

Abstract
Obesity is a risk factor for estrogen receptor-positive (ER+) breast cancer after menopause. The pro-proliferative effects of estrogens are well characterized and there is a growing body of evidence to also suggest an important role in tumorigenesis. Importantly, obesity not only increases the risk of breast cancer, but it also increases the risk of recurrence and cancer-associated death. Aromatase is the rate-limiting enzyme in estrogen biosynthesis and its expression in breast adipose stromal cells is hypothesized to drive the growth of breast tumors and confer resistance to endocrine therapy in obese postmenopausal women. The molecular regulation of aromatase has been characterized in response to many obesity-related molecules, including inflammatory mediators and adipokines. This review is aimed at providing an overview of our current knowledge in relation to the regulation of estrogens in adipose tissue and their role in driving breast tumor development, growth and progression.
Graphical Abstract
1. Sources of estrogens in pre- and postmenopausal women
Estrogens play an important role in a number of physiological processes, including regulating energy metabolism, stress responses, mineral balance, as well as sexual development [1]. In premenopausal women, estrogens are predominantly produced by the ovary [2]. The hypothalamus releases gonadotropin-releasing hormone (GnRH), which stimulates the secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). FSH stimulates the biosynthesis of estrogens in growing ovarian follicles, which then act on the hypothalamus to induce the production of LH. An acute rise in LH triggers ovulation and the development of the corpus luteum. After menopause, the ovaries produce negligible levels of estrogens. The importance of gonadal steroidogenesis in normal breast development and in the origin of breast cancer is emphasized by the fact that early menstruation and late menopause are linked to a higher risk of breast cancer [3]. Similarly, late menarche and early menopause (before the age of 40) result in a significant reduction in the risk of developing breast cancer [4]. It is somewhat paradoxical, therefore, that the majority of breast cancers occur after menopause, when circulating estrogen levels are low.
The de novo biosynthesis of sex hormones necessitates cholesterol, which is the precursor to all adrenal and gonadal steroid hormones [5]. The first process in steroidogenesis is the transport of cholesterol to the inner mitochondrial membrane by the steroidogenic acute regulator (StAR). Next, cholesterol is converted to pregnenolone by the cytochrome P450 side-chain cleavage enzyme. The formation of the testosterone precursor androstenedione from pregnenolone is dependent on the action of 3β-HSD to produce progesterone and CYP17A1, which converts progesterone to androstenedione via a two-step mechanism. Androstenedione is then converted to testosterone by 17βHSD enzymes, and can then be aromatized to estradiol (17β-estradiol/E2). In postmenopausal women, however, it is circulating dehydroepiandrosterone sulfate (DHEA-S) from the adrenals that is the source of androgen for estrogen formation at peripheral sites. The local biosynthesis of estrogens within the breast [6, 7] and circulating levels of estrogens in blood [8, 9], believed to be a reflection of adipose-derived steroid production, are directly associated with driving breast tumor cell proliferation [10]. The intracrinology that occurs in the breast as a result of the complex interaction of enzymes responsible for the activation and inactivation of steroid hormones has been the focus of many studies to explain the increased risk of breast cancer after menopause, when gonadal estrogen biosynthesis has ceased [11, 12]. Specifically, the breast expresses all enzymes required for the conversion of DHEA-S to E2, including steroid sulfatase, 3β-HSD, 17βHSD1 and aromatase [13, 14]. Of these enzymes, the best characterized in terms of its regulation in obesity is the enzyme involved in the rate-limiting step in estrogen biosynthesis, aromatase.
2. Aromatase
Cytochrome P450 aromatase (P450arom) is a microsomal enzyme that is expressed in the endoplasmic reticulum and catalyzes one of the final steps in estrogen biosynthesis by converting 19-carbon steroids (androgens, e.g. androstenedione and testosterone) to 18-carbon steroids (estrogens, e.g. estrone and estradiol) [15]. Aromatase is found in many tissues, including the gonads, brain, adipose tissue, placenta, blood vessels, skin, bone and in breast cancer tissue [16]. Its expression in breast adipose is hypothesized to be a major driver of estrogen-dependent breast cancer after menopause. The aromatase (CYP19A1) gene is located on chromosome 15q21.2 and is approximately 123kb long with nine coding exons (II-X) and a 93kb regulatory region.
Eight tissue-specific promoters regulate the expression of the CYP19A1 gene yielding transcripts with unique 5’-untranslated regions [17]. These are promoters I.1 (placenta major, « 93kb), I.2a (placenta minor, « 78kb), I.4 (skin, adipose tissue and bone, « 73kb), I.7 (endothelial cell and breast cancer, « 36kb), I.f (brain, « 33kb), I.6 (bone, « 0.7kb), I.3 (adipose tissue and breast cancer, « 0.2kb) and II (ovary, adipose tissue, breast cancer and endometriosis, within 1kb) [15, 17]. In normal breast adipose tissue, low levels of aromatase are derived from activation of the distal promoter I.4, known to be regulated by glucocorticoids and class 1 cytokines, such as interleukin 6 (IL-6), interleukin 11 (IL-11), leukemia inhibitory factor (LIF) and oncostatin M (OSM), via the Janus kinase-1/signal transducer and activator of transcription 3 (JAK1/STAT3) pathway [18]. Promoter I.4 is also activated by the inflammatory mediator tumor necrosis factor alpha (TNFα), via the mitogen-activated protein (MAP) kinase-AP1 pathway [18]. However, in the breast adipose tissue of women with breast cancer, the majority of transcripts are derived from the coordinated activation of promoters I.3 and II in a cAMP-dependent manner [18, 19].
2.1. Aromatase regulation in obesity
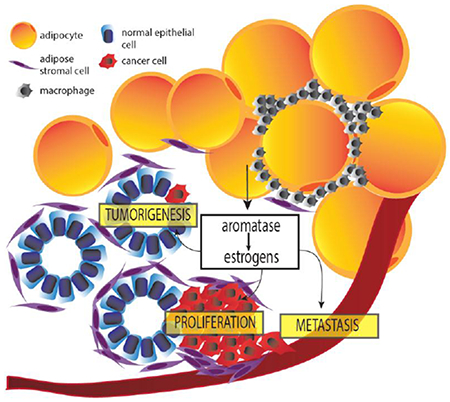
The prevalence of obesity has been steadily rising worldwide, and there is now strong evidence to support a causal link between obesity and the development of many cancers, including breast, ovarian, renal, pancreatic, leukemia, multiple myeloma, and esophageal cancers [20–25]. For breast cancer, the link is strongest for postmenopausal women and for the development of ER+ breast cancer, suggesting an important role of estrogens in driving obesity-associated breast cancer growth and development [20, 24]. After menopause, adipose tissue is the primary source of estrogen production in the body [26–28]. Interestingly, BMI is found to be positively associated with tissue levels of estrogens [29, 30]. Therefore, as fat mass increases with increasing body weight, aromatase expression and consequently estrogen levels, are also elevated, an effect that is more prominent in postmenopausal women [31–37].
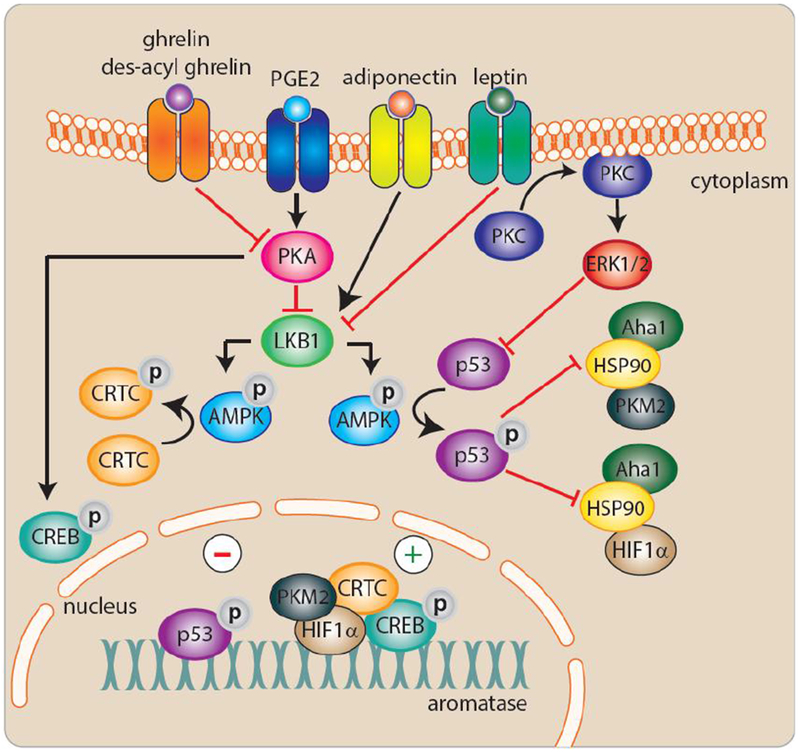
A number of studies have further explored the positive association between obesity and local estrogen production by highlighting specific factors dysregulated in obese adipose tissue that induce aromatase expression in adipose stromal cells (ASCs) (Figure 1). Following up on the seminal findings that proinflammatory mediators and cytokines (e.g. PGE2, TNFα, IL-1, IL-6, COX-2) play key roles in regulating estrogen production in ASCs [18, 19, 38–42], several recent mechanistic studies have provided greater insight into the regulation of aromatase by some of these factors in obese adipose tissue. For example, using both cell culture and clinical samples Wang et al. showed that p53 is a negative regulator of aromatase expression in ASCs and treatment with PGE2, which is elevated in obesity, inhibits p53 resulting in elevation in aromatase [43]. PGE2 was also shown to stabilize HIF1α, leading to the binding and stimulation of aromatase PII [44]. More recently, Subbaramaiah et al. found that PGE2 downregulates SIRT1 in a human ASC cell line leading to the upregulation of HIF1α [45]. Further supporting the role of PGE2 in induction of aromatase, IL-6 in sera from obese subjects was found to induce breast cancer cell PGE2 secretion, which in turn induced aromatase expression in primary ASCs. This effect was nullified by both depletion of IL-6 from sera or treatment with celecoxib, an inhibitor of the enzyme COX-2 which catalyzes the conversion of arachidonic acid to PGE2 [46]. These findings provide a new level of complexity regarding the role of IL-6 in regulating aromatase expression in ASCs, as previous findings demonstrated that it can increase the activity of promoter I.4 in the presence of the IL-6 soluble receptor [18]. Adipokines have also been examined for their role in regulating aromatase.
