

Abstract
Mitochondria and cytochrome c release play a role in the death of neurons and glia after cerebral ischemia. In the present study, we investigated whether BID, a proapoptotic promoter of cytochrome c release and caspase 8 substrate, was expressed in brain, activated after an ischemic insult in vivo and in vitro, and contributed to ischemic cell death. We detected BID in the cytosol of mouse brain and primary cultured mouse neurons and demonstrated, by using recombinant caspase 8, that neuronal BID also is a caspase 8 substrate. After 2 h of oxygen/glucose deprivation, BID cleavage was detected in neurons concurrent with caspase 8 activation but before caspase 3 cleavage. Bid−/− neurons were resistant to death after oxygen/glucose deprivation, and caspase 3 cleavage was significantly reduced; however, caspase 8 cleavage did not differ from wild type. In vivo, BID was cleaved 4 h after transient middle cerebral artery occlusion. Infarct volumes and cytochrome c release also were less in Bid−/− mice (−67% and −41%, respectively) after mild focal ischemia. These findings suggest that BID and the mitochondrial-amplification pathway promoting caspase activation contributes importantly to neuronal cell death after ischemic insult.
Programmed cell death is a feature of chronic and acute neurodegenerative diseases, including Alzheimer's and Huntington's disease, amyotrophic lateral sclerosis (ALS), and stroke (1, 2). Programmed cell death is mediated, in part, by caspases, a family of cysteine proteases that cleave and disassemble proteins essential for cell survival (3). Caspases are activated during cerebral ischemia (4–6), and ischemic cell death is significantly attenuated by caspase inhibition (7) or caspase gene deletion (8).
The sequence of events leading to caspase activation has been well characterized in nonneuronal cells (9). In these cells, BID, a 22-kDa cytosolic member of the Bcl-2 family of proapoptotic proteins (10) provides one mechanism by which TNF/Fas family death-receptor activation is linked to downstream events (11). These death receptors activate caspase 8, which cleaves BID to its truncated active form (tBID; 15kDa; ref. 12). tBID targets the outer mitochondrial membrane and induces conformational changes in BAK and BAX (13) and, by so doing, triggers cytochrome c release into the cytosol (14). There cytochrome c together with APAF-1 and caspase 9 form the apoptosome complex, which results in activation of caspase 3 and other effector caspases and ultimately cause cell death (15). The importance of BID in death receptor-mediated programmed cell death and in amplification of upstream cell-death signals is demonstrated by the fact that Bid knockout mice are strongly resistant to death receptor-induced hepatocellular apoptosis (16).
Very little is known about BID expression and its role in brain, or in fact, whether a similar death pathway involving BID exists in neurons. However, expression of cell-surface death receptors of the TNF/Fas family (17–19), cleavage of caspase 8 (5) and caspase 3 (6), and release of cytochrome c from mitochondria (20) have all been described individually in ischemia. Given these similarities, we hypothesized that BID may figure as a mediator upstream of mitochondria in neuronal death after cerebral ischemia. To explore this hypothesis, we used well characterized models of ischemic cell death to show that BID is cleaved after hypoxia/ischemia in vitro [by oxygen/glucose deprivation (OGD)] and in vivo [by middle cerebral artery occlusion (MCAo)]. We determined whether Bid knockout mice, or neurons isolated from them, were protected against ischemia and examined the relationship between the presence of tBID, cytochrome c release, and caspase 3 and 8 activation in adult mouse brain.
Materials and Methods
Isolation of Cytosolic and Mitochondrial Proteins.
Tissues from adult C57BL/6 mice (20–25 g) were homogenized in 10 volumes of hypertonic buffer (21). Supernatants containing cytosolic proteins were isolated by centrifugation at 20,000 × g. tBID was enriched by immunoprecipitation (150 μg of protein incubated with anti-BID/tBID antibody at 1:500 dilution over night). Mitochondria were isolated as described (21), sonicated for 2 × 30 s (Sonic Dismembrenator, Model 550, Fischer Scientific, Pittsburgh, PA), and stored at −80°C. All procedures were performed at 4°C.
