects. Both TRPV4 agonist and antagonists elicited similar neural crest cell-related defects under normothermic conditions. It is not uncommon for a gain or loss of function of a signaling pathway to result in similar defects, particularly in secondary heart field defects like DORV. For example, in previous studies we have shown that too much or too little FGF signaling in the pharynx (which is mediated by neural crest cells) results in proliferation of the secondary heart field progenitors or premature differentiation of the progenitors respectively(18, 40, 41). Both result in a failure of the secondary heart cells to lengthen the outflow tract which is necessary for proper outflow tract alignment. Similarly, Noonan and Leopard Syndromes have overlapping cardiac and craniofacial phenotypes. Noonan syndrome is attributed to an activating mutation in PTPN11 in 50% of patients whereas Leopard Syndrome is associated with an inactivating mutation in the same gene.
In our model, the timing of the hyperthermia and or TRP channel activation in neural crest cells coincided with their migration into the pharyngeal arches. While the immediate consequences of transient TRP channel activation in neural crest cells will be the focus of future studies, many studies have already shown that increased Ca2+ signaling impacts cell migration, survival and proliferation, all of which are linked to neural crest-associated heart and facial defects. Disruption of neural crest cell polarity is linked to conotruncal and craniofacial defects(42, 43). Increased maternal homocysteine is associated with neural crest-associated defects and homocysteine enhances cardiac neural crest cell attachment and alters migration in vitro through a Ca2+-dependent mechanism(44, 45). Further, increased Ca2+ homeostasis in neural crest cells is linked to human diseases including fetal alcohol syndrome and Timothy syndrome, both of which have neural crest-related craniofacial and or conotruncal defects(46, 47). Together, these studies suggest that intracellular Ca2+ homeostasis is critical for normal neural crest-dependent development.
The study of environmentally-induced transient alterations in genetically normal TRP channel activation has not been described in the context of congenital birth defects. In part, this may be due to the lack of widely available technology enabling precise temporal control of targeted cell-specific activation. Purely pharmacologic approaches lack cell specificity and can be influenced by bioavailability through placental interfaces and maternal or fetal metabolism. Genetically modified channels with constitutive activity, or those that lack activity, can be targeted to cell-specific populations but cannot facilitate transient changes needed to study the impact of passing environmental factors such as fever. Technologies using synthetic designer ligands such as DREADD receptor approaches can provide cell specificity and some temporal resolution(48, 49). However, in our study we needed much shorter restrictions on activation times to mimic the temperature spikes such as those that occur in febrile states. Once synthetic compounds used to activate DREADD receptors are injected into the egg, we would not be able to remove the compounds and therefore their effects could last hours to days depending on metabolism within the chick embryo. Our approach using RF offered the ability to precisely control the presence or absence of channel stimulus remotely. Here, we developed a simplified technology, FeRIC, that utilized the cell’s endogenous iron stores to convert the modified channel into a remotely controlled channel with high temporal resolution and cellular specificity.
The physical mechanisms of ferritin-dependent electromagnetic control over ion channels is under debate (50, 51). The functionality of FeRIC channels is in line with those of similar technologies independently developed by other laboratories(33, 35, 52). Here, we showed that FeRIC channels depended on the presence of endogenous intracellular ferritin. When the FTH1 gene was deleted in vitro, TRPV1FeRIC was unresponsive to RF. Reintroduction of FTH1 in these cells rescued the remote capabilities of TRPV1FeRIC. In another experiment, we showed that the introduction of temperature-sensitive amino acid substitutions in the outer pore region of TRPV1 also abolished RF-dependent channel activity. These data are suggestive but not conclusive evidence for a temperature-dependent mechanism. However, one important theoretical consideration suggests that RF waves at the power level reported in the literature are not sufficient to induce significant heating of ferritin(50). Thus, whether FeRIC-modified channels are activated by RF by a heat-dependent mechanism will require further measurements, such as nanoscale-resolution mapping of iron distribution and temperature change in unexposed and RF-exposed cells expressing these channels. Should this evidence become available, we anticipate that FeRIC modifications could be expanded to study other temperature-sensitive channels, both in the context of development as well as in relation to other spatiotemporally sensitive disease mechanisms.
Lastly, maternal infections with associated fevers in the first trimester are linked to clinically important craniofacial and cardiac defects. Craniofacial defects range from midface hypoplasia to clefts and cardiac defects include obstructive lesions and conotruncal defects. Hyperthermia has been proposed as a direct teratogen; however, the mechanisms linking temperature to birth defects are unknown(53, 54). It is critical to distinguish between infection-based teratogenicity and simple hyperthermia because the latter is a modifiable risk factor. Acetaminophen is drug commonly used by pregnant women and is safe and protects against some fever-associated birth defects(8, 55, 56). However, new concerns have been raised over its repetitive use in pregnancy and the risk for behavioral problems in childhood(57). Our data provide a rational molecular mechanism for hyperthermia-induced teratogenicity mediated through transient gating of temperature-activated ion channels in neural crest cells. Because hyperthermia is a modifiable risk factor in pregnant women, these fever-associated birth defects may be preventable through public awareness and judicious use of anti-pyretics in febrile pregnant women. We hope that these observations will stimulate detailed clinical studies with a view to implement new policies in prenatal care and allow pregnant women to fully weigh the risks and benefits of antipyretic therapy during pregnancy.
Materials and Methods:
Animals
All animal experiments were approved by the Duke University Animal Care and Use Committee, and were performed in accordance with the institutional and NIH ethical guidelines. Fertilized Ross Hubert chick eggs (Gallus gallus domesticus, Mountaire Farms, Siler City, NC) were incubated for 1–10 days at 37°C and 70% humidity. Embryos were staged according to Hamburger and Hamilton(58). Adult zebrafish (Danio rerio) and the Tg-1.4col1a1:egfp line were maintained at 28°C on a 14/10 h light and dark cycle. Embryos were raised at 28°C and staged according to hours post fertilization (hpf) and morphology. Embryos were growth in E3 medium (NaCl, 5mM; KCl, 0.17mM; CaCl2, 0.33mM; MgSO4, 0.33mM, 0.00001% Methylene Blue). All embryos (chick or zebrafish) were stage matched and randomized to the specific treatment groups described below. The investigator was not blinded to the various treatment groups.
Cell Lines
Where indicated, the human embryonic kidney cell line (HEK293T, Clontech Cat#632180) or Chinese Hamster Ovary cell line (CHO, ATCC CCL-61) were used. Both cell lines have tested negative for Mycoplasma contamination by Clontech or ATCC respectively. The identity of HEK293T cell line was verified by STR analysis at Clontech. The identity of CHO cell lines was confirmed at the ATCC using an Isoenzyme (Interspecies) assay for hamster.
