

Abstract
Activation of the renin-angiotensin system (RAS) plays a pivotal role in mediating hypertension, chronic kidney and cardiovascular diseases. As Wnt/β-catenin regulates multiple RAS genes, we speculated that this developmental signaling pathway might also participate in blood pressure (BP) regulation. To test this, we utilized two rat models of experimental hypertension: chronic angiotensin II infusion and remnant kidney after 5/6 nephrectomy. Inhibition of Wnt/β-catenin by ICG-001 blunted angiotensin II-induced hypertension. Interestingly, angiotensin II was able to induce the expression of multiple Wnt genes in vivo and in vitro, thereby creating a vicious cycle between Wnt/β-catenin and RAS activation. In the remnant kidney model, renal β-catenin was upregulated, and delayed administration of ICG-001 also blunted BP elevation and abolished the induction of angiotensinogen, renin, angiotensin-converting enzyme and angiotensin II type 1 receptor. ICG-001 also reduced albuminuria, serum creatinine and blood urea nitrogen, and inhibited renal expression of fibronectin, collagen I and plasminogen activator inhibitor-1, and suppressed the infiltration of CD3+ T cells and CD68+ monocytes/macrophages. In vitro, incubation with losartan prevented Wnt/β-catenin-mediated fibronectin, α-smooth muscle actin and Snail1 expression, suggesting that the fibrogenic action of Wnt/β-catenin is dependent on RAS activation. Taken together, these results suggest an intrinsic linkage of Wnt/β-catenin signaling with BP regulation. Our studies also demonstrate that hyperactive Wnt/β-catenin can drive hypertension and kidney damage via RAS activation.
Keywords: Wnt, β-catenin, renin-angiotensin system, blood pressure, renal fibrosis
1. Introduction
The renin-angiotensin system (RAS) plays a fundamental role in controlling blood pressure (BP), body fluid balance and tissue homeostasis [1–3]. As such, dysregulation of RAS leads to BP elevation and plays a causative role in the development of chronic kidney and cardiovascular diseases [4–6]. RAS consists of several key components including angiotensinogen (AGT), renin, angiotensin converting enzyme (ACE), angiotensin II type 1 and type 2 receptors (AT1 and AT2) [7, 8]. In normal physiological conditions, RAS components are tightly regulated and separately produced by different organs such as liver, kidney and lung, which ensures precise control of their activities in the circulation. After kidney injury, however, intra-renal RAS is markedly activated, as a result of the simultaneous upregulation of all RAS components in diseased kidneys [9, 10]. Intra-renal RAS components exert numerous biological actions that extend far beyond BP control, and they play a crucial role in mediating renal inflammation and fibrosis by a diverse array of mechanisms [11–13]. However, how the local expression of RAS components in the kidneys is regulated remains incompletely understood.
We recently demonstrated that β-catenin, the principal intracellular mediator of canonical Wnt signaling [14–16], is a master transcriptional regulator controlling the expression of all RAS genes in diseased kidneys, including AGT, renin, ACE, AT1 and AT2 [9]. This finding is consistent with numerous observations that Wnt/β-catenin signaling is activated in a wide variety of chronic kidney diseases (CKD), in which RAS activation is a common pathologic finding [17–21]. In this context, it is conceivable to speculate that Wnt/β-catenin could promote BP elevation and kidney injury by the virtue of its ability to promote RAS activation. Consistently, blockade of Wnt/β-catenin signaling by a range of approaches is able to ameliorate kidney fibrotic lesions in animal models of unilateral ureteral obstruction (UUO) and adriamycin nephropathy [17, 22–24]. However, despite a definitive role for Wnt/β-catenin in mediating intra-renal RAS activation, whether this developmental signaling actually participates in BP regulation is totally unknown.
In this study, we tested the hypothesis that Wnt/β-catenin signaling, through activation of RAS, participates in BP regulation in rats. We also investigated whether a Wnt/β-catenin/RAS axis plays a critical role in promoting renal inflammation, fibrosis and progressive decline of kidney function in CKD. By using two well-characterized rat models of experimental hypertension induced by chronic angiotensin II (Ang II) infusion or 5/6 nephrectomy (5/6NX), we demonstrated that blockade of Wnt/β-catenin signaling normalizes BP and ameliorates kidney injury by targeting intrarenal RAS activation. Our studies for the first time suggest an intrinsic linkage of a developmental signaling pathway to BP regulation in vivo.
2. Results
2.1. Inhibition of Wnt/β-catenin blunts Ang II-mediated hypertension
To investigate whether Wnt/β-catenin signaling regulates BP, we utilized a rat model of chronic Ang II infusion via osmotic mini-pump. This is a widely used, well characterized experimental model of hypertension [3, 25, 26]. As shown in Figure 1, Ang II infusion caused significant BP elevation in rats. At 3 or 7 days after continuous Ang II infusion, both systolic blood pressure (SBP) and mean blood pressure (MBP) were markedly elevated. However, injections of ICG-001, a peptidomimetic small molecule that specifically inhibits β-catenin-mediated gene transcription [24, 27–29], alleviated the elevation in BP induced by Ang II (Figure 1). These results strongly suggest a potential involvement of Wnt/β-catenin signaling in BP regulation in vivo.
Figure 1.

Inhibition of Wnt/β-catenin signaling by ICG-001 blocks blood pressure elevation induced by chronic angiotensin II (Ang II) infusion in rats. (A) Diagram shows experimental design. Red bar denotes Ang II treatment. Green bar shows the treatment schedule of ICG-001. Arrows indicate the timing for BP assessment. Blood pressure was measured using the tail-cuff technique at 3 and 7 days after Ang II infusion. Both systolic blood pressure (B, D) and mean blood pressure (C, E) are shown, respectively. **P < 0.01 versus sham controls; †P < 0.05 versus vehicle (n=5).