In Vitro BID Cleavage Assay.
Tissues from male C57BL/6 mice (20–25 g) were homogenized in five volumes of homogenization buffer (22). The homogenate (100 μl) was incubated with 10 μl of 100 units/μl active recombinant caspase 8 (Biomol, Plymouth Meeting, PA) for 60 min at 37°C and thereafter transferred to −80°C.
Western Blot Analysis.
Protein (10–20 μg per lane) was separated and blotted as described (23). The blot was probed with anti-BID (rabbit polyclonal, 1:1,000) or anti-caspase 8 proform (SK 441, rabbit polyclonal, 1:1,000) or cleaved caspase 8 (SK 439, rabbit polyclonal, 1:1,000), or anti-cytochrome c (clone 7H8.2C12, 1:200, PharMingen), caspase 3 proform (LAP4, 1:1,000), or anti-active caspase 3 (D175, 1:1,000, Cell Signaling Technology, Beverly, MA) at 4°C overnight. Membranes then were exposed to horseradish-peroxidase-conjugated anti-rabbit IgG for 1 h. Antibody binding was detected by using the enhanced chemiluminescence (ECL) system (Amersham Pharmacia). Equal protein loading of gels was assured by Coomassie blue or α-tubulin staining. Relative optical densities of protein bands were quantitated by using the Bio-Rad MULTIANALYST VERSION 1.0.
Murine Neuronal Cell Cultures and OGD.
Primary mouse neurons were prepared from E14–E16 embryos as modified (24) and plated at a density of 200,000 cells per cm2 in serum-free neurobasal medium (NBM) with 2% (vol/vol) B27 supplement/2 mM glutamine/100 units/ml penicillin/streptomycin (GIBCO/BRL). Cells were used for experiments on day 9–10 of culture. Under these conditions, ≈90% of cells were neurons as determined by MAP-2 staining (data not shown). OGD on murine neocortical cell cultures was performed as described (24) with minor modifications. Briefly, the culture medium was replaced by glucose-free Earle's balanced salt solution purged by nitrogen gas for 10 min (pO2 ≈ 5–6%). Then the cells were placed for 2 h in a chamber filled with 5% CO2 and 95% N2. Control cells were incubated in glucose-free Earle's balanced salt solution in a normoxic incubator for the same period. OGD was terminated by switching back to normal culture conditions. By using these conditions and neurons grown in NBM, we found that the apoptotic component of ischemic cell death was unmasked, and that cell death was abrogated to basal level by adding zDEVD.fmk (100 μM) during the period of OGD (in three experiments). After 24 h, cells were stained for 30 min with 33 μg/ml Hoechst 33342 (Molecular Probes), fixed in 4% (vol/vol) paraformaldehyde, and examined morphologically by fluorescence microscopy (25). Six culture dishes were analyzed from each individual culture. From each dish, five random microscopic fields (400×) were chosen. Between 150 and 300 cells were counted for each field by two independent researchers, one of whom was naïve to the genotype, and the percentage of apoptotic cells/total cell number was calculated. We relied on nuclear morphology (condensation or fragmentation) to score apoptotic cell death. The density of plated cells was similar between groups (not shown). More than 95% of Hoechst positive cells showing nuclear condensation or fragmentation were also terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling positive (not shown). In a preliminary study, we also stained cells with propidium iodide (PI) to assess cell membrane integrity before and after OGD and found no difference in the percentage of positive PI-stained cells (≈5–10%).
Transgenic Mice.
Bid-deficient mice (Bid−/−) were generated as described (16) and backcrossed 7 to 8 generations into the C57BL/6 background strain. Bid−/− mice were born at the expected Mendelian frequency and showed no apparent developmental abnormalities. As controls, wild-type (wt) animals (Bid+/+) from the same litter (littermates) were used for all experiments. The macro- and microscopic brain morphology of uninjured Bid−/− mice did not differ from wt controls. Only male mice matched for age (6 weeks) and weight (20–25 g) were used for experiments.