Figure 1:
Molecular regulation of aromatase in obesity.
Leptin, an adipokine increased in obesity, inhibits p53 in human breast derived ASCs, leading to an increase in aromatase expression [47]. This finding complemented prior work demonstrating that PGE2 and leptin stimulate aromatase expression by suppressing the activity of energy sensors LKB1/AMPK, thereby alleviating their suppressive effects on CREB-regulated transcriptional co-activators (CRTCs) which stimulate aromatase [48, 49]. Interestingly, LKB1 and AMPK are stimulated by adiponectin, an adipokine produced by healthy adipocytes, leading to suppression of the PII-specific expression of aromatase. This suggests that AMPK-activating drugs may selectively inhibit aromatase in adipose tissue, including breast. More recently, the orexigenic hormone ghrelin and its unacylated form, des-acyl ghrelin, which are reduced in obesity, were also shown to suppress PII-driven aromatase expression mediated via suppression of cAMP [50].
The regulation of PI.4 has also been examined in the context of obesity. Promoter I.4-specific transcripts, present in adipose tissue, can be stimulated by inflammatory mediators, including IL-6, IL-11, leukemia inhibitory factor, oncostatin M, as well as TNFα [51, 52]. Using preclinical models and human breast tissue, the TNFα-mediated induction of aromatase was shown to require ERK1/2 activation, an effect that was blocked by the anti-inflammatory cytokine IL-10 [53]. Taken together, these studies propose new mechanisms that explain the elevation in estrogen produced by breast adipose tissue in obese postmenopausal women.
It is important to highlight that since aromatase is produced by dysfunctional breast adipose tissue, obesity as defined by BMI may not be the best predictor of local estrogen levels since it does not account for volume of body fat or quality of fat. For example, a recent study found that breast white adipose tissue inflammation and other systemic correlates of metabolic syndrome (e.g. leptin) were strongly correlated with aromatase expression and activity in women with a normal BMI (≤24.9 kg/m2)[54]. In a prospective study of 12,159 postmenopausal women in Sweden, body fat % was a better predictor of breast cancer incidence than BMI [55]. It is possible that this discrepancy is due to body fat % being a superior readout of estrogen levels which are closely tied to breast cancer risk.
3. Estrogens, estrogen receptors and breast cancer
Estrogens have been shown to function predominantly by interacting with two estrogen receptors (ERs), ERα and ERβ [56]. Estrogen receptors are fundamental for mammary gland maturation and physiological events such as puberty and pregnancy. ERα is found in nearly 50-80% of breast cancers, and its expression correlates with better prognosis and a lower chance of recurrence [57, 58]. ERβ has also been detected in breast tumors, and is suspected to contribute to hormonal sensitivity and resistance [59, 60]. Studies show decreased ERβ RNA levels in invasive breast cancers in comparison with the normal mammary gland [60]. Although the role and mechanism through which decreased ERβ expression results in tumorigenesis is unknown, results from several studies suggest a stimulatory role of ERα and an inhibitory role of ERβ in relation to proliferation of estrogen-dependent cells [61, 62].
3.1. Estrogens as mutagens and effects on breast epithelium
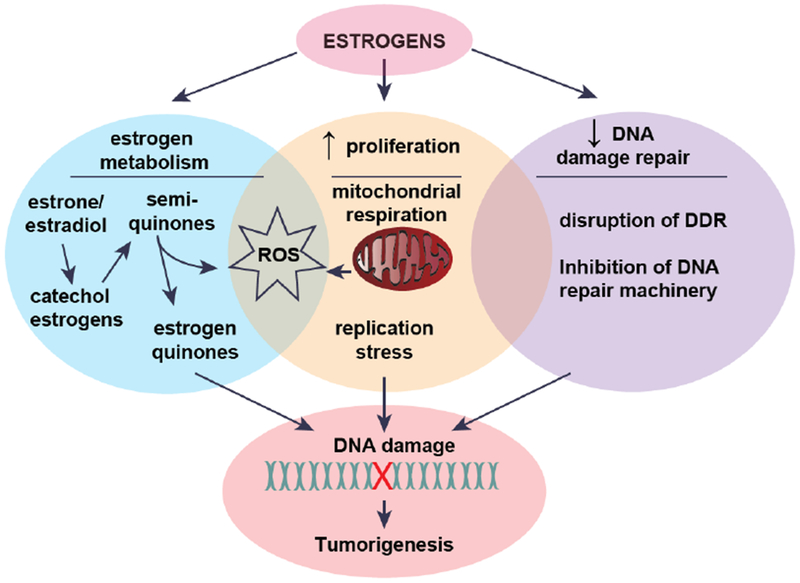
Estrogens are a significant driver of ER+ breast cancer, with studies suggesting a role in ER− breast cancer as well [63–65]. Neighboring the estrogen-producing ASCs are breast epithelial cells, which are hormone-sensitive and express the ER. Estrogens play an important in role in the normal development of breast epithelium by stimulating proliferation and ductal morphogenesis [66]. However, when exposed to high levels of estrogens such as in the setting of obesity, the pro-proliferative effect of these steroids may cause accumulation of replication errors leading to mutations and the development of breast cancer (Figure 2). Proliferating cells also have higher energy demands that require increased mitochondrial activity, which could potentially lead to an elevation in reactive oxygen species (ROS) as a byproduct of cellular respiration. Felty et al. found that estradiol can directly stimulate the production of intracellular ROS from mitochondria in several breast cancer cell lines [67].
Figure 2:
Estrogens and tumorigenesis.
Additionally, estrogens can be metabolized to catechols followed by further oxidation to semi-quinones and quinones through a process of redox cycling that produces ROS. This is important in the context of tumorigenesis because estrogen quinones are mutagenic and can interact directly with DNA to form adducts, a form of DNA damage [68–70]. Several studies have shown that treating normal breast epithelial cells (MCF-10A) with estrogen metabolites induces elevation in intracellular ROS leading to oxidative DNA damage [71–73]. By interacting directly with DNA, estrogen metabolites do not require the estrogen receptor to exert their mutagenic effects, which may explain the role of estrogen in promoting some ER− breast cancers. Indeed, Savage et al. found that estrogen and estrogen metabolites caused DNA double strand breaks (DSB) in both normal breast epithelial cells and ER− breast cancer cells [74]. Given the abundance of evidence for mutagenic and mitogenic effects of estrogens, the obesity-induced elevation in local estrogen production is likely to drive DNA damage in breast epithelial cells leading to a greater risk of tumorigenesis.
Interestingly, there is a growing body of literature implicating estrogens in the disruption of the DNA damage response (DDR) and DNA repair machinery. For example, some studies have indicated that ERα downregulates ATM and ATR, important initiators of the DDR [75–77]. This is hypothesized to result in defective processing of DNA damage. The mechanisms by which estrogen signaling alters DDR and DNA repair has been reviewed in detail [78] and provides a novel theory for how estrogens promote breast cancer, i.e. not only by inducing DNA damage but also potentially diminishing the cell’s ability to sense and repair damage.
3.2. Mechanism of breast cancer growth in response to estrogen
In breast cancer, estrogens can act via genomic and non-genomic mechanisms. Genomic actions of ERs are associated with the regulation of estrogen-response element (ERE)-dependent and ERE-independent gene expression [79–81]. In ERE-dependent genomic activation, estrogen binding to its receptor is associated with increased interaction with coactivator proteins in order to bind to the ERE in DNA, resulting in changes in gene expression that regulate growth, differentiation, apoptosis and angiogenesis [80]. However, estrogens can also facilitate gene transcription via pathways that do not require EREs. In ERE-independent genomic activation, the estrogen-ER complex can also interact with other DNA bound transcription factors such as Fos/Jun in order to bind to AP-1 or SP1 sites in the promoter regions of target genes, thereby resulting in activation of gene transcription [79, 81].
Non-genomic effects are actions mediated via activation of ER localized closely to or at the plasma membrane [82–84]. Membrane-associated ER may interact with many proteins including adaptor proteins, G-proteins, Src, growth factor receptors (EGFR, IGFR1, HER2), cytoplasmic kinases (MAPKs, PI3K, AKT) as well as signaling enzymes (adenyl cyclase) [85–89]. The actions mediated by this mechanism are independent of gene transcriptional changes [85–89].
Recent findings show that non-genomic effects also involve the orphan GPCR-like protein, GPR30 (G protein coupled receptor 30) [90, 91]; also named GPER [92]. It has been reported that estrogens can act on membrane GPER in order to stimulate release EGF or EGF-related ligands leading to a transient activation of the EGFR, which in turn activates MAPK and PI3K signaling pathways [93, 94]. Interestingly, activation of GPER leads to the stimulation of adenylyl cyclase activity and increases in cAMP formation, which then leads to a marked inhibition of cell proliferation [95]. It is hypothesized that GPR30 inhibits MAPK activity and can also increase intracellular calcium stores, resulting in apoptosis and inhibition of MCF7 cell proliferation [96, 97]. Thus, GPR30 could be a novel potential target for ER+ breast cancers.