2.2. Angiotensin II induces Wnt expression in vivo and in vitro
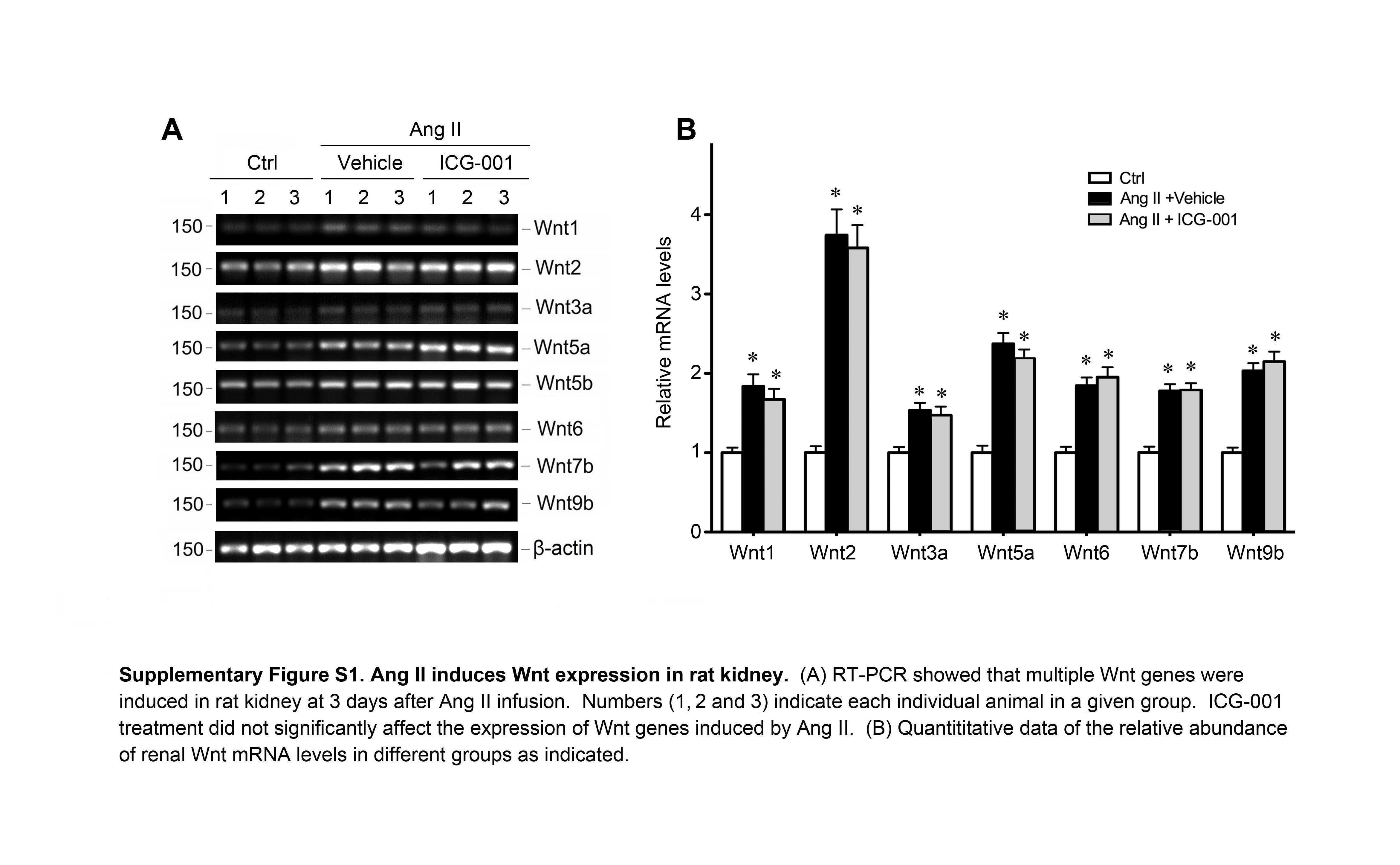
To delineate the mechanism by which ICG-001 mitigates Ang II-mediated hypertension, we studied the interplay between Ang II and Wnt/β-catenin in vivo and in vitro. As shown in Figure 2, chronic Ang II infusion induced renal expression of multiple Wnt genes. The mRNA levels of Wnt1, Wnt2, Wnt3a, Wnt5a, Wnt6, Wnt7b, Wnt9b and Wnt16 were considerably upregulated at 7 days after Ang II infusion, compared with sham controls (Figure 2, A and B). Similar induction of Wnt mRNA was also found in rat kidney at 3 days after Ang II infusion (Supplementary Figure S1). Western blot analyses also revealed that Ang II induced several Wnt proteins (Figure 2, C and D). Not surprisingly, injections of ICG-001 did not significantly affect Wnt induction by Ang II in rat kidneys (Figure 2A), because it primarily blocks β-catenin activity [27, 30].
Figure 2.

Angiotensin II induces Wnt expression and RAS activation in vivo and in vitro. (A) RT-PCR analysis shows that multiple Wnt genes were induced in rat kidneys at 7 days after Ang II infusion. Numbers (1, 2, and 3) indicate each individual animal in a given group. (B) Quantitative data of the relative Wnt mRNA levels among different groups. *P < 0.05 versus control group (n=5). (C, D) Western blot analysis shows the abundance of Wnt proteins at 7 days after Ang II infusion in the absence or presence of ICG-001. Representative Western blot (C) and quantitative data (D) are presented. *P < 0.05 versus control group (n=5). (E, F) Western blot analysis shows a dramatic increase in renal β-catenin, ACE and AT1 abundance at 7 days after Ang II infusion, which could be blocked by ICG-001. Representative Western blot (E) and quantitative data (F) are presented. *P < 0.05 versus sham controls, †P < 0.05 versus vehicle controls (n=5). (G) ICG-001 ameliorated Ang II-mediated proteinuria in rats. Urinary albumin levels were assessed by a specific ELISA, and reported after correction with urinary creatinine. *P < 0.05 versus sham controls, †P < 0.05 versus vehicle controls (n=5). (H) Ang II induces multiple Wnt expression in rat kidney interstitial fibroblasts (NRK-49F) in vitro. NRK-49F cells were incubated with different doses of Ang II as indicated for 24 h, and Wnt mRNA expression was assessed by RT-PCR. (I, J) Ang II induces protein expression of β-catenin and RAS components in a dose-dependent manner. NRK-49F cells were treated with different doses of Ang II for 24 hours. Representative Western blot (I) and quantitative data (J) are presented. *P < 0.05 versus controls (n=3). (K) Representative Western blot shows that Ang II induced β-catenin, AT1 and ACE expression in a time-dependent manner. NRK-49F cells were treated with 100 nM Ang II for various periods of time as indicated.
Consistent with induction of multiple Wnts, Ang II also caused a dramatic induction of β-catenin in rat kidneys. As shown in Figure 2, E and F, renal β-catenin protein was markedly upregulated at 7 days after Ang II infusion. Accordingly, ACE and AT1, the downstream target genes of β-catenin, were also induced in the kidneys (Figure 2, E and F). However, administration of ICG-001 reduced the protein expression of β-catenin, ACE and AT1 (Figure 2, E and F). Of note, ICG-001 also reduced albuminuria in rats at 7 days after Ang II infusion (Figure 2G). These data suggest that inhibition of β-catenin by ICG-001 could block hypertension, RAS activation and kidney injury induced by Ang II infusion.