Transient Focal Cerebral Ischemia.
Cerebral ischemia was induced by an intraluminal filament as described (26). Successful occlusion and reperfusion was demonstrated by laser-Doppler flowmetry (PF2B; Perimed, Stockholm) in each animal. Functional outcome was assessed on a five-point scale (0 = no deficit; 1 = weakness of the contralateral forepaw; 2 = circling; 3 = loss of righting reflex; 4 = no motor activity).
Cytochrome c Measurement.
Cytochrome c in cytosolic fractions of brain homogenates was measured by an ELISA system specific for mouse cytochrome c (R & D Systems). Homogenates were prepared from an equivalent lesioned area (striatum) in both strains. Results are expressed as ng of cytochrome c per mg of protein. Values obtained from the nonischemic hemisphere were subtracted from the values for the ischemic hemisphere to obtain a measure for cytochrome c release specific to ischemia. The cytosolic origin of the samples was proven by the fact that cytochrome oxidase, a mitochondrial protein, was not detected in these brain fractions (data not shown).
Statistical Analysis.
Data are presented as mean ± SEM. All statistical analyses were made with the SIGMASTAT 2.0 software package (Jandel Scientific, Erkrath, Germany). For comparison between wt and Bid−/− groups, the Mann–Whitney Rank Sum test was used. P < 0.05 was considered statistically significant.
Results
Phenotypes of Bid−/− Mice.
Macroscopically, we observed no differences in brain size or weight and no differences in brain histology [by hematoxylin/eosin (H&E) and cresyl violet] between mutants and wt strains. There were no differences in numbers of neurons when other sites, like superior cervical ganglia and facial nerve nuclei, were counted (S.J.K., unpublished observation). The Bid−/− mice showed no other obvious abnormal neuronal phenotypes, and the development of neuronal lineages was apparently normal (16). Regarding neuronal cell cultures, we found that the yield of neurons from Bid−/− mice and wt littermates and the total numbers of cells in cultures before and after OGD were similar.
Expression of BID.
We characterized BID expression by demonstrating its presence and subcellular localization in brain. A 22-kDa band corresponding to BID was observed in murine whole-brain homogenates (n = 2; Fig. 1a). The 22-kDa band was absent in homogenates from Bid−/− mice (n = 2; Fig. 1a). We detected this band in the cytosolic fraction of homogenates but not in isolated mitochondria (Fig. 1b). Liver tissue, where the same subcellular distribution has been described (16), was used as a positive control and showed the expected findings (Fig. 1b).
Figure 1.

BID is expressed in mouse brain homogenate and is cleaved by recombinant caspase 8. (a) Mouse (C57BL/6) brain homogenate was probed for BID with a rabbit polyclonal antibody (Western blotting). A 22-kDa band, which corresponds to recombinant mouse BID, was detected (12). This band was not detected in brain homogenate from Bid−/− mice (n = 3). (b) The cytosolic fraction (C) and isolated mitochondria (M) were prepared from brain and liver of wt C57BL/6 mice and probed for BID with a rabbit polyclonal antibody (Western blotting). (c) To detect cleavage of BID by caspase 8, fresh brain and liver homogenates from wt mice were incubated with recombinant caspase 8 (+8) and compared with control homogenate incubated for the same time without caspase 8 (C) (n = 2; Western blotting).
To establish that neuronal BID is a substrate of caspase 8 as in the liver (16), whole-brain and liver homogenates were incubated with recombinant caspase 8. Immunoblotting revealed BID and tBID (15 kDa) in both organs (n = 2; Fig. 1c).
BID Cleavage After OGD in Vitro.