There is also an important cross-talk between ER genomic and non-genomic signaling pathways. Estrogen binding to nuclear ER can increase transforming growth factor (TGFα) and amphiregulin expression. Once TGFα and amphiregulin are bound, they stimulate EGFR in order to activate MAPK and AKT [79, 81, 98]. Conversely, several cytokines, growth factors, EGFR ligands and IGF1R-related pathways activate MAPK/ERK, PI3K/AKT, p90rsk and p38 MAPK, which lead to ER phosphorylation. The phosphorylation of the AF-1 serines 118,167 and threonine 311, or other domains, results in the activation of ER [99–103].
Both genomic and non-genomic signaling pathways of the ER play a critical role in breast cancer development, progression, and survival. This is due in part to the regulation of the anti-apoptotic gene BcI2, pro-apoptotic gene caspase and cell cycle regulator cyclin D1 [104–106]. Additionally, estrogens increase the growth of breast tumors by increasing the number of G0/G1 cells entering into the cell cycle, therefore resulting in greater proliferation [107, 108]. Moreover, PI3K interacts with Src in order to promote S-phase entry of MCF7 cells in the presence of estradiol [109].
3.3. Role of estrogens in breast cancer metastasis
Estrogens have also been shown to influence breast cancer progression. For example, estrogen treatment has been shown to cause cytoskeletal remodeling, and contribute to cancer cell migration and invasion in vitro [110]. As mentioned above, estrogens not only influence the etiology of ER+ breast tumors, but also of ER− breast cancers. In ER− breast cancer cells, estrogen actions on GPER1 lead to increased invasion and migration, promoting a prometastatic phenotype [111]. ERα splice variants have also been shown to mediate extra-nuclear effects of estrogens through activation of PKC signaling pathway and promote metastasis in ERα-positive and ERα-negative breast cancer cell lines [112]. Estradiol-induced proteins like G1P3 are able to rescue cells undergoing anoikis [113], favoring prometastatic estrogenic effects. Estradiol-induced expression of Proteinase Inhibitor-9 (PI-9) with inhibitory activity against Granzyme-B contributes to breast cancer immune escape [114]. Another estrogen contribution to metastasis is related to premetastatic niche formation and bone marrow derived myeloid (BMD) cells recruitment. ERα expression in BMDs is necessary for estrogen-mediated tumor promotion of ER− breast cancer [115]. Estrogens might contribute to BMD recruitment through VEGF-A and/or SDF1α that were previously described as downstream mediators of estradiol/ER-induced angiogenesis and macrophage chemotaxis, respectively. Sartorius et al. demonstrated that estradiol promotes brain metastasis of ER− breast cancer cells by modulating astrocyte function, suggesting that existing endocrine therapies may provide some clinical benefit towards reducing and managing brain metastases in patients with ER− breast tumors [116].
A number of epidemiological studies supports these laboratory-based studies. For example, endocrine therapy has been shown to decrease the risk of developing ER+ contralateral breast cancer [117]. Interestingly, in this study, the authors found that patients on antiestrogen therapy presented a higher risk of contralateral ER− breast cancer. In a retrospective analysis of metastatic behavior of breast cancer subtypes [118], data demonstrate that women with ER+ tumors are more likely to develop bone metastases and have improved disease-free survival compared to women who have ER− tumors. Their study support a close relationship between ER+ tumors and metastasis-specific survival, a finding that is consistent with data from previous studies [119].
4. Endocrine therapy
ERα and progesterone receptor (PR) expression have the greatest predictive value for response to hormonal therapy [120]. Despite breast cancer being the most commonly diagnosed cancer among women, cancer-related mortality rates are declining, in part, due to advances in adjuvant therapy [121]. Endocrine or hormonal therapies, including aromatase inhibitors, have revolutionized treatment for breast cancer patients. Current endocrine therapy holds major therapeutic value, especially for ER+ breast cancer patients.
4.1. Tamoxifen
The initial findings by George Beatson regarding the important role estrogen plays in breast development have served as the basis for research, development, and discovery of tamoxifen in 1967 by Harper and Walpole [122, 123]. Tamoxifen is a selective ER modulator (SERM) used to treat estrogen-dependent breast cancer and reduce the risk of cancer recurrence in premenopausal and postmenopausal women [124]. This drug was approved by the food and drug administration (FDA) in 1998 and is known by the brand names Nolvadex and Soltamox. Tamoxifen is a non-steroidal antiestrogen with triphenylethylene structure [125]. As a prodrug, tamoxifen has little affinity for the ER. It is metabolized in the liver into an active metabolite, 4-hydroxytamoxifen [126] which acts as an antagonist of the ER in breast tissue, leading to the inhibition of binding with coactivator proteins, thereby blocking the G1 phase of the cell cycle and preventing cell proliferation. Another tamoxifen metabolite 4-hydroxy-N-desmethyl tamoxifen (endoxifen) is present at greater concentrations in plasma than 4-hydroxytamoxifen, and thus may be just as important, if not more, to the anti-estrogenic action of tamoxifen [127, 128].
In ER-positive breast cancer, most clinical studies demonstrate that tamoxifen should be taken continuously for five years or ten years. According to the worldwide Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) trial, it was suggested that patients who are treated with tamoxifen for ten years have reduced risk of breast cancer recurrence and cancer-associated death compared with patients who are treated with tamoxifen for only 5 years [129]. Another clinical trial, the aTTom trial, showed that patients who were continuously treated with tamoxifen for ten years also had significantly reduced risk of breast cancer recurrence compared with patients who took tamoxifen for 5 years [130]. Although tamoxifen treatment helps to reduce the risk of cancer recurrence, side effects, including endometrial and uterine cancers [131, 132], loss of blood flow to parts of the brain and significant loss of bone mineral density in premenopausal women [125, 133], have been reported, thereby highlighting a need for safer alternative therapies.
4.2. Fulvestrant
Fulvestrant (ICI 164384) is a selective ER degrader (SERD). It is classified as a pure steroidal antiestrogen (European medicines agency, brand name Faslodex) [134]. It is a class of drug that targets estrogen receptors approximately 100 times faster than tamoxifen [134] and immediately causes degradation of the ER in breast cancer cells which reduces their ability to be activated by estrogen as well as the ability of ER to be activated via estrogen-independent mechanisms [135]. The loss of the estrogen receptor in breast cancer results in inhibition of breast cancer cell growth. In vitro, fulvestrant inhibits the growth of tamoxifen-resistant ER positive MCF7 breast cancer cells [136]. In vivo, fulvestrant also inhibits tamoxifen-resistant and ER positive MCF7 breast cancer xenografts [137]. In clinical studies, a correlative downregulation of ER with increasing dose was observed in postmenopausal women with primary breast cancer treated with single doses of fulvestrant for 15-22 days before surgery [138].
The FDA has approved this drug for the treatment of hormone receptor-positive metastatic breast cancer. Breast cancer patients receive this therapy once a month via intramuscular injection [139]. However, side effects such as diarrhea, hot flushes and throat inflammation have been reported that can affect the quality of life of breast cancer patients [140]. It is recommended for use in postmenopausal women who cannot be effectively treated with tamoxifen or aromatase inhibitors, as fulvestrant acts independently of estrogens [141–144].
4.3. Aromatase inhibitors
Aromatase inhibitors (AIs) are a class of drugs that block the action of the aromatase enzyme [145] in order to reduce the amount of estrogen in the body. There are two types of AIs, irreversible steroidal inhibitors such as exemestane (brand name, Aromasin®) [146], and non-steroidal inhibitors such as anastrozole (brand name, Arimidex®) [147] and letrozole (brand name, Femara®) [148]. These three inhibitors were all approved by the FDA and most clinical studies prove that treating breast cancer with exemestane [149], anastrozole [150] or letrozole [151] highly reduces breast cancer recurrence compared with tamoxifen treatment. The levels of FSH increase as a consequence of estrogen suppression. For this reason, AIs are contra-indicated in premenopausal women as use can lead to the development of polycystic ovaries and incomplete inhibition of ovarian estrogen biosynthesis. AI use is also associated with well-documented side effects such as osteoporosis [152], joint and muscle pain [153, 154] and hot flushes [155] because of the whole-body inhibition of aromatase and estrogen production.
5. Endocrine resistance
While endocrine therapy has enhanced the lives of many breast cancer patients, emergence of resistance is inevitable with advanced breast cancer and is to be expected over time. Endocrine resistance in cancer cells can be divided broadly into two main categories – de novo and acquired resistance. Breast tumors that show no response to first line hormonal therapy are examples of de novo resistance. On the other hand, tumors that exhibit a response initially to endocrine therapy, but then later recur are examples of acquired or secondary resistance.
Endocrine resistance may be considered to reflect four possible mechanisms: 1) Pharmacological resistance; 2) Changes in expression of ERα and its co-regulators; 3) Alterations in expression of cell cycle signaling molecules; 4) Alternate growth receptor pathways, which is especially relevant in the context of obesity.
The first possible theory for developing hormonal resistance is by means of pharmacological mechanisms. As previously mentioned, tamoxifen is a pro-drug that undergoes extensive oxidation in the liver, primarily by CYP3A and CYP2D6 [126]. However, polymorphisms in tamoxifen-metabolizing genes affect the efficacy of the enzymes and thus plasma concentrations of the active metabolites of tamoxifen. Retrospective clinical data suggests that women with CYP2D6 4*/4* genotype have null or reduced enzyme activity, resulting in higher risk of tumor relapse [156].