To further confirm whether Ang II induces Wnt expression, we investigated the regulation of Wnt ligands by Ang II using an in vitro cell culture system. As shown in Figure 2H, incubation of normal rat kidney interstitial fibroblasts (NRK-49F) with Ang II induced the mRNA expression of numerous Wnt genes. Similarly, Ang II also induced β-catenin, AT1 and ACE proteins in a dose- and time-dependent manner in NRK-49F cells (Figure 2, I-K, and supplementary Figure S2).
2.3. Inhibition of Wnt/β-catenin blunts BP elevation and RAS activation in remnant kidney
To support a role of Wnt/β-catenin in BP regulation, we extended our studies to the rat remnant kidney model after 5/6 nephrectomy (5/6NX). This subtotal renal ablation model is closely analogous to the nature and course of human CKD [31, 32], and is characterized by hypertension, proteinuria, kidney dysfunction and glomerulosclerosis and interstitial fibrosis. As shown in Figure 3A, β-catenin protein was induced in remnant kidney at 12 weeks after 5/6NX, compared with sham controls. Immunostaining exhibited that β-catenin was predominantly localized in the degenerated renal tubules with dilated lumens (Figure 3A, arrows). Similarly, Western blot analyses of whole kidney lysates also revealed that β-catenin protein was significantly induced in remnant kidney (Figure 3, B and C). These results indicate that, similar to Ang II infusion and other CKD models [17, 23], canonical Wnt/β-catenin signaling is activated in the remnant kidney model.
Figure 3.

Inhibition of Wnt/β-catenin signaling by ICG-001 normalizes blood pressure and activation of renin-angiotensin system in rat remnant kidney model. (A) Wnt/β-catenin is activated in rat remnant kidney model in vivo. Representative micrographs demonstrate the expression and localization of β-catenin in remnant kidney. Rat kidney sections from sham control and 5/6NX at 12 weeks were stained immunohistochemically for β-catenin protein. Arrows indicate β-catenin–positive staining in renal tubules. Scale bar, 50 μm. The images were representative from 6 rats per group. (B and C) Western blot analyses show a dramatic increase in renal β-catenin abundance after 5/6NX. Representative Western blot (B) and quantitative data (C) are presented. Data are means ± SEM of 6 animals per group. *P < 0.05 versus sham controls (n=6). (D) Diagram shows experimental design. Green bar shows the treatment schedule of ICG-001 or vehicle. (E, F) Blood pressure was measured using the tail-cuff technique at 12 weeks after 5/6NX. Both systolic blood pressure (E) and mean blood pressure (F) are shown, respectively. *P < 0.05 versus sham controls; †P < 0.05 versus vehicle (n=6). Scale bar, 50 μm. (G) Kidney paraffin sections from different groups were stained for angiotensinogen (AGT), renin, angiotensin-converting enzyme (ACE) and angiotensin II type 1 receptor (AT1), respectively. Arrows indicate positive staining in renal tubules, and arrowhead denotes AT1-positive interstitial cells. The images were representative from 6 animals per group.
We next examined the effects of inhibition of Wnt/β-catenin signaling by ICG-001 on BP and RAS activation in remnant kidney model. The experimental protocol is presented in Figure 3D. We chose to administer ICG-001 at 6 weeks after 5/6NX, a time point when kidney injury was already established in this model [31, 32]. This closely imitates the clinical setting in which patients often seek an effective treatment of established CKD. As shown in Figure 3, E and F, both SBP and MBP levels were significantly elevated in rats at 12 weeks after 5/6NX, compared with sham controls. Interestingly, ICG-001 virtually normalized both SBP and MBP (Figure 3, E and F). The finding that ICG-001 blunted BP elevation in remnant kidney prompted us to investigate its effect on RAS activation. As shown in Figure 3G, subtotal renal ablation after 5/6NX induced AGT and renin proteins predominantly in renal tubular epithelium, whereas ACE was mainly induced in the brush border of tubular cells. Of note, tubules with positive staining for AGT and renin displayed enlarged lumens, suggesting a close correlation between activation of RAS components and tubular injury. Similarly, 5/6NX also drastically induced AT1 protein, compared with sham controls. AT1 was induced not only in renal tubules (Figure 3G, arrow) but also in the interstitial cells (Figure 3G, arrowhead) in remnant kidney. These results demonstrate that ICG-001 has an attenuating effect on both BP elevation and RAS activation.
2.4. ICG-001 ameliorates kidney dysfunction and renal fibrosis
We next examined the effects of ICG-001 on renal function and proteinuria in remnant kidney model, as normal rats have low level of serum creatinine and minimal level of albumin in the urine. As shown in Figure 4, A and B, ICG-001 reduced both serum creatinine and blood urea nitrogen. SDS-PAGE analysis of urine samples revealed that a protein at the size of 65 kDa, presumably albumin [33], was elevated at 12 weeks after 5/6NX (Figure 4C). Compared with vehicle controls, this protein was greatly deceased in rats treated with ICG-001 (Figure 4C). Similar results were obtained when urinary albumin levels were assessed by quantitative ELISA (Figure 4D).
Figure 4.

Blockade of Wnt/β-catenin signaling by ICG-001 ameliorates kidney dysfunctions and renal interstitial fibrosis after 5/6NX. (A, B) ICG-001 treatment improves kidney function. Serum creatinine (A) and blood urea nitrogen level (B) were assessed at 12 weeks after 5/6NX. *P < 0.05 versus sham controls; †P < 0.05 versus vehicle (n=6). (C) SDS-PAGE analysis shows the abundance and composition of urinary proteins in rats from different groups. Urine samples after normalization to creatinine were analyzed on SDS-PAGE and stained with Comarossi Blue reagent. Numbers (1, 2 and 3) indicate each individual animal in a given group. A protein at the size of 65 kDa, presumably albumin, is indicated by asterisk. (D) Urinary albumin levels in rats at 12 weeks after 5/6NX. Urinary albumin was expressed as μg/mg creatinine. (E) Kidney sections were stained with periodic acid-Schiff (PAS) and Masson-trichrome (MTS) reagents, respectively. Representative micrographs from different groups as indicated are shown. Arrows indicate positive staining. Scale bar, 50 μm. (F) Kidney weights in different groups after ICG-001 treatment. The kidney weight to body weight ratio (KW/BW) at 12 weeks after 5/6NX was calculated and presented. *P < 0.05 versus sham controls, †P < 0.05 versus vehicle controls (n=6). (G) Quantitative determination of renal fibrotic lesions in different groups. *P < 0.05 versus sham controls; †P < 0.05 versus vehicle (n=6). (H) Immunohistochemical staining shows that ICG-001 inhibited myofibroblast activation in remnant kidney. Rat kidney sections at 12 weeks after 5/6NX were immunostained with specific antibody against α-SMA. Arrows indicate positive staining. Scale bar, 50 μm. The images were representative from 6 animals per group. Except for vessel, there was no α-SMA-positive interstitial cell in normal rat kidney (not shown).