We investigated whether ischemia triggers BID cleavage and activates caspases (caspase 8 and 3), which mediate BID cleavage in nonneuronal cells. We exposed cultured cortical neurons to 2 h of OGD, and assessed cell death 24 h later by nuclear staining with Hoechst 33342 dye. Cell death was first observed 6–8 h after OGD; most dead cells had densely stained, shrunken, and compacted nuclei, which is consistent with morphological criteria for nuclear apoptosis (data not shown). A 43-kDa cleavage product corresponding to cleaved caspase 8 was noted 3 h after OGD and was expressed at approximately the same level over the next 24 h (n = 2; Fig. 2a). Evidence of BID processing also was observed by 2 h after OGD and was maintained for the duration of the experiment (n = 2; Fig. 2b). The active fragment of caspase 3 (20 kDa) was noted 3–6 h after OGD and seemed to increase over the next 9 h (n = 2; Fig. 2a). Therefore, BID cleavage and caspase 3 and 8 activation preceded the onset of nuclear signs of apoptosis.
Figure 2.

BID, caspase 8, and caspase 3 are cleaved after OGD. Primary cortical neuronal cell cultures were subjected to 2 h of OGD. (Results are representative of 2–3 independent experiments per time point.) (a) At different time points after restoring oxygen and glucose, cell lysates were probed by Western blot analysis for caspase 8 proform (55 kDa, antibody SK 441), cleavage product (43 kDa, antibody SK 440), caspase 3 proform (32 kDa, antibody LAP4), and active form (20 kDa, antibody D175). (b) At different time points, samples were probed for BID (OGD; Upper) and compared with cultures not subjected to OGD (Control; Lower).
Primary Cortical Neurons from Bid−/− Mice Are Protected from OGD.
To test the hypothesis that BID not only accompanies but also mediates neuronal cell death from ischemia, we studied the effects of OGD on wt and Bid−/− cultured cortical neurons. Examination by light microscopy did not show any differences between the brains of Bid−/− and wt embryos, and the cell cultures derived from these animals had similar morphologies before OGD (data not shown). Twenty four hours after OGD, wt cells showed condensed and brightly stained nuclei consistent with apoptotic cell death (Fig. 3a), whereas in cells cultured from Bid−/− mice, this staining pattern was less frequently found (Fig. 3b). Cell counting showed a 24 ± 4% (n = 4) increase in the number of condensed nuclei in wt cells compared with no OGD treatment, whereas the corresponding increase in Bid−/− cells was only 11 ± 3%, constituting a 54% reduction in cell death attributable to OGD in Bid−/− cells (P < 0.01; Fig. 4a).
Figure 3.
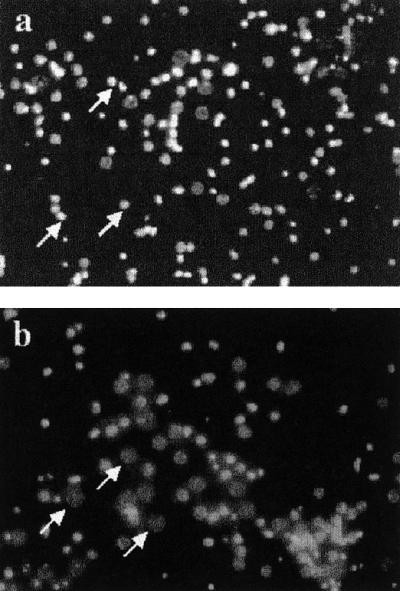
Morphology of Bid−/− and wt neurons after OGD. Enriched primary neuron cultures were subjected to 2 h of OGD and assessed 24 h later for apoptotic nuclear morphology with Hoechst 33342. (a) In wt cultures (n = 4), many cells show condensed, brightly stained apoptotic nuclei (arrows). (b) In the Bid−/− cultured neurons (n = 4), living cells (arrows; dim, nonhomogeneous nuclear staining) are more frequently observed (Magnification: ×200). Total cell number for both a and b was approximately 90 per panel.
Figure 4.