Hyperthermia experiments
Chick embryos were incubated at 37°C until the desired age (HH 9–15). The eggs were then placed in a 40–42°C incubator. Internal egg temperature was monitored every 15 min using a digital thermometer probe. It generally took 45–60 min for the eggs to reach the targeted temperature. Once the target temperature was achieved, the eggs were incubated for another 60 min. Following hyperthermia, embryos were re-incubated at 37°C and allowed to develop until day 10 (HH36). The embryos were harvested and gross congenital defects were documented. All hearts and heads were further processed and analyzed for structural defects (see below). Hyperthermic exposures of 40–41°C at HH11–13 produced the most consistent defects. For the hyperthermia rescue experiments embryos were either pretreated with 10μM SB366791 or 0.1% DMSO in PBS, incubated at 41°C for 1hr as described above (see below for details of drug delivery), returned to a 37°C incubator and harvested at HH36 for analysis of heart and craniofacial defects.
Pharmacological treatments of chick embryos
Eggs were windowed at HH11–14 and treated with 20μL of varying concentrations of TRPV channel agonists or antagonist (see below) to determine an effective dose range. Controls included untreated embryos and DMSO-vehicle controls. The eggs were sealed with tape, incubated until HH36 and analyzed for heart and craniofacial defects. All drug stocks were made in DMSO. Stocks were diluted in PBS such that the DMSO concentration was never higher than 0.1% and all treatments were delivered in 20μL volume to the embryo. We determined the effective GSK101 (TRPV4 agonist) dose to be 20μL of a 10–50μM solution per egg (~3.3nM-16.7nM final concentration in an average 60mL egg volume). The incidence and severity of defects increased at the higher doses. Lower doses did not induce defects and no concentration tested caused lethality. Eggs dosed with the 10–100μM RN1734 (1.2nM-12nM final concentration) caused significant incidence of heart and craniofacial defects. For the hyperthermia and TRPV1FeRIC rescue experiments embryos were treated with 10μM SB366791 (1.0 nM final concentration).
TRPV RT-PCR
Total RNA was extracted from HH14, HH22, or dissected HH9 neural folds. cDNA was used is subsequent PCR reactions using the following published primer sets: Chick TRPV1 (F) GTCCTGCATAGACACATGT Chick TRPV1 (R) GCACAAAATACTGTATCCC (59); Chick TRPV2(F) CCCTTGGAGTCACCTTACC Chick TRPV2(R) CTTCCCAGTCTTTGCATCT (60); Chick TRPV3(F) CCCCTCAATTCACTCCTGC Chick TRPV3(R) GGAAAGGCATTCACCACCA (60); Chick TRPV4(F) TTCAAGGATTGGGCATACG Chick TRPV4(R) ATTAACCCTCACTAAAGGGCAACTTCCAGATGTGTTTG (59); Chick SLUG(F) GCCTGCCTTCAAAATGCCACGCTCCTTCCTGG Chick SLUG(R) GGCTGCTGCGTAGCACACTGAGTCATGCAGTC (61).
For the flow sorted chick neural crest explant studies, a plasmid encoding GFP was electroporated at HH9 into the neural tube. The electroporated embryos were removed and the neural tubes were enzymatically dissociated from the surrounding tissue. Specifically, the cranial and cardiac neural crest region extending from the midbrain to the third somite was placed in a solution containing 0.5μg/ml dispase and 1μ,g/ml collagenase for 1–2min. The embryos were then placed in a stop/wash solution of DMEM with 20% fetal bovine serum (FBS) and the surrounding tissues were dissected away from the neural tube using fine needles. The isolated neural tubes were cultured on fibronectin-coated plates overnight at 37°C in DMEM with 20% FBS supplemented with 2% chick embryo extract. 24 hours after plating, the neural tubes were mechanically removed with fine forceps. The remaining neural crest cells were treated with 0.25% trypsin for 2 minutes at 37°C, collected, and washed in DMEM with 20% FBS. The cells were sorted in the Duke Flow Cytometry Core Facility on a MoFlo XDP (Beckman Coulter Life Sciences).
In situ hybridization and immunohistochemistry
Whole mount in situ hybridization was carried out using a standard protocol with DIG-labeled probes(62). Previously published probes were used for TRPV1 and TRPV4 in situ hybridizations(59). PCR amplified probe templates were cloned and sequenced at the Duke Cancer Biology Sequencing Facility. Plasmids containing probe sequences were used to generate RNA probes from the T7 or SP6 promoters in vitro. After visualization of DIG, the embryos were embedded in paraffin, sectioned and photographed to document the cellular localization of TRPV1 and TRPV4 transcripts. To determine co-localization of TRPV1 and TRPV4 transcripts with neural crest cells, the coverslips were soaked in PBS to remove from the sectioned in situ hybridizations. Tissue sections were stained with mouse IgM HNK1 antibody (ATCC) overnight at 4°C and developed with 3,3′ -Diaminobenzidine tetrahydrochloride. All images were acquired on a Leica DMRA2 compound microscope.
Plasmids
To generate the TRPV1wt construct, full length murine wild-type TRPV1 was PCR amplified from cDNA generated from spinal cord tissue from C57/Bl6 mice. 5’ SpeI sites and 3’ NotI sites were introduced into their respective locations outside of the open reading frame using PCR. This product was subcloned into SpeI and NotI sites within the multiple cloning site of the PLVX-IRES-mCherry vector to generate TRPV1wt (Clontech, for detailed map see fig. S12). To generate the TRPV1FeRIC construct, PCR primers were designed to eliminate the 3’ stop site in wild-type TRPV1 and introduce a novel 3’ XbaI site. PCR primers introducing a 5’ XbaI site and a 3’ NotI site and a stop codon were used to amplify human Kininogen1 domain 5 (FeRIC) from whole blood DNA. This FeRIC fragment was subcloned into the XbaI Not1 sites within the PLVX-IRES-mCherry vector (for detailed map see fig. S12). All completed constructs were sequenced verified by the Duke Cancer Biology Sequencing Facility and analyzed using MacVector 13.0.
To generate the TRPV1ΔT FeRIC construct, a synthesized cassette of a 798 bp region was designed that included a native TRPV1 PsHA1 restriction site, introduced the code for triple mutant amino acids N628K, N652T, and Y653T, and removed the native stop codon. We added a novel Xba1 restriction site for ligation in frame with FeRIC sequence. Synthesis of the cassette was done by BioBasic Inc. Original TRPV1FeRIC construct was digested by psHA1 and Xba1, which removed the 3’ terminal end of TRPV1 ending at the FeRIC start. The psHA1-Xba1 mutant cassette was then ligated into the corresponding sites in the TRPV1FeRIC construct. Complete construct with these 3 mutations was sequenced verified by the Duke Cancer Biology Sequencing Facility and analyzed using MacVector 13.0.