We further investigated the effects of ICG-001 on renal hypertrophy and fibrosis in remnant kidney, as glomerular or tubular hypertrophy is an early response of the kidney to injury. As shown in Figure 4E, Periodic acid-Schiff (PAS) staining demonstrated thickened glomerular and tubular basement membranes, with enlarged glomeruli and tubules (Figure 4E), indicating renal hypertrophy. Consistently, ICG-001 ameliorated renal hypertrophy and normalized the kidney-to-body weight ratio at 12 weeks after 5/6NX, compared with vehicle controls (Figure 4F). Masson’s trichrome staining (MTS) revealed significant collagen deposition in the tubulointerstitial compartment of remnant kidney at 12 weeks after 5/6NX, and ICG-001 substantially attenuated these fibrotic lesions (Figure4E). Quantitative determination of renal fibrotic lesions by using morphometric analysis showed similar results (Figure 4G). Consistently, ICG-001 also inhibited the activation of myofibroblasts in remnant kidney, as illustrated by immunohistochemical staining for α-SMA (Figure 4H).
We further examined the effect of ICG-001 on expression of major interstitial matrix genes in remnant kidney. As shown in Figure 5, A-C, compared with sham controls, renal mRNA expression of fibronectin, type I and type III collagens was markedly induced at 12 weeks after 5/6NX, which was inhibited by ICG-001. In addition, ICG-001 also repressed mRNA expression of plasminogen activator inhibitor-1 (PAI-1) (Figure 5D). Similarly, Western blot analyses illustrated a reduced protein expression of fibronectin, type I collagen, α-SMA and PAI-1 in remnant kidney treated with ICG-001 (Figure 5, E-I). Immunostaining also confirmed that ICG-001 reduced the deposition of fibronectin and type I collagen in remnant kidney (Figure 5J).
Figure 5.

ICG-001 inhibits matrix gene expression in remnant kidney. (A-D) Quantitative, real-time RT-PCR results reveal that ICG-001 inhibited renal expression of fibronectin (A), type I collagen (B), type III collagen (C), and PAI-1 (D) mRNA at 12 weeks after 5/6NX. Relative mRNA levels are reported after normalization with β-actin. Ctrl, control. *P < 0.05 versus sham controls; †P < 0.05 versus vehicle (n=6). (E-I) Western blot analyses show the expression of fibronectin, type I collagen, α-SMA and PAI-1 in different groups. Numbers (1, 2 and 3) indicate each individual animal in a given group. Representative Western blot (E, H) and quantitative data for fibronectin (F), type I collagen (G), α-SMA and PAI-1 (I) are shown. Relative protein levels (fold induction versus control group) are presented after normalization with actin. *P < 0.05 versus sham controls; †P < 0.05 versus vehicle (n=6). (J) Representative micrographs show that ICG-001 inhibited fibronectin and type I collagen deposition in rat remnant kidney at 12 weeks after 5/6NX. Frozen kidney sections were stained with specific antibodies against fibronectin and type I collagen, respectively. Arrows indicate positive staining. Scale bar, 50 μm. The images were representative from 6 animals per group.
2.5. ICG-001 inhibits renal inflammation
We next assessed renal infiltration of inflammatory cells in remnant kidney. As shown in Figure 6A, immunostaining for CD3 and CD68 antigens demonstrated that 5/6NX led to significant infiltration of T cells and monocytes/macrophages, and this was inhibited by ICG-001. Computer-aided quantitative analysis confirmed a dramatic suppression of renal infiltration of inflammatory CD3+ T cell and CD68+ macrophages by ICG-001 at 12 weeks after 5/6NX (Figure 6, B and C). As shown in Figure 6D, qRT-PCR revealed an induction of proinflammatory RANTES and TNF-α mRNA in remnant kidney, compared with sham controls. Interestingly, ICG-001 completely inhibited both RANTES and TNF-α mRNA induction, suggesting that blockade of Wnt/β-catenin signaling inhibits 5/6NX-induced renal inflammation.
Figure 6.

ICG-001 attenuates inflammatory infiltration and represses proinflammatory cytokines expression. (A) Representative micrographs show renal infiltration of CD3+ T cells and CD68+ monocytes/macrophages. Kidney sections from different groups were immunohistochemically stained with specific antibodies against CD3 and CD68 antigens, respectively. Arrows indicate positive staining. Scale bar, 50 μm. (B, C) Quantitative determination of CD3− (B) and CD68− (C) positive cells in various groups as indicated. *P < 0.05 versus sham controls, †P < 0.05 versus vehicle (n=6). (D) Graphic presentation shows the relative mRNA levels of RANTES and TNF-α mRNA determined by qRT-PCR. Relative mRNA levels were determined after normalization with β-actin and expressed as fold induction over sham controls. Data are expressed as mean ± SEM. *P < 0.05 versus sham controls, †P < 0.05 versus vehicle (n=6).
2.6. RAS activation is required for the Wnt/β-catenin-mediated fibrogenic response in vitro
To investigate whether RAS activation is important for Wnt/β-catenin-mediated fibrogenesis, we examined the effects of losartan, an AT1 receptor blocker (ARB), on the regulation of fibrosis-related genes in kidney tubular epithelial cells. To this end, human kidney proximal tubular epithelial cells (HKC-8) were transfected with constitutively activated β-catenin expression vector (pDel-β-cat) [18, 34]. Shown in Figure 7A, over-expression of β-catenin induced the expression of fibronectin, α-SMA and Snail1 proteins in HKC-8 cells. As expected, incubation with ICG-001 abolished the induction of these proteins (Figure 7, A and B). Interestingly, blockade of RAS signaling by losartan also abolished the β-catenin-triggered induction of fibronectin, α-SMA and Snail1 in HKC-8 cells (Figure 7, C and D). Similarly, losartan also inhibited the expression of fibronectin, α-SMA and Snail1 induced by overexpression of Wnt1 (Figure 7E). As illustrated in Figure 7F, these results indicate that Wnt/β-catenin-mediated fibrogenesis are dependent on RAS activation in kidney cells in vitro.