Neurons cultured from Bid−/− mouse brain are resistant to OGD. (a) Cell death at 24 h was reduced in the Bid−/− cells (n = 4; P < 0.01) exposed to OGD as compared with cells obtained from wt littermates (n = 4). The total cell number was not different in both groups and did not change significantly after OGD. Data are mean ±SEM. (b) Cell homogenates from these cultures (wt and Bid−/−) were probed for cleaved caspase 8 (43 kDa, antibody SK 439, Upper) and cleaved caspase 3 (20 kDa, antibody D175, Lower) at 12 and 24 h after OGD. The ratio of the optical density of the 20-kDa caspase 3 cleavage band to that of a loading control (α-tubulin) was 0.71 ± 0.04 in wt cells vs. 0.20 ± 0.06 in mutants (n = 3; P < 0.001)
To determine BID's role in caspase activation and its position in the molecular pathways that lead to ischemic cell death, we compared the presence of cleaved caspases 3 and 8 in wt and Bid−/− cells after OGD. There was no difference between wt and Bid−/− cultures in the cleavage of caspase 8 (to a 43-kDa product) after OGD (Fig. 4b Upper). In contrast, a band corresponding to caspase 3 cleavage product (20 kDa) was significantly reduced in Bid−/− cultures, compared with wt cultures as determined by densitometry. The ratio of the optical density of the 20-kDa band to that of a loading control (α-tubulin) was 0.71 ± 0.04 in wt cells vs. 0.20 ± 0.06 in mutants (n = 3, P < 0.001; Fig. 4b Lower). These data demonstrate that BID deletion reduces caspase 3 but not caspase 8 activation, suggesting that BID acts at an intermediate position between these caspases in the molecular pathway of neuronal cell death.
Neuroprotection in Bid−/− Mice After Focal Cerebral Ischemia.
Next, we determined whether the resistance of cultured neurons to OGD reflected similar cell-death processing in central nervous system (CNS) ischemic injury in vivo. In wt mice, we demonstrated that BID was cleaved to its 15-kDa active fragment (tBID) after MCAo (n = 2; Fig. 5). We investigated whether BID was part of apoptotic signaling pathways in vivo by comparing the levels of cytosolic cytochrome c in homogenates of ischemic brains from wt and Bid−/− mice subjected to 2 h of MCAo. A significant reduction (−41%) was found in the mutant brain by an ELISA method (3.9 ± 1.1 ng/mg protein vs. 6.6 ± 1.1 ng/mg protein for mutant and wt, respectively; n = 4; P < 0.03; Fig. 6a) and by immunoblotting (Fig. 6b). As an important precondition, we verified that there were no premorbid differences between wt and Bid−/− groups in brain volume, vascular anatomy, resting absolute blood flow (209 ± 24 ml/100 g/min vs. 220 ± 17 ml/100 g/min in wt and mutants, respectively) or overt behavior (data not shown).
Figure 5.

BID cleavage after focal ischemia in vivo. Two hours after 2 h of MCAo, brain homogenates from the ischemic (I) and contralateral (C) hemisphere were subjected to immunoprecipitation with an anti-tBID antibody followed by Western blot analysis. Recombinant tBid obtained by incubation of recombinant Bid with recombinant active caspase 8 was used as positive control (tBID; n = 2).
Figure 6.

Cytochrome c release after focal ischemia in vivo. (a) After ischemia, 41% less cytochrome c was measured by ELISA in the cytosol of Bid−/− mice (n = 4) as compared with wt littermates (n = 4, P < 0.03). (b) Cytosolic cytochrome c in ischemic striatum (I) was increased over contralateral values (C). This increase was attenuated in Bid−/− mice (Western blot).