The second possible mechanism for developing endocrine resistance is through mutations in the ESR1 gene, which encodes ERα. Expression of ERα has long been used to predict clinical response to anti-estrogen therapy. The mutations mainly occur at two residues in the ligand-binding domain, which replaces tyrosine with serine or asparagine at residue 537 and replaces aspartic acid with glycine at residue 538 [157]. A study found a mutation rate of 12% among 76 patients with metastatic ER+ breast cancer, and a 20% mutation rate among individuals with heavily pretreated disease [158]. Analysis of ESR1 mutations was performed on plasma samples from patients participating in the PALOMA3 and SoFEA studies, and a mutation rate of 25% was observed in patients with breast cancer progression on endocrine therapy, and an even higher mutation rate of 29% in patients who received prior AI therapy [159]. Similarly, a mutation rate of 39% was detected in patients with prior AI sensitivity. These findings suggest that although ESR1 mutations are a rare cause of primary endocrine resistance, the mutations arise more commonly with acquired secondary resistance to AI therapy [159]. Several studies have sought to clarify the role of ERβ, if any, in relation to response and resistance to endocrine therapy. Despite some conflicting evidence, it has been suggested that low levels of ERβ expression are related to tamoxifen resistance [59].
Additionally, overexpression of the ER co-activator AIB1 (or SRC3 or NCoA3) and down regulation of the co-repressor NCoR is associated with tamoxifen resistance [160–162]. Finally, increased levels of transcription factors such as NFkB and AP-1, which increase the interaction of ER with specific gene promoters, have also been linked to endocrine resistance [163, 164].
Endocrine resistance can also occur as a result of alterations in key cell cycle checkpoints [165]. The cell cycle involves a complex sequence of events through which a cell duplicates, and involves many regulatory proteins such as cyclin proteins, and cyclin-dependent kinases, oncogenes and tumor-suppressor genes, and mitotic checkpoint proteins. The balance of proliferative and anti-proliferative signals determines if a cell will progress from the G1 phase to the S phase, or withdraw into the dormant phase [166]. Anti-proliferative signals are communicated via the retinoblastoma (Rb) tumor suppressor protein, while Rb itself is regulated via complexes of cyclin and cyclin-dependent kinases [167]. Cyclin-dependent kinase 4, in complex with cyclin D1, D2 or D3, controls the phosphorylation of Rb, which in turn regulates the progression of the cell from G1 to S phase [168]. By means of CDK4 inactivation, or cyclin D1 and E1 amplification, tumor cells are able to circumvent cell cycle regulation [169, 170]. Interestingly, cyclin D1 amplification is a common occurrence in estrogen-receptor positive breast cancers with 58% and 29% incidence rate in luminal B and luminal A cancers, respectively (Cancer Genome Atlas Network, 2012). Similarly, reduced expression of p21 and p27 (negative cell cycle regulators) and inactivation of Rb are also associated with poor response to hormonal therapy, especially tamoxifen [171, 172]. As a result, endocrine therapy and CDK4/6 inhibitors, in combination or sequentially, are now a viable option for the treatment of hormone receptor-positive breast cancer as a first line therapy and following development of resistance to endocrine therapy [173].
Another possible mechanism for developing endocrine resistance is by the means of enhanced autophagy, which is an intracellular process that recycles damaged or unnecessary organelles (macroautophagy) or proteins (microautophagy). Under conditions of compromised autophagy, it was noted that cytotoxic effects of 4-hydroxytamoxifen (4-OHT) were significantly increased [174]. Inhibition of autophagosome function stimulated a strong caspase-dependent cell death in the 4-OHT treated, anti-estrogen resistant cells [174, 175]. Thus, impaired autophagy increases sensitivity to endocrine therapy, and inhibition of the autophagosome may be a potential target to overcome resistance and improve efficacy of hormonal treatment of ER+ breast cancers.
Finally, overexpression and amplification of growth factor receptors, such as FGFR1 (fibroblast growth factor receptor -1), IGF1R (insulin growth factor -1 receptor), HER2 (human epidermal growth receptor -2), HER3 (human epidermal growth receptor -3), and EGFR (epidermal growth factor receptor) [176–179], which converge on the PI3K/Akt/mTOR and Raf/Mek/Erk pathways, have been shown to be associated with sustained tumor proliferation and survival independent of estrogen [176–179]. These pathways provide alternative survival stimuli to the tumors, and can emerge to act as ER-independent drivers of tumor growth, thus conferring resistance to endocrine therapy. In addition, these pathways can be activated by amplification of the receptors and/or their respective ligands. Alternatively, deregulation of downstream signaling molecules such as an activating mutation in the PI3K p110 catalytic subunit or the loss of expression of PTEN tumor suppressor can also lead in activation of the pathways [180].
5.1. Endocrine resistance in obesity
Several studies have shown that obese women are more likely to face a poor breast cancer prognosis as compared to lean women, and a greater chance of breast cancer recurrence [181–183]. In the Anastrozole, Tamoxifen Alone or in Combination (ATAC) [150, 184, 185] and Australian Breast Cancer Study Group (ABCSG)[186] trials there was evidence that the relative benefit of anastrozole vs. tamoxifen was greatest when BMI was normal vs. elevated. A 2014 meta-analysis concluded that there is an association between BMI and outcome, and that there is increased risk of mortality in individuals with BMI over 25 kg/m2 at diagnosis [187]. Different studies have evaluated the efficacy of AI inhibitors in suppressing circulating estrogen levels and its relation with BMI. Interestingly, Elliott et al. found that BMI and estradiol were higher in metastatic patients [188], and Lonning et al. showed that estrone levels positively correlate with BMI in women receiving AIs [189]. In a study by Sestak et al., obese women were less likely to observe benefits of AI treatment despite a significant reduction in circulating estrogens [190]. Therefore, it seems that higher BMI may contribute to reduced efficacy of AIs.
Many circulating factors found in the serum of obese postmenopausal women can amplify crosstalk between nongenomic ERα signaling and PI3K/Akt/mTOR and Raf/Mek/Erk pathways which promotes breast cancer progression [191]. For example, cells grown in media supplemented with sera from obese patients have greater IGF-1R activation in comparison to control sera [191]. As mentioned previously, FGFR1 is a known mediator of endocrine therapy resistance [177, 179, 192]. The FGFR1 regulatory pathway is one of the pathways that shows shared activation in patients with obesity and those with resistance to AI therapy [193]. Phosphorylation of FGFR1 and FGFR1 ligand expression is increased with obesity, metabolic dysfunction, weight gain and adipocyte hypertrophy [193]. Weight gain leads to a positive energy balance, and in the context of obesity/metabolic dysfunction, promotes FGFR ligand production from adipose tissue, which may also result in the activation of receptors in nearby breast cancer cells to promote growth after estrogen deprivation [193]. Leptin has also been shown to induce cell proliferation through activation of MAPK signaling, and its receptors are expressed in both normal breast tissue and solid tumors [194]. Additionally, leptin can mimic the effects of ERα transactivation in an ER+ breast cancer cell line, including down-regulation of ER mRNA and protein levels and up-regulation of the estrogen-dependent gene pS2 [195]. Since serum leptin concentrations are correlated with the percentage of body fat [196], obese individuals have a higher chance of developing endocrine resistance due to the mechanism described above. Moreover, leptin stimulates the expression of aromatase through enhanced binding of CREB, CRTC and AP-1 to specific sites in the promoter region [48, 197], thus amplifying in situ production of E2 and driving breast tumor growth.
Nevertheless, a number of studies found no association between BMI and breast cancer outcomes. Coscia et al. showed a reduction in plasma estrone and estradiol following anastrozol treatment, but no significant changes in steroid concentration in association with BMI [198]. Two recent studies showed no significant relationship between high BMI and the efficacy of two different aromatase inhibitors [199, 200]. In a cohort where more than two thirds of postmenopausal women receiving adjuvant letrozole therapy were obese, there was no association for worse outcome in the obese women compared with lean women receiving the same treatment [200]. Zewenghiel et al. found no statistically significant difference between the three BMI categories (normal weight, overweight and obesity) and time to progression during fulvestrant treatment in their 173 patient cohort [199]. Therefore, additional studies are required to determine which patient group and/or treatment may be impacted by BMI before altering disease management.
6. Conclusions and future directions
Obesity is now an established risk factor for breast cancer in postmenopausal women. The local production of estrogens in the breast adipose tissue is suspected to be a key driver of breast cancer development and growth, and a mediator of resistance to endocrine therapy. Additional factors in obesity are also important for disease development and progression, and a better understanding of the mechanisms at play will inform management of breast cancer in an increasingly obese female population. Additional studies are also required to determine whether pharmacological or lifestyle interventions will reduce the risk of breast cancer development and progression in this obese population.
SBMB_2019_10 Manuscript Highlights.