Figure 7.

RAS activation is required for Wnt/β-catenin-mediated fibrogenic response in vitro. (A, B) Western blot analyses and quantitative data showed that ICG-001 abolished β-catenin-mediated fibronectin, α-SMA and Snail1 expression in tubular epithelial cells. HKC-8 cells were transfected with empty vector (pcDNA3) or constitutively activated β-catenin expression plasmid (pDel-β-cat) for 24 hours, following by incubation with ICG-001 (10 μM). *P < 0.05 versus controls (n=3); †P < 0.05 versus pDel-β-cat alone (n=3). (C, D) Losartan abolished β-catenin-mediated fibrogenic action. HKC-8 cells were treated as indicated. Whole cell lysates were immunoblotted for the protein expression of fibronectin, α-SMA and Snail1, respectively. Representative Western blot analyses (C) and quantitative data (D) are presented. *P < 0.05 versus controls (n=3); †P < 0.05 versus pDel-β-cat alone (n=3). (E) Losartan also inhibited the expression of fibrosis-related genes induced by Wnt1. HKC-8 cells were transfected with pcDNA3 or pHA-Wnt1 plasmid for 24 hours, followed by incubation with losartan (10−6 M). Quantitative data on the relative abundance of fibronectin, α-SMA and Snail1 are presented. *P < 0.05 versus controls (n=3); †P < 0.05 versus pDel-β-cat alone (n=3). (F) Diagram denotes the potential role and mechanism of Wnt/β-catenin signaling in the pathogenesis of CKD induced by 5/6NX.
3. Discussion
Hypertension affects more than 1.5 billion people worldwide and is a major independent risk factor for chronic kidney and cardiovascular diseases [10]. Although the precise cause of elevated BP in each affected individual remains to be determined, at least some of these cases are presumably associated with RAS hyperactivity. It is estimated that about 80% of CKD patients are hypertensive [7], suggesting that renal injury and hypertension form a vicious cycle. We recently demonstrated that β-catenin is a master transcriptional regulator that directly controls intrarenal RAS expression [9], underscoring a potential connection between a hyperactive Wnt signaling and RAS activation. In the present study, we further confirm that blockade of Wnt/β-catenin signaling lowers BP, inhibits intra-renal RAS activation and ameliorates kidney damage in rat models of chronic Ang II infusion and remnant kidney. To our knowledge, this is the first study that links a developmental signaling pathway to BP regulation in intact animals.
Of the regulatory systems for BP control, RAS activation plays a predominant role in the development of hypertension. This view is corroborated by the impressive anti-hypertensive benefits of the pharmacologic blockade of RAS using either ACE inhibitors or AT1 blockers in the clinical setting [10, 35]. Although RAS is a hormonal system that could affect BP by acting on many tissues such as the central nervous system, heart, kidney and vasculature, genetic studies have established a critical role for intra-renal RAS activation in the pathogenesis of hypertension [3, 36, 37]. For example, an earlier study demonstrated that the mean arterial BP is elevated in the double-transgenic mice expressing human AGT and renin specifically in renal proximal tubule [36]. Furthermore, when the AT1 receptor is deleted from the proximal tubule of the kidney, the mice are protected against the Ang II-dependent hypertension [3, 37]. These observations are in harmony with the concurrent induction of all RAS components in kidney tubules (Figure 3G) and BP elevation (Figure 3, E and F) in remnant kidney after 5/6NX. Given that Wnt/β-catenin regulates multiple RAS genes [9, 16], it is conceivable that the intra-renal RAS activation could account for the major mechanism of action of this developmental signaling in promoting hypertension and kidney damage.
Apart from the BP regulation, Wnt/β-catenin-mediated RAS activation could provoke kidney injury via a variety of other mechanisms. Ang II, the active effector of RAS, is known to activate NF-κB and TGF-β1 signaling [38, 39], and thereby directly triggers renal inflammation and fibrogenesis. Accordingly, blockade of this signaling by ICG-001 is quite effective in blunting renal infiltration of inflammatory T cells and monocytes/macrophages, as well as chemokines expression (Figure 6). Similarly, ICG-001 also inhibits the expression of various fibrosis-related genes both in vivo (Figure 5) and in vitro (Figure 7), and attenuates the accumulation and deposition of extracellular matrix. Of particular interest, Wnt/β-catenin-mediated induction of various fibrogenic genes is completely hampered by losartan (Figure 7), suggesting that RAS activation is required for mediating the profibrotic action of Wnt/β-catenin. Therefore, as shown in Figure 7F, Wnt/β-catenin/RAS constitutes a pathologic axis that plays an essential role in mediating hypertension, inflammation and fibrosis.
One of the novel findings in the present study is that Ang II induced Wnt genes expression and β-catenin activation (Figure 2). Since previous studies demonstrate that Wnt/β-catenin activates multiple RAS genes, it becomes clear that Ang II and Wnt/β-catenin can mutually stimulate each other, thereby creating a vicious cycle of Wnt/β-catenin and RAS activation in diseased kidneys (Supplementary Figure S3). This finding also explains why ICG-001 can blunt Ang II-induced hypertension, because the action of Ang II could depend on the chronic activation of the Wnt/β-catenin/RAS axis (Supplementary Figure S3). It should be pointed out that although several studies have shown Wnt/β-catenin activation in different CKD models [18, 20, 22], its regulation has not been investigated in remnant kidney models. The results that β-catenin is induced in both Ang II infusion and remnant kidney models (Figures 2 and 3), together with earlier reports in other animal models [17, 23], suggest that activation of Wnt/β-catenin signaling is a common pathologic finding in a wide variety of CKD, regardless of their etiologies. In this context, it is tempting to speculate that the Wnt/β-catenin/RAS axis could be an unprecedented therapeutic target in the treatment of hypertension and CKD.
Consistent with the notion that β-catenin controls multiple RAS genes, its inhibition by ICG-001 simultaneously abrogates the induction of several RAS components (Figure 3G). Unlike the current anti-RAS therapy using ACE inhibitor or AT1 blockers, which targets one RAS component a time, targeting β-catenin would concurrently repress multiple RAS genes, and therefore is presumed to have an improved therapeutic efficacy. Indeed, inhibition of β-catenin by ICG-001 appears quite effective in normalizing BP (Figure 1 and 3), reducing proteinuria (Figure 4) and alleviating kidney fibrosis and inflammation (Figures 4–6). Because ICG-001 binds specifically to CBP and blocks CBP/β-catenin interaction, its action is presumably mediated by disrupting CBP/β-catenin-regulated gene transcription [24, 27–29]. It is also noted, however, that renal β-catenin protein level is reduced in Ang II-infused rats after ICG-001 therapy (Figure 2), which raises the possibility that ICG-001 may affect β-catenin expression either directly or as a consequence of its destabilization. It should be stressed that ICG-001 was given at 6 weeks after 5/6NX (Figure 3D), a time point when kidney injury is already established in this model. In that regard, our present study reinforces the notion that inhibition of Wnt/β-catenin/RAS axis by ICG-001 is able to ameliorate an established kidney injury.