To generate the TRPV4WT construct, full-length rat TRPV4 cDNA was a generous gift from Dr. Robert Lefkowitz (Duke University). Spe1 and Not1 restriction sites were introduced using PCR as above. The full-length wild-type TRPV4 was subcloned into PLVX-IRES-mCherry vector to generate TRPV4WT (Clontech, for detailed map see fig. S13). To generate the TRPV4FeRIC construct, PCR primers were designed to eliminate the 3’ stop site in wild-type TRPV4 and introduce a 3’ Not1 site. PCR primers introducing a 5’ Not1 site and a 3’ BamHI site and a stop codon were used to amplify human Kininogen1 domain 5 (FeRIC). This FeRIC fragment was subcloned into the XbaI BamH1 sites within the PLVX-IRES-mCherry vector containing TRPV4 (for detailed map see fig. S13). All completed constructs were sequenced verified by the Duke Cancer Biology Sequencing Facility and analyzed using MacVector 13.0.
Full-length chick TRPV4 was synthesized and sequence verified by Biobasic Inc. The full-length construct was subcloned into the PLVX expression vector using novel Spe1 (5’) and Not1 (3’) sites introduced outside of the open reading frame. Full-length zebrafish TRPV4 was cloned from pooled zebrafish larval cDNA generated from embryos at 24–96hpf using the following 5’ primer containing a novel Spe1 site and the 3’ primer containing a novel xba1 site: zfTRPV4(F) CTATTTCCGGTGAATTCCTCGAGACTAGTCTGGCCATGACAGAGTCCTTGTCTG and zfTRPV4® CGGGATCCGCGGCCGCTCTAGATTAGCTTTCAGACTTGAGTCGG. All constructs were sequenced verified by the Duke Cancer Biology Sequencing Facility and analyzed using MacVector 13.0.
To generate the FTH1 constructs, the full-length human FTH1 was cloned using RT-PCR and cDNA generated from FeCl2 stimulated HEK293T cells. 5′-GCCGCCATGACGACCGCGT and 3′-CCGAGGCTTAGCTTTCATT primers flank the entire open reading frame. The entire open reading frame was cloned into pcDNA 3.1. For FTH1mCherry fusion construct, PCR was used to abolish the stop codon and subclone FTH1 into in the multiple cloning site of pLVX-mCherry-N1 vector (Clontech) in frame with mCherry. Sequences were verified by the Duke Cancer Biology Sequencing Facility and analyzed using MacVector 13.0.
CRISPR Cas9 FTH1 deletion
Ferritin heavy chain (FTH1) was deleted in HEK293T cells using commercially available double nickase plasmids (Santa Cruz Biotechnology Inc.). This system uses two 20nt gRNA sequences targeting exon 2 of the human FTH1 gene. Briefly, cells were transfected with plasmids encoding the gRNA sequences, Cas9, and the puromycin resistance gene. 24 hours later, transfected cells were selected in DMEM media containing 5mg/ml puromycin for 5 days. Surviving cells were harvested and washed and serially diluted into a 96-well plate. Wells containing single cells were expanded and screened by RT-PCR for full length FTH1 mRNA using primers listed above. Full-length FTH1 PCR products were cloned and the expected 147bp sequence deletion was confirmed in exon 2 of FTH1. 88% of the individual clones generated were FTH1 negative by RT-PCR and western blot. HEKFTH1KO clone 2 was expanded and used for subsequent TRPV1FeRIC RF analysis in this study.
RF coil
The RF emitting coil consisted of a double loop wire with a loop diameter of approximately 4 cm. The coil was connected in parallel with tuning capacitors forming an LC circuit. The circuit was tuned to a resonance frequency at approximately 175 MHz. RF signal was generated by a broadband (0–400 MHz) signal generator (Boonton Electronics, NJ, Model 102A) and amplified using a 50-W linear RF amplifier (Electronic Navigation Industries, TX, Model 550L). Electric current across the coil was measured using a current monitor (Pearson Electronics, CA, Model 2877). The peak-to-peak current was measured to be 1.5 A. During experiments, the coil sits roughly 1.5 cm above the cells or chick embryo.
RF simulation
The electromagnetic fields emitted by the RF coil were computed by cylindrical finite-difference time-domain simulation in Matlab© (Mathworks, MA, version R2014a) as described by Dib and Weller(63). Perfectly-matched-layer boundary conditions were implemented as described by Schneider(64) and the simulation resolution was 0.5 cm in the z direction, 0.25 cm in the radial direction and π/28 radians in the φ direction. The computed magnetic field was verified by comparison with the analytical expression for the magnetic field along the axis of a static current loop. The electromagnetic fields were simulated for a coil loaded with a dish of 0.5-cm thickness and 2-cm radius. The dish electrical conductivity was 0.5 S/m, similar to human tissue (65). The AC input was 1.5 A peak-to-peak at 175 MHz.
Bilateral electroporations and RF delivery to eggs
For the cranial and cardiac neural crest cell electroporation studies, eggs were incubated to HH9–11. The eggs were windowed and the vitelline membrane was torn. One or two drops of Ringers solution was applied so that the embryo fell away from the vitelline membrane. Platinum electrodes were placed at the cranial end on either side of the embryo. TRPV1WT, TRPV1FeRIC, TRPV4WT, or TRPV4FeRIC constructs were injected into the neural tube using a glass micropipette. DNA concentration was 4μg/μl. Four 50ms square wave pulses were immediately applied at 20 V each. A drop of Ringers was again applied to the embryo, and injection and electroporation were repeated for the other side of the neural tube. 24 hours after electroporation, expression of the plasmid was confirmed by visualizing mCherry expression. Then the egg was placed in a 37°C chamber, the RF coil was placed on the egg surrounding the embryo and 10 minutes of RF stimulation (see above) was delivered. Control (RFNeg) eggs were placed in the same 37°C chamber for 10 minutes without RF stimulation. For the TRPV1FeRIC rescue experiments, TRPV1FeRIC-expressing embryos were pretreated with 10μM SB366791 for 1hr prior to RF exposure.
Primary neural crest cell cultures
A mixture of GCaMP6(66) (obtained from Addgene#40753) was electroporated alone or in combination with the TRPV constructs described above into chick neural tubes at HH9. The electroporated embryos were removed from the egg and the neural tubes were enzymatically dissociated from the surrounding tissue. Specifically, the cranial and cardiac neural crest region extending from the midbrain to the third somite was placed in a solution containing 0.5μg/ml dispase and 1μg/ml collagenase for 1–2min. The embryos were then placed in a stop/wash solution of DMEM with 20% fetal bovine serum (FBS) and the surrounding tissues were dissected away from the neural tube using fine needles. The isolated neural tubes were cultured on fibronectin-coated plates overnight at 37°C in DMEM with 20% FBS supplemented with 2% chick embryo extract and Ca2+ imaging was performed as described below. Mouse neural crest cell explants were generated as above using E8.5 embryos from CD1 mice. Mouse neural tubes were cultured at 37°C in DMEM with 20% FBS.