To investigate if BID mediates ischemic cell death, Bid−/− and wt littermate mice were subjected to 30 min of MCAo and 48 h of reperfusion. Blood flow distal to the occlusion dropped below 20% in the two groups and returned to preocclusion levels after recirculation. The mean infarct volume was 67% smaller in Bid−/− mice (55.5 ± 29.0 mm3 and 18.3 ± 6.4 mm3 in wt and mutant, respectively; n = 8 per group; P < 0.002; Fig. 7A). In wt animals, the lateral striatum and the adjacent cerebral cortex showed extensive ischemic injury, whereas the cortex was spared in Bid−/− mice (Fig. 7B). Animals had only slight neurological deficits 24 and 48 h after ischemia (0.4 ± 0.2), and there was no difference between groups. After ischemia, brain edema was minimal and did not differ between groups; the volumes of the ischemic hemispheres were 102 ± 3% and 104 ± 5% of the contralateral hemispheres in wt and mutants, respectively.
Figure 7.

(A) Ischemic damage is reduced in Bid−/− mice. (a) Infarct volume was reduced by 67% in Bid−/− mice as compared with wt mice (n = 8 per group; P < 0.01) after 30 min of MCAo and 48 h of reperfusion. (b) Protection in Bid−/− mice was significant at the level of the striatum (section 3; P < 0.01) with a trend of infarct size difference at other brain section levels. (B) Ischemic tissue damage in Bid−/− and Bid+/+ mice. As shown by representative H&E-stained coronal-brain sections, Bid−/− mice had significantly smaller infarcts after 30 min of MCAO and 48 h of reperfusion (a) as compared with Bid mice (b). High-power micrographs (×200) taken adjacent to penumbral areas (cortical layer III) showing little evidence of cellular damage in Bid−/− mice (c), and extensive injury in wt mice (d).
Discussion
By using an in vivo model of mild focal cerebral ischemia and in vitro neuronal OGD that favors apoptotic cell death, we established that BID is a critical mediator of ischemic cell death within the CNS. We observed less cell death in highly enriched neuronal cultures from Bid−/− mice after OGD (Fig. 4) and reduced ischemic brain injury in mutant mice with a deletion in Bid (Fig. 7). We also found BID cleavage and caspase 3 cleavage in ischemic brain and in cultured neurons after OGD. In both paradigms, treatment with caspase inhibitors attenuated cell death and tissue injury (21, 22). Because in vitro neuroprotection by zDEVD.fmk (100 μM) was more pronounced in Bid+/+ neurons than protection in Bid−/− neurons (without treatment), our findings suggest that hypoxia and glucose deprivation may trigger other caspase pathway(s) that are independent of BID activation. We conclude that BID as well as caspases are critical cell-death mediators of ischemic brain injury in these models.
In nonneuronal cells, caspase activation can occur by means of a sequence of events known as type II death-receptor signaling (27), in which stimulation of death receptors activates caspase 8. Caspase 8 in turn cleaves BID to tBID. tBID causes cytochrome c release from mitochondria, which results in the activation of caspase 3 and other caspases. Our data show that the molecular pathway for ischemic cell death in neurons resembles the type II pathway. Caspase 8 likely acts upstream of BID in neurons, as suggested by the unchanged levels of cleaved caspase 8 between Bid−/− and wt cultures after hypoxia/ischemia in vitro (Fig. 4b). Furthermore, similar to focal ischemia in vivo (5), hypoxia/ischemia in vitro triggers activation of caspase 8 (Fig. 2a), and this activation occurred at least simultaneously with BID cleavage (Fig. 2b). We also have evidence that caspase 8 cleaved BID to tBID from brain homogenates (Fig. 1c), and that caspase 8 is constitutively expressed in brain (5) and in cultured cortical neurons (Fig. 2a). Although caspase 8 may not be the only enzyme that can cleave BID in ischemia (28), taken together, caspase 8 mediates BID cleavage in the CNS being the cysteine protease with the strongest BID-cleaving activity (12).
Regarding cytochrome c release, BID acts upstream of both cytochrome c release and caspase-3 activation in nonneuronal cells (12). In the CNS, we found attenuated cytochrome c release in ischemic striatum from Bid−/− mice after MCAo (Fig. 6a) and less caspase 3 activation after OGD in Bid−/− cortical neurons (Fig. 4b). Furthermore, BID cleavage anticipated caspase 3 activation in culture (Fig. 2) as reported in fas-induced hepatocellular apoptosis (16). In addition, Bid deletion reduces cytochrome c release, and protects against fas-mediated liver injury (16) and against hypoxia-induced CNS injury in vitro and in vivo, as shown in our present study.