The present study has some limitations. One potential pitfall is the lack of genetic inhibition of Wnt/β-catenin signaling through knockout or knockdown approaches, in addition to pharmacologic inhibition using ICG-001. Because rat models were used in the present study, it is technically difficult to manipulate gene expression in this species in vivo. Future studies are warranted to use genetic approaches in mice to directly link Wnt/β-catenin to BP regulation. Another limitation of this study was using serum creatinine level, rather than the estimated glomerular filtration rate (eGFR), as an indicator of renal functionality. As creatinine clearance is more accurate than serum creatinine levels for assessing renal function in CKD, more studies are needed in the future to fully address this issue.
In summary, we have shown herein that Wnt/β-catenin is activated in two rat models of experimental hypertension: chronic Ang II infusion and remnant kidney after subtotal renal ablation. Such Wnt/β-catenin activation is accompanied by intra-renal RAS activation and development of hypertension and kidney injury. Consistently, inhibition of β-catenin with a small-molecule inhibitor ICG-001 lowers BP, represses RAS expression, reduces proteinuria, inhibits renal inflammation and fibrosis, and preserves kidney function. These results suggest an intrinsic linkage of this developmental signaling to BP regulation. Our studies also indicate that Wnt/β-catenin/RAS constitutes a pathologic axis, and disruption of this axis holds promise as an effective strategy for treatment of hypertension and CKD.
4. Materials and methods
4.1. Animal models
All animal studies were performed according to the NIH Guide for the Care and Use of Laboratory Animals and by use of the procedures approved by the Committee of Laboratory Animals in the Southern Medical University, and the Institutional Animal Care and Use Committee at the University of Pittsburgh, respectively. Two rat models of hypertension, namely chronic angiotensin II (Ang II) infusion and remnant kidney after 5/6 nephrectomy (5/6NX), were used in this study. For the Ang II infusion model, male Sprague-Dawley (SD) rats (age 8 weeks), weighting 200–220g, were purchased from Harlan Laboratories (Indianapolis, IN). SD rats were subjected to Ang II infusion model using osmotic mini-pump by an established protocol, as described previously [25, 40]. Briefly, under general anesthesia with pentobarbital sodium (35~45 mg/kg, intraperitoneal injection), an osmotic mini-pump (Alzet, 2ML4, Cupertino, CA) was implanted subcutaneously for the chronic administration of Ang II at the rate 325 ng/kg/min. Rats were divided into three groups: (i) sham control (n=5), (ii) Ang II infusion rats treated with vehicle (n=5), and (iii) Ang II infusion rats injected with ICG-001 (n=5). ICG-001 (Chembest, Shanghai, China) was administered daily by intraperitoneal injection at 5 mg/kg body wt. The body weights and blood pressure of the rats were measured as indicated. At 7 days after Ang II infusion, all rats were sacrificed. Urine, blood and kidney tissue were collected for various analyses.
For the 5/6NX model, male SD rats, weighting 200–220g, were subjected to 5/6NX using two-step protocol (Figure 3D), as described previously [32]. Briefly, under general anesthesia with pentobarbital sodium, rats were subjected to surgical resection of two thirds of the left kidney. One week later, rats underwent right nephrectomy. Sham-operated rats had their kidneys exposed without nephrectomy. Six weeks after operation, the 5/6NX rats were assigned into groups after randomization with serum creatinine level and blood pressure. Three groups of rats were used: (i) sham control (n=6), (ii) 5/6NX rats treated with vehicle (n=6), and (iii) 5/6NX rats injected with ICG-001 (n=6). ICG-001 was administered by intraperitoneal injection at 5 mg/kg body wt, three times per week. At 12 weeks after 5/6NX, all animals were sacrificed. Urine, blood and kidney tissues were collected for analysis.
4.2. Cell culture and treatment
Normal rat kidney interstitial fibroblast cells (NRK-49F) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in Dulbecco’s modified Eagle’s medium/F12 medium supplemented with 10% fetal bovine serum. The NRK-49F cells within 15 passages were seeded on six-well culture plates to 60 to 70% confluence in the complete medium containing 10% fetal bovine serum for 16 hours, and then changed to serum-free medium after washing twice with medium. Ang II was added to the culture at the concentrations as indicated and incubated for 24 h. Cells were then analyzed for Wnt expression. Human proximal tubular epithelial cells (HKC, clone-8) were provided by Dr. L. Racusen (Johns Hopkins University, Baltimore, Maryland). Cell culture was carried out according to the procedures described previously [41]. HKC-8 cells within 15 passages were transfected with Wnt1 expressing vectors (pHA-Wnt1), N-terminally truncated, constitutively activated β-catenin expression vector (pDel-β-cat) or empty vector (pcDNA3) by lipofectamine 2000 (Invitrogen, Carlsbad, CA), according to the manufacturer’s protocol. In some experiments, cells were incubated with ICG-001 at 10 μM or losartan at 10−6 M (#61188; Sigma). Whole-cell lysates were prepared and subjected to Western blot analyses.
4.3. Renal function and BP assessment
Serum and urine creatinine as well as blood urea nitrogen levels were measured by using automatic biochemical analyzer (AU480, Beckman-Coulter Inc., Brea, CA). Urinary albumin level was determined using rat Albumin ELISA Quantization kit, according to the manufacturer’s protocol (Bethyl Laboratories Inc., Montgomery, TX). Urinary albumin was normalized to urine creatinine and expressed as μg/mg urinary creatinine. BP measurements were performed by the tail-cuff technique using the CODA blood pressure monitor (Kent Scientific, Torrington, CT). Rats were acclimated to the BP measurements by measuring their BP daily for 3 days prior to the measurements used in the study.