Ca2+ imaging
HEK293T or CHO cells were plated on collagen-coated glass bottom 35 mm dishes and cultured in 10% FBS and 1% PenStrep in DMEM. After 24 hours, cells were co-transfected using the Lipofectamine LTX Plus reagent with GCaMP6 and either TRPV1WT, TRPV1FeRIC, TRPV4WT, or TRPV4FeRIC. For Ca2+ imaging of chick neural crest cells, neural crest cells were electroporated, isolated and cultured as described above. Twenty-four hours after electroporation, cytosolic Ca2+ were monitored by fluorescence imaging of cells positive for GCaMP6+ and TRPV channels (mCherry+) using a 20x objective on an inverted Olympus IX50 microscope equipped with a camera (Q-Imaging-Retiga 1300i) controlled by Metamorph software 7.8 (Molecular Devices). Images were captured at 1 frame per sec. Unless stated otherwise, all experiments were carried out at room temperature. Neural crest cell recordings were carried out in Leibovitz’s L-15 Medium (ThermoFisher) supplemented with Ca2+ to a final concentration of 2mM. For HEK293T cells, the imaging buffer solution contained 140mM NaCl, 2.8mM KCl, 1mM MgCl2, 2mM CaCl2, 10mM glucose, and 10mM HEPES at pH 7.4. RF was delivered for 10 min using a custom built RF emitting coil designed to fit the 35mm tissue culture dish as described above. After RF, cells were then exposed to TRP channel agonists, 1μM capsaicin or 1μM GSK1016790A. In a second series of experiments, HEK293T or neural crest cells were exposed to TRPV1 or TRPV4 channel inhibitors (10μM SB366791 or 10μM RN1734) for 10 min and then imaged for changes in intracellular Ca2+ with or without RF exposure. Images were analyzed using Metamorph software; cytosolic regions of interest (ROIs) were placed over those cells which co-expressed GCaMP6 and TRPV channels. GCaMP6 fluorescence intensity was measured for each image of the time-lapse acquisition (650 sec). The data were fit with a double exponential decay time and corrected for photobleaching using the Microcal Origin 7 software (OriginLab, MA). Responses are presented as ΔF/F0, where F0 is the resting fluorescence averaged over 60 s before the start of stimulation and ΔF is the change in fluorescence over resting values. For each experimental condition, 50–150 cells were analyzed and each experiment was conducted at least 3 times. Plots in the individual figures are representative tracings.
To image Ca2+ changes in mouse neural crest cells, neural tubes from E8.5 embryos were dissected and plated as described above. 8–24 hours after culture, cells were washed with imaging buffer solution and incubated with 5 μM fluo-4 AM (Molecular Probes) for 40 min at room temperature. Fluo-4 loaded cells were imaged using the imaging system described above. Fluo-4 was excited using a blue filter and green emission was collected at 1 frame per second. Functional TRPV1 channels were analyzed by exposing the explants to 1μM capsaicin 8–18 hours after plating. 10μM SB366791 was used to block capsaicin-mediated Ca2+ responses. For assessment of TRPV4 channels, at 8–24 hours neural crest cells explants were exposed to GSK1016790A (0.1 – 1000 nM). The TRPV4 inhibitor RN1734 (10μM) was used to inhibit the GSK1016790A mediated response. Data were analyzed off-line with MetaMorph Image Analysis software. Changes in cytosolic Ca2+ were estimated as the relative increase of fluorescence intensity (F) from baseline fluorescence (F0).
Temperature Activation Ca2+ Imaging Assays
Temperature-sensitivity of TRPV channels was examined at a hyperthermic (40°C) temperature in neural crest cells or HEK293T cells transfected with GCAMP6 and either TRPV1FeRIC or TRPV1ΔTFeRIC, using Lipofectamine LTX Plus reagent. Twenty-four later, transfected cells were identified by expression of both GCaMP6 and TRPV1 channels (mCherry+). Primary GCaMP6+ chick neural crest cells were also generated as described above to examine the thermosensitivity of endogenous TRPV channels. For Ca2+ imaging, neural crest cells or HEK293T cells were placed on a recording chamber in the stage of Nikon-eclipse Ti-s inverted micropscope. Cells were epi-illuminated using a blue filter and continuously perfused with the imaging buffer solution through a gravity-driven perfusion system (~2mL per min). Drugs were applied using same gravity system. The temperature was controlled with a CL-100 temperature controller (Warner Instruments) and a SC-20 dual In-Line Heater/Cooler (Harvard Apparatus). During experiments, temperature was monitored with a thermistor (TA-29, Warner Instruments) within the recording chamber. Heat stimulus was delivered stepwise to increase the temperature from 25°C to 40°C over a 3-minute period. After removal of the heat stimulus, cells were exposed to TRPV channel agonists such as pH 5 or 1 μM GSK1016790A. Neural crest cells were exposed to TRPV4 channel inhibitor 10 μM RN1734 for 10 min and then imaged for changes in intracellular Ca2+ with heat or GSK1016790A exposure. GCaMP6 green emission was collected from cells at 1 frame per second. Images were analyzed offline using Nikon Element software; ROIs were placed over those cells which co-expressed GCaMP6 and TRPV channels, or over the GCAMP6 positive neural crest cells for endogenous TRPV channel analysis. GCaMP6 fluorescence intensity was measured for each image of the time-lapse acquisition. Data were analyzed using the Microcal Origin 7 software (OriginLab, MA). Responses are presented as ΔF/F0 and plots correspond to average ± SEM.
Membrane integrity and cell viability assays
Membrane integrity assays were conducted in HEK293T cells in vitro. Membrane integrity was assayed using LIVE/DEAD Fixable Green Dead Cell Stain Kit (Life Technologies) following manufacturer’s instructions. In this assay, the green dye is excluded from cells with intact membranes. However, cells with disrupted membranes, the dye accumulates in the cell and reacts with cytoplasmic proteins. Briefly, mCherry or TRPv1FeRIC expressing HEK293T cells were exposed to either no RF, RF for 10 minutes, or 10 minutes at 55°C. After 5 minutes, cells were stained with manufacturer’s green fluorescent amine reactive dye then fixed with 1% paraformaldehyde. Cells were analyzed using a 488nm line of an argon-ion laser and green fluorescence was detected in the green channel of the flow cytometer (530/30 nm).
Cell viability was measured with the CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT, Promega). HEK293T cells (40–50% confluent) transfected with TRPV 1FeRIC or mCherry were exposed to RF for 10 minutes and cultured at 37°C for 30 hours before performing the assay according to the manufacturer’s instructions. RF-exposed cells from each group was compared to transfected cells without RF exposure.
Immunoprecipitation
N-terminally FLAG tagged wild-type and FeRIC modified TRPV1 and TRPV4 were generated by PCR. HEK293T cells in a 6-well plate were transfected with 3μg of plasmid DNA using Lipofectamine PLUS reagent (Invitrogen). 8 hours after transfection, culture media was exchanged with fresh media containing 100μM FeCl2 for endogenous ferritin induction. 18 hours later, protein was extracted and immunoprecipitation experiments were performed as described(67) with the following modification: CHAPS lysis buffer contained 5 mM CHAPS, 50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM CaCl, 5% sucrose, 0.5% Triton X-100, protease inhibitor cocktail (Roche). Anti-FLAG M2 affinity gel (Sigma A2220) was used to immunoprecipitate FLAG tagged TRPV1 or TRPV4 constructs following the manufacturer’s instructions. Samples were probed by western blot using antibodies against DDDDK (1:2,000, Abcam, ab1170) or FTH1 (1:1,000, Cell Signaling #3998).