Obesity is linked to an increased risk of developing hormone receptor positive breast cancer
Estrogens stimulate cancer development, growth and progression
The local production of estrogen in adipose tissue drives the growth of breast cancer after menopause
7. Acknowledgments
This work was supported by NIH/NCI R01CA215797-01 (to K.A. Brown) and by the Dr. Robert C. and Veronica Atkins Foundation Curriculum in Metabolic Diseases at Weill Cornell Medical College (to R. Moges).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saladin KS, Anatomy & physiology: the unity of form and function 6. ed. 2012, New York, NY: McGraw-Hill. xxvi, 1136, A-14, G-20, C-3, I-49. [Google Scholar]
- 2.Cui J, Shen Y, and Li R, Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends in molecular medicine, 2013. 19(3): p. 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelsey JL, Gammon MD, and John EM, Reproductive factors and breast cancer. Epidemiol Rev, 1993. 15(1): p. 36–47. [DOI] [PubMed] [Google Scholar]
- 4.Pike MC, et al. , Estrogens, progestogens, normal breast cell proliferation, and breast cancer risk. Epidemiol Rev, 1993. 15(1): p. 17–35. [DOI] [PubMed] [Google Scholar]
- 5.Ghayee HK and Auchus RJ, Basic concepts and recent developments in human steroid hormone biosynthesis. Rev Endocr Metab Disord, 2007. 8(4): p. 289–300. [DOI] [PubMed] [Google Scholar]
- 6.Santner SJ, Feil PD, and Santen RJ, In situ estrogen production via the estrone sulfatase pathway in breast tumors: relative importance versus the aromatase pathway. J Clin Endocrinol Metab, 1984. 59(1): p. 29–33. [DOI] [PubMed] [Google Scholar]
- 7.Gunnarsson C, Hellqvist E, and Stal O, 17beta-Hydroxysteroid dehydrogenases involved in local oestrogen synthesis have prognostic significance in breast cancer. Br J Cancer, 2005. 92(3): p. 547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas HV, et al. , A prospective study of endogenous serum hormone concentrations and breast cancer risk in post-menopausal women on the island of Guernsey. Br J Cancer, 1997. 76(3): p. 401–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toniolo PG, et al. , A prospective study of endogenous estrogens and breast cancer in postmenopausal women. J Natl Cancer Inst, 1995. 87(3): p. 190–7. [DOI] [PubMed] [Google Scholar]
- 10.Larionov AA, Berstein LM, and Miller WR, Local uptake and synthesis of oestrone in normal and malignant postmenopausal breast tissues. J Steroid Biochem Mol Biol, 2002. 81(1): p. 57–64. [DOI] [PubMed] [Google Scholar]
- 11.Labrie F, Intracrinology in action: importance of extragonadal sex steroid biosynthesis and inactivation in peripheral tissues in both women and men. J Steroid Biochem Mol Biol, 2015. 145: p. 131–2. [DOI] [PubMed] [Google Scholar]
- 12.Labrie F, All sex steroids are made intracellularly in peripheral tissues by the mechanisms of intracrinology after menopause. J Steroid Biochem Mol Biol, 2015. 145: p. 133–8. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki T, et al. , Sex steroid-producing enzymes in human breast cancer. Endocr Relat Cancer, 2005. 12(4): p. 701–20. [DOI] [PubMed] [Google Scholar]
- 14.Belanger C, et al. , Adipose tissue intracrinology: potential importance of local androgen/estrogen metabolism in the regulation of adiposity. Horm Metab Res, 2002. 34(11–12): p. 737–45. [DOI] [PubMed] [Google Scholar]
- 15.Sebastian S and Bulun SE, A highly complex organization of the regulatory region of the human CYP19 (aromatase) gene revealed by the Human Genome Project. J Clin Endocrinol Metab, 2001. 86(10): p. 4600–2. [DOI] [PubMed] [Google Scholar]
- 16.Simpson ER, et al. , Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr Rev, 1994. 15(3): p. 342–55. [DOI] [PubMed] [Google Scholar]
- 17.Mahendroo MS, Mendelson CR, and Simpson ER, Tissue-specific and hormonally controlled alternative promoters regulate aromatase cytochrome P450gene expression in human adipose tissue. J Biol Chem, 1993. 268(26): p. 19463–70. [PubMed] [Google Scholar]
- 18.Zhao Y, et al. , Aromatase P450 gene expression in human adipose tissue. Role of a Jak/STAT pathway in regulation of the adipose-specific promoter. J Biol Chem, 1995. 270(27): p. 16449–57. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Y, et al. , Tumor necrosis factor-alpha stimulates aromatase gene expression in human adipose stromal cells through use of an activating protein-1 binding site upstream of promoter I.4. Mol Endocrinol, 1996. 10(11): p. 1350–7. [DOI] [PubMed] [Google Scholar]
- 20.Huang Z, et al. , Dual effects of weight and weight gain on breast cancer risk. JAMA, 1997. 278(17): p. 1407–11. [PubMed] [Google Scholar]
- 21.van den Brandt PA, et al. , Pooled analysis of prospective cohort studies on height, weight, and breast cancer risk. Am J Epidemiol, 2000. 152(6): p. 514–27. [DOI] [PubMed] [Google Scholar]
- 22.Reeves GK, et al. , Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ, 2007. 335(7630): p. 1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calle EE and Kaaks R, Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer, 2004. 4(8): p. 579–91. [DOI] [PubMed] [Google Scholar]
- 24.Renehan AG, et al. , Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet, 2008. 371(9612): p. 569–78. [DOI] [PubMed] [Google Scholar]
- 25.Bae HS, et al. , Obesity and epithelial ovarian cancer survival: a systematic review and meta-analysis. J Ovarian Res, 2014. 7: p. 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown KA and Simpson ER, Obesity and breast cancer: mechanisms and therapeutic implications. Front Biosci (Elite Ed), 2012. 4: p. 2515–24. [DOI] [PubMed] [Google Scholar]
- 27.Grodin JM, Siiteri PK, and MacDonald PC, Source of estrogen production in postmenopausal women. J Clin Endocrinol Metab, 1973. 36(2): p. 207–14. [DOI] [PubMed] [Google Scholar]
- 28.Santen RJ, et al. , History of aromatase: saga of an important biological mediator and therapeutic target. Endocr Rev, 2009. 30(4): p. 343–75. [DOI] [PubMed] [Google Scholar]
- 29.Kakugawa Y, et al. , Associations of obesity and physical activity with serum and intratumoral sex steroid hormone levels among postmenopausal women with breast cancer: analysis of paired serum and tumor tissue samples. Breast Cancer Res Treat, 2017. 162(1): p. 115–125. [DOI] [PubMed] [Google Scholar]
- 30.Schairer C, et al. , Quantifying the Role of Circulating Unconjugated Estradiol in Mediating the Body Mass Index-Breast Cancer Association. Cancer Epidemiol Biomarkers Prev, 2016. 25(1): p. 105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baglietto L, et al. , Circulating steroid hormone concentrations in postmenopausal women in relation to body size and composition. Breast Cancer Res Treat, 2009. 115(1): p. 171–9. [DOI] [PubMed] [Google Scholar]
- 32.Rose DP, Komninou D, and Stephenson GD, Obesity, adipocytokines, and insulin resistance in breast cancer. Obes Rev, 2004. 5(3): p. 153–65. [DOI] [PubMed] [Google Scholar]
- 33.Morris PG, et al. , Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev Res (Phila), 2011. 4(7): p. 1021–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Key TJ, et al. , Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J Natl Cancer Inst, 2003. 95(16): p. 1218–26. [DOI] [PubMed] [Google Scholar]
- 35.Brown KA, et al. , Menopause Is a Determinant of Breast Aromatase Expression and Its Associations With BMI, Inflammation, and Systemic Markers. J Clin Endocrinol Metab, 2017. 102(5): p. 1692–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lukanova A, et al. , Body mass index, circulating levels of sex-steroid hormones, IGF-I and IGF-binding protein-3: a cross-sectional study in healthy women. Eur J Endocrinol, 2004. 150(2): p. 161–71. [DOI] [PubMed] [Google Scholar]
- 37.McTiernan A, et al. , Relation of BMI and physical activity to sex hormones in postmenopausal women. Obesity (Silver Spring), 2006. 14(9): p. 1662–77. [DOI] [PubMed] [Google Scholar]
- 38.Singh A, et al. , The regulation of aromatase activity in breast fibroblasts: the role of interleukin-6 and prostaglandin E2. Endocr Relat Cancer, 1999. 6(2): p. 139–47. [DOI] [PubMed] [Google Scholar]
- 39.Richards JA and Brueggemeier RW, Prostaglandin E2 regulates aromatase activity and expression in human adipose stromal cells via two distinct receptor subtypes. J Clin Endocrinol Metab, 2003. 88(6): p. 2810–6. [DOI] [PubMed] [Google Scholar]
- 40.Purohit A, Newman SP, and Reed MJ, The role of cytokines in regulating estrogen synthesis: implications for the etiology of breast cancer. Breast Cancer Res, 2002. 4(2): p. 65–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reed MJ, et al. , Interleukin-1 and interleukin-6 in breast cyst fluid: their role in regulating aromatase activity in breast cancer cells. J Endocrinol, 1992. 132(3): p. R5–8. [DOI] [PubMed] [Google Scholar]
- 42.Subbaramaiah K, et al. , Increased levels of COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov, 2012. 2(4): p. 356–65. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Wang X, et al. , Prostaglandin E2 inhibits p53 in human breast adipose stromal cells: a novel mechanism for the regulation of aromatase in obesity and breast cancer. Cancer Res, 2015. 75(4): p. 645–55. [DOI] [PubMed] [Google Scholar]
- 44.Samarajeewa NU, et al. , HIF-1alpha stimulates aromatase expression driven by prostaglandin E2 in breast adipose stroma. Breast Cancer Res, 2013. 15(2): p. R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subbaramaiah K, et al. , Prostaglandin E2 down-regulates sirtuin 1 (SIRT1) leading to elevated levels of aromatase, providing insights into the obesity-breast cancer connection. J Biol Chem, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowers LW, et al. , Obesity-associated systemic interleukin-6 promotes preadipocyte aromatase expression via increased breast cancer cell prostaglandin E2 production. Breast Cancer Res Treat, 2015. 149(1): p. 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zahid H, et al. , Leptin regulation of the p53-HIF1alpha/PKM2-aromatase axis in breast adipose stromal cells: a novel mechanism for the obesity-breast cancer link. Int J Obes (Lond), 2018. 42(4): p. 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown KA, et al. , Subcellular localization of cyclic AMP-responsive element binding protein-regulated transcription coactivator 2 provides a link between obesity and breast cancer in postmenopausal women. Cancer Res, 2009. 69(13): p. 5392–9. [DOI] [PubMed] [Google Scholar]