4.4. Histology and Immunohistochemical Staining
Paraffin-embedded rat kidney sections (4 μm thickness) were prepared by a routine procedure [17]. Sections were stained with periodic acid-Schiff reagent by standard protocol. Kidney sections were also subjected to Masson’s Trichrome staining for assessing collagen deposition and fibrotic lesions. Semi-quantitative determination of renal fibrosis score was carried out by previously reported methods [32]. Briefly, Masson trichrome-stained kidney sections were graded for the presence of interstitial fibrosis according to the following scale: 0, no evidence of interstitial fibrosis; 1, <25% involvement; 2, 25to 50% involvement, and 3, >50% involvement. The scale for each rat was reported as the mean of 10 random high-power (X400) fields per section [32]. Immunohistochemical staining was performed using established protocol, as described previously [9]. Antibodies used were as follow: rabbit polyclonal anti-α-smooth muscle actin antibody (ab5694), rabbit monoclonal anti-β-catenin antibody (ab32572), rabbit monoclonal anti-CD3 antibody (ab16669; Abcam, Cambridge, MA), mouse monoclonal anti-CD68 antibody (MCA341GA; Serotec), goat polyclonal anti-AGT (sc-7419), goat polyclonal anti-renin (sc-27320, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-AT1 receptor (AB15552; Millipore, Billerica, MA).
4.5. Immunofluorescence staining
Kidney cryosections were fixed with 3.7% paraformalin for 15 minutes at room temperature. After blocking with 10% donkey serum for 30 minutes, the slides were immunostained with primary antibodies against fibronectin (sc-9068, Santa Cruz Biotechnology), rabbit polyclonal anti-collagen I (234167, EMD Millipore).
4.6. Western Blot Analysis
Protein expression in kidney homogenates was analyzed by Western blot analysis as described previously [17]. The primary antibodies used were as follow: rabbit polyclonal anti-fibronectin (sc-9068; Santa Cruz Biotechnology), rabbit polyclonal anti-collagen I (234167, EMD Millipore), rabbit monoclonal anti-β-catenin antibody (ab32572; Abcam, Cambridge, MA), and rabbit monoclonal anti-β-actin (4970, Cell Signaling Technology), rabbit polyclonal anti-AT1 receptor (ab15552; Millipore, Billerica, MA), rabbit monoclonal anti-ACE (ab75762; abcam, Cambridge, MA).
4.7. RT-PCR and Real-time PCR
Total RNA isolation was carried out using the TRIzol RNA isolation system (Invitrogen, Carlsbad, CA) according to the manufacturer’s instruction. The first strand of complementary DNA was synthesized using 1 μg of RNA in 20 μl of reaction buffer containing MMLV-RT and random primers at 37°C for 50 minutes. RT-PCR analyses of Wnt mRNA expression were carried out as described previously [17]. Real-time RT-PCR was performed in a total volume of 25μl in duplicate by using the SYBR green reagents (Life Technologies, Carlsbad, CA). The sequences of the primer pairs are given in Supplemental Table 1. Relative mRNA level of various genes was determined after normalization with β-actin.
4.8. Statistical analyses
All data examined are expressed as mean ± SEM. Statistical analyses of the data were performed using SPSS 13.0 for Windows (SPSS Inc, Chicago, IL). Comparisons between groups were made using one-way ANOVA, followed by Student-Newman-Keuls test or Dunnett’s T3 procedure when the assumption of equal variances did not hold. P<0.05 was considered to be significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Highlights.
Inhibition of Wnt/β-catenin blunts hypertension induced by angiotensin II and subtotal renal ablation in rats.
Angiotensin II induces multiple Wnt genes and activates β-catenin and renin-angiotensin system (RAS).
The fibrogenic action of Wnt/β-catenin is dependent on RAS activation in vitro.
These studies suggest an intrinsic linkage of Wnt/β-catenin to hypertension via RAS activation.
Acknowledgments
This work was supported by the National Science Foundation of China Grants 81521003 and 81770715, State Key Laboratory of Organ Failure Research 201801, Fujian Provincial Health and Family Planning of Young and Middle-aged Talents Program Grant 2017-ZQN-92, and National Institutes of Health Grants DK064005, DK091239 and DK079307.
Footnotes
Conflict of interest
The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Crowley SD, Coffman TM, Recent advances involving the renin-angiotensin system, Exp Cell Res, 318 (2012) 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nguyen G, Renin and prorenin receptor in hypertension: what’s new?, Curr Hypertens Rep, 13 (2011) 79–85. [DOI] [PubMed] [Google Scholar]
- [3].Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM, AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure, Cell Metab, 13 (2011) 469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Herzog CA, Asinger RW, Berger AK, Charytan DM, Diez J, Hart RG, Eckardt KU, Kasiske BL, McCullough PA, Passman RS, DeLoach SS, Pun PH, Ritz E, Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO), Kidney Int, 80 (2011) 572–586. [DOI] [PubMed] [Google Scholar]
- [5].Kobori H, Nangaku M, Navar LG, Nishiyama A, The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease, Pharmacol. Rev, 59 (2007) 251–287. [DOI] [PubMed] [Google Scholar]
- [6].Floege J, Antagonism of canonical Wnt/β-catenin signaling: Taking RAS blockade to the next level?, J Am Soc Nephrol, 26 (2015) 3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Santos PC, Krieger JE, Pereira AC, Renin-angiotensin system, hypertension, and chronic kidney disease: pharmacogenetic implications, J Pharmacol Sci, 120 (2012) 77–88. [DOI] [PubMed] [Google Scholar]
- [8].Locatelli F, Del Vecchio L, Cavalli A, Inhibition of the renin-angiotensin system in chronic kidney disease: a critical look to single and dual blockade, Nephron, 113 (2009) c286–293. [DOI] [PubMed] [Google Scholar]
- [9].Zhou L, Li Y, Hao S, Zhou D, Tan RJ, Nie J, Hou FF, Kahn M, Liu Y, Multiple genes of the renin-angiotensin system are novel targets of Wnt/β-catenin signaling, J Am Soc Nephrol, 26 (2014) 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ruggenenti P, Cravedi P, Remuzzi G, Mechanisms and treatment of CKD, J Am Soc Nephrol, 23 (2012) 1917–1928. [DOI] [PubMed] [Google Scholar]
- [11].Reiser J, Mundel P, Dual effects of RAS blockade on blood pressure and podocyte function, Curr Hypertens Rep, 9 (2007) 403–408. [DOI] [PubMed] [Google Scholar]