Heart analysis
Hearts from the various treatment groups were harvested at day 10, photographed, fixed overnight in 10% formalin, paraffin embedded, serially sectioned and stained with hematoxylin and eosin and assessed for defects as previously described(41). Briefly, the hearts was scored as DORV if the aorta or the aortic portion of the outflow was not wedged between the tricuspid and mitral valve and shifted to the right in relation to the pulmonary trunk. All hearts categorized as DORV had a concomitant VSD. Hearts were classified as overriding aorta if the outflow VSD was positioned such that the aortic vestibule was in communication with the pulmonary infundibulum and the aorta was overriding the septal defect. In embryos with abnormal persistence of arch artery vessels, only vessels with patent lumens were scored. Outflow obstructions lesions were qualitatively assessed compared to stage-matched controls. Quantitative measurements were carried out as described below (see Stereology). Hearts from embryos with ectopia cordis were excluded form analysis because the etiology of the alignment defects cannot be distinguished between the experimental treatment or the open chest wall.
Stereology
Hematoxylin and eosin staining was performed on serial 10-μm sections through the entire heart from the ventricular apex through the aortic arch as previously described (41). The aortic and pulmonary valves were identified. For the aorta, we began with the last section containing coronary artery and moving superiorly, the lumens in 3–4 adjacent tissue sections were analyzed. For the pulmonary trunk, we identified the last section with pulmonary valve tissue attached to the vessel wall and analyzed 3–4 tissue sections above that. Analysis was conducted in bright field using the Cavalieri probe in Stereo Investigator 11 (MBF Biosciences, VT) on a Zeiss AxioImager2 microscope. Analysis parameters include: tissue cut thickness, 10μm; analysis objective, 10x; probe grid size, 50μm x 50μm; grid orientation, randomized. The results are presented as the average luminal area in mm2 for each vessel. The average coefficient of error (CE, Gundersen m+1) is a standard statistical value that is used extensively in the stereological studies. The definition of the CE is defined as the standard error of the mean of repeated estimates divided by the mean. An average CE was reported for each of the experiments within the individual figures and was less than 0.05.
MRI thermometry
MRI thermometry was used to measure internal heating in chicken eggs due to RF. This technique uses the temperature-dependent chemical shift of water to measure internal temperature changes during the time between two MRI scans of a stationary object(39). TRPV1FeRIC was electroporated into the neural tube of chick embryos at HH9. Twenty-four hours later, eggs were imaged using a 7.0 T small-bore animal MRI scanner controlled by Agilent software version vnmr4 (Santa Clara, CA). A multi-echo gradient echo sequence was used to measure frequency maps. The sequence parameters were: field of view (FOV) = 7×7 cm, matrix =128×128, repetition time (TR) = 200 ms, first echo time (TE1) = 5 ms, second echo time (TE2) = 10 ms, flip angle = 10°, slice thickness = 2 mm. Each scan took 20 s and was repeated every 10 minutes during 140 minutes, alternating 10 minute periods of RF and no RF. The first two echoes of each scan were used to create 14 maps that measured changes in temperature during periods of RF and 14 maps that measured temperature changes without RF.
Anatomical MRI and heart visualization
Prior to MRI, embryo specimens were immersion fixed in 2.5 mM ProHance (Gadoteridol, Bracco Diagnostics Inc., Princeton, NJ) dissolved in 10% formalin. ProHance, a gadolinium contrast agent, was used to decrease the T1 (longitudinal relaxation time) and to improve SNR(68). Images were acquired on a 9.4T system (400 MHz vertical bore Oxford superconducting magnet). The system consists of an 89-mm vertical bore magnet controlled by an Agilent VnmrJ 4.0 console (Agilent Technologies, Santa Clara, CA). The specimens were firmly secured in an acrylic specimen cartridge and immersed in Fomblin (Ausimont USA, Inc., Thorofare, NJ) to limit susceptibility artifacts at the tissue boundary. The cartridge was placed in a copper solenoid RF resonator (14-mm diameter, 21-mm length). A set of 3D multi-echo gradient echo images were acquired (10 echoes, TE1/spacing/TE10 = 3.9/7.6/71.9 ms) with a field of view of 22×11×11 mm3 and a matrix size of 768×384×384, resulting in isotropic resolution of 28.6×28.6×28.6 μm3. Final images were produced using a weighted sum of the individual echo images(69, 70).
One specimen was rotated to a position such that all four chambers of the heart were visible on a cross section. This position served as a reference. The coronal plane of this reference space was orthogonal to the atrial septum. Images were registered to this space using rigid body transformation, correlation cost function, and sinc interpolation (FMRIB Software Library, http://www.fmrib.ox.ac.uk/fsl). The heart wall, chambers, and vessels were segmented using seeded region growing in Avizo software (Visualization Sciences Group, Burlington, MA). The individual chambers (right atrium, right ventricle, left atrium, left ventricle) and vessels (pulmonary artery, aorta) were further segmented manually at the respective valves.
qPCR for cellular Fe2+ homeostasis
HEK293T cells were transfected with control mCherry, TRPV1WT, TRPV1FeRIC, TRPV4WT, or TRPV4FeRIC. Seventy-two hours after transfection, total RNA was extracted and cDNA was prepared using High Capacity cDNA RT (Applied Biosciences). qPCR was performed using SYBR green (IQ SYBR green Supermix; Biorad) on a Biorad C1000 Touch Thermal Cycler. mRNA expression was calculated using the ΔΔCt method and normalized to the relative expression of β-actin. mRNA levels for mCherry were adjusted to 1. Primers used have been previously published(71). Primer sequences are: Fpn(F) TTGCAGGAGTCATTGCTGCTA Fpn(R) TGGAGTTCTGCACACCATTGAT; Fth(F) CCATCAACCGCCAGATCAAC Fth(R) GCCACATCATCTCGGTCAAA; Ftl(F) GGGCCTCCTACACCTACCTC Ftl(R) CTCCTGGGTTTTACCCCATT; Slc11a2+1RE(F) TGTTTGATTGCATTGGGTCTG Slc11a2+1RE(R) CGCTCAGCAGGACTTTCGAG; Tfrl ex 2,3(F) TCAAGCCAGATCAGCATTCTC Tfrl ex 2,3(R) AGCCAGTTTCATCTCCACATG.
Avian craniofacial analysis
To visualize the development of the skeletal structures, day 10–11 chicks from the various treatment groups were fixed in 95% ethanol and processed for alcian blue and alizarin red staining as described by(72). Once cleared in glycerol, the length of the jaw structures (in mm) was measured using a dissecting microscope fitted with an ocular micrometer. The upper beak measurement extended from the quadratojugal to the tip of the upper beak. To ensure comparable staging across treatment groups, the upper beak length for each chick were normalized to individual femur lengths.