- 49.Samarajeewa NU, et al. , CREB-regulated transcription co-activator family stimulates promoter II-driven aromatase expression in preadipocytes. Horm Cancer, 2013. 4(4): p. 233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Docanto MM, et al. , Ghrelin and des-acyl ghrelin inhibit aromatase expression and activity in human adipose stromal cells: suppression of cAMP as a possible mechanism. Breast Cancer Res Treat, 2014. 147(1): p. 193–201. [DOI] [PubMed] [Google Scholar]
- 51.Agarwal VR, et al. , Alternatively spliced transcripts of the aromatase cytochrome P450 (CYP19) gene in adipose tissue of women. J Clin Endocrinol Metab, 1997. 82(1): p. 70–4. [DOI] [PubMed] [Google Scholar]
- 52.Zhao Y, et al. , Transcriptional regulation of CYP19 gene (aromatase) expression in adipose stromal cells in primary culture. J Steroid Biochem Mol Biol, 1997. 61(3–6): p. 203–10. [DOI] [PubMed] [Google Scholar]
- 53.Martinez-Chacon G, et al. , IL-10 suppresses TNF-alpha-induced expression of human aromatase gene in mammary adipose tissue. FASEB J, 2018. 32(6): p. 3361–3370. [DOI] [PubMed] [Google Scholar]
- 54.Iyengar NM, et al. , Metabolic Obesity, Adipose Inflammation and Elevated Breast Aromatase in Women with Normal Body Mass Index. Cancer Prev Res (Phila), 2017. 10(4): p. 235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lahmann PH, et al. , A prospective study of adiposity and postmenopausal breast cancer risk: the Malmo Diet and Cancer Study. Int J Cancer, 2003. 103(2): p. 246–52. [DOI] [PubMed] [Google Scholar]
- 56.Kuiper GG, et al. , Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A, 1996. 93(12): p. 5925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Osborne CK, Steroid hormone receptors in breast cancer management. Breast Cancer Res Treat, 1998. 51(3): p. 227–38. [DOI] [PubMed] [Google Scholar]
- 58.McGuire WL, Hormone receptors: their role in predicting prognosis and response to endocrine therapy. Semin Oncol, 1978. 5(4): p. 428–33. [PubMed] [Google Scholar]
- 59.Esslimani-Sahla M, et al. , Estrogen receptor beta (ER beta) level but not its ER beta cx variant helps to predict tamoxifen resistance in breast cancer. Clin Cancer Res, 2004. 10(17): p. 5769–76. [DOI] [PubMed] [Google Scholar]
- 60.Palmieri C, et al. , Estrogen receptor beta in breast cancer. Endocr Relat Cancer, 2002. 9(1): p. 1–13. [DOI] [PubMed] [Google Scholar]
- 61.Dupont S, et al. , Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development, 2000. 127(19): p. 4277–91. [DOI] [PubMed] [Google Scholar]
- 62.Korach KS, Insights from the study of animals lacking functional estrogen receptor. Science, 1994. 266(5190): p. 1524–7. [DOI] [PubMed] [Google Scholar]
- 63.Pequeux C, et al. , Stromal estrogen receptor-alpha promotes tumor growth by normalizing an increased angiogenesis. Cancer Res, 2012. 72(12): p. 3010–9. [DOI] [PubMed] [Google Scholar]
- 64.Yue W, et al. , Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids, 2013. 78(2): p. 161–70. [DOI] [PubMed] [Google Scholar]
- 65.Yager JD and Davidson NE, Estrogen carcinogenesis in breast cancer. N Engl J Med, 2006. 354(3): p. 270–82. [DOI] [PubMed] [Google Scholar]
- 66.Sternlicht MD, Key stages in mammary gland development: the cues that regulate ductal branching morphogenesis. Breast Cancer Res, 2006. 8(1): p. 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Felty Q, et al. , Estrogen-induced mitochondrial reactive oxygen species as signal-transducing messengers. Biochemistry, 2005. 44(18): p. 6900–9. [DOI] [PubMed] [Google Scholar]
- 68.Fernandez SV, Russo IH, and Russo J, Estradiol and its metabolites 4-hydroxyestradiol and 2-hydroxyestradiol induce mutations in human breast epithelial cells. Int J Cancer, 2006. 118(8): p. 1862–8. [DOI] [PubMed] [Google Scholar]
- 69.Lu F, et al. , Estrogen metabolism and formation of estrogen-DNA adducts in estradiol-treated MCF-10F cells. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin induction and catechol-O-methyltransferase inhibition. J Steroid Biochem Mol Biol, 2007. 105(1–5): p. 150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cavalieri EL, et al. , Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc Natl Acad Sci U S A, 1997. 94(20): p. 10937–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Park SA, et al. , 4-hydroxyestradiol induces anchorage-independent growth of human mammary epithelial cells via activation of IkappaB kinase: potential role of reactive oxygen species. Cancer Res, 2009. 69(6): p. 2416–24. [DOI] [PubMed] [Google Scholar]
- 72.Fussell KC, et al. , Catechol metabolites of endogenous estrogens induce redox cycling and generate reactive oxygen species in breast epithelial cells. Carcinogenesis, 2011. 32(8): p. 1285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singh B and Bhat HK, Superoxide dismutase 3 is induced by antioxidants, inhibits oxidative DNA damage and is associated with inhibition of estrogen-induced breast cancer. Carcinogenesis, 2012. 33(12): p. 2601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Savage KI, et al. , BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res, 2014. 74(10): p. 2773–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guo X, et al. , Estrogen receptor alpha regulates ATM Expression through miRNAs in breast cancer. Clin Cancer Res, 2013. 19(18): p. 4994–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Song L, et al. , miR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS One, 2011. 6(9): p. e25454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pedram A, et al. , Estrogen inhibits ATR signaling to cell cycle checkpoints and DNA repair. Mol Biol Cell, 2009. 20(14): p. 3374–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caldon CE, Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front Oncol, 2014. 4: p. 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deroo BJ and Korach KS, Estrogen receptors and human disease. J Clin Invest, 2006. 116(3): p. 561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hall JM, Couse JF, and Korach KS, The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem, 2001. 276(40): p. 36869–72. [DOI] [PubMed] [Google Scholar]
- 81.McKenna NJ, Lanz RB, and O’Malley BW, Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev, 1999. 20(3): p. 321–44. [DOI] [PubMed] [Google Scholar]
- 82.Razandi M, et al. , ERs associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol Endocrinol, 2002. 16(1): p. 100–15. [DOI] [PubMed] [Google Scholar]
- 83.Razandi M, et al. , Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem, 2003. 278(4): p. 2701–12. [DOI] [PubMed] [Google Scholar]
- 84.Marino M and Ascenzi P, Membrane association of estrogen receptor alpha and beta influences 17beta-estradiol-mediated cancer cell proliferation. Steroids, 2008. 73(9–10): p. 853–8. [DOI] [PubMed] [Google Scholar]
- 85.Wong CW, et al. , Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci U S A, 2002. 99(23): p. 14783–8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86.Schiff R, et al. , Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res, 2004. 10(1 Pt 2): p. 331S–6S. [DOI] [PubMed] [Google Scholar]
- 87.Migliaccio A, et al. , Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J, 1996. 15(6): p. 1292–300. [PMC free article] [PubMed] [Google Scholar]
- 88.Migliaccio A, et al. , Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J, 1998. 17(7): p. 2008–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kahlert S, et al. , Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem, 2000. 275(24): p. 18447–53. [DOI] [PubMed] [Google Scholar]
- 90.Filardo EJ, et al. , Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol, 2000. 14(10): p. 1649–60. [DOI] [PubMed] [Google Scholar]
- 91.Thomas P, et al. , Conserved estrogen binding and signaling functions of the G protein-coupled estrogen receptor 1 (GPER) in mammals and fish. Steroids, 2010. 75(8–9): p. 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alexander SP, Mathie A, and Peters JA, Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol, 2011. 164 Suppl 1: p. S1–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thomas P, et al. , Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology, 2005. 146(2): p. 624–32. [DOI] [PubMed] [Google Scholar]
- 94.Filardo EJ, et al. , Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol, 2002. 16(1): p. 70–84. [DOI] [PubMed] [Google Scholar]
- 95.Ahola TM, et al. , G protein-coupled receptor 30 is critical for a progestin-induced growth inhibition in MCF-7 breast cancer cells. Endocrinology, 2002. 143(9): p. 3376–84. [DOI] [PubMed] [Google Scholar]
- 96.Ahola TM, et al. , Progestin and G protein-coupled receptor 30 inhibit mitogen-activated protein kinase activity in MCF-7 breast cancer cells. Endocrinology, 2002. 143(12): p. 4620–6. [DOI] [PubMed] [Google Scholar]
- 97.Ariazi EA, et al. , The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res, 2010. 70(3): p. 1184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Frasor J, et al. , Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology, 2003. 144(10): p. 4562–4574. [DOI] [PubMed] [Google Scholar]
- 99.Sun M, et al. , Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res, 2001. 61(16): p. 5985–91. [PubMed] [Google Scholar]
- 100.Kato S, et al. , Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science, 1995. 270(5241): p. 1491–4. [DOI] [PubMed] [Google Scholar]
- 101.Joel PB, et al. , pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol, 1998. 18(4): p. 1978–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Campbell RA, et al. , Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem, 2001. 276(13): p. 9817–24. [DOI] [PubMed] [Google Scholar]