Despite strong evidence linking BID to pore formation in the outer mitochondrial membrane, and to cytosolic release of cytochrome c, cytochrome c release was not completely abrogated in cells and tissues of Bid−/− mice (Fig. 6). Release was apparently not caused by rupture of mitochondrial membranes as we did not detect cytochrome oxidase, a mitochondrial protein, in cytosolic fractions at the same time (not shown). Our data suggest that BID is not the sole mechanism for cytochrome c release after cerebral ischemia. Other mechanisms and molecules, such as unique mechanisms related to Bax, Bad, Bcl-2, Bcl-XL, oxygen radicals, MnSOD, or opening of the mitochondrial permeability transition, may contribute to the release process (29–32). Furthermore, Bcl-2, and Bcl-w modulate cytochrome c release; an increase in their expression, for example, could protect tissues from injury during ischemia (33, 34). BID and other mechanisms also may effect survival in nonneuronal cell types (e.g., astrocytes, endothelial cells, microglia) which were spared in ischemic Bid−/− mouse brain. Further work is necessary to clarify this point. Nevertheless, the results generated by using Bid−/− mice demonstrate convincingly a prominent role for BID in acute CNS injury. Phenotypically, these mutant mice do not have any CNS developmental defect (16) or any difference in microscopic or macroscopic CNS morphologies, as compared with wt mice.
In summary, BID promotes death in neurons after OGD in vitro and in brain after focal cerebral ischemia in vivo. Because BID is strategically located upstream of mitochondria and caspase 3 processing (35), BID presents an attractive target for CNS diseases in which apoptotic cell death is prominent.
Acknowledgments
This work was supported by National Institutes of Health Grants NS10828 and NS374141–02 (to M.A.M.) and K08NS02162 (to D.L.) and by the Deutsche Forschungsgemeinschaft Pl 249/5–1 (to N.P.). The antibodies LAP4, SK 439, and SK 441 were a generous gift from Dr. Frank Barone (SmithKline Beecham). We also thank Dr. H. Li for the plasmid used in the in vitro generation of BID.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- tBID
truncated BID
- OGD
oxygen/glucose deprivation
- MCAo
middle cerebral artery occlusion
- wt
wild type
- CNS
central nervous system
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Mattson M P. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 2.Yuan J, Yankner B A. Nature (London) 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 3.Robertson G S, Crocker S J, Nicholson D W, Schulz J B. Brain Pathol. 2000;10:283–292. doi: 10.1111/j.1750-3639.2000.tb00262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krajewski S, Krajewska M, Ellerby L M, Welsh K, Xie Z, Deveraux Q L, Salvesen G S, Bredesen D E, Rosenthal R E, Fiskum G, Reed J C. Proc Natl Acad Sci USA. 1999;96:5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velier J J, Ellison J A, Kikly K K, Spera P A, Barone F C, Feuerstein G Z. J Neurosci. 1999;19:5932–5941. doi: 10.1523/JNEUROSCI.19-14-05932.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli K J, Yuan J, Moskowitz M A. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hara H, Friedlander R M, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz M A. Proc Natl Acad Sci USA. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schielke G P, Yang G Y, Shivers B D, Betz A L. J Cereb Blood Flow Metab. 1998;18:180–185. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Strasser A, O'Connor L, Dixit V M. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 10.Wang K, Yin X M, Chao D T, Milliman C L, Korsmeyer S J. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 11.Nagata S. Nat Cell Biol. 1999;1:E143–E145. doi: 10.1038/14094. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Zhu H, Xu C J, Yuan J. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 13.Zha J, Weiler S, Oh K J, Wei M C, Korsmeyer S J. Science. 2000;290:1761–1765. doi: 10.1126/science.290.5497.1761. [DOI] [PubMed] [Google Scholar]