Zebrafish pharmacological treatment and imaging
We treated zebrafish larvae at 48 hours post fertilization (hpf) to assess the impact of TRPV4 stimulation on cranial neural crest cell convergent and extension. Zebrafish embryos were generated by natural pairwise matings of −1.4col1a1:egfp transgenic adults (AB background)(31) Embryos were collected 30 minutes after spawning, stored at 28°C in embryo media (0.3g/L NaCl, 75mg/L CaSO4, 37.5mg/L NaHCO3, 0.003% methylene blue), and distributed randomly into separate petri dishes (50 embryos per dish). At 48hpf, embryos were treated with either 20uM GSK1016790A (GSK101) in 0.1% DMSO, 0.1% DMSO (vehicle control), or 20uM GSK1153218A, a compound that is structurally similar to GSK101 but lacks detectible activity (Supplemental Table. 1). We obtained ventral images of cartilaginous craniofacial structures at 3 and 4 days post fertilization (dpf) using the Vertebrate Automated Screening Technology (VAST; software version 1.2.2.8.) platform (Union Biometrica) as described(73). Embryos were collected after imaging and returned to their petri dish with fresh experimental media and stored at 28°C until the 4dpf imaging time point. Craniofacial defects were quantified by measuring the distance between Meckel’s cartilage and the ceratohyal using ImageJ. All experiments were repeated at least 3 times.
Statistical analysis
All experiments unless otherwise noted were repeated a minimum of 3 times. Differences in categorical data sets (percentage of animals with congenital defects) were analyzed using Fisher’s exact testing. Differences in continuous data sets were analyzed using a one-way ANOVA followed by Bonferroni post-hoc testing or unpaired t testing, where appropriate. Data are expressed as mean ± SEM where applicable. A P value less than 0.05 was considered a statistically significant difference.
Supplementary Material
Acknowledgements:
The authors thank Stacy A. Duffy and Harriett A. Stadt for technical assistance. The authors thank Dan Price and Heather Madsen (GlaxoSmithKline) for GSK1153218A and their expertise. C.L. and E.J.B. are grateful to Dr. Nancy Andrews (Duke University) for her valuable insight on intracellular cellular iron homeostasis. The authors wish to thank Russell Dibb of Duke University Center for In Vivo Microscopy for assisting with MRI experiments. We thank Drs. Ronald Goldberg, Vann Bennett, and Wolfgang Liedtke (Duke University) for critical review of the manuscript, and Dr. Robert Lefkowitz (Duke University) for providing plasmids.
Funding: E.J.B. and M.R.H. and research reported here were supported by Jean and George Brumley Jr. Neonatal Perinatal Research Institute and the Zeist Foundation.
E.J.B. was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under award number K12HD 043494 and T32HD043728 and Duke Health Scholars Award (DUHS). M.R.H. was supported by The Hartwell Foundation, Mandel Foundation and AHA 16GRNT30980012. E.G. was supported by NIBIB T32EB001040. C.L. was supported in part by the National Institutes of Health grants NIMH R01MH096979, NHLBI R21HL122759 and NIBIB P41EB015897.
Footnotes
Data and materials availability: TRPV 1FeRIC, TRPV1WT, TRPV4FeRIC, TRPV4WT and TRPV1Δt FeRIC plasmids will be made available upon request and will require a materials transfer agreement from E.J.B or C.L.
Competing Interests: C.L. and E.J.B. have filed a patent application (WO2016004281 A1 PCT/US2015/038948) relating to the use of FeRIC for cell modulation and treatments.
References and Notes
- 1.Hoffman JIE, Kaplan S, The incidence of congenital heart disease, Journal of the American College of Cardiology 39, 1890–1900 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A, for the National Birth Defects Prevention Network, Kirby RS, Collins JS, Eds. Updated national birth prevalence estimates for selected birth defects in the United States, 2004–2006, Birth Defect Res A 88, 1008–1016 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Botto LD, Correa A, Decreasing the burden of congenital heart anomalies: an epidemiologic evaluation of risk factors and survival, Progress in Pediatric Cardiology 18, 111–121 (2003). [Google Scholar]
- 4.Dixon MJ, Marazita ML, Beaty TH, Murray JC, Cleft lip and palate: understanding genetic and environmental influences, Nature Publishing Group 12, 167–178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dreier JW, Andersen AMN, Berg-Beckhoff G, Systematic Review and Meta-analyses: Fever in Pregnancy and Health Impacts in the Offspring, PEDIATRICS 133, e674–e688 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Csáky-Szunyogh M, Vereczkey A, Kósa Z, Gerencsér B, Czeizel AE, Risk and protective factors in the origin of conotruncal defects of heart--a population-based case-control study, Am. J. Med. Genet. 161A, 2444–2452 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Botto LD, Panichello JD, Browne ML, Krikov S, Feldkamp ML, Lammer E, Shaw GM, Congenital heart defects after maternal fever, YMOB 210, 359.e1–359.e11 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Oster ME, Riehle-Colarusso T, Alverson CJ, Correa A, Associations between maternal fever and influenza and congenital heart defects, The Journal of Pediatrics 158, 990–995 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Smith DW, Clarren SK, Harvey MA, Hyperthermia as a possible teratogenic agent, The Journal of Pediatrics 92, 878–883 (1978). [DOI] [PubMed] [Google Scholar]
- 10.Botto LD, Panichello JD, Browne ML, Krikov S, Feldkamp ML, Lammer E, Shaw GM, Congenital heart defects after maternal fever, YMOB 210, 359.e1–359.e11 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Milunsky A, Maternal Heat Exposure and Neural Tube Defects, JAMA 268, 882–885 (1992). [PubMed] [Google Scholar]
- 12.Kirby ML, Gale TF, Stewart DE, Neural crest cells contribute to normal aorticopulmonary septation, Science 220, 1059–1061 (1983). [DOI] [PubMed] [Google Scholar]
- 13.Waldo KL, Kumiski D, Kirby ML, Cardiac neural crest is essential for the persistence rather than the formation of an arch artery, Dev. Dyn. 205, 281–292 (1996). [DOI] [PubMed] [Google Scholar]
- 14.Santagati F, Rijli FM, Cranial neural crest and the building of the vertebrate head, Nat Rev Neurosci 4, 806–818 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Keyte A, Hutson MR, The neural crest in cardiac congenital anomalies, Differentiation 84, 25–40 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leatherbury L, Gauldin HE, Waldo K, Kirby ML, Microcinephotography of the developing heart in neural crest-ablated chick embryos, Circulation 81, 1047–1057 (1990). [DOI] [PubMed] [Google Scholar]