- 103.Bunone G, et al. , Activation of the uniiganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J, 1996. 15(9): p. 2174–83. [PMC free article] [PubMed] [Google Scholar]
- 104.Acconcia F, et al. , Survival versus apoptotic 17beta-estradiol effect: role of ER alpha and ER beta activated non-genomic signaling. J Cell Physiol, 2005. 203(1): p. 193–201. [DOI] [PubMed] [Google Scholar]
- 105.Marino M, et al. , Distinct nongenomic signal transduction pathways controlled by 17beta-estradiol regulate DNA synthesis and cyclin D(1) gene transcription in HepG2 cells. Mol Biol Cell, 2002. 13(10): p. 3720–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marino M, Acconcia F, and Trentalance A, Biphasic estradiol-induced AKT phosphorylation is modulated by PTEN via MAP kinase in HepG2 cells. Mol Biol Cell, 2003. 14(6): p. 2583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dickson RB and Lippman ME, Growth factors in breast cancer. Endocr Rev, 1995. 16(5): p. 559–89. [DOI] [PubMed] [Google Scholar]
- 108.Doisneau-Sixou SF, et al. , Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer, 2003. 10(2): p. 179–86. [DOI] [PubMed] [Google Scholar]
- 109.Castoria G, et al. , PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. Embo Journal, 2001. 20(21): p. 6050–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Giretti MS, et al. , Extra-nuclear signalling of estrogen receptor to breast cancer cytoskeletal remodelling, migration and invasion. PLoS One, 2008. 3(5): p. e2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jiang QF, et al. , 17beta-estradiol promotes the invasion and migration of nuclear estrogen receptor-negative breast cancer cells through cross-talk between GPER1 and CXCR1. J Steroid Biochem Mol Biol, 2013. 138: p. 314–24. [DOI] [PubMed] [Google Scholar]
- 112.Chaudhri RA, et al. , Membrane estrogen signaling enhances tumorigenesis and metastatic potential of breast cancer cells via estrogen receptor-alpha36 (ERalpha36). J Biol Chem, 2012. 287(10): p. 7169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cheriyath V, et al. , G1P3, an interferon- and estrogen-induced survival protein contributes to hyperplasia, tamoxifen resistance and poor outcomes in breast cancer. Oncogene, 2012. 31(17): p. 2222–36. [DOI] [PubMed] [Google Scholar]
- 114.Lauricella M, et al. , The analysis of estrogen receptor-alpha positive breast cancer stem-like cells unveils a high expression of the serpin proteinase inhibitor PI-9: Possible regulatory mechanisms. Int J Oncol, 2016. 49(1): p. 352–60. [DOI] [PubMed] [Google Scholar]
- 115.Iyer V, et al. , Estrogen promotes ER-negative tumor growth and angiogenesis through mobilization of bone marrow-derived monocytes. Cancer Res, 2012. 72(11): p. 2705–13. [DOI] [PubMed] [Google Scholar]
- 116.Sartorius CA, et al. , Estrogen promotes the brain metastatic colonization of triple negative breast cancer cells via an astrocyte-mediated paracrine mechanism. Oncogene, 2016. 35(22): p. 2881–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li CI, et al. , Tamoxifen therapy for primary breast cancer and risk of contralateral breast cancer. J Natl Cancer Inst, 2001. 93(13): p. 1008–13. [DOI] [PubMed] [Google Scholar]
- 118.Savci-Heijink CD, et al. , Retrospective analysis of metastatic behaviour of breast cancer subtypes. Breast Cancer Res Treat, 2015. 150(3): p. 547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hess KR, et al. , Estrogen receptors and distinct patterns of breast cancer relapse. Breast Cancer Res Treat, 2003. 78(1): p. 105–18. [DOI] [PubMed] [Google Scholar]
- 120.Bardou VJ, et al. , Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J Clin Oncol, 2003. 21(10): p. 1973–9. [DOI] [PubMed] [Google Scholar]
- 121.Kohler BA, et al. , Annual Report to the Nation on the Status of Cancer, 1975-2011, Featuring Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J Natl Cancer Inst, 2015. 107(6): p. djv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Beatson GT, On the Treatment of Inoperable Cases of Carcinoma of the Mamma: Suggestions for a New Method of Treatment, with Illustrative Cases. Trans Med Chir Soc Edinb, 1896. 15: p. 153–179. [PMC free article] [PubMed] [Google Scholar]
- 123.Harper MJ and Walpole AL, A new derivative of triphenylethylene: effect on implantation and mode of action in rats. J Reprod Fertil, 1967. 13(1): p. 101–19. [DOI] [PubMed] [Google Scholar]
- 124.Jordan VC, Fourteenth Gaddum Memorial Lecture. A current view of tamoxifen for the treatment and prevention of breast cancer. Br J Pharmacol, 1993. 110(2): p. 507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Clemons M, Danson S, and Howell A, Tamoxifen (“Nolvadex”): a review. Cancer Treat Rev, 2002. 28(4): p. 165–80. [DOI] [PubMed] [Google Scholar]
- 126.Desta Z, et al. , Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther, 2004. 310(3): p. 1062–75. [DOI] [PubMed] [Google Scholar]
- 127.Johnson MD, et al. , Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res Treat, 2004. 85(2): p. 151–9. [DOI] [PubMed] [Google Scholar]
- 128.Lim YC, et al. , Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemother Pharmacol, 2005. 55(5): p. 471–8. [DOI] [PubMed] [Google Scholar]
- 129.Davies C, et al. , Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet, 2013. 381(9869): p. 805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gray RG, Rea D, and Handley K, ATTom: Long-term effects of continouing adjuvant tamoxifen to 10years versus stopping at 5years in 6,953 women with early breast cancer. J Clin Oncol (suppl; abstr 5), 2013. [Google Scholar]
- 131.Fisher B, et al. , Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst, 1998. 90(18): p. 1371–88. [DOI] [PubMed] [Google Scholar]
- 132.Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet, 1998. 351(9114): p. 1451–67. [PubMed] [Google Scholar]
- 133.Powles TJ, et al. , Effect of tamoxifen on bone mineral density measured by dual-energy x-ray absorptiometry in healthy premenopausal and postmenopausal women. J Clin Oncol, 1996. 14(1): p. 78–84. [DOI] [PubMed] [Google Scholar]
- 134.Wakeling AE, Dukes M, and Bowler J, A potent specific pure antiestrogen with clinical potential. Cancer Res, 1991. 51(15): p. 3867–73. [PubMed] [Google Scholar]
- 135.Wakeling AE, Similarities and distinctions in the mode of action of different classes of antioestrogens. Endocr Relat Cancer, 2000. 7(1): p. 17–28. [DOI] [PubMed] [Google Scholar]
- 136.DeFriend DJ, et al. , Effects of 4-hydroxytamoxifen and a novel pure antioestrogen (ICI182780) on the clonogenic growth of human breast cancer cells in vitro. Br J Cancer, 1994. 70(2): p. 204–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Osborne CK, et al. , Comparison of the effects of a pure steroidal antiestrogen with those of tamoxifen in a model of human breast cancer. J Natl Cancer Inst, 1995. 87(10): p. 746–50. [DOI] [PubMed] [Google Scholar]
- 138.DeFriend DJ, et al. , Investigation of a new pure antiestrogen (ICI 182780) in women with primary breast cancer. Cancer Res, 1994. 54(2): p. 408–14. [PubMed] [Google Scholar]
- 139.Robertson JF, et al. , Comparison of the short-term biological effects of 7alpha-[9-(4,4,5,5,5-pentafluoropentylsulfinyl)-nonyl]estra-1,3,5, (10)-triene-3,17beta-diol (Faslodex) versus tamoxifen in postmenopausal women with primary breast cancer. Cancer Res, 2001. 61(18): p. 6739–46. [PubMed] [Google Scholar]
- 140.Di Leo A, et al. , Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol, 2010. 28(30): p. 4594–600. [DOI] [PubMed] [Google Scholar]
- 141.Perey L, et al. , Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK21/00). Ann Oncol, 2007. 18(1): p. 64–9. [DOI] [PubMed] [Google Scholar]
- 142.Ingle JN, et al. , Fulvestrant in women with advanced breast cancer after progression on prior aromatase inhibitor therapy: North Central Cancer Treatment Group Trial N0032. J Clin Oncol, 2006. 24(7): p. 1052–6. [DOI] [PubMed] [Google Scholar]
- 143.Chia S, et al. , Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol, 2008. 26(10): p. 1664–70. [DOI] [PubMed] [Google Scholar]
- 144.Howell A, et al. , Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol, 2002. 20(16): p. 3396–403. [DOI] [PubMed] [Google Scholar]
- 145.Mokbel K, The evolving role of aromatase inhibitors in breast cancer. Int J Clin Oncol, 2002. 7(5): p. 279–83. [DOI] [PubMed] [Google Scholar]
- 146.Evans TR, et al. , Phase I and endocrine study of exemestane (FCE 24304), a new aromatase inhibitor, in postmenopausal women. Cancer Res, 1992. 52(21): p. 5933–9. [PubMed] [Google Scholar]
- 147.Yates RA, et al. , Arimidex (ZD1033): a selective, potent inhibitor of aromatase in postmenopausal female volunteers. Br J Cancer, 1996. 73(4): p. 543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lipton A, et al. , Letrozole (CGS 20267). A phase I study of a new potent oral aromatase inhibitor of breast cancer. Cancer, 1995. 75(8): p. 2132–8. [DOI] [PubMed] [Google Scholar]
- 149.Coombes RC, et al. , A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med, 2004. 350(11): p. 1081–92. [DOI] [PubMed] [Google Scholar]
- 150.Howell A, et al. , Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet, 2005. 365(9453): p. 60–2. [DOI] [PubMed] [Google Scholar]
- 151.Goss PE, et al. , A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med, 2003. 349(19): p. 1793–802. [DOI] [PubMed] [Google Scholar]
- 152.Mouridsen HT, Incidence and management of side effects associated with aromatase inhibitors in the adjuvant treatment of breast cancer in postmenopausal women. Curr Med Res Opin, 2006. 22(8): p. 1609–21. [DOI] [PubMed] [Google Scholar]
- 153.Amir E, et al. , Toxicity of adjuvant endocrine therapy in postmenopausal breast cancer patients: a systematic review and meta-analysis. J Natl Cancer Inst, 2011. 103(17): p. 1299–309. [DOI] [PubMed] [Google Scholar]
- 154.Crew KD, et al. , Prevalence of joint symptoms in postmenopausal women taking aromatase inhibitors for early-stage breast cancer. J Clin Oncol, 2007. 25(25): p. 3877–83. [DOI] [PubMed] [Google Scholar]
- 155.National_Cancer_Institute. Hormone therapy for the breast. 2012; Available from: http://www.cancer.gov/cancertopics/factsheet/Therapy/hormone-therapy-breast.