- 14.Wei M C, Lindsten T, Mootha V K, Weiler S, Gross A, Ashiya M, Thompson C B, Korsmeyer S J. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 15.Zou H, Li Y, Liu X, Wang X. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 16.Yin X M, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth K A, Korsmeyer S J. Nature (London) 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- 17.Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J, Schenkel J, Herdegen T, Debatin K M. J Neurosci. 1999;19:3809–3817. doi: 10.1523/JNEUROSCI.19-10-03809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrison D C, Roberts J, Campbell C A, Crook B, Davis R, Deen K, Meakin J, Michalovich D, Price J, Stammers M, Maycox P R. Neuroscience. 2000;96:147–160. doi: 10.1016/s0306-4522(99)00502-3. [DOI] [PubMed] [Google Scholar]
- 19.Rosenbaum D M, Gupta G, D'Amore J, Singh M, Weidenheim K, Zhang H, Kessler J A. J Neurosci Res. 2000;61:686–692. doi: 10.1002/1097-4547(20000915)61:6<686::AID-JNR12>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 20.Fujimura M, Morita-Fujimura Y, Murakami K, Kawase M, Chan P H. J Cereb Blood Flow Metab. 1998;18:1239–1247. doi: 10.1097/00004647-199811000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Chiarugi A, Moroni F. J Neurochem. 1999;72:1125–1132. doi: 10.1046/j.1471-4159.1999.0721125.x. [DOI] [PubMed] [Google Scholar]
- 22.Bossy-Wetzel E, Newmeyer D D, Green D R. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsushita K, Wu Y, Qiu J, Lang-Lazdunski L, Hirt L, Waeber C, Hyman B T, Yuan J, Moskowitz M A. J Neurosci. 2000;20:6879–6887. doi: 10.1523/JNEUROSCI.20-18-06879.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldberg M P, Choi D W. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whiteside G, Munglani R. Brain Res Brain Res Protoc. 1998;3:52–53. doi: 10.1016/s1385-299x(98)00020-8. [DOI] [PubMed] [Google Scholar]
- 26.Huang Z, Huang P L, Panahian N, Dalkara T, Fishman M C, Moskowitz M A. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 27.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli K J, Debatin K M, Krammer P H, Peter M E. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen M, He H P, Zhan S X, Krajewski S, Reed J C, Gotlieb R R. J Biol Chem. 2001;276:30724–30728. doi: 10.1074/jbc.M103701200. [DOI] [PubMed] [Google Scholar]
- 29.Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M, Chan P H. J Neurosci. 2000;20:2817–2824. doi: 10.1523/JNEUROSCI.20-08-02817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siesjo B K, Elmer E, Janelidze S, Keep M, Kristian T, Ouyang Y B, Uchino H. Acta Neurochir Suppl. 1999;73:7–13. doi: 10.1007/978-3-7091-6391-7_2. [DOI] [PubMed] [Google Scholar]
- 31.Fiskum G, Murphy A N, Beal M F. J Cereb Blood Flow Metab. 1999;19:351–369. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Springer J E, Azbill R D, Nottingham S A, Kennedy S E. J Neurosci. 2000;20:7246–7251. doi: 10.1523/JNEUROSCI.20-19-07246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinou J C, Dubois-Dauphin M, Staple J K, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 34.Yan C, Chen J, Chen D, Minami M, Pei W, Yin X M, Simon R P. J Cereb Blood Flow Metab. 2000;20:620–630. doi: 10.1097/00004647-200003000-00020. [DOI] [PubMed] [Google Scholar]
- 35.Fink K, Zhu J, Namura S, Shimizu-Sasamata M, Endres M, Ma J, Dalkara T, Yuan J, Moskowitz M A. J Cereb Blood Flow Metab. 1998;18:1071–1076. doi: 10.1097/00004647-199810000-00003. [DOI] [PubMed] [Google Scholar]