- 17.Li Y-X, Zdanowicz M, Young L, Kumiski D, Leatherbury L, Kirby ML, Cardiac neural crest in zebrafish embryos contributes to myocardial cell lineage and early heart function, Dev. Dyn. 226, 540–550 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, Abu-Issa R, Kirby ML, Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart, Developmental Biology 281, 78–90 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Julius D, Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, The capsaicin receptor: a heat-activated ion channel in the pain pathway, Nature 389, 816–824 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D, A capsaicin-receptor homologue with a high threshold for noxious heat, Nature 398, 436–441 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Dhaka A, Murray AN, Mathur J, Earley TJ, Petrus MJ, Patapoutian A, TRPM8 is required for cold sensation in mice, Neuron 54, 371–378 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Waning J, Vriens J, Owsianik G, Stuwe L, Mally S, Fabian A, Frippiat C, Nilius B, Schwab A, A novel function of capsaicin-sensitive TRPV 1 channels: Involvement in cell migration, Cell Calcium 42, 17–25 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Martin E, Dahan D, Cardouat G, Gillibert-Duplantier J, Marthan R, Savineau J-P, Ducret T, Involvement of TRPV1 and TRPV4 channels in migration of rat pulmonary arterial smooth muscle cells, Pflugers Arch - Eur J Physiol 464, 261–272 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Ye L, Kleiner S, Wu J, Sah R, Gupta RK, Banks AS, Cohen P, Khandekar MJ, Boström P, Mepani RJ, Laznik D, Kamenecka TM, Song X, Liedtke W, Mootha VK, Puigserver P, Griffin PR, Clapham DE, Spiegelman BM, TRPV4 Is a Regulator of Adipose Oxidative Metabolism, Inflammation, and Energy Homeostasis, Cell 151, 96–110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi QY, Zhang JB, Mi YQ, Song Y, Ma J, Zhang YL, Congenital heart defects and maternal fever: systematic review and meta-analysis, J Perinatol 34, 677–682 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Güler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M, Heat-evoked activation of the ion channel, TRPV4, J. Neurosci. 22, 6408–6414 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordt S-E, Julius D, Molecular basis for species-specific sensitivity to “hot” chili peppers, Cell 108, 421–430 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Siemens J, Zhou S, Piskorowski R, Nikai T, Lumpkin EA, Basbaum AI, King D, Julius D, Spider toxins activate the capsaicin receptor to produce inflammatory pain, Nature 444, 208–212 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Vincent F, Acevedo A, Nguyen MT, Dourado M, DeFalco J, Gustafson A, Spiro P, Emerling DE, Kelly MG, Duncton MAJ, Identification and characterization of novel TRPV4 modulators, BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS 389, 490–494 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Mangos S, Liu Y, Drummond IA, Dynamic expression of the osmosensory channel trpv4 in multiple developing organs in zebrafish, Gene Expression Patterns 7, 480–484 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kague E, Gallagher M, Burke S, Parsons M, Franz-Odendaal T, Fisher S, Roehl HH, Ed. Skeletogenic Fate of Zebrafish Cranial and Trunk Neural Crest, PLoS ONE 7, e47394–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang H, Delikanli S, Zeng H, Ferkey DM, Pralle A, Remote control of ion channels and neurons through magnetic-field heating of nanoparticles, Nature Nanotech 5,1–5 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Stanley SA, Sauer J, Kane RS, Dordick JS, Friedman JM, Remote regulation of glucose homeostasis in mice using genetically encoded nanoparticles, Nature Medicine 21,1–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanley SA, Gagner JE, Damanpour S, Yoshida M, Dordick JS, Friedman JM, Radio-wave heating of iron oxide nanoparticles can regulate plasma glucose in mice, Science 336, 604–608 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wheeler MA, Smith CJ, Ottolini M, Barker BS, Purohit AM, Grippo RM, Gaykema RP, Spano AJ, Beenhakker MP, Kucenas S, Patel MK, Deppmann CD, Güler AD, Genetically targeted magnetic control of the nervous system, Nature Neuroscience 19, 756–761 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coffman LG, Parsonage D, D’Agostino RJ, Torti FM, Torti SV, Regulatory effects of ferritin on angiogenesis, Proc Natl Acad Sci U S A 106, 570–575 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen R, Romero G, Christiansen MG, Mohr A, Anikeeva P, Wireless magnetothermal deep brain stimulation, Science, 1–7 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Grandl J, Kim SE, Uzzell V, Bursulaya B, Petrus M, Bandell M, Patapoutian A, Temperature-induced opening of TRPV1 ion channel is stabilized by the pore domain, Nature Neuroscience 13, 708–714 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishihara Y, Calderon A, Watanabe H, Okamoto K, Suzuki Y, Kuroda K, Suzuki Y, A precise and fast temperature mapping using water proton chemical shift, Magn Reson Med 34, 814–823 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Hutson MR, Zeng XL, Kim AJ, Antoon E, Harward S, Kirby ML, Arterial pole progenitors interpret opposing FGF/BMP signals to proliferate or differentiate, Development 137, 3001–3011 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hutson MR, Zhang P, Stadt HA, Sato AK, Li Y-X, Burch J, Creazzo TL, Kirby ML, Cardiac arterial pole alignment is sensitive to FGF8 signaling in the pharynx, Developmental Biology 295, 486–497 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Xu X, Francis R, Wei CJ, Linask KL, Lo CW, Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells, Development 133, 3629–3639 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Jin Y, Li J, Seto E, Kuo E, Yu W, Schwartz RJ, Blazo M, Zhang SL, Peng X, Inactivation of Cdc42 in neural crest cells causes craniofacial and cardiovascular morphogenesis defects, Developmental Biology 383, 239–252 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Heidenreich DJ, Reedy MV, Brauer PR, Homocysteine enhances cardiac neural crest cell attachment in vitro by increasing intracellular calcium levels, Dev. Dyn. 237, 2117–2128 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Brauer PR, Rosenquist TH, Effect of elevated homocysteine on cardiac neural crest migration in vitro, Dev. Dyn. 224, 222–230 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Garic A, Flentke GR, Amberger E, Hernandez M, Smith SM, CaMKII activation is a novel effector of alcohol’s neurotoxicity in neural crest stem/progenitor cells, J. Neurochem. 118, 646–657 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramachandran KV, Hennessey JA, Barnett AS, Yin X, Stadt HA, Foster E, Shah RA, Yazawa M, Dolmetsch RE, Kirby ML, Pitt GS, Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development, J. Clin. Invest. 123, 1638–1646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strader CD, Gaffney T, Sugg EE, Candelore MR, Keys R, Patchett AA, Dixon RA, Allele-specific activation of genetically engineered receptors, Journal of Biological Chemistry 266, 5–8 (1991). [PubMed] [Google Scholar]