- 156.Goetz MP, et al. , Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol, 2005. 23(36): p. 9312–8. [DOI] [PubMed] [Google Scholar]
- 157.Toy W, et al. , ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet, 2013. 45(12): p. 1439–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jeselsohn R, et al. , Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res, 2014. 20(7): p. 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fribbens C, et al. , Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J Clin Oncol, 2016. 34(25): p. 2961–8. [DOI] [PubMed] [Google Scholar]
- 160.Lavinsky RM, et al. , Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci U S A, 1998. 95(6): p. 2920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Osborne CK, et al. , Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst, 2003. 95(5): p. 353–61. [DOI] [PubMed] [Google Scholar]
- 162.Shou J, et al. , Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst, 2004. 96(12): p. 926–35. [DOI] [PubMed] [Google Scholar]
- 163.Johnston SR, et al. , Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clin Cancer Res, 1999. 5(2): p. 251–6. [PubMed] [Google Scholar]
- 164.Zhou Y, et al. , Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer, 2007. 7: p. 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Murphy CG and Dickler MN, The Role of CDK4/6 Inhibition in Breast Cancer. Oncologist, 2015. 20(5): p. 483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pardee AB, G1 events and regulation of cell proliferation. Science, 1989. 246(4930): p. 603–8. [DOI] [PubMed] [Google Scholar]
- 167.Morgan DO, Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol, 1997. 13: p. 261–91. [DOI] [PubMed] [Google Scholar]
- 168.Sherr CJ, D-type cyclins. Trends Biochem Sci, 1995. 20(5): p. 187–90. [DOI] [PubMed] [Google Scholar]
- 169.Shapiro GI, Cyclin-dependent kinase pathways as targets for cancer treatment. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 2006. 24(11): p. 1770–83. [DOI] [PubMed] [Google Scholar]
- 170.Span PN, et al. , Cyclin-E is a strong predictor of endocrine therapy failure in human breast cancer. Oncogene, 2003. 22(31): p. 4898–904. [DOI] [PubMed] [Google Scholar]
- 171.Chu IM, Hengst L, and Slingerland JM, The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer, 2008. 8(4): p. 253–67. [DOI] [PubMed] [Google Scholar]
- 172.Perez-Tenorio G, et al. , Cytoplasmic p21WAF1/CIP1 correlates with Akt activation and poor response to tamoxifen in breast cancer. Int J Oncol, 2006. 28(5): p. 1031–42. [PubMed] [Google Scholar]
- 173.Errico A, Breast cancer: PALOMA-3 confirms that CDK4/6 is a key therapeutic target. Nat Rev Clin Oncol, 2015. 12(8): p. 436. [DOI] [PubMed] [Google Scholar]
- 174.Schoenlein PV, et al. , Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy, 2009. 5(3): p. 400–3. [DOI] [PubMed] [Google Scholar]
- 175.Samaddar JS, et al. , A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol Cancer Ther, 2008. 7(9): p. 2977–87. [DOI] [PubMed] [Google Scholar]
- 176.Ellis MJ, et al. , Estrogen-independent proliferation is present in estrogen-receptor HER2-positive primary breast cancer after neoadjuvant letrozole. J Clin Oncol, 2006. 24(19): p. 3019–25. [DOI] [PubMed] [Google Scholar]
- 177.Fox EM, et al. , A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res, 2011. 71(21): p. 6773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Frogne T, et al. , Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat, 2009. 114(2): p. 263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Turner N, et al. , FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res, 2010. 70(5): p. 2085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Shoman N, et al. , Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod Pathol, 2005. 18(2): p. 250–9. [DOI] [PubMed] [Google Scholar]
- 181.Ligibel JA, et al. , Body Mass Index, PAM50 Subtype, and Outcomes in Node-Positive Breast Cancer: CALGB 9741 (Alliance). J Natl Cancer Inst, 2015. 107(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Protani M, Coory M, and Martin JH, Effect of obesity on survival of women with breast cancer: systematic review and meta-analysis. Breast Cancer Res Treat, 2010. 123(3): p. 627–35. [DOI] [PubMed] [Google Scholar]
- 183.Sahin S, et al. , The association between body mass index and immunohistochemical subtypes in breast cancer. Breast, 2017. 32: p. 227–236. [DOI] [PubMed] [Google Scholar]
- 184.Baum M, et al. , Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer: results of the ATAC (Arimidex, Tamoxifen Alone or in Combination) trial efficacy and safety update analyses. Cancer, 2003. 98(9): p. 1802–10. [DOI] [PubMed] [Google Scholar]
- 185.Baum M, et al. , Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet, 2002. 359(9324): p. 2131–9. [DOI] [PubMed] [Google Scholar]
- 186.Gnant M, et al. , The predictive impact of body mass index on the efficacy of extended adjuvant endocrine treatment with anastrozole in postmenopausal patients with breast cancer: an analysis of the randomised ABCSG-6a trial. Br J Cancer, 2013. 109(3): p. 589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chan DS, et al. , Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Ann Oncol, 2014. 25(10): p. 1901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Elliott KM, et al. , Effects of aromatase inhibitors and body mass index on steroid hormone levels in women with early and advanced breast cancer. Br J Surg, 2014. 101(8): p. 939–48. [DOI] [PubMed] [Google Scholar]
- 189.Lonning PE, Haynes BP, and Dowsett M, Relationship of body mass index with aromatisation and plasma and tissue oestrogen levels in postmenopausal breast cancer patients treated with aromatase inhibitors. Eur J Cancer, 2014. 50(6): p. 1055–64. [DOI] [PubMed] [Google Scholar]
- 190.Sestak I, et al. , Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: an exploratory analysis from the ATAC trial. J Clin Oncol, 2010. 28(21): p. 3411–5. [DOI] [PubMed] [Google Scholar]
- 191.Bowers LW, et al. , Obesity enhances nongenomic estrogen receptor crosstalk with the PI3K/Akt and MAPK pathways to promote in vitro measures of breast cancer progression. Breast Cancer Res, 2013. 15(4): p. R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Formisano L, et al. , Association of FGFR1 with ERalpha Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER(+) Breast Cancer. Clin Cancer Res, 2017. 23(20): p. 6138–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wellberg EA, et al. , FGFR1 underlies obesity-associated progression of estrogen receptor-positive breast cancer after estrogen deprivation. JCI Insight, 2018. 3(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Laud K, et al. , Identification of leptin receptors in human breast cancer: functional activity in the T47-D breast cancer cell line. Mol Cell Endocrinol, 2002. 188(1–2): p. 219–26. [DOI] [PubMed] [Google Scholar]
- 195.Catalano S, et al. , Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J Biol Chem, 2004. 279(19): p. 19908–15. [DOI] [PubMed] [Google Scholar]
- 196.Considine RV, et al. , Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med, 1996. 334(5): p. 292–5. [DOI] [PubMed] [Google Scholar]
- 197.Catalano S, et al. , Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J Biol Chem, 2003. 278(31): p. 28668–76. [DOI] [PubMed] [Google Scholar]
- 198.Coscia EB, et al. , Estrone and Estradiol Levels in Breast Cancer Patients Using Anastrozole Are Not Related to Body Mass Index. Rev Bras Ginecol Obstet, 2017. 39(1): p. 14–20. [DOI] [PubMed] [Google Scholar]
- 199.Zewenghiel L, Lindman H, and Valachis A, Impact of body mass index on the efficacy of endocrine therapy in patients with metastatic breast cancer - A retrospective two-center cohort study. Breast, 2018. 40: p. 136–140. [DOI] [PubMed] [Google Scholar]
- 200.Zekri J, Farag K, and Allithy A, Obesity and outcome of post-menopausal women receiving adjuvant letrozole for breast cancer. Ecancermedicalscience, 2018. 12: p. 821. [DOI] [PMC free article] [PubMed] [Google Scholar]