- [12].Muller DN, Luft FC, Direct renin inhibition with aliskiren in hypertension and target organ damage, Clin J Am Soc Nephrol, 1 (2006) 221–228. [DOI] [PubMed] [Google Scholar]
- [13].Nguyen G, Muller DN, The biology of the (pro)renin receptor, J Am Soc Nephrol, 21 (2010) 18–23. [DOI] [PubMed] [Google Scholar]
- [14].Angers S, Moon RT, Proximal events in Wnt signal transduction, Nat Rev Mol Cell Biol, 10 (2009) 468–477. [DOI] [PubMed] [Google Scholar]
- [15].Clevers H, Nusse R, Wnt/β-catenin signaling and disease, Cell, 149 (2012) 1192–1205. [DOI] [PubMed] [Google Scholar]
- [16].Zuo Y, Liu Y, New insights into the role and mechanism of Wnt/beta-catenin signalling in kidney fibrosis, Nephrology, 23 Suppl 4 (2018) 38–43. [DOI] [PubMed] [Google Scholar]
- [17].He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y, Wnt/β-catenin signaling promotes renal interstitial fibrosis, J Am Soc Nephrol, 20 (2009) 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y, Wnt/β-catenin signaling promotes podocyte dysfunction and albuminuria, J Am Soc Nephrol, 20 (2009) 1997–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Heikkila E, Juhila J, Lassila M, Messing M, Perala N, Lehtonen E, Lehtonen S, Sjef Verbeek J, Holthofer H, β-Catenin mediates adriamycin-induced albuminuria and podocyte injury in the adult mouse kidneys, Nephrol Dial Transplant, 25 (2010) 2437–2446. [DOI] [PubMed] [Google Scholar]
- [20].von Toerne C, Schmidt C, Adams J, Kiss E, Bedke J, Porubsky S, Gretz N, Lindenmeyer MT, Cohen CD, Grone HJ, Nelson PJ, Wnt pathway regulation in chronic renal allograft damage, Am J Transplant, 9 (2009) 2223–2239. [DOI] [PubMed] [Google Scholar]
- [21].Zhou T, He X, Cheng R, Zhang B, Zhang RR, Chen Y, Takahashi Y, Murray AR, Lee K, Gao G, Ma JX, Implication of dysregulation of the canonical wingless-type MMTV integration site (WNT) pathway in diabetic nephropathy, Diabetologia, 55 (2012) 255–266. [DOI] [PubMed] [Google Scholar]
- [22].Surendran K, Schiavi S, Hruska KA, Wnt-dependent beta-catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis, J Am Soc Nephrol, 16 (2005) 2373–2384. [DOI] [PubMed] [Google Scholar]
- [23].He W, Kang YS, Dai C, Liu Y, Blockade of Wnt/β-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury, J Am Soc Nephrol, 22 (2011) 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hao S, He W, Li Y, Ding H, Hou Y, Nie J, Hou FF, Kahn M, Liu Y, Targeted inhibition of β-catenin/CBP signaling ameliorates renal interstitial fibrosis, J Am Soc Nephrol, 22 (2011) 1642–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nunes FC, Braga VA, Chronic angiotensin II infusion modulates angiotensin II type I receptor expression in the subfornical organ and the rostral ventrolateral medulla in hypertensive rats, J Renin Angiotensin Aldosterone Syst, 12 (2011) 440–445. [DOI] [PubMed] [Google Scholar]
- [26].Jia Z, Zhang A, Zhang H, Dong Z, Yang T, Deletion of microsomal prostaglandin E synthase-1 increases sensitivity to salt loading and angiotensin II infusion, Circulation research, 99 (2006) 1243–1251. [DOI] [PubMed] [Google Scholar]
- [27].Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Kim HY, Moon SH, Ha JR, Kahn M, A small molecule inhibitor of β-catenin/CREB-binding protein transcription, Proc Natl Acad Sci U S A, 101 (2004) 12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Henderson WR Jr., Chi EY, Ye X, Nguyen C, Tien YT, Zhou B, Borok Z, Knight DA, Kahn M, Inhibition of Wnt/β-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis, Proc Natl Acad Sci U S A, 107 (2010) 14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Higuchi Y, Nguyen C, Yasuda SY, McMillan M, Hasegawa K, Kahn M, Specific direct small molecule p300/beta-catenin antagonists maintain stem cell potency, Curr Mol Pharmacol, 9 (2016) 272–279. [DOI] [PubMed] [Google Scholar]
- [30].Eguchi M, Nguyen C, Lee SC, Kahn M, ICG-001, a novel small molecule regulator of TCF/β-catenin transcription, Med Chem, 1 (2005) 467–472. [DOI] [PubMed] [Google Scholar]
- [31].Yang HC, Zuo Y, Fogo AB, Models of chronic kidney disease, Drug Discov Today Dis Models, 7 (2010) 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li HY, Hou FF, Zhang X, Chen PY, Liu SX, Feng JX, Liu ZQ, Shan YX, Wang GB, Zhou ZM, Tian JW, Xie D, Advanced oxidation protein products accelerate renal fibrosis in a remnant kidney model, J Am Soc Nephrol, 18 (2007) 528–538. [DOI] [PubMed] [Google Scholar]
- [33].Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y, Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria, J Am Soc Nephrol, 20 (2009) 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].He W, Tan R, Dai C, Li Y, Wang D, Hao S, Kahn M, Liu Y, Plasminogen activator inhibitor-1 is a transcriptional target of the canonical pathway of Wnt/β-catenin signaling, J Biol Chem, 285 (2010) 24665–24675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hou FF, Zhang X, Zhang GH, Xie D, Chen PY, Zhang WR, Jiang JP, Liang M, Wang GB, Liu ZR, Geng RW, Efficacy and safety of benazepril for advanced chronic renal insufficiency, N Engl J Med, 354 (2006) 131–140. [DOI] [PubMed] [Google Scholar]
- [36].Lavoie JL, Lake-Bruse KD, Sigmund CD, Increased blood pressure in transgenic mice expressing both human renin and angiotensinogen in the renal proximal tubule, Am J Physiol Renal Physiol, 286 (2004) F965–971. [DOI] [PubMed] [Google Scholar]
- [37].Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM, Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney, Pro. Natl. Acad. Sci. USA, 103 (2006) 17985–17990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zeisberg M, Neilson EG, Mechanisms of tubulointerstitial fibrosis, J Am Soc Nephrol, 21 (2010) 1819–1834. [DOI] [PubMed] [Google Scholar]
- [39].Liu Y, Cellular and molecular mechanisms of renal fibrosis, Nat. Rev. Nephrol, 7 (2011) 684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kemp BA, Howell NL, Gildea JJ, Keller SR, Padia SH, Carey RM, AT2 receptor activation induces natriuresis and lowers blood pressure, Circulation research, 115 (2014) 388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tan R, Zhang X, Yang J, Li Y, Liu Y, Molecular basis for the cell type specific induction of SnoN expression by hepatocyte growth factor, J Am Soc Nephrol, 18 (2007) 2340–2349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.