- 49.Nawaratne V, Leach K, Suratman N, Loiacono RE, Felder CC, Armbruster BN, Roth BL, Sexton PM, Christopoulos A, New insights into the function of M4 muscarinic acetylcholine receptors gained using a novel allosteric modulator and a DREADD (designer receptor exclusively activated by a designer drug), Molecular Pharmacology 74, 1119–1131 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Meister M, Physical limits to magnetogenetics, eLife 5, e17210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anikeeva P, Jasanoff A, Problems on the back of an envelope, eLife 5 (2016), doi: 10.7554/eLife.19569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanley SA, Kelly L, Latcha KN, Schmidt SF, Yu X, Nectow AR, Sauer J, Dyke JP, Dordick JS, Friedman JM, Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism, Nature 531, 647–650 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edwards MJ, Hyperthermia as a teratogen: a review of experimental studies and their clinical significance, Teratog Carcinog Mutagen 6, 563–582 (1986). [DOI] [PubMed] [Google Scholar]
- 54.Edwards MJ, Review: Hyperthermia and fever during pregnancy, Birth Defect Res A 76, 507–516 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Thorpe PG, Gilboa SM, Hernandez-Diaz S, Lind J, Cragan JD, Briggs G, Kweder S, Friedman JM, Mitchell AA, Honein MA, The National Birth Defects Prevention Study, Medications in the first trimester of pregnancy: most common exposures and critical gaps in understanding fetal risk, Pharmacoepidemiol Drug Saf, n/a-n/a (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feldkamp ML, Meyer RE, Krikov S, Botto LD, Acetaminophen use in pregnancy and risk of birth defects: findings from the National Birth Defects Prevention Study, Obstet Gynecol 115, 109–115 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Stergiakouli E, Thapar A, Davey Smith G, Association of Acetaminophen Use During Pregnancy With Behavioral Problems in Childhood, JAMA Pediatr 170, 964–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.HAMBURGER V, HAMILTON HL, A series of normal stages in the development of the chick embryo, J Morphol 88, 49–92 (1951). [PubMed] [Google Scholar]
- 59.Bell GW, Yatskievych TA, Antin PB, GEISHA, a whole-mount in situ hybridization gene expression screen in chicken embryos, Dev. Dyn. 229, 677–687 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Somogyi CS, Matta C, Foldvari Z, Juhasz T, Katona E, Takacs AR, Hajdu T, Dobrosi N, Gergely P, Zakany R, Polymodal Transient Receptor Potential Vanilloid (TRPV) Ion Channels in Chondrogenic Cells, Int J Mol Sci 16, 18412–18438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marin F, Nieto MA, Expression of chicken slug and snail in mesenchymal components of the developing central nervous system, Dev. Dyn. 230, 144–148 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Wilkinson DG, Nieto MA, Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts, Methods Enzymol 225, 361–373 (1993). [DOI] [PubMed] [Google Scholar]
- 63.Dib N, Weller T, Two-dimensional finite difference time domain analysis of cylindrical transmission lines, International Journal of Electronics 87, 1065–1081 (2000). [Google Scholar]
- 64.Berenger J-P, A perfectly matched layer for the absorption of electromagnetic waves, Journal of Computational Physics 114, 185–200 (1994). [Google Scholar]
- 65.Gabriel C, Gabriel S, Corthout E, The dielectric properties of biological tissues: I. Literature survey, Phys Med Biol 41, 2231–2249 (1996). [DOI] [PubMed] [Google Scholar]
- 66.Chen T-W, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS, Ultrasensitive fluorescent proteins for imaging neuronal activity, Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benner EJ, Luciano D, Jo R, Abdi K, Paez-Gonzalez P, Sheng H, Warner DS,Liu C, Eroglu C, Kuo CT, Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4, Nature 497, 369–373 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson GA, Benveniste H, Black RD, Hedlund LW, Maronpot RR, Smith BR, Histology by magnetic resonance microscopy, Magn Reson Q 9, 1–30 (1993). [PubMed] [Google Scholar]
- 69.Wu B, Li W, Avram AV, Gho S-M, Liu C, Fast and tissue-optimized mapping of magnetic susceptibility and T2* with multi-echo and multi-shot spirals, Neuroimage 59 297–305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dibb R, Qi Y, Liu C, Magnetic susceptibility anisotropy of myocardium imaged by cardiovascular magnetic resonance reflects the anisotropy of myocardial filament alphahelix polypeptide bonds, J Cardiovasc Magn Reson 17, 60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matak P, Matak A, Moustafa S, Aryal DK, Benner EJ, Wetsel W, Andrews NC, Disrupted iron homeostasis causes dopaminergic neurodegeneration in mice, Proc Natl Acad Sci U S A, 201519473–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wassersug RJ, A procedure for differential staining of cartilage and bone in whole formalin-fixed vertebrates, Stain Technol 51, 131–134 (1976). [DOI] [PubMed] [Google Scholar]
- 73.Isrie M, Breuss M, Tian G, Hansen AH, Cristofoli F, Morandell J, Kupchinsky ZA, Sifrim A, Rodriguez-Rodriguez CM, Dapena EP, Doonanco K, Leonard N, Tinsa F, Moortgat S, Ulucan H, Koparir E, Karaca E, Katsanis N, Marton V, Vermeesch JR, Davis EE, Cowan NJ, Keays DA, Van Esch H, AR TICLE Mutations in Either TUBB or MAPRE2 Cause Circumferential Skin Creases Kunze Type, The American Journal of Human Genetics 97, 790–800 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kontges G, Lumsden A, Rhombencephalic neural crest segmentation is preserved throughout craniofacial ontogeny, Development 122, 3229–3242 (1996). [DOI] [PubMed] [Google Scholar]
- 75.Noden DM, The role of the neural crest in patterning of avian cranial skeletal, connective, and muscle tissues, Developmental Biology 96, 144–165 (1983). [DOI] [PubMed] [Google Scholar]
- 76.Couly GF, Coltey PM, Le Douarin NM, The triple origin of skull in higher vertebrates: a study in quail-chick chimeras, Development 117, 409–429 (1993). [DOI] [PubMed] [Google Scholar]
- 77.Wada N, Hedgehog signaling is required for cranial neural crest morphogenesis and chondrogenesis at the midline in the zebrafish skull, Development 132, 3977–3988 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Dreier JW, Andersen A-MN, Berg-Beckhoff G, Systematic review and metaanalyses: fever in pregnancy and health impacts in the offspring, PEDIATRICS 133, e674–88 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Acs N, Banhidy F, Puho E, Czeizel AE, Maternal influenza during pregnancy and risk of congenital abnormalities in offspring, Birth Defect Res A 73, 989–996 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Hentze MW, Muckenthaler MU, Galy B, Camaschella C, Two to tango: regulation of Mammalian iron metabolism, Cell 142, 24–38 (2010). [DOI] [PubMed] [Google Scholar]
- 81.Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP, Chendrimada TP, Lashinger ESR, Gordon E, Evans L, Misajet BA, Demarini DJ, Nation JH, Casillas LN, Marquis RW, Votta BJ, Sheardown SA, Xu X, Brooks DP, Laping NJ, Westfall TD, N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}−3-hydroxypropanoyl)-1-piperazinyl]carbonyl}−3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I, J Pharmacol Exp Ther 326, 432–442